

ABSTRACT
Immune evasion of tumors poses a major challenge for immunotherapy. For human papillomavirus (HPV)-induced malignancies, multiple immune evasion mechanisms have been described, including altered expression of antigen processing machinery (APM) components. These changes can directly influence epitope presentation and thus T-cell responses against tumor cells. To date, the APM had not been studied systematically in a large array of HPV+ tumor samples. Therefore in this study, systematic expression analysis of the APM was performed on the mRNA and protein level in a comprehensive collection of HPV16+ cell lines. Subsequently, HPV+ cervical tissue samples were examined by immunohistochemistry. ERAP1 (endoplasmic reticulum aminopeptidase 1) was the only APM component consistently altered – namely overexpressed – in HPV16+ tumor cell lines. ERAP1 was also found to be overexpressed in cervical intraepithelial neoplasia and cervical cancer samples; expression levels were increasing with disease stage. On the functional level, the influence of ERAP1 expression levels on HPV16 E7-derived epitope presentation was investigated by mass spectrometry and in cytotoxicity assays with HPV16-specific T-cell lines. ERAP1 overexpression did not cause a complete destruction of any of the HPV epitopes analyzed, however, an influence of ERAP1 overexpression on the presentation levels of certain HPV epitopes could be demonstrated by HPV16-specific CD8+ T-cells. These showed enhanced killing toward HPV16+ CaSki cells whose ERAP1 expression had been attenuated to normal levels. ERAP1 overexpression may thus represent a novel immune evasion mechanism in HPV-induced malignancies, in cases when presentation of clinically relevant epitopes is reduced by overactivity of this peptidase.
KEYWORDS: Antigen processing machinery (APM), cervical cancer, endoplasmic reticulum aminopeptidase 1 (ERAP1), human papillomavirus (HPV), T-cell epitopes
Abbreviations
- HPV
human papillomavirus
- APM
antigen processing machinery
- ERAP1
endoplasmic reticulum aminopeptidase 1
- MHC
major histocompatibility complex
- HLA
human leukocyte antigen
- HNSCC
head and neck squamous cell carcinoma
- CIN
cervical intraepithelial neoplasia
- MS
mass spectrometry
- aa
amino acid
- CTL
cytotoxic T-cell
Introduction
Human papillomaviruses (HPVs) are the most common sexually transmitted agents in the world, with over 200 types known to date.1 High-risk HPVs can cause cervical cancer, among which 60% are caused by HPV16 and 15% by HPV182. Additionally, high-risk HPVs also cause other anogenital cancers and oropharyngeal cancers.2,3 Usually, HPV infections are cleared by the immune system within 6 to 18 months. In less than 5% of people, persistent infection with a high-risk type develops, which is a necessary prerequisite for subsequent malignant transformation.4
Although prophylactic vaccines against the most common HPV high-risk types have been developed and are highly effective, vaccination coverage is not optimal everywhere.5,6 In addition, people already infected by HPV cannot be cured by prophylactic vaccination. Since the time span from initial infection to cancer is 10–20 years, therapeutic anti-HPV vaccines are intensively investigated.7-9 Unfortunately, overall clinical success has so far been limited. To increase clinical success of therapeutic HPV vaccines it is crucial to gain further insights into immune evasion strategies of HPV-induced malignancies. Natural immune control of HPV infections is achieved by innate and adaptive immune responses, including effector T-cells and specific antibodies. Multiple immune evasion mechanisms to escape host immune control have been described,10-12 such as alteration of antigen processing and presentation on major histocompatibility complex I (MHC-I) at multiple steps. These changes can directly interfere with effective cytotoxic T-cell (CTL) epitope generation. The immunoproteasome subunits PSMB8 and PSMB9, the transporter associated with antigen processing (TAP), MHC-I, the antigen processing machinery (APM) components calreticulin, calnexin, ERAP1, PDIA3 and tapasin have been reported to be decreased in HPV infected cells.13-16
However, a systematic analysis of all APM components in a large collection of HPV-positive tumor cells has not been conducted to date. Importantly, data of direct immunological consequences of these changes in the APM, i.e. alterations in the HPV epitope repertoire, is limited, since only a few epitopes have been analyzed.13,17 Therefore, we have systematically investigated the expression of the APM in a comprehensive collection of HPV16-positive cell lines on the mRNA and protein levels. The endoplasmic reticulum aminopeptidase 1 (ERAP1), which plays a key role in editing the MHC-I peptidome,18,19 was the only APM component that was consistently altered. Surprisingly, it was found to be overexpressed in HPV16-positive cell lines and in cervical intraepithelial neoplasia (CIN) and cervical cancer samples. In recent years, it has been demonstrated that ERAP1 overexpression leads to destruction of immunodominant epitopes from melanoma20 and colorectal carcinoma.21,22 In these studies, tumor-specific T-cell responses were increased upon attenuation of ERAP1 expression in target cells. Therefore, we focused on the question whether ERAP1 overexpression influences epitope presentation in HPV16-mediated carcinomas, and may thus be a novel immune evasion mechanism of HPV-induced malignancies.
Results
APM components are differentially expressed in HPV-negative and HPV16-positive cells on the mRNA level
A qRT-PCR screen of 20 APM components was performed (Fig. 1A). For this screen, 6 HPV-negative and 23 HPV16-positive cell lines were analyzed, which consisted of 12 cervical cancer cell lines, 6 head and neck squamous cell carcinoma (HNSCC) cell lines, and 5 cell lines transfected with the whole HPV16 genome. The HPV-negative cell lines had a strikingly uniform expression pattern, making them suitable for meaningful comparisons. Interestingly, cell lines that were not naturally transformed by HPV, but transfected with the HPV16 genome, were similar to the HPV-negative group. In contrast, gene expression levels of naturally HPV16-transformed cell lines differed from HPV-negative cells for many APM components. Gene expression of HLA-A and -B was elevated in the majority of tumor cell lines, as were β2m (β2-microglobulin) levels. Many peptide loading complex associated genes, namely tapasin, calnexin, calreticulin, ERAP1 and ERAP2, and all tested components of the immunoproteasome, PSMB8, PSMB9, PSMB10, PSME1 and PSME2, had higher mRNA expression levels.
Figure 1.
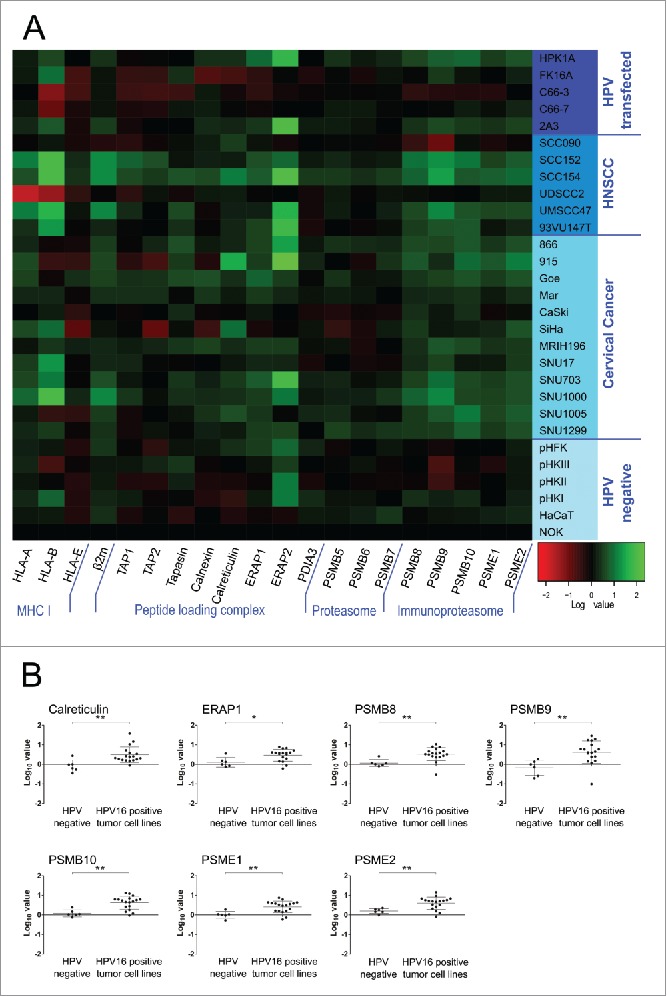
(A) Constitutive expression of the APM in keratinocyte cell lines on the mRNA level. Total RNA was purified from cells, reverse transcribed to cDNA and analyzed for APM component expression by qRT-PCR. PCR reactions were run in triplicates. The geometric mean of at least three of the following genes was used as internal reference: GAPDH, PGK1, PPIA and IPO8. Gene expression was calibrated to the NOK cell line. All quantitative qRT-PCR data were processed by LinRegPCR and data management was done by qbasePLUS 2. Log10 data were plotted as a heatmap using R. Expression of 20 APM components is shown. Each column represents a single gene and each row represents one cell line. Increased expression is shown in green and decreased expression is shown in red (see scale bar in the bottom right corner). HNSCC = head and neck squamous cell carcinoma. (B) APM components that showed a significantly elevated expression in HPV16-positive tumor cell lines compared to HPV-negative cells. Log10 expression data is plotted for each cell line, mean ± SD of each group is shown. *p ≤ 0.05, **p ≤ 0.01 (unpaired t-test).
As many APM components are inducible by IFNγ, the same qRT-PCR screen was performed after IFNγ treatment of cells. The analysis showed that APM components both in HPV-negative and HPV16-positive cell lines are inducible by IFNγ, suggesting that all cell lines of this screen had intact IFNγ signaling. No differential expression of APM components between HPV-negative and HPV16-positive cell lines was observed anymore after IFNγ-treatment (data not shown).
To assess if any of the observed upregulations were statistically significant, cell lines were grouped into HPV-negative cell lines and HPV16-positive tumor cell lines, including cervical cancer and HNSCC. Of note, the HPV16-transfected group was omitted from this analysis as this group had a similar expression pattern as HPV-negative cell lines. Fig. 1B shows all genes that were found to be significantly upregulated in HPV16-positive tumor cell lines. These genes were the chaperone calreticulin, the aminopeptidase ERAP1 and all tested components of the immunoproteasome (PSMB8, PSMB9, PSMB10, PSME1 and PSME2). All other analyzed genes did not show significantly different expression from the HPV-negative group (Fig. S1).
ERAP1 is overexpressed on the protein level in HPV16-positive tumor cells
All APM components were tested for protein expression by Western blot. On the protein level, ERAP1 was the only APM component that showed overexpression (Fig. 2). In a representative Western Blot, ERAP1 was expressed higher in 13 out of 16 HPV16-positive cell lines in comparison to the HPV-negative cell lines (Fig. 2A). In Fig. 2C, pooled data of three independent experiments is shown, confirming the consistent overexpression. ERAP1 is IFNγ-inducible. After IFNγ-treatment, we did not observe differential expression of ERAP1 between HPV16-positive and HPV-negative cells any more, suggesting that IFNγ-signaling is intact in all our cell lines (Fig. 2B and D). This is in line with the qRT-PCR results, which also showed no difference in expression levels after IFNγ treatment. Taken together, the mRNA screen and the protein analysis confirmed that ERAP1 is overexpressed in HPV16-positive tumor cells.
Figure 2.
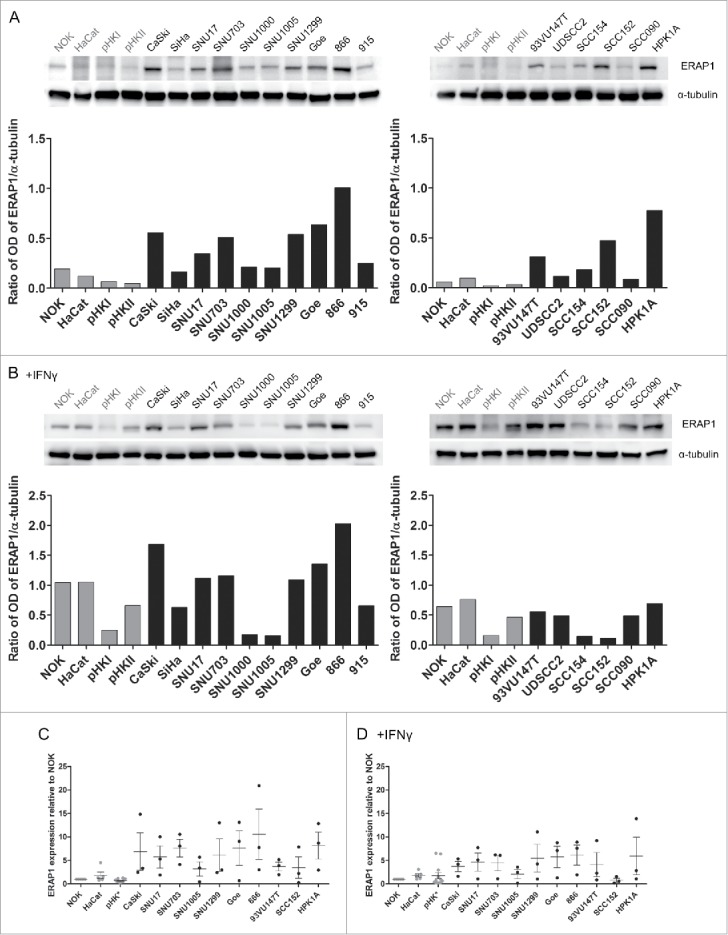
ERAP1 protein expression with and without IFNγ treatment. Cells were either left untreated (A) or treated with IFNγ (B) for 48 h. 50µg of each cellular lysate was analyzed for ERAP1 expression by Western blot. Optical density (OD) was quantified using the ImageJ software and ERAP1 expression was normalized to α-tubulin for each cell line, as depicted below the Western blots. The results are representative of three independent experiments. C and D represent pooled data from these three independent experiments. ERAP1 expression was calculated relative to NOK. Results are plotted as means ± SEM. Data marked in gray corresponds to HPV-negative cell lines. pHK* = data from primary human keratinocytes derived from three donors and pooled primary human foreskin keratinocytes from multiple donors.
ERAP1 is overexpressed in CIN and cervical cancer samples
After analysis of HPV16-positive tumor cell lines, immunohistochemical analysis of ERAP1 was performed in human cervical tissue sections. ERAP1 expression in dysplastic tissue was compared to histologically normal cervical epithelium. Different degrees of cervical intraepithelial neoplasia (CIN): CIN I (n = 9), CIN II (n = 6), CIN III (n = 9), and squamous cell carcinomas (SCC, n = 10) were analyzed and compared to normal cervical epithelium (n = 23). As a surrogate marker for HPV oncogene expression, p16 was used.23 p16-negative normal cervical epithelium showed only low levels of ERAP1 protein expression (Fig. 3A). In contrast, epithelial ERAP1 levels were clearly elevated in dysplastic CIN I to III lesions and in established cervical cancers. To quantify ERAP1 expression, a score was applied that considered both the percentage of ERAP1 expressing cells and the intensity of ERAP1 staining. Analysis of this histological score showed that epithelial ERAP1 levels were significantly increased in CIN and SCC when compared to normal epithelium (Fig. 3B and C). All analyzed CIN II, CIN III and SCC tissue sections had elevated ERAP1 expression levels. In Fig. 3C, histological scores were equally categorized into low (score range: 0–95), intermediate (score range: 96–190) and high expression (score range: 191–285). This analysis clearly demonstrated that ERAP1 expression increases with disease progression, being lowest in CIN I and highest in SCC. Furthermore, the analysis revealed that histologically normal tissue expressed ERAP1 only at low levels, as do 67% of CIN I lesions. The percentage of tissue sections that expressed ERAP1 at high levels was rising from CIN II to SCC. All analyzed sections were transformed with HPV high-risk types (HPV genotyping information is given in Fig. S2), HPV16-positive tissue sections are shown in blue in Fig. 3B. Notably, HPV16-positive sections did not behave differently regarding ERAP1 expression than other high-risk HPV-positive sections.
Figure 3.
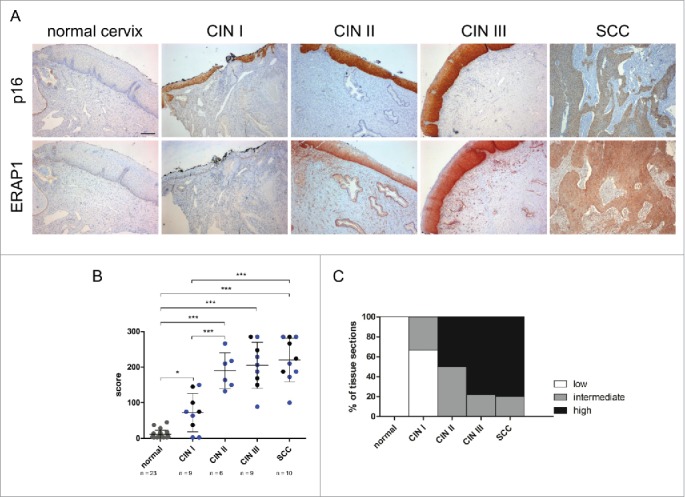
Immunohistochemical analysis of ERAP1 expression in cervical tissue. A, Human tissue sections were stained for ERAP1 and p16, a surrogate marker for HPV oncogene expression. Protein expression was analyzed in histologically normal cervical epithelium (normal cervix), in HPV16-positive dysplastic CIN (cervical intraepithelial neoplasia) I to CIN III lesions and in SCC (cervical squamous cell carcinoma). Scale bar corresponds to 200µm. B, Immunohistochemical scores were determined by multiplying the frequency score of ERAP1 expressing cells with the maximum intensity score of ERAP1 expression. Each symbol represents one sample, symbols in gray represent HPV-negative samples, symbols in blue represent HPV16-transformed tissue, and symbols in black represent tissue transformed with other HPV high-risk types. Means ± SD are indicated. *p ≤ 0.05, ***p ≤ 0.001 (ANOVA multiple comparison test). C, Immunohistological scores were equally categorized into low (0–95), intermediate (96–190) and high expression (191–285).
The effect of ERAP1 overexpression on the HPV16 epitope repertoire
To be able to assess the functional consequences of ERAP1 overexpression on the HPV16 epitope repertoire, a siRNA-mediated knockdown of ERAP1 was established in HPV16-positive tumor cell lines (Fig. 4). We deliberately chose a knockdown and not a knockout approach, as our goal was to compare the effects of normal ERAP1 expression levels (as present in non-malignant keratinocytes) to the effects of ERAP1 overexpression. The knockdown was stable for at least 72h and allowed comparison of the same tumor cell line either with the inherent ERAP1 overexpression, or with ERAP1 expression attenuated to levels observed in HPV-negative keratinocytes.
Figure 4.
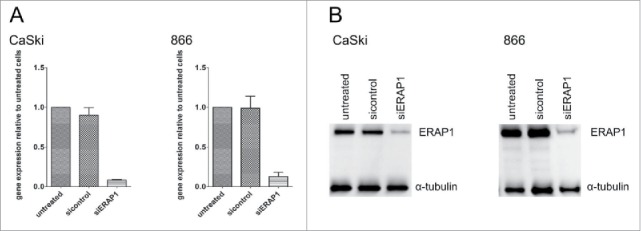
Knockdown of ERAP1 using siRNAs in HPV16-positive cells. Cells were left untreated or were treated either with an ERAP1-targeting (siERAP1) or a non-targeting control siRNA pool (sicontrol) for 72h. A, Total RNA was purified from cells, reverse transcribed to cDNA and analyzed for ERAP1 expression by qRT-PCR. PCR reactions were performed in triplicates. At least one or the geometric mean of the following genes was used as internal reference gene: GAPDH and PGK1. ERAP1 expression was calculated relative to untreated cells. The results are plotted as means ± SD from at least three independent experiments. B, 50µg of each cellular lysate was loaded and ERAP1 expression analyzed by Western blot with α-tubulin detection as a loading control. The results are representative of at least three independent experiments.
As a first approach to analyze presentation of HPV16-derived epitopes on the cell surface, a set of eight HLA-A2-restricted HPV16 E7-derived epitopes was analyzed by mass spectrometry. HLA-A2/epitope complexes were immunoprecipitated from CaSki cells, epitopes isolated and the epitopes analyzed by targeted LC-MS.3 This analysis allows to assess the absolute presence or absence of a certain epitope on MHC-I molecules on the cell surface. The presence of six of the eight epitopes on the cell surface was confirmed (Table S4). No absolute difference in presentation of these epitopes was observed between CaSki cells with high and attenuated ERAP1 expression. Thus, we can conclude that ERAP1 overexpression did not result in a complete loss of presentation of any of these six epitopes on the cell surface. However, there may still be differences in the amount of the presented peptides, which needs to be addressed with an assay system that is sensitive enough to detect variations in MHC-I presentation levels.
To this end, we elicited specific cytotoxic T-cell lines. We chose an 11-mer peptide (E781–91) as a target peptide for T-cell line generation, as ERAP1 has been reported to preferentially trim peptides of 10 and more amino acids in length.24,25 We used CaSki and 866 cells that endogenously overexpress ERAP1 as target cells and a cell line that does not naturally overexpress ERAP1 (FK16A) for control purposes. The HLA-matched but HPV16-negative cell line C33A was used as negative control. We employed a flow cytometry-based cytotoxicity assay that allows the simultaneous co-culture of effector CD8+ T-cells with two target cell populations.26 The experiment revealed that CaSki with attenuated ERAP1 expression were killed better by E681–91-specific T-cells than CaSki cells overexpressing ERAP1 (Fig. 5A). In contrast, no difference in killing was observed with 866 target cells (Fig. 5B). Only unspecific killing was observed for the HPV16-negative control cell line (Fig. 5A and B). As expected, no difference in killing between ERAP1 siRNA-treated and control siRNA-treated cells was observed with FK16A target cells that had a low ERAP1 expression level from the start (data not shown).
Figure 5.
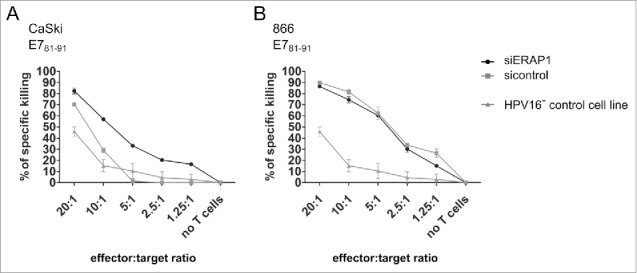
The effect of ERAP1 attenuation in target cells on cytotoxicity mediated by HPV16 E781–91-specific CD8+ T-cells. Vital-FR assay using HPV16 E781–91-specific CD8+ effector T-cells from a HLA-A2-positive donor. The HLA-A2-positive and HPV16-positive cell lines CaSki (A) and 866 (B) were used as target cells, whereas the HLA-matched HPV16-negative cell line C33A (HPV16- control cell line) served as background control. 24 h after siRNA transfection, target cells and titrated numbers of effector cells were incubated for 48 h and were then analyzed by flow cytometry. One representative experiment of two independent experiments is shown. The results are plotted as mean ± SD from at least duplicates.
Discussion
The development of a therapeutic anti-HPV vaccine is important to allow treatment of people who are already HPV-infected. In order to develop a clinically successful vaccine it is crucial to elucidate immune evasion strategies of HPV-mediated cancers. Therefore, in this study, we have systematically investigated APM component expression on the mRNA and protein level in a comprehensive collection of HPV16-positive cell lines. The collection contained cell lines from HPV-positive cervical cancers and HNSCC, from primary tumors and metastases; and included long-standing and newly established cell lines. HPV-negative cells originating from primary or immortalized keratinocytes served as negative controls. The mRNA screen showed upregulation of different APM components. This was surprising, as in most former studies a downregulation of APM components was reported in connection with HPV-induced diseases.15,16 Our observations, however, are not in conflict with these studies because they usually described a downregulation of APM components together with the downregulation of surface HLA, which we did not observe in our cell line collection. On the protein level, ERAP1 was the only APM component that showed significant upregulation in HPV16-positive tumor cells, as well as in immunohistochemistry of CIN lesions and cervical cancer. The expression of ERAP1 in human tumors has been reported to be highly variable, ranging from low to high expression. Ovarian, breast and lung cancers tend to downregulate ERAP1, whereas colorectal adenocarcinoma, renal cell carcinoma, skin cancer and melanoma tend to have high ERAP1 expression levels.27-29 In discordance with our results, Mehta and colleagues reported a downregulation of ERAP1 in cervical cancer, but only in 15% of cases.16 They also found that downregulation of ERAP1 is an independent bad prognostic factor. Conversely, the Human Protein Atlas specifically mentions cervical cancer as one of the cancers where ERAP1 is found to be upregulated.30 These different observations may be explained by variable expression of ERAP1 within one tumor entity and the use of different experimental methods and controls.
ERAP1 is an important editor of the MHC-I peptidome by generating or destroying MHC-I-binding peptides.18,19 In humans, there is a homolog of ERAP1, called ERAP2. However, ERAP2 is not expressed in 25% of the population because one of the two alleles leads to nonsense-mediated decay.31 Furthermore, ERAP2 has different cutting preferences. Thus, ERAP2 cannot be considered to compensate for ERAP1 and therefore was not analyzed in this study. In our analysis of the functional consequences of the observed ERAP1 overexpression in HPV16-positive cells, we have shown that ERAP1 attenuation in CaSki cells did not change absolute presentation of a panel of eight HPV16 E7-derived peptides, as analyzed by MS. It has to be noted that the MS approach was primarily designed to compare absolute differences, i.e., to investigate whether ERAP1 overexpression results in a complete loss of any peptides. This was not the case in the present study.
One of the distinct enzymatic properties of ERAP1 is its molecular ruler function, which has been described by Chang et al..24 They propose that ERAP1 spares short peptides (8–9aa) whereas longer peptides (≥10aa) are trimmed efficiently.19,24 This mechanism could be one explanation why we did not observe differences in absolute presentation in the panel of eight peptides analyzed by MS. Half of these epitopes were 9-mers, so possibly not prone to further trimming by ERAP1 anymore. A recent study demonstrated that a large proportion of the MHC-I peptidome is not dependent on ERAP1,32 which may also explain why we did not observe differences in presentation of the MS-analyzed epitopes.
Epitope-specific T-cells are very sensitive and even react to small variances in levels of peptide presentation. Indeed, cytotoxicity assays using HPV16-specific CTLs specific for an E7-derived 11-mer peptide revealed that epitope-specific CTL responses are altered by ERAP1 expression levels. When using CaSki cells, which naturally overexpress ERAP1, as a target cell line, we observed enhanced killing toward cells with attenuated ERAP1 expression, indicating that the target epitope is presented to a lesser extent in the ERAP1-overexpressing cells, and at higher levels upon ERAP1 attenuation. However, there was no difference in killing by the same T-cell line using ERAP1-attenuated or overexpressing 866 target cells. Differences in killing of these two target cell lines by the same T-cell line may at least be partly explained by different ERAP1 variants within these cells, which we determined by sequencing (Table S5). The enzymatic function of ERAP1 can be influenced by single nucleotide polymorphisms.33-36
In recent years, ERAP1 gained much interest as a possible target molecule in the treatment of autoimmune diseases and cancer. ERAP1 variants in combination with distinct HLA types predispose for the development of the autoimmune diseases ankylosing spondylitis, birdshot chorioretinopathy and Behçet's disease. The exact pathogenic mechanisms are not fully understood but it is proposed that an altered HLA class I epitope repertoire contributes to pathogenesis.34,35,37 In cancer, the hypothesis that an overactive ERAP1 can destroy tumor-associated epitopes is supported by a study on the melanoma epitope MART-126–35. Melanomas overexpress ERAP1, and MART-126–35 presentation was enhanced by ERAP1 downregulation, reflected by enhanced anti-tumor CTL responses.20 Similarly, presentation of the immunodominant colon carcinoma epitope GSW11 is strongly enhanced when ERAP1 is attenuated.21,22 Interestingly, both GSW11 and MART26–35 have low binding affinities for MHC-I, supporting the proposed hypothesis of a “bind-trim-release” mechanism for ERAP1.20,21 According to this model, ERAP1 trims one amino acid at a time and releases its substrate. The free peptide can then either bind to MHC-I or bind to ERAP1 again and the next amino acid will be cleaved. Thus, MHC-I and ERAP1 compete for the available peptide pool and this competition decides about the fate of a peptide. In case of a high affinity peptide, the peptide would bind stably to MHC-I and is protected from further trimming. On the contrary, low affinity peptides briefly bind to MHC-I and are released again, rendering them prone to another round of trimming by ERAP1 before they are efficiently bound to the MHC-I binding groove. The fact that the 11-mer HPV16 epitope E781–91 also has a low binding affinity to HLA-A2 may indicate that this mechanism could also play a role in our findings.
Of note, our work is based on blood from healthy donors, suggesting that the memory T-cells used for our assays were induced during a previous infection with HPV, which was successfully cleared. Our immunohistochemical data demonstrate that the expression of ERAP1 increases with disease stage. Thus, it is tempting to speculate that, while ERAP1 expression increases with progressing disease, the presentation of E781–91 and other longer epitopes may decrease – resulting in the removal of target antigens of an existing immune response.
Taking into account that ERAP1 plays a role in autoimmune diseases and cancer, the interest in a chemical inhibitor for this peptidase is rising. Currently, several groups are working on the identification of a specific chemical inhibitor for ERAP1, but these inhibitors still lack experimental evidence concerning efficiency and toxicity in vitro and in vivo.22,38 When specific ERAP1 inhibitors become available, it would be interesting to test them not only in already known ERAP1-overexpressing tumors such as melanoma and colon cancer, but also in ERAP1-overexpressing HPV-mediated malignancies. Inhibition of ERAP1 could save some epitopes from overtrimming, and thus result in more immunogenic tumors.
In conclusion, this study demonstrated that ERAP1 is upregulated in HPV16-positive tumor cells, CIN lesions and established cervical cancer. Of note, a T-cell line against an 11-mer HPV16 E7-derived peptide showed increased killing upon ERAP1 attenuation in a HPV16-positive tumor cell line, indicating that presentation of this epitope is reduced when ERAP1 is expressed at high levels. Thus, the observed overexpression of ERAP1 in advanced HPV disease stages can alter the presentation of certain HPV16-derived epitopes.
Methods
Cell lines
This study included 3 HPV-negative keratinocyte lines (NOK, HaCat, C33A [ATCC HTB-31]) and HPV-negative primary human keratinocytes [pHK] and primary human foreskin keratinocytes [pHFK]), 12 HPV16-positive cervical cancer cell lines (866, 915, Goe, Mar, CaSki [ATCC CRL-1550], SiHa [ATCC HTB-35], MRI-H196, SNU17, SNU703, SNU1000, SNU1005, SNU1299), 6 HPV16-positive head and neck squamous cell carcinoma (HNSCC) cell lines (SCC090, SCC152, SCC154, UDSCC2, UMSCC47, 93VU147T) and 5 cell lines transfected with the HPV16 genome (HPK1A, FK16A, C66-3, C66-7 and 2A3). Details on all cell lines and their respective media are listed Tables S1.1 and S1.2. Cell authentication (based on SNP-profiling and comparison to a data bank of 828 STR-profiling authenticated human reference cell lines)39 and contamination testing (based on multiplex PCR and detection of products by specific oligonucleotide probes for 14 Mycoplasma species, SMRV, EBV, and contaminating cells from 12 species)40 was conducted for every newly acquired cell line. Contamination testing was repeated every 8 weeks, authentication testing every 4 months. Contamination and authentication testing was performed by the DKFZ core facility for functional genomics. After initial testing, cells were expanded and many identical vials of the lowest possible passage number were frozen. For use in experiments, cells were passaged for a maximum of 10 passages before a new vial of the low passage number stock was thawn.
Quantitative real-time PCR
RNA was isolated using the RNeasy Mini Kit (Qiagen #74106) and 1µg was reverse transcribed into cDNA using the QuantiTect Reverse Transcription Kit (Qiagen #205311). Quantitative PCR was performed in triplicates using TaqMan-specific probes (Applied Biosystems, Waltham, MA, USA; listed in Table S2) according to the manufacturer's instructions. The geometric mean of at least 3 out of 4 reference genes (GAPDH, PPIA, IPO8 and PGK1) was used for normalization.41 Data was rigorously controlled for PCR efficiency, linear dynamic range and intra-assay repeatability (precision) and processed using LinRegPCR (Heart Failure Research Center, Amsterdam, Netherlands) and qBase Plus (Biogazelle, Zwijnaarde, Belgium).
Cell extracts and Western blot analysis
Cells were lysed in RIPA buffer: 1% sodium deoxycholate (Applichem #302-95-4), 0.1% SDS (Roth #0183.1), 0.15M NaCl (Sigma #31434), 0.01M disodium hydrogenphosphate (Roth #T879.1), 2mM EDTA (Gerbu #1034), 1% NP-40 (Sigma #9016-45-9), 1mM PMSF (Roth #6367.1) and 2x cOmplete mini protease inhibitor cocktail (Roche #11836153001). Clear lysates were boiled in 4x Laemmli sample buffer for 5min: 222mM Tris pH 6.8 (Sigma #T1503-1kg), 3.5% SDS, 35% glycerol (Sigma #G5516-500ML), 0.016% bromophenol blue (Roth #A512.2), 10% β-mercaptoethanol (Roth #4227.1). SDS-PAGE and Western blot were performed according to standard procedures, antibodies and dilutions are listed in Table S3.
Immunohistochemical analysis
Sections of formalin-fixed, paraffin-embedded CIN and cervical cancer cone biopsy specimens were prepared for staining following standard procedures, for antibodies see Table S3. Immunohistological scores were calculated by multiplying staining frequency and maximal intensity. Staining frequency ranged from 0 (no ERAP1 expressing cells) to 5 (< 10%), 30 (10–50%), 70 (50–90%) or 95 (> 90% ERAP1 expressing cells) and the maximal intensity was set to 0 = no, 1 = low, 2 = intermediate and 3 = high expression.
ERAP1 small interfering RNA
ERAP1 (NCBI-ID: 51752) was attenuated with a siPOOL containing 30 ERAP1-specific siRNAs purchased from siTOOLs BIOTECH (Martinsried, Germany). A non-targeting siPOOL served as a negative control. siRNA (5–10nM) was transfected into cells using RNAiMAX (Life Technologies #13778150) according to the manufacturer's instructions.
Immunoprecipitation and mass spectrometry
Immunoprecipitation (IP) of HLA-A2 molecules from CaSki cells was performed using GammaBind Plus Sepharose beads (GE Healthcare #17-0886-01) coupled to mouse-anti-human-HLA-A2 (clone BB7.2), as described in.42 IP samples were washed and acidified with 0.3% TFA (Sigma #91709) to elute peptides from HLA-A2-peptide complexes. The eluates were purified with a Seppak cartridge (Waters #WAT023625) and concentrated with a vacuum centrifuge. Peptides were analyzed by targeted nanoLC-MS3 using a NanoAcquity UPLC system (Waters) on-line connected to a QTRAP6500 (AB SCIEX, Darmstadt, Germany) mass spectrometer. A minimum of three fragments of at least 5aa in length with the best signal-to-noise ratios were measured per peptide. To confirm the presence of a target peptide, MS spectra of IP samples were compared to the spectra of respective synthetic reference peptides.
T-cell lines
Peripheral blood mononuclear cells (PBMC) were isolated from fresh blood of volunteers (ethical approval S-393/2011) using Ficoll-Paque™ Plus (GE Healthcare Bio-Sciences #17-1440-02) and Leucosep tubes (Greiner Bio-One #227290). T-cell lines were generated by three weekly stimulations using autologous DCs or autologous B-cells.43 107 cells/ml PBMCs were plated with 20:1–200:1 HPV16-peptide-pulsed (10µg/ml peptide) autologous matured dendritic cells (DCs) in 24-well culture plates in T-cell medium supplemented with 10ng/ml IL-7 (PeproTech #207-IL). Peptides were synthesized in the in-house DKFZ peptide facility with a purity of >95% and were dissolved in DMSO (Sigma #D8418-50ml). 5 days after the first stimulation, 20IU/ml IL-2 (PeproTech #200–02) were added and 50IU/ml IL-2 were added 5 days after the second and third stimulation. Cultures were restimulated with peptide-pulsed DCs one week after the first stimulation, and with peptide-pulsed and 32Gy-irradiated autologous CD40-activated B-cells one week later. B-cells were used as APCs in a ratio of 1:5 with the T-cells. One week after the last stimulation, CD8+ T-cells were purified using the untouched CD8+ T-cell isolation kit (Miltenyi Biotec #130-096-495).
Vital-FR cytotoxicity assay
1×106/ml target cells were stained with 5µM carboxyfluorescein (CFSE, LifeTechnologies #C1157) and transfected with the siRNA Pool targeting ERAP1 or stained with 0.25µM FarRed (dimethyldodecylamine oxide-succinimidyl ester, LifeTechnologies #C34564) and transfected with the siRNA control. The assay was performed as described previously.26 In brief, labeled cells were used for siRNA-mediated knockdown. After 24h, 3×103 CFSE-labeled and 3×103 FarRed-labeled target cells were mixed with purified CD8+ effector T-cells (effector:target ratios 1.25:1 to 20:1). Target cells only served as negative controls. Cells were plated in T-cell medium supplemented with 10U/ml IL-2 in a final volume of 200µl and incubated for 48h at 37°C. Specific killing of labeled cells was analyzed by flow cytometry. Data analysis was performed with FlowJo (TreeStar, Ashland, OR, USA). Specific lysis was calculated as 100-[(% target cells with T-cells/% target cells only)x100].
Statistics
For statistical analyses, PRISM®5 (GraphPad, La Jolla, CA, USA) was used. Statistical test types used are indicated in the figure legends.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors thank Jonathan Dörre for performing the immunohistochemical stainings, and Dr. Caroline Goldstein for proofreading of the manuscript. We thank all blood donors for their voluntary support of our study. Support by the DKFZ Flow Cytometry Core Facility and the Biostatistics Department is gratefully acknowledged.
Funding
This work was supported by a grant of the Manfred Lautenschläger foundation to A.B. Riemer, a Marie Curie Career Integration Grant of the European Commission to A.B. Riemer, a PhD scholarship to R. Blatnik and a postdoctoral fellowship to A.K. Grabowska by the Eduard and Melanie zur Hausen Foundation, a PhD scholarship to S. Hoppe by the Helmholtz International Graduate School of the German Cancer Research Center (DKFZ), a grant by the Hector II foundation to A.B. Riemer and a grant to A.B. Riemer by the German Center for Infection Research (DZIF).
References
- 1.Van Doorslaer K, Tan Q, Xirasagar S, Bandaru S, Gopalan V, Mohamoud Y, Huyen Y, McBride AA. The papillomavirus episteme: A central resource for papillomavirus sequence data and analysis. Nucleic Acids Res 2013; 41(Database issue):D571-8; PMID:23093593; https://doi.org/ 10.1093/nar/gks984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schiffman M, Doorbar J, Wentzensen N, de Sanjose S, Fakhry C, Monk BJ, Stanley MA, Franceschi S. Carcinogenic human papillomavirus infection. Nat Rev Dis Primers 2016; 2:16086; PMID:27905473; https://doi.org/ 10.1038/nrdp.2016.86 [DOI] [PubMed] [Google Scholar]
- 3.zur Hausen H. Papillomaviruses and cancer: From basic studies to clinical application. Nat Rev Cancer 2002; 2(5):342-50; PMID:12044010; https://doi.org/ 10.1038/nrc798 [DOI] [PubMed] [Google Scholar]
- 4.Castle PE, Rodriguez AC, Burk RD, Herrero R, Wacholder S, Hildesheim A, Morales J, Rydzak G, Schiffmann M. (Proyecto Epidemiologico Guanacaste Group). Long-term persistence of prevalently detected human papillomavirus infections in the absence of detectable cervical precancer and cancer. J Infect Dis 2011; 203(6):814-22; PMID:21343148; https://doi.org/ 10.1093/infdis/jiq116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bonanni P, Bechini A, Donato R, Capei R, Sacco C, Levi M, Boccalini S. Human papilloma virus vaccination: Impact and recommendations across the world. Ther Adv Vaccines 2015; 3(1):3-12; PMID:25553242; https://doi.org/ 10.1177/2051013614557476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Garland SM. The Australian experience with the human papillomavirus vaccine. Clin Ther 2014; 36(1):17-23; PMID:24417782; https://doi.org/ 10.1016/j.clinthera.2013.12.005 [DOI] [PubMed] [Google Scholar]
- 7.Khallouf H, Grabowska AK, Riemer AB. Therapeutic vaccine strategies against human papillomavirus. Vaccines (Basel) 2014; 2(2):422-62; PMID:26344626; https://doi.org/ 10.3390/vaccines2020422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Skeate JG, Woodham AW, Einstein MH, Da Silva DM, Kast WM. Current therapeutic vaccination and immunotherapy strategies for HPV-related diseases. Hum Vacc Immunother 2016; 12(6):1418-29; PMID:26835746; https://doi.org/ 10.1080/21645515.2015.1136039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van der Sluis TC, van der Burg SH, Arens R, Melief CJ. New approaches in vaccine-based immunotherapy for human papillomavirus-induced cancer. Curr Opin Immunol 2015; 35:9-14; PMID:26001120; https://doi.org/ 10.1016/j.coi.2015.05.002 [DOI] [PubMed] [Google Scholar]
- 10.Grabowska AK, Riemer AB. The invisible enemy - how human papillomaviruses avoid recognition and clearance by the host immune system. Open Virol J 2012; 6:249-56; PMID:23341860; https://doi.org/ 10.2174/1874357901206010249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Westrich JA, Warren CJ, Pyeon D. Evasion of host immune defenses by human papillomavirus. Virus Res 2017; 231:21-33; PMID:27890631; https://doi.org/ 10.1016/j.virusres.2016.11.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van der Burg SH, Arens R, Melief CJ. Immunotherapy for persistent viral infections and associated disease. Trends Immunol 2011; 32(3):97-103; PMID:21227751; https://doi.org/ 10.1016/j.it.2010.12.006 [DOI] [PubMed] [Google Scholar]
- 13.Evans M, Borysiewicz LK, Evans AS, Rowe M, Jones M, Gileadi U, Cerundolo V, Man S. Antigen processing defects in cervical carcinomas limit the presentation of a CTL epitope from human papillomavirus 16 E6. J Immunol 2001; 167(9):5420-8; PMID:11673561; https://doi.org/ 10.4049/jimmunol.167.9.5420 [DOI] [PubMed] [Google Scholar]
- 14.Georgopoulos NT, Proffitt JL, Blair GE. Transcriptional regulation of the major histocompatibility complex (MHC) class I heavy chain, TAP1 and LMP2 genes by the human papillomavirus (HPV) type 6b, 16 and 18 E7 oncoproteins. Oncogene 2000; 19(42):4930-5; PMID:11039910; https://doi.org/ 10.1038/sj.onc.1203860 [DOI] [PubMed] [Google Scholar]
- 15.Hasim A, Abudula M, Aimiduo R, Ma JQ, Jiao Z, Akula G, Wang T, Abudula A. Post-transcriptional and epigenetic regulation of antigen processing machinery (APM) components and HLA-I in cervical cancers from Uighur women. PLoS One 2012; 7(9):e44952; PMID:23024775; https://doi.org/ 10.1371/journal.pone.0044952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mehta AM, Jordanova ES, Kenter GG, Ferrone S, Fleuren GJ. Association of antigen processing machinery and HLA class I defects with clinicopathological outcome in cervical carcinoma. Cancer Immunol Immunother 2008; 57(2):197-206; PMID:17622526; https://doi.org/ 10.1007/s00262-007-0362-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Albers A, Abe K, Hunt J, Wang J, Lopez-Albaitero A, Schaefer C, Gooding W, Whiteside TL, Ferrone S, DeLeo A, et al.. Antitumor activity of human papillomavirus type 16 E7-specific T cells against virally infected squamous cell carcinoma of the head and neck. Cancer Res 2005; 65(23):11146-55; PMID:16322265; https://doi.org/ 10.1158/0008-5472.CAN-05-0772 [DOI] [PubMed] [Google Scholar]
- 18.York IA, Brehm MA, Zendzian S, Towne CF, Rock KL. Endoplasmic reticulum aminopeptidase 1 (ERAP1) trims MHC class I-presented peptides in vivo and plays an important role in immunodominance. Proc Natl Acad Sci U S A 2006; 103(24):9202-7; PMID:16754858; https://doi.org/ 10.1073/pnas.0603095103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.York IA, Chang SC, Saric T, Keys JA, Favreau JM, Goldberg AL, Rock KL. The ER aminopeptidase ERAP1 enhances or limits antigen presentation by trimming epitopes to 8–9 residues. Nat Immunol 2002; 3(12):1177-84; PMID:12436110; https://doi.org/ 10.1038/ni860 [DOI] [PubMed] [Google Scholar]
- 20.Keller M, Ebstein F, Burger E, Textoris-Taube K, Gorny X, Urban S, Zhao F, Dannenberg T, Sucker A, Keller C, et al.. The proteasome immunosubunits, PA28 and ER-aminopeptidase 1 protect melanoma cells from efficient MART-126–35 -specific T-cell recognition. Eur J Immunol 2015; 45(12):3257-68; PMID:26399368; https://doi.org/ 10.1002/eji.201445243 [DOI] [PubMed] [Google Scholar]
- 21.James E, Bailey I, Sugiyarto G, Elliott T. Induction of protective antitumor immunity through attenuation of ERAAP function. J Immunol 2013; 190(11):5839-46; PMID:23610143; https://doi.org/ 10.4049/jimmunol.1300220 [DOI] [PubMed] [Google Scholar]
- 22.Zervoudi E, Saridakis E, Birtley JR, Seregin SS, Reeves E, Kokkala P, Aldhamen YA, Amalfitano A, Mavridis IM, James E, et al.. Rationally designed inhibitor targeting antigen-trimming aminopeptidases enhances antigen presentation and cytotoxic T-cell responses. Proc Natl Acad Sci U S A 2013; 110(49):19890-5; PMID:24248368; https://doi.org/ 10.1073/pnas.1309781110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klaes R, Friedrich T, Spitkovsky D, Ridder R, Rudy W, Petry U, Dallenbach-Hellweg G, Schmidt D, von Knebel Doeberitz M. Overexpression of p16(INK4A) as a specific marker for dysplastic and neoplastic epithelial cells of the cervix uteri. Int J Cancer 2001; 92(2):276-84; PMID:11291057; https://doi.org/ 10.1002/ijc.1174 [DOI] [PubMed] [Google Scholar]
- 24.Chang SC, Momburg F, Bhutani N, Goldberg AL. The ER aminopeptidase, ERAP1, trims precursors to lengths of MHC class I peptides by a “molecular ruler” mechanism. Proc Natl Acad Sci U S A 2005; 102(47):17107-12; PMID:16286653; https://doi.org/ 10.1073/pnas.0500721102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hearn A, York IA, Rock KL. The specificity of trimming of MHC class I-presented peptides in the endoplasmic reticulum. J Immunol 2009; 183(9):5526-36; PMID:19828632; https://doi.org/ 10.4049/jimmunol.0803663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stanke J, Hoffmann C, Erben U, von Keyserling H, Stevanovic S, Cichon G, Schneider A, Kaufmann AM. A flow cytometry-based assay to assess minute frequencies of CD8+ T cells by their cytolytic function. J Immunol Methods 2010; 360(1–2):56-65; PMID:20558172; https://doi.org/ 10.1016/j.jim.2010.06.005 [DOI] [PubMed] [Google Scholar]
- 27.Fruci D, Ferracuti S, Limongi MZ, Cunsolo V, Giorda E, Fraioli R, Sibilio L, Carroll O, Hattori A, van Endert PM, et al.. Expression of endoplasmic reticulum aminopeptidases in EBV-B cell lines from healthy donors and in leukemia/lymphoma, carcinoma, and melanoma cell lines. J Immunol 2006; 176(8):4869-79; PMID:16585582; https://doi.org/ 10.4049/jimmunol.176.8.4869 [DOI] [PubMed] [Google Scholar]
- 28.Fruci D, Giacomini P, Nicotra MR, Forloni M, Fraioli R, Saveanu L, van Endert P, Natali PG. Altered expression of endoplasmic reticulum aminopeptidases ERAP1 and ERAP2 in transformed non-lymphoid human tissues. J Cell Physiol 2008; 216(3):742-9; PMID:18393273; https://doi.org/ 10.1002/jcp.21454 [DOI] [PubMed] [Google Scholar]
- 29.Kamphausen E, Kellert C, Abbas T, Akkad N, Tenzer S, Pawelec G, Schild H, van Endert P, Seliger B. Distinct molecular mechanisms leading to deficient expression of ER-resident aminopeptidases in melanoma. Cancer Immunol Immunother 2010; 59(8):1273-84; PMID:20419298; https://doi.org/ 10.1007/s00262-010-0856-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A, et al.. Proteomics. Tissue-based map of the human proteome. Science 2015; 347(6220):1260419; PMID:25613900; https://doi.org/ 10.1126/science.1260419 [DOI] [PubMed] [Google Scholar]
- 31.Andres AM, Dennis MY, Kretzschmar WW, Cannons JL, Lee-Lin SQ, Hurle B, Program NCS, Schwartzberg PL, Williamson SH, Bustamante CD, et al.. Balancing selection maintains a form of ERAP2 that undergoes nonsense-mediated decay and affects antigen presentation. PLoS Genet 2010; 6(10):e1001157; PMID:20976248; https://doi.org/ 10.1371/journal.pgen.1001157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nagarajan NA, de Verteuil DA, Sriranganadane D, Yahyaoui W, Thibault P, Perreault C, Shastri N. ERAAP shapes the peptidome associated with classical and nonclassical MHC class I molecules. J Immunol 2016; 197(4):1035-43; PMID:27371725; https://doi.org/ 10.4049/jimmunol.1500654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Costantino F, Talpin A, Evnouchidou I, Kadi A, Leboime A, Said-Nahal R, Bonilla N, Letourneur F, Leturcq T, Ka Z, et al.. ERAP1 gene expression is influenced by nonsynonymous polymorphisms associated with predisposition to spondyloarthritis. Arthritis Rheumatol 2015; 67(6):1525-34; PMID:25740711; https://doi.org/ 10.1002/art.39072 [DOI] [PubMed] [Google Scholar]
- 34.Alvarez-Navarro C, Martin-Esteban A, Barnea E, Admon A, Lopez de Castro JA. Endoplasmic reticulum aminopeptidase 1 (ERAP1) polymorphism relevant to inflammatory disease shapes the peptidome of the birdshot chorioretinopathy-associated HLA-A*29:02 antigen. Mol Cell Proteomics 2015; 14(7):1770-80; PMID:25892735; https://doi.org/ 10.1074/mcp.M115.048959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reeves E, Colebatch-Bourn A, Elliott T, Edwards CJ, James E. Functionally distinct ERAP1 allotype combinations distinguish individuals with ankylosing spondylitis. Proc Natl Acad Sci U S A 2014; 111(49):17594-9; PMID:25422414; https://doi.org/ 10.1073/pnas.1408882111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reeves E, Edwards CJ, Elliott T, James E. Naturally occurring ERAP1 haplotypes encode functionally distinct alleles with fine substrate specificity. J Immunol 2013; 191(1):35-43; PMID:23733883; https://doi.org/ 10.4049/jimmunol.1300598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guasp P, Alvarez-Navarro C, Gomez-Molina P, Martin-Esteban A, Marcilla M, Barnea E, Admon A, Lopez de Castro JA. The peptidome of Behcet's disease-associated HLA-B*51:01 includes two subpeptidomes differentially shaped by endoplasmic reticulum aminopeptidase 1. Arthritis Rheumatol 2016; 68(2):505-15; PMID:26360328; https://doi.org/ 10.1002/art.39430 [DOI] [PubMed] [Google Scholar]
- 38.Stamogiannos A, Papakyriakou A, Mauvais FX, van Endert P, Stratikos E. Screening identifies thimerosal as a selective inhibitor of endoplasmic reticulum aminopeptidase 1. ACS Med Chem Lett 2016; 7(7):681-5; PMID:27437077; https://doi.org/ 10.1021/acsmedchemlett.6b00084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Castro F, Dirks WG, Fahnrich S, Hotz-Wagenblatt A, Pawlita M, Schmitt M. High-throughput SNP-based authentication of human cell lines. Int J Cancer 2013; 132(2):308-14; PMID:22700458; https://doi.org/ 10.1002/ijc.27675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schmitt M, Pawlita M. High-throughput detection and multiplex identification of cell contaminations. Nucleic Acids Res 2009; 37(18):e119; PMID:19589807; https://doi.org/ 10.1093/nar/gkp581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Riemer AB, Keskin DB, Reinherz EL. Identification and validation of reference genes for expression studies in human keratinocyte cell lines treated with and without interferon-gamma - a method for qRT-PCR reference gene determination. Exp Dermatol 2012; 21(8):625-9; PMID:22775998; https://doi.org/ 10.1111/j.1600-0625.2012.01537.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rucevic M, Kourjian G, Boucau J, Blatnik R, Garcia Bertran W, Berberich MJ, Walker BD, Riemer AB, Le Gall S. Analysis of major histocompatibility complex-bound HIV peptides identified from various cell types reveals common nested peptides and novel T cell responses. J Virol 2016; 90(19):8605-20; PMID:27440904; https://doi.org/ 10.1128/JVI.00599-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liebig TM, Fiedler A, Zoghi S, Shimabukuro-Vornhagen A, von Bergwelt-Baildon MS. Generation of human CD40-activated B cells. J Vis Exp 2009; 32:pii:1373; PMID:19838159; https://doi.org/ 10.3791/1373 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.