

Abstract
The regulation of gene expression by steroid hormones plays an important role in the normal development and function of many organs, as well as in the pathogenesis of endocrine‐related cancers, especially breast cancer. However, clinical data suggest that combined testosterone and estrogen treatments on post‐menopausal women increase the risk of breast cancer. Experiments have shown that many, if not all kallikreins are under steroid hormone regulation in breast cancer cell lines. Their implication as prognostic and diagnostic markers has also been well‐documented. Thus, we investigated the effect of combined hormone stimulation with androgens and 17β‐estradiol on the ductal caricinoma cell line BT474. This cell line has been shown to be sensitive to both, androgens (secreting PSA) and estrogens (secreting a number of kallikreins including KLK10, 11, and KLK14). We found that PSA expression was downregulated upon combined hormone stimulation, confirming reports that estrogen can antagonize and block the activity of the androgen receptor. Upon analysis of estrogen‐sensitive kallikreins 10, 11, and 14, all showed to be synergistically enhanced in their expression three‐ to fourfold, upon joint hormone treatment versus individual hormone stimulation. The enhancement is dependent upon the action of androgens as treatment with the androgen receptor antagonist cyproterone actetate normalized the expression of KLK10, 11, and KLK14 to estrogen‐stimulation levels. The synergistic effects between estrogens and androgens on estrogen‐sensitive genes may have implications on the role of the kallikreins in associated risk of breast cancer and progression.
Keywords: Kallikreins, Breast cancer, Gene expression, Steroid hormones, Hormone-dependent gene expression, Hormone receptors, Cell signaling
Non‐standard abbreviations
- KLK
kallikrein gene
- KLK
kallikrein protein
- PSA
prostate-specific antigen
- DHT
dihydrotestosterone
- AR
androgen receptor
- ER
estrogen receptor
- PGR
progesterone receptor
- GR
glucocorticoid receptor
- IGFBP4
insulin-like growth factor binding protein-4
- ADAMTS-1
A Disintegrin And Metalloproteinase with ThromboSpondin repeats-1
1. Introduction
Steroid hormones play a critical role in breast cancer development and have been associated with an increased epithelial cell proliferation and in turn facilitating malignant transformation. In particular, two sex hormones that have been very well characterized both in vitro and in vivo are estrogen and progesterone (Somboonporn and Davis, 2004a; Stein and McDonnell, 2006). The serum concentrations of these hormones together with their respective receptors are also used as epidemiological markers in assessing breast cancer risk. Studies have also shown that the direct action of these steroid hormones on different breast tissues is dependent upon their specific receptors. Another category of sex hormones that has been extensively studied in breast cancer in human and mice are androgens. Androgens have been shown to have both stimulatory and inhibitory actions on the growth of several breast cancer cell lines (Maggiolini et al., 1999; Hackenberg et al., 1988; Poulin et al., 1988; Zhou et al., 2000). However, their etiological role in breast cancer has been unclear. Unclear is whether the action of androgens is direct through their cognate receptor or via their metabolization into estrogen‐like byproducts by aromatase activity. Also, recent studies suggest that subnormal levels of androgens may adversely affect a women's health, while on the other hand other studies indicate that supranormal levels may also have adverse effects on the female reproductive system including abnormal growth and tumorgenesis.
When women reach menopausal age, there is a decrease in endogenous levels of sex hormones, particularly testosterone and estrogen, and have been associated with menopausal symptoms. Clinical trials have demonstrated that the exogenous administration of these hormones can ameliorate these symptoms partially. However, there have been several studies that have associated endogenous elevated serum levels of estrogen and free testosterone hormone with breast cancer risk. This increased risk is of particular significance in post‐menopausal women receiving HRT (Somboonporn and Davis, 1999, 2005, 2005, 2004, 2004, 2005).
The molecular mechanism of the action of sex hormones is that they exert their effect by binding to their cognate hormone receptor. Upon binding to the receptor, the hormone–receptor complex translocates into the nucleus, binds to DNA cis‐elements known as hormone response elements (HREs) in the upstream proximal promoter, and interacting with several other coactivating proteins and the general transcriptional machinery to modulate transcriptional activation. The consensus HRE sequence consists of a palindromic sequence separated by a unique nucleotide sequence. Hormone receptors, in particular the glucocorticoid, androgen and progesterone receptors (GR, AR and PGR, respectively) recognize very similar DNA cis‐elements, however, the estrogen receptor (ER) binds to a quite unique sequence (Klinge, 2001; Aranda and Pascual, 2001; Claessens et al., 2001). Therefore, the sensitivity/expression of a particular hormone‐dependent regulated gene in a cell line to any given steroid hormone is dependent upon both the presence of the hormone receptor and consensus HRE binding sites. By far, the gene whose regulation by steroid hormones has been most thoroughly studied is the human tissue kallikrein gene, prostate‐specific antigen (PSA). ThePSA gene possesses three androgen response elements (ARE‐I, ARE‐II, and ARE‐III). ARE‐I and ARE‐II were identified in the upstream promoter region (−170bp and −400bp), functionally tested and found to be active in LNCaP, a prostate cancer cell line (Cleutjens et al., 1996; Cinar et al., 2004). ARE‐III was found at −4316bp, which induced a dramatic increase in PSA transcription, in comparison to ARE‐I and ARE‐II (Cleutjens et al., 1997). AREs have been found in other genes, including other members of the kallikrein gene family. We are currently in the process of elucidating hormone responsive elements for other kallikreins. More recently, literature is accumulating for non‐genotropic actions of steroid hormones via another category of hormone receptors, which are associated with the plasma membrane. Instead the actions of these steroid hormone receptors are characterized by activation of a variety of signal transduction pathways including, MEK/ERK, PI3K/AKT, and JNK pathways (Zivadinovic and Watson, 2005; Peterziel et al., 1999; Kang et al., 2004; Papakonstanti et al., 2003; Stoica et al., 2003a).
All 15 kallikrein genes show differential expression patterns in many cancers at the mRNA and protein levels and many kallikreins have been examined as prognostic indicators in breast cancer including, PSA, KLK5, 6, 10, and KLK14 (Yousef et al., 2003, 2005, 2007, 2006, 2005, 2002, 2002, 1996). Previous studies have found that there is a close association between steroid hormone stimulation of breast cancer cell lines and coordinated kallikrein gene expression (Borgono et al., 2003; Luo et al., 2000; Paliouras and Diamandis, 2006a; Magklara et al., 2000). However, it has never been examined if the expression profiles would change upon multiple hormone stimulations. Therefore, would significant changes in kallikrein gene expression be of clinical importance within the context that HRT with estrogen and testosterone and increases in breast cancer risk? Thus, in this paper we examined a number of androgen and estrogen hormone‐regulated kallikrein genes in the breast cancer cell line BT474, to determine if these two steroid hormones can act synergistically to enhance kallikrein gene expression.
2. Materials and methods
2.1. Cell lines
The breast cancer cell line BT474, used in the following experiments was obtained from the American Type Culture Collection (ATCC), Rockville MD, and was selected as to their well‐defined kallikrein expression.
2.2. Steroids and inhibitor compounds
All steroid hormones, the steroid antagonist cyproterone acetate, and BSA‐conjugated testosterone (testosterone 3‐(O‐carboxymethyl)oxime:BSA) were obtained from Sigma Chemical Co., St. Louis, MO. The aromatase inhibitor xanthenone (4‐(imidazolylmethyl)‐1‐nitro‐9H‐9‐xanthenone) was purchased from EMD Biosciences Inc., San Diego, CA. Steroid and inhibitor stock solutions and dilutions were prepared in 100% ethanol and the aromatase inhibitor in DMSO.
2.3. Cell culture: hormone stimulations and blocking studies
BT474 cell line was cultured in phenol‐red‐free RPMI 1640 media supplemented with FBS (11%), at 37°C, 5% CO2 in plastic culture flasks. Once confluent, 8×105cells were seeded into 6‐well plates with the same medium to allow the cells to adhere. Twenty‐four hours after plating the medium was changed to RMPI supplemented with 10% charcoal–dextran stripped FBS and incubated for an additional 24h. The following day, the medium was changed to fresh RMPI/charcoal–dextran stripped FBS for stimulation and inhibitor studies.
2.4. Stimulation experiments
The following steroid hormones were used for all stimulations: dihydrotestosterone (DHT), 17β‐estradiol (Est), testosterone (Test), and BSA‐conjugated testosterone (BSA:Test). Cells were incubated with each hormone added once for 24h for RNA analysis and for 6 days for measuring secreted kallikrein protein production in cell supernatants. All stimulations were performed in triplicate.
2.5. Blocking and inhibitor studies
The cell line BT474 was cultured as described in the stimulation experiments. To block steroid hormone receptors, blockers for different hormones (1μM final concentration) were added for 1h into the culture media, to which the cells were then stimulated with hormones. After 24h, the cells were harvested for total mRNA extraction or 6 days for analysis of secreted kallikrein proteins. Blocking experiments were repeated in triplicate.
Aromatase inhibitor treatments were carried out similarly, with multiple doses every second day at a final concentration of 400nM, as recommended by the manufacturer, for this particular cell line.
2.6. RNA extraction and RT‐PCR
Total RNA was extracted from breast cancer cells using TRIzol reagent (Invitrogen, Carlsbad, CA) following the manufacturer's instructions. RNA concentration was determined spectrophotometrically and 5μg of total RNA was reverse‐transcribed into first strand cDNA using the Superscript™ First Strand Synthesis kit (Invitrogen) using an Oligo(dT) primer. PCRs were carried out using Qiagen HotStar Taq Polymerase (Qiagen, Valencia, CA) on first strand cDNA for multiple kallikreins. Table 1 lists the primers and expected product size for each kallikrein and other transcript analyzed. An equal amount of each PCR product was run out on 0.9% agarose gels and visualized by ethidium bromide staining. Primer sequences for RT‐PCRs can be found in Table 1.
2.7. Quantification of KLKs in cell culture supernatants
The concentration of each KLK was measured with specific and quantitative immunofluorometric ELISA assays developed in our laboratory. In brief, 96‐well polystyrene plates were first coated with 500ng/well of an KLK‐specific capture antibody. After overnight incubation, the plates were washed, 50μL of culture supernatant or standards and equal volume of assay buffer were added and incubated at room temperature for 2h. Plates were washed and biotinylated antibodies were subsequently added. Following incubation with biotinylated antibodies, alkaline phosphatase‐conjugated streptavidin was added. Finally, diflunisal phosphate (DFP) and terbium‐based detection solutions were added and fluorescence was measured with the Cyberfluor 615 Immunoanalyzer (MDS Nordion, Kanata, ON, Canada). The calibration and data reduction were performed automatically. More details for the ELISA assays used have been described elsewhere as follows: PSA, hK10, hK11, and hK14.
2.8. Western blot analysis
BT474 cell lysates from 24h hormone stimulated and inhibitor treated cells were prepared for Western blot analysis. The antibodies used for Western blot analysis included, AR (N‐20), ERα (D‐12), and PGR (AB‐52), and β‐ACTIN (C4) from EMD Biosciences Inc., San Diego, CA and GR (Clone 41) from BD Biosciences, San Jose, CA. Phospho‐specific Western blots were carried out from lysates derived from 30min, 8h, and 24h hormone stimulated BT474 cells with phospho‐Thr308‐AKT and phosphor‐p44/42 (Thr202/Tyr204) ERK1/2 (E10) along with anti‐AKT (pan‐11E7) and p44/42 ERK1/2 used as loading controls Westerns and purchased from Cell Signaling Technology, Davers, MA.
3. Results
3.1. Estrogen‐sensitive kallikreins are synergistically enhanced by androgens
Previous hormone studies on the kallikreins in breast cancer cell lines have focused on individual hormone stimulations, with many kallikreins showing either estrogen‐specific expression such as, KLK5, 6, 10, 11, 13, and KLK14, while others show androgen‐dependent expressions like PSA and KLK2 (Paliouras and Diamandis, 2006a). Translation studies have assessed the kallikreins as prognostic indicators in breast cancer, and as combined hormone treatments that are carried out in post‐menopausal women have been associated with increased risk for breast cancer we decided to investigate the mechanism of regulation of a number of hormone‐regulated kallikreins. We selected the breast cancer cell line, BT474, to carry out these studies for several reasons. First, the cell line expresses the androgen, estrogen, progesterone and glucocorticoid receptors at relatively the same level (Luo et al., 2003). Second, the kallikrein hormone‐dependent expression patterns for this cell lines have been well characterized in previous publications (Borgono et al., 2003; Paliouras and Diamandis, 2006a; Magklara et al., 2000; Luo et al., 2003). Finally, the cell line was isolated from localized ductal carcinoma, representative of Stage II cancer of a post‐menopausal woman (Lacroix and Leclercq, 2004). Thus, these above criteria were optimal for clinical association of the potential increased risk of combined hormone therapies and kallikrein expression.
BT474 were stimulated with two concentrations of DHT (100nM and 10nM) and 17β‐estradiol and possible combinations of these hormones and concentrations. These concentrations fall within the normal physiological levels of these hormones. We first analyzed the expression of PSA protein levels, as BT474 is sensitive to androgen to produce PSA. It has been shown that estradiol can antagonize the androgen receptor by directly competing for the androgen binding site on the AR and block PSA expression in the presence of androgen (Zhu et al., 2005; Stover et al., 1987). We observed a similar blocking of PSA expression upon combined DHT and estradiol stimulation (Figure 1A).
Figure 1.
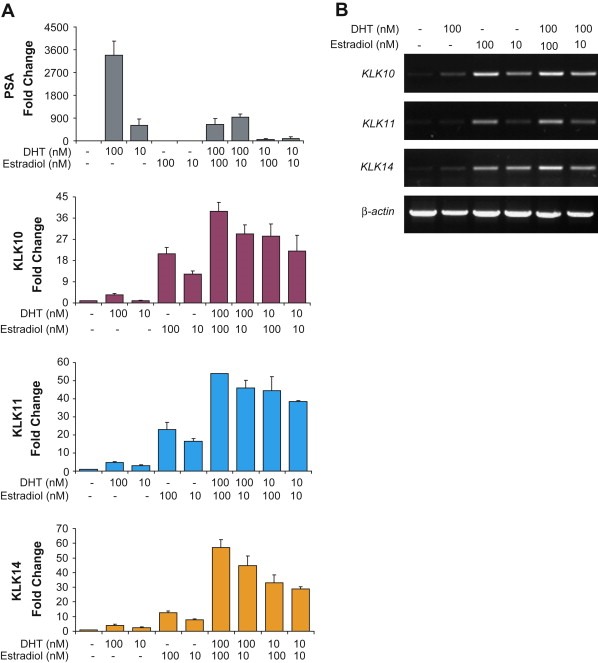
Enhanced kallikrein expression upon combined DHT and estradiol stimulation. Cells were treated with either 100nM or 10nM DHT and 100nM or 10nM estradiol and combinations of the two hormone concentrations and quantified by ELISA of condition media supernatants. (A) Kallikrein protein expression profiles. PSA shows specific dose‐dependent DHT dependent expression, and is reduced significantly in the presence of estradiol. KLK10, 11, and KLK14 show specific estrogen‐dependent expression and are enhanced by the addition of DHT. (B) RT‐PCR analysis of KLK10, 11, and KLK14 genes also show enhanced transcriptional activation with joint DHT and estradiol stimulation. Actin expression was used as a control for all RT‐PCR analysis.
The expression of KLK10, 11, and KLK14 proteins in BT474 have been shown to be estrogen‐dependent, however, upon combined hormone stimulation the expression of these kallikreins were synergistically enhanced, rather than an additive effect of the presence of DHT in the presence of estradiol. KLK10 is enhanced approximately twofold by either concentration of DHT. When BT474 cells are jointly stimulated with estradiol and DHT, KLK11 and KLK14 expression levels are enhanced by almost three‐ and fourfold, respectively. These enhanced changes in kallikreins' levels are specific to androgen and estradiol stimulations as combined stimulations with the glucocorticoid dexamathesone did not show any effect (data not shown). The increase in secreted kallikrein protein levels observed in the condition media is a transcriptional event as RT‐PCR analysis of KLK10, 11, and KLK14 mRNA expression shows a pattern of increased transcript levels upon joint hormone stimulation (Figure 1B).
3.2. Aromatase activity does not contribute to synergistic enhancement of kallikreins' expression
It has been proposed that the link of combined testosterone and estrogen HRT and increased breast cancer risk is a result of aromatase activity of the conversion of testosterone into estrogen‐like products (James et al., 1987; Thijssen et al., 1991; Perel et al., 1980). Although our initial enhancement experiments were performed with non‐aromatizable DHT, we performed combined hormone stimulations using estradiol with an aromatizable androgen, testosterone. Again, we saw a similar synergistic enhancement pattern of PSA, KLK10, 11, and KLK14 protein expression upon joint stimulation of estradiol with testosterone as we observed with DHT (Figure 2). The addition of the aromatase inhibitor xanthenone (Recanatini et al., 2001) did not alter the expression patterns, suggesting that result of the aromatization of testosterone is not the contributing process by which estrogen‐dependent expression of KLK10, 11, and KLK14 are enhanced upon androgen stimulation. Similarly, it has been reported that the aromatization results in a conversion of less than 2% of androgen compounds into estrogen‐like products (Karaer et al., 2004), a factor that is insignificant to the enhanced kallikrein expression we are observing.
Figure 2.

Aromatase inhibitor treatment. PSA, KLK10, 11, and KLK14 protein expression profiles were analyzed for synergistic enhancement upon stimulation with the aromatizable androgen, testosterone, and estradiol. BT474 cells were also subsequently treated with the aromatase inhibitor xanthenone in the presence of both steroid hormones.
3.3. No significant changes in hormone receptor levels upon combined hormone stimulation
We next asked whether the enhanced changes in KLK10, 11 and KLK14 that were observed upon combined DHT and estradiol treatment may be resulting from changes in hormone receptor levels. Therefore we carried out Western blot analysis of the androgen, estrogen and progesterone receptors (Figure 3). We did not observe any significant changes in AR, ER, and GR steroid hormone receptor levels that may attribute to the dramatic changes we see in kallikrein expression. However, the estrogen‐responsive expression of the progesterone receptor (PGR) was antagonized by the additional treatment of DHT.
Figure 3.
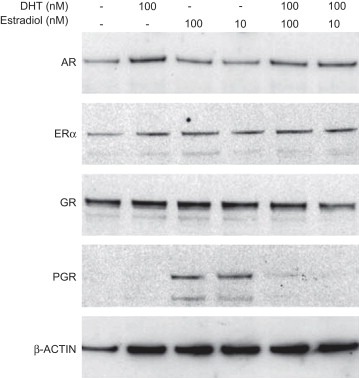
Western blot analysis of hormone receptors. Cell lysates from BT474 were analyzed for changes in androgen (AR), estrogen (ER) and glucocorticoid (GR) and progesterone (PGR) receptor levels. β‐ACTIN is used as the loading control for this analysis.
3.4. Identifying other estrogen‐sensitive genes with similar androgenic enhanced expression
We then analyzed the expression profiles of other known estrogen‐regulated genes to determine whether the synergistic enhancement we are observing with the KLK10, 11 and KLK14 upon joint hormone stimulations with DHT and estradiol is shared phenomena or exclusive to these kallikreins. Using unpublished microarray data of steroid hormone stimulated BT474 cells, we selected a number of estrogen and androgen‐sensitive genes to identify other genes whose transcription may also be affected by combined estrogen and androgen hormone treatments. The known estrogen‐sensitive genes we analyzed by RT‐PCR were the included, pS2, c‐fos, c‐myc, and IGFBP4 and an androgen‐responsive gene, ADAMTS‐1 (A Disintegrin And Metalloproteinase with ThromboSpondin repeats‐1) (Figure 4). Similar to estrogen‐dependent PGR protein expression, RT‐PCR analysis indicated that neither, pS2, c‐fos or c‐myc transcripts are stimulated by estradiol, but antagonized by the addition of DHT. Only IGFBP4 expression was enhanced by the treatment of both DHT and estrogen. As for the one other androgen‐sensitive gene ADAMTS‐1, it also showed a similar expression pattern to PSA with its androgen responsiveness repressed by the presence of estradiol. Thus, it appears that the synergistic enhancement observed amongst the kallikreins is exclusive to a small number of estrogen‐regulated genes. Due to the scale of our initial screening we have not attempted to exhaust other transcripts but focused only on the best‐characterized estrogen‐regulated genes, this does not exclude the possibility that many other estrogen‐dependent genes do not share such a similar regulatory mechanism.
Figure 4.
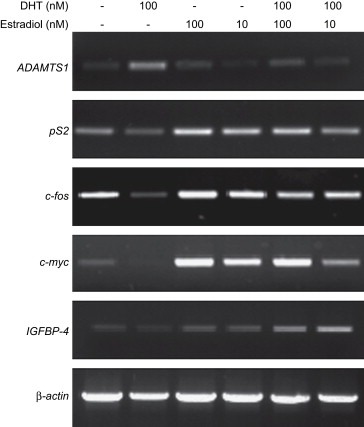
RT‐PCR analysis of other estrogen upregulated genes. PS2, PGR, c‐fos, c‐myc, IGFBP4, and ADAMTS‐1 were analyzed by RT‐PCR for changes in expression upon joint DHT and estradiol hormone stimulations.
3.5. The synergistic enhancement of the estrogen‐regulated kallikreins is dependent androgen receptor activity
To further elucidate a mechanism we wanted to determine whether the enhancing effect observed by joint hormone stimulations is dependent on hormone receptor activity. Thus, we carried out blocking experiments using antagonists of estrogens (ICI 182,780) and androgens (cyproterone acetate). A single final dose of 1μM of each antagonist was added with the stimulating hormones, followed by ELISAs for KLK10, 11, and KLK14, 6 days later. As shown in Figure 5, the estrogen antagonist ICI 182,780 completely blocks the expression of these kallikreins in estradiol and DHT stimulated cells, confirming the expression of these kallikreins as estrogen‐dependent. However, the addition of the androgen receptor antagonist cyproterone acetate lowers the expression levels to near estradiol‐only stimulated. Therefore, the synergistic enhancement of the expression of these kallikreins observed upon the stimulation of DHT and estradiol is dependent upon androgens and androgen receptor activity.
Figure 5.
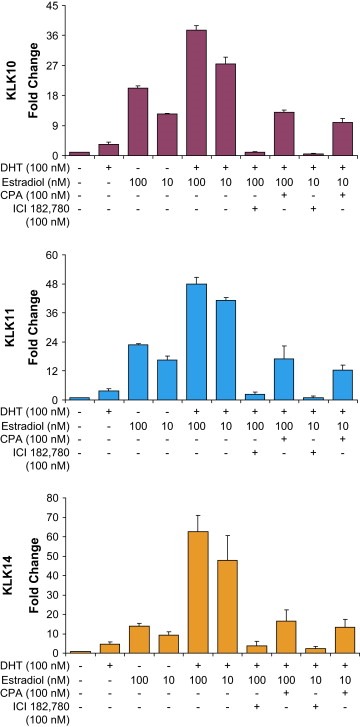
Hormone receptor antagonist treatment of combined hormone stimulated cells. Jointly hormone stimulated BT474 cells were treated with a single dose of 1μM final concentration of either ICI 182,780 (ICI) or cyproterone acetate (CPA). After 6 days, the cell supernatants were collected and ELISAs were carried out on, KLK10, KLK11, and KLK14.
3.6. Membrane androgen receptor is responsible for the enhanced expression of estrogen‐regulated kallikreins
Our results were indicating two contrasting actions of androgen treatment on estrogen‐sensitive gene expression. First, that the expression of estrogen‐dependent kallikreins 10, 11, and 14 were synergistically enhanced by the combined stimulation of DHT and estradiol. Second, some estrogen‐sensitive genes, including pS2 and PGR, were antagonized by androgens. Therefore, we asked whether two different forms of the androgen receptor may be responsible for our observations. Our approach was to use a BSA‐conjugated testosterone (BSA:Test) in conjunction with estradiol stimulation. The BSA:Test cannot be internalized to the cell and can only therefore act upon the membrane bound androgen receptor (Erlanger et al., 1957, 1959). It has been shown that BSA:Test can stimulate PSA secretion in LNCaP cells (Papakonstanti et al., 2003; Kampa et al., 2005; Stathopoulos et al., 2003), and our results were able to similarly stimulate PSA in BT474 cells (Figure 6A). Moreover, BSA:Test‐dependent PSA expression was also antagonized by estradiol treatments. When we analyzed levels of KLK10, 11, and KLK14 proteins of combined BSA:Test and estradiol stimulation, we once again observed a synergistic enhancement in their expression, as previously shown with DHT and testosterone.
Figure 6.
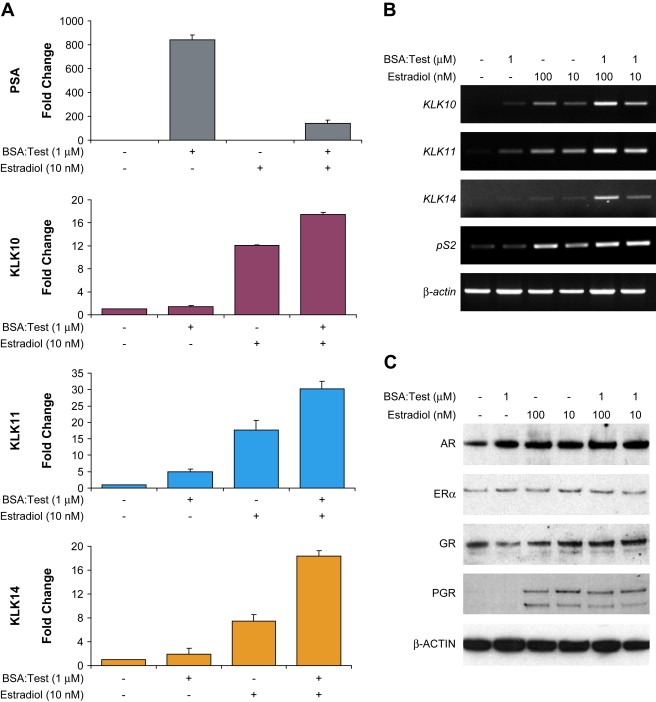
BSA‐conjugated testosterone treatment also enhances estrogen‐dependent KLK expression. (A) BT474 cells were treated with BSA:Test and also together with estradiol and PSA, KLK10, 11 and KLK14 protein expression profiles were analyzed by ELISA. (B) RT‐PCR profiling of KLK10, 11, 14, and pS2 upon BSA:Test–estradiol co‐stimulations. (C) Western blot analysis of AR, ERα, GR and PGR of cell lysates from BT474 cells co‐treated with BSA:Test and estradiol.
RT‐PCR analysis of KLK10, 11, and KLK14 mRNA also indicated that the enhancement was a transcriptional event (Figure 6B). Also important was that our results were able to segregate the two androgen functions that we had observed in our initial characterization of other estrogen‐sensitive genes. As kallikrein gene expression was enhanced by BSA:Test, the expression of pS2 and PGR were no longer antagonized by the action of an androgenic compound functioning through a membrane bound receptor (Figure 6B, C).
3.7. Membrane bound androgen receptor activates PI3K/AKT pathway
Previous reports indicated that membrane associated hormone receptors can act through a variety of intracellular signaling pathways together with our observations that a membrane bound androgen receptor can affect estrogen‐dependent kallikrein gene expression. Therefore, we looked for changes in the active states of both MEK/ERK and PI3K/AKT pathways. BT474 cells were stimulated with DHT, BSA:Test and estradiol alone and together for a time course, corresponding to time points used in our previous experiments, and changes in ERK and AKT activity were analyzed by phosphor‐specific Western blot analysis (Figure 7). DHT and BSA:Test were able to stimulate and sustain AKT but not ERK activity over the three time points (30min, 8h and 24h). The results that AKT activation by either DHT and BSA:Test can be sustained for a 24h period, together with the protein expression data for KLK10, 11, and KLK14, illustrate that the action of the membrane bound androgen receptor is not a rapid event as several reports would suggest.
Figure 7.
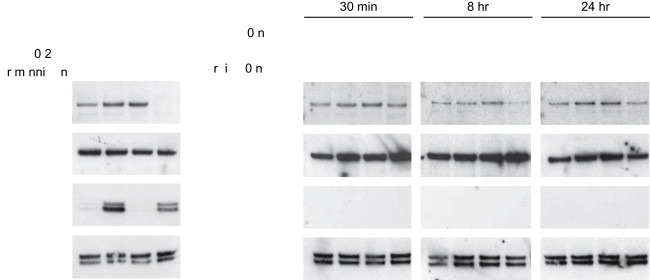
AKT activation by androgens. Activity of ERK1/2 and AKT were analyzed by phospho‐specific Western blot analysis, over a time course, of androgen and estradiol stimulated BT474 cells. As a control of AKT and ERK1/2 activation BT474 cells were treated with 10ng/mL of EGF for 10min with and without the specific pathway inhibitors Wortmannin (500nM) and U0126 (10μM).
4. Discussion
We have extensively published the steroid hormone‐dependent regulation of the kallikrein gene family members in several cancer cell lines, especially breast cancer cells, whose expression patterns have been linked as cancer biomarkers. This report provides an innovative insight into the mechanism required for the genetic regulation of estrogen‐dependent kallikrein gene expression in breast cancer cells. The observations of androgens to synergistically enhance estrogen‐sensitive kallikrein gene expression of a breast cancer cell line representative of an early stage ductal adenocarcinoma also provide a unique pathway of the action of steroid hormone receptors.
Our data show that the expression of estrogen‐responsive kallikreins is synergistically enhanced by androgens. The addition of DHT to estrogen stimulated BT474 cells increased the expression of KLK10, 11, and KLK14 greater than the additive effects of individual hormone stimulation (Figure 1). The synergistic effect of androgens was also dose‐dependent, as higher concentrations of DHT enhanced lower concentrations. Analysis of other observed and independently characterized estrogen‐responsive genes, pS2, PGR, c‐myc, and c‐fos, did not show a similar enhancing phenotype and were downregulated by the presence of androgens. However, of the non‐kallikrein estrogen‐responsive genes, IGFBP4 was the only one also enhanced by DHT (Figure 4). The observation that IGFBP4 is also synergistically enhanced by androgens is of some significance as other members of the IGFBP family have been associated with cancer progression (Marshman and Streuli, 2002). Most recently, it has been shown that IGFBP3, IGFBP4 and IGFBP5 are modulated in ER‐positive by estrogens in ovarian cancer cells and provide a predictive signature of outcome of a subset of ovarian cancers (Walker et al., 2007). Moreover, contrast to androgen enhancement of estrogen‐responsive kallikreins, the androgen stimulation of PSA was antagonized by estrogens. A similar result was observed for the androgen‐responsive gene, ADAMTS‐1. The antagonizing of PSA expression by estrogen has also been observed by other authors and reported that estrogens' ligands with androgens can compete for the androgen receptor. Furthermore, this enhancement is a result of increased transcriptional activity and not a result of increased secretion or protein stability in the condition media. The synergistic action of androgens is also not a result of aromatase conversion of androgens, like testosterone, into estrogen‐like products (Figure 2). However, microarray analysis is a means to discern all estrogen‐responsive genes that are transcriptionally enhanced by co‐stimulation of androgens. Western blot analysis indicated that enhanced changes in kallikrein expression, from combined hormone treatments, were not a result of changes in hormone receptor expression levels but directed by androgens through changes in androgen receptor activity (Figure 5). Inhibition of the activity of the androgen receptor by the use of cyproterone acetate blocked enhanced expression of all the kallikreins to a near estrogen‐alone stimulated expression level. It has been shown that cyproterone acetate can inhibit androgen binding to both cytosolic and membrane androgen receptors.
Membrane associated hormone receptors have been shown to activate several signaling pathways and in turn augment gene expression. A membrane associated ER receptor has been shown to stimulate ERK and AKT phosphorylation upon estrogen‐stimulation in MCF7 breast cancer cells (Zivadinovic and Watson, 2003, 2003, 2005). Moreover, a membrane bound androgen receptor has also been shown to stimulate AKT activity and PSA expression in prostate cancer cell lines upon stimulation with a BSA‐conjugated testosterone analog (Papakonstanti et al., 2003; Stathopoulos et al., 2003). Stimulation with BSA:Test also increased secretion of PSA in BT474 cancer cells, but more importantly we could also enhance estrogen‐dependent kallikrein gene expression through activation of the membrane bound androgen receptor (Figure 6). The experiment also distinguished between the two activities of the androgen receptor isoforms. We no longer observed the repression of pS2 or PGR expression, upon stimulation with BSA:Test. However, what remains unclear is a mechanism by which other estrogen‐regulated genes may be repressed by androgens. Thus, the activation of a membrane bound androgen receptor contributing to enhanced estrogen‐induced gene expression provides a unique mechanism of hormone‐dependent kallikrein regulation. The possibility of androgens, via a membrane bound receptor, to activate AKT signaling also has implications on other cellular events and downstream targets that may be associated with tumorgenesis.
Androgen but not estrogen‐stimulation corresponded with an increased AKT phosphorylation. AKT activation occurs specifically through the androgen ligand activation of the membrane bound receptor. The downstream targets of AKT are many; however, it has been reported that AKT can phosphorylate ERα at several serine residues with Ser167 as an AKT‐consensus phosphorylation site (Vilgelm et al., 2006; Sun et al., 2001; Campbell et al., 2001). The phosphorylation of ERα via AKT has been correlated with an increased receptor activity and estrogen‐induced gene expression. Moreover, increased AKT activity is associated with tamoxifen resistance in breast cancer cells (McCubrey et al., 2006). Altogether our results would suggest that the association of AKT activation by androgens and subsequent increase of ER activity plus estrogen ligand binding may attribute to the synergistic enhancement in gene expression that we are observing for the estrogen‐dependent kallikrein genes and IGFBP4, and possibly other estrogen‐regulated genes (Figure 8). Such activity of the receptor and downstream signal pathways would have repercussions on gene expression and cellular transformations associated with cancer progression. Finally, most often membrane bound hormone receptor activity often is associated with rapid non‐genotropic events. However, our results clearly show that kallikrein expression and AKT activity by way of androgen activation of the membrane hormone receptor can be sustained for prolonged periods of time.
Figure 8.
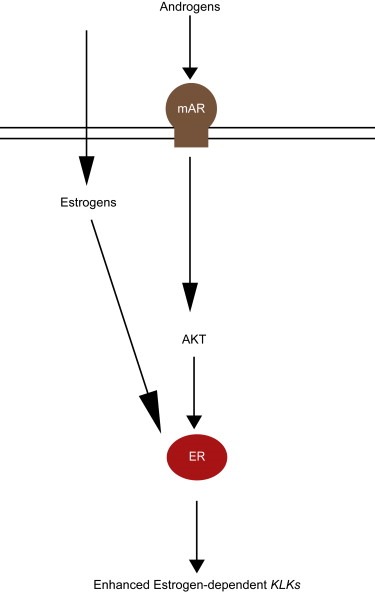
Schematic of enhanced estrogen‐dependent KLK expression by androgens via AKT pathway.
The correlation of hormone‐dependent kallikrein regulation and the clinical observation of the major contribution of estrogens associated to their expression and disease manifestation is well understood. In vitro, we have also identified several kallikrein proteolytic targets that further provide a physiological function for the kallikreins in tumorgenesis. Many of the substrates that have been identified include the extracellular matrix (ECM) proteins, laminin α‐5 chain precursor, matrilin‐4, and collagen IV. KLK5, 6 and KLK13 are also able to hydrolyze a variety of ECM proteins including, laminin, fibronectin and collagen I, II, and III. The targeting of ECM proteins by the kallikreins is theorized to be associated with an increase in aggressiveness of tumor cells. In prostate cancer cells overexpression of both PSA and KLK4 showed increases in cell migration, linked to loss of E‐cadherin (Borgono et al., 2004; Yousef and Diamandis, 2002; Paliouras and Diamandis, 2006b). In addition, we have published extensive clinical research articles correlating this association (Obiezu and Diamandis, 2005; Paliouras et al., 2007; Borgono and Diamandis, 2004). Therefore, our results showing that androgens can significantly increase expression of estrogen‐dependent kallikrein gene expression under estrogen‐stimulating conditions would have major clinical implications especially as it relates to HRT strategies. Furthermore, our results would also indicate a need to return to expanding our screening of kallikreins as it specifically relates to samples of individuals receiving combined testosterone and estrogen treatments. Such screening will be emphasized in post‐menopausal women where the association of combined HRT and breast cancer risk is greatest.
Supporting information
Supplementary data
Supplementary data 1.
1.1.
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.molonc.2008.01.001.
Paliouras Miltiadis, Diamandis Eleftherios P., (2008), Androgens act synergistically to enhance estrogen‐induced upregulation of human tissue kallikreins 10, 11, and 14 in breast cancer cells via a membrane bound androgen receptor, Molecular Oncology, 1, doi: 10.1016/j.molonc.2008.01.001.
References
- Aranda, A. , Pascual, A. , 2001. Nuclear hormone receptors and gene expression. Physiol. Rev.. 81, 1269–1304. [DOI] [PubMed] [Google Scholar]
- Borgono, C.A. , Diamandis, E.P. , 2004. The emerging roles of human tissue kallikreins in cancer. Nat. Rev. Cancer. 4, 876–890. [DOI] [PubMed] [Google Scholar]
- Borgono, C.A. , Grass, L. , Soosaipillai, A. , Yousef, G.M. , Petraki, C.D. , Howarth, D.H. , Fracchioli, S. , Katsaros, D. , Diamandis, E.P. , 2003. Human kallikrein 14: a new potential biomarker for ovarian and breast cancer. Cancer Res.. 63, 9032–9041. [PubMed] [Google Scholar]
- Borgono, C.A. , Michael, I.P. , Diamandis, E.P. , 2004. Human tissue kallikreins: physiologic roles and applications in cancer. Mol. Cancer Res.. 2, 257–280. [PubMed] [Google Scholar]
- Campbell, R.A. , Bhat-Nakshatri, P. , Patel, N.M. , Constantinidou, D. , Ali, S. , Nakshatri, H. , 2001. Phosphatidylinositol 3-kinase/AKT-mediated activation of estrogen receptor alpha: a new model for anti-estrogen resistance. J. Biol. Chem.. 276, 9817–9824. [DOI] [PubMed] [Google Scholar]
- Cauley, J.A. , Lucas, F.L. , Kuller, L.H. , Stone, K. , Browner, W. , Cummings, S.R. , 1999. Elevated serum estradiol and testosterone concentrations are associated with a high risk for breast cancer. Study of osteoporotic fractures research group. Ann. Intern. Med.. 130, 270–277. [DOI] [PubMed] [Google Scholar]
- Cinar, B. , Yeung, F. , Konaka, H. , Mayo, M.W. , Freeman, M.R. , Zhau, H.E. , Chung, L.W. , 2004. Identification of a negative regulatory cis-element in the enhancer core region of the prostate-specific antigen promoter: implications for intersection of androgen receptor and nuclear factor-kappaB signalling in prostate cancer cells. Biochem. J.. 379, 421–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claessens, F. , Verrijdt, G. , Schoenmakers, E. , Haelens, A. , Peeters, B. , Verhoeven, G. , Rombauts, W. , 2001. Selective DNA binding by the androgen receptor as a mechanism for hormone-specific gene regulation. J. Steroid Biochem. Mol. Biol.. 76, 23–30. [DOI] [PubMed] [Google Scholar]
- Cleutjens, K.B. , van Eekelen, C.C. , van der Korput, H.A. , Brinkmann, A.O. , Trapman, J. , 1996. Two androgen response regions cooperate in steroid hormone regulated activity of the prostate-specific antigen promoter. J. Biol. Chem.. 271, 6379–6388. [DOI] [PubMed] [Google Scholar]
- Cleutjens, K.B. , van der Korput, H.A. , van Eekelen, C.C. , van Rooij, H.C. , Faber, P.W. , Trapman, J. , 1997. An androgen response element in a far upstream enhancer region is essential for high, androgen-regulated activity of the prostate-specific antigen promoter. Mol. Endocrinol.. 11, 148–161. [DOI] [PubMed] [Google Scholar]
- Cummings, S.R. , Lee, J.S. , Lui, L.Y. , Stone, K. , Ljung, B.M. , Cauleys, J.A. , 2005. Sex hormones, risk factors, and risk of estrogen receptor-positive breast cancer in older women: a long-term prospective study. Cancer Epidemiol. Biomarker Prev.. 14, 1047–1051. [DOI] [PubMed] [Google Scholar]
- Erlanger, B.F. , Borek, F. , Beiser, S.M. , Lieberman, S. , 1957. Steroid-protein conjugates. I. Preparation and characterization of conjugates of bovine serum albumin with testosterone and with cortisone. J. Biol. Chem.. 228, 713–727. [PubMed] [Google Scholar]
- Erlanger, B.F. , Borek, F. , Beiser, S.M. , Lieberman, S. , 1959. Steroid-protein conjugates. II. Preparation and characterization of conjugates of bovine serum albumin with progesterone, deoxycorticosterone, and estrone. J. Biol. Chem.. 234, 1090–1094. [PubMed] [Google Scholar]
- Hackenberg, R. , Hofmann, J. , Holzel, F. , Schulz, K.D. , 1988. Stimulatory effects of androgen and antiandrogen on the in vitro proliferation of human mammary carcinoma cells. J. Cancer Res. Clin. Oncol.. 114, 593–601. [DOI] [PubMed] [Google Scholar]
- James, V.H. , McNeill, J.M. , Lai, L.C. , Newton, C.J. , Ghilchik, M.W. , Reed, M.J. , 1987. Aromatase activity in normal breast and breast tumor tissues: in vivo and in vitro studies. Steroids. 50, 269–279. [DOI] [PubMed] [Google Scholar]
- Kaaks, R. , Rinaldi, S. , Key, T.J. , Berrino, F. , Peeters, P.H. , Biessy, C. , Dossus, L. , Lukanova, A. , Bingham, S. , Khaw, K.T. , Allen, N.E. , Bueno-de-Mesquita, H.B. , van Gils, C.H. , Grobbee, D. , Boeing, H. , Lahmann, P.H. , Nagel, G. , Chang-Claude, J. , Clavel-Chapelon, F. , Fournier, A. , Thiebaut, A. , Gonzalez, C.A. , Quiros, J.R. , Tormo, M.J. , Ardanaz, E. , Amiano, P. , Krogh, V. , Palli, D. , Panico, S. , Tumino, R. , Vineis, P. , Trichopoulou, A. , Kalapothaki, V. , Trichopoulos, D. , Ferrari, P. , Norat, T. , Saracci, R. , Riboli, E. , 2005. Postmenopausal serum androgens, oestrogens and breast cancer risk: the European prospective investigation into cancer and nutrition. Endocr. Relat. Cancer. 12, 1071–1082. [DOI] [PubMed] [Google Scholar]
- Kampa, M. , Nifli, A.P. , Charalampopoulos, I. , Alexaki, V.I. , Theodoropoulos, P.A. , Stathopoulos, E.N. , Gravanis, A. , Castanas, E. , 2005. Opposing effects of estradiol- and testosterone-membrane binding sites on T47D breast cancer cell apoptosis. Exp. Cell Res.. 307, 41–51. [DOI] [PubMed] [Google Scholar]
- Kang, H.Y. , Cho, C.L. , Huang, K.L. , Wang, J.C. , Hu, Y.C. , Lin, H.K. , Chang, C. , Huang, K.E. , 2004. Nongenomic androgen activation of phosphatidylinositol 3-kinase/Akt signaling pathway in MC3T3-E1 osteoblasts. J. Bone. Miner. Res.. 19, 1181–1190. [DOI] [PubMed] [Google Scholar]
- Karaer, O. , Oruc, S. , Koyuncu, F.M. , 2004. Aromatase inhibitors: possible future applications. Acta Obstet. Gynecol. Scand.. 83, 699–706. [DOI] [PubMed] [Google Scholar]
- Klinge, C.M. , 2001. Estrogen receptor interaction with estrogen response elements. Nucleic Acids Res.. 29, 2905–2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacroix, M. , Leclercq, G. , 2004. Relevance of breast cancer cell lines as models for breast tumours: an update. Breast Cancer Res. Treat.. 83, 249–289. [DOI] [PubMed] [Google Scholar]
- Luo, L.Y. , Grass, L. , Diamandis, E.P. , 2000. The normal epithelial cell-specific 1 (NES1) gene is up-regulated by steroid hormones in the breast carcinoma cell line BT-474. Anticancer Res.. 20, 981–986. [PubMed] [Google Scholar]
- Luo, L.Y. , Grass, L. , Diamandis, E.P. , 2003. Steroid hormone regulation of the human kallikrein 10 (KLK10) gene in cancer cell lines and functional characterization of the KLK10 gene promoter. Clin. Chim. Acta. 337, 115–126. [DOI] [PubMed] [Google Scholar]
- Maggiolini, M. , Donze, O. , Jeannin, E. , Ando, S. , Picard, D. , 1999. Adrenal androgens stimulate the proliferation of breast cancer cells as direct activators of estrogen receptor alpha. Cancer Res.. 59, 4864–4869. [PubMed] [Google Scholar]
- Magklara, A. , Grass, L. , Diamandis, E.P. , 2000. Differential steroid hormone regulation of human glandular kallikrein (hK2) and prostate-specific antigen (PSA) in breast cancer cell lines. Breast Cancer Res. Treat.. 59, 263–270. [DOI] [PubMed] [Google Scholar]
- Marshman, E. , Streuli, C.H. , 2002. Insulin-like growth factors and insulin-like growth factor binding proteins in mammary gland function. Breast Cancer Res.. 4, 231–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCubrey, J.A. , Steelman, L.S. , Abrams, S.L. , Lee, J.T. , Chang, F. , Bertrand, F.E. , Navolanic, P.M. , Terrian, D.M. , Franklin, R.A. , D'Assoro, A.B. , Salisbury, J.L. , Mazzarino, M.C. , Stivala, F. , Libra, M. , 2006. Roles of the RAF/MEK/ERK and PI3K/PTEN/AKT pathways in malignant transformation and drug resistance. Adv. Enzyme Regul.. 46, 249–279. [DOI] [PubMed] [Google Scholar]
- Obiezu, C.V. , Diamandis, E.P. , 2005. Human tissue kallikrein gene family: applications in cancer. Cancer Lett.. 224, 1–22. [DOI] [PubMed] [Google Scholar]
- Paliouras, M. , Diamandis, E.P. , 2006. Coordinated steroid hormone-dependent and independent expression of multiple kallikreins in breast cancer cell lines. Breast Cancer Res. Treat.. [DOI] [PubMed] [Google Scholar]
- Paliouras, M. , Diamandis, E.P. , 2006. The kallikrein world: an update on the human tissue kallikreins. Biol. Chem.. 387, 643–652. [DOI] [PubMed] [Google Scholar]
- Paliouras, M. , Borgono, C. , Diamandis, E.P. , 2007. Human tissue kallikreins: the cancer biomarker family. Cancer Lett.. 249, 61–79. [DOI] [PubMed] [Google Scholar]
- Pampalakis, G. , Sotiropoulou, G. , 2006. Multiple mechanisms underlie the aberrant expression of the human kallikrein 6 gene in breast cancer. Biol. Chem.. 387, 773–782. [DOI] [PubMed] [Google Scholar]
- Papakonstanti, E.A. , Kampa, M. , Castanas, E. , Stournaras, C. , 2003. A rapid, nongenomic, signaling pathway regulates the actin reorganization induced by activation of membrane testosterone receptors. Mol. Endocrinol.. 17, 870–881. [DOI] [PubMed] [Google Scholar]
- Perel, E. , Wilkins, D. , Killinger, D.W. , 1980. The conversion of androstenedione to estrone, estradiol, and testosterone in breast tissue. J. Steroid Biochem.. 13, 89–94. [DOI] [PubMed] [Google Scholar]
- Peterziel, H. , Mink, S. , Schonert, A. , Becker, M. , Klocker, H. , Cato, A.C. , 1999. Rapid signalling by androgen receptor in prostate cancer cells. Oncogene. 18, 6322–6329. [DOI] [PubMed] [Google Scholar]
- Poulin, R. , Baker, D. , Labrie, F. , 1988. Androgens inhibit basal and estrogen-induced cell proliferation in the ZR-75-1 human breast cancer cell line. Breast Cancer Res. Treat.. 12, 213–225. [DOI] [PubMed] [Google Scholar]
- Recanatini, M. , Bisi, A. , Cavalli, A. , Belluti, F. , Gobbi, S. , Rampa, A. , Valenti, P. , Palzer, M. , Palusczak, A. , Hartmann, R.W. , 2001. A new class of nonsteroidal aromatase inhibitors: design and synthesis of chromone and xanthone derivatives and inhibition of the P450 enzymes aromatase and 17 alpha-hydroxylase/C17, 20-lyase. J. Med. Chem.. 44, 672–680. [DOI] [PubMed] [Google Scholar]
- Sidiropoulos, M. , Pampalakis, G. , Sotiropoulou, G. , Katsaros, D. , Diamandis, E.P. , 2005. Downregulation of human kallikrein 10 (KLK10/NES1) by CpG island hypermethylation in breast, ovarian and prostate cancers. Tumor Biol.. 26, 324–336. [DOI] [PubMed] [Google Scholar]
- Somboonporn, W. , Davis, S.R. , 2004. Postmenopausal testosterone therapy and breast cancer risk. Maturitas. 49, 267–275. [DOI] [PubMed] [Google Scholar]
- Somboonporn, W. , Davis, S.R. , 2004. Testosterone effects on the breast: implications for testosterone therapy for women. Endocr. Rev.. 25, 374–388. [DOI] [PubMed] [Google Scholar]
- Stathopoulos, E.N. , Dambaki, C. , Kampa, M. , Theodoropoulos, P.A. , Anezinis, P. , Delakas, D. , Delides, G.S. , Castanas, E. , 2003. Membrane androgen binding sites are preferentially expressed in human prostate carcinoma cells. BMC. Clin. Pathol.. 3, (1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein, R.A. , McDonnell, D.P. , 2006. Estrogen-related receptor alpha as a therapeutic target in cancer. Endocr. Relat. Cancer. 13, (Suppl. 1) S25–S32. [DOI] [PubMed] [Google Scholar]
- Stoica, G.E. , Franke, T.F. , Wellstein, A. , Czubayko, F. , List, H.J. , Reiter, R. , Morgan, E. , Martin, M.B. , Stoica, A. , 2003. Estradiol rapidly activates Akt via the ErbB2 signaling pathway. Mol. Endocrinol.. 17, 818–830. [DOI] [PubMed] [Google Scholar]
- Stoica, G.E. , Franke, T.F. , Moroni, M. , Mueller, S. , Morgan, E. , Iann, M.C. , Winder, A.D. , Reiter, R. , Wellstein, A. , Martin, M.B. , Stoica, A. , 2003. Effect of estradiol on estrogen receptor-alpha gene expression and activity can be modulated by the ErbB2/PI 3-K/Akt pathway. Oncogene. 22, 7998–8011. [DOI] [PubMed] [Google Scholar]
- Stover, E.P. , Krishnan, A.V. , Feldman, D. , 1987. Estrogen down-regulation of androgen receptors in cultured human mammary cancer cells (MCF-7). Endocrinology. 120, 2597–2603. [DOI] [PubMed] [Google Scholar]
- Sun, M. , Paciga, J.E. , Feldman, R.I. , Yuan, Z. , Coppola, D. , Lu, Y.Y. , Shelley, S.A. , Nicosia, S.V. , Cheng, J.Q. , 2001. Phosphatidylinositol-3-OH Kinase (PI3K)/AKT2, activated in breast cancer, regulates and is induced by estrogen receptor alpha (ERalpha) via interaction between ERalpha and PI3K. Cancer Res.. 61, 5985–5991. [PubMed] [Google Scholar]
- Thijssen, J.H. , Blankenstein, M.A. , Donker, G.H. , Daroszewski, J. , 1991. Endogenous steroid hormones and local aromatase activity in the breast. J. Steroid Biochem. Mol. Biol.. 39, 799–804. [DOI] [PubMed] [Google Scholar]
- Tworoger, S.S. , Missmer, S.A. , Barbieri, R.L. , Willett, W.C. , Colditz, G.A. , Hankinson, S.E. , 2005. Plasma sex hormone concentrations and subsequent risk of breast cancer among women using postmenopausal hormones. J. Natl. Cancer Inst.. 97, 595–602. [DOI] [PubMed] [Google Scholar]
- Vilgelm, A. , Lian, Z. , Wang, H. , Beauparlant, S.L. , Klein-Szanto, A. , Ellenson, L.H. , Di Cristofano, A. , 2006. Akt-mediated phosphorylation and activation of estrogen receptor alpha is required for endometrial neoplastic transformation in Pten± mice. Cancer Res.. 66, 3375–3380. [DOI] [PubMed] [Google Scholar]
- Walker, G. , MacLeod, K. , Williams, A.R. , Cameron, D.A. , Smyth, J.F. , Langdon, S.P. , 2007. Insulin-like growth factor binding proteins IGFBP3, IGFBP4, and IGFBP5 predict endocrine responsiveness in patients with ovarian cancer. Clin. Cancer Res.. 13, 1438–1444. [DOI] [PubMed] [Google Scholar]
- Yousef, G.M. , Diamandis, E.P. , 2002. Human tissue kallikreins: a new enzymatic cascade pathway?. Biol. Chem.. 383, 1045–1057. [DOI] [PubMed] [Google Scholar]
- Yousef, G.M. , Borgono, C.A. , Scorilas, A. , Ponzone, R. , Biglia, N. , Iskander, L. , Polymeris, M.E. , Roagna, R. , Sismondi, P. , Diamandis, E.P. , 2002. Quantitative analysis of human kallikrein gene 14 expression in breast tumours indicates association with poor prognosis. Br. J. Cancer.. 87, 1287–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yousef, G.M. , Scorilas, A. , Kyriakopoulou, L.G. , Rendl, L. , Diamandis, M. , Ponzone, R. , Biglia, N. , Giai, M. , Roagna, R. , Sismondi, P. , Diamandis, E.P. , 2002. Human kallikrein gene 5 (KLK5) expression by quantitative PCR: an independent indicator of poor prognosis in breast cancer. Clin. Chem.. 48, 1241–1250. [PubMed] [Google Scholar]
- Yu, H. , Diamandis, E.P. , Levesque, M. , Giai, M. , Roagna, R. , Ponzone, R. , Sismondi, P. , Monne, M. , Croce, C.M. , 1996. Prostate specific antigen in breast cancer, benign breast disease and normal breast tissue. Breast Cancer Res. Treat.. 40, 171–178. [DOI] [PubMed] [Google Scholar]
- Zhou, J. , Ng, S. , Adesanya-Famuiya, O. , Anderson, K. , Bondy, C.A. , 2000. Testosterone inhibits estrogen-induced mammary epithelial proliferation and suppresses estrogen receptor expression. FASEB. J.. 14, 1725–1730. [DOI] [PubMed] [Google Scholar]
- Zhu, Y.S. , Cai, L.Q. , Huang, Y. , Fish, J. , Wang, L. , Zhang, Z.K. , Imperato-McGinley, J.L. , 2005. Receptor isoform and ligand-specific modulation of dihydrotestosterone-induced prostate specific antigen gene expression and prostate tumor cell growth by estrogens. J. Androl.. 26, 500–508. discussion 509–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivadinovic, D. , Watson, C.S. , 2005. Membrane estrogen receptor-alpha levels predict estrogen-induced ERK1/2 activation in MCF-7 cells. Breast Cancer Res.. 7, R130–R144. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data