

ABSTRACT
Our previous phase I/IIA study showed that autologous dendritic cells (DCs) pulsed with tumor-associated antigens are well tolerated in patients with hepatocellular carcinoma (HCC). In this randomized, multicenter, open-label, phase II trial, we investigated the efficacy and safety of this DC-based adjuvant immunotherapy with 156 patients, who treated for HCC with no evidence of residual tumor after standard treatment modalities. Patients were randomly assigned to immunotherapy (n = 77; injection of 3 × 107 DC cells, six times over 14 weeks) or control (n = 79; no treatment). The primary end point was recurrence-free survival (RFS), and the secondary endpoints were immune response and safety. The RFS between the immunotherapy and control groups was not significantly different (hazard ratio [HR], 0.97; 95% confidence interval [CI], 0.60–1.56; p = 0.90). However, post-hoc subgroup analyses revealed that DC immunotherapy significantly reduced the risk of tumor recurrence of non-radiofrequency ablation (non-RFA) group patients (n = 83, HR, 0.49; 95% CI, 0.26–0.94; p = 0.03), whereas unexpectedly increased the risk of recurrence in RFA group (n = 61, p = 0.01). Tumor-specific immune responses were significantly enhanced (both p < 0.01) in the immunotherapy group. Baseline serum interleukin (IL)-15 was statistically correlated with RFS prolongation (HR, 0.16; 95% CI, 0.03–1.58; p = 0.001) within the immunotherapy groups. Overall adverse events were more frequent in the immunotherapy group (p < 0.001) but were mainly mild to moderate in severity. In conclusion, adjuvant immunotherapy with DC vaccine reduces the risk of tumor recurrence in HCC patients who underwent standard treatment modalities other than RFA. Baseline IL-15 might be a candidate biomarker for DC-based HCC immunotherapy.
KEYWORDS: Adjuvant immunotherapy, biomarker, dendritic cell vaccine, hepatocellular carcinoma, recurrence-free survival
Abbreviations
- AE
adverse event
- AFP
α-fetoprotein
- CI
confidence interval
- DC
dendritic cell
- HCC
hepatocellular carcinoma
- ELISPOT
enzyme-linked immunospot
- GPC-3
glypican-3
- HR
hazard ratio
- ICF
Informed consent form
- IL-15
interleukin 15
- ITT
intention-to-treat
- MAGE-1
melanoma-associated antigen-1
- OS
overall survival
- PEI
percutaneous ethanol injection
- PP
per-protocol
- RFA
radiofrequency ablation
- RFS
recurrence-free survival
- TAA
tumor-associated antigen
- TACE
transarterial chemoembolization
- TEAE
treatment-emergent adverse event
Introduction
Although surveillance programs for early detection of hepatocellular carcinoma (HCC) in high-risk populations increase the likelihood of curative treatment,1,2 long-term prognosis is poor; more than 750,000 patients per year die of HCC globally.3 A major cause of poor outcome is frequent tumor recurrence even after curative treatment.4 There are many efforts to develop adjuvant therapies to reduce recurrence, such as a recent study from our group that demonstrated the efficacy and safety of adjuvant immunotherapy with autologous cytokine-induced killer (CIK) cells in HCC patients after potentially curative treatment.5 However, the benefits of most adjuvant agents remain in question, and there are no adjuvant therapies recommended by current international guidelines.6-8
Dendritic cells (DCs) are professional antigen-presenting cells that play a critical role in the cell-mediated immune response by stimulating antigen-specific cytotoxic T cells.9,10 Autologous DCs offer a practical basis for a tumor vaccine,11,12 and DC-based adoptive immunotherapy has been tested in clinical trials in various malignancies, including prostate cancer, melanoma, renal cell carcinoma, and HCC.13-19 A previous phase II study of 35 patients used intravenous vaccination with ex vivo DCs pulsed with HepG2 lysate and showed evidence of antitumor efficacy in a few patients with advanced HCC.18 A phase I/II study reported that strong T cell responses against α-fetoprotein (AFP) were generated by immunization with DCs pulsed with four AFP peptides, as immunogenic tumor-associated antigens (TAAs) in HCC patients. However, no clinical responses were observed in the treated patients.17 To enhance the efficacy of DC vaccination, multiple TAAs (i.e., α-fetoprotein (AFP), glypican-3 (GPC-3), and melanoma-associated antigen-1 (MAGE-1)) were used to pulse DCs in a phase I/II study from Japan, and DC vaccination effectively and safely induced T cell responses in patients with advanced HCC.19 Encouraged by these promising results, manufacturing techniques were refined and standardized, and an individualized autologous multiple TAA-pulsed DC vaccine (CreaVax-HCC; JW CreaGene Inc., Seongnam-si, Gyeonggi-do, Korea) was developed. Our preceding phase I/IIA study demonstrated that this vaccine was well tolerated in patients with HCC and prolonged time-to-recurrence (TTR) and recurrence-free survival (RFS) compared with the historical control group.20
In the present study, we assessed the efficacy and safety of adjuvant immunotherapy with TAA-pulsed DC vaccine in patients treated for HCC with complete remission and aimed to find pre-treatment biomarker that could predict treatment response.
Results
Patients
Between 15 June, 2010 and 23 September, 2011, 179 participants were screened. Vaccination and evaluation were performed following the protocol (Fig. 1A). Patient flow from registration to analysis is summarized in the CONSORT diagram of patient flowchart (Fig. 1B). A total of 156 eligible patients were randomly assigned to either the immunotherapy (n = 77) or the control group (n = 79). Among these randomized patients, 144 (69 immunotherapy and 75 control) were included in the efficacy analysis; seven patients who withdrew informed consent and did not receive vaccination in the immunotherapy group were excluded from the efficacy analysis according to pre-specified intention-to-treat (ITT) analysis criteria. Four control patients received no follow-up and were excluded since then. Five patients in the immunotherapy group discontinued intervention and four patients in the control group discontinued observation. The safety analysis included 146 patients (70 immunotherapy and 76 control). At the time of data cut-off, the median follow-up duration was 30.5 mo in the immunotherapy group and 30.0 mo in the control group.
Figure 1.

Study design and CONSORT diagram of patient flow. (A) Study design for vaccination and evaluation. CR, complete remission; Pts, patients; SR, surgical recession; RFA, radiofrequency ablation; PEI, percutaneous ethanol injection; TACE, transarterial chemoembolization; ICF, Informed consent form; LKP, leukaperisis; DC, dendritic cell; RFS, recurrence-free survival; CT, computed tomography; MRI, magnetic resonance imaging; TTR, time-to-recurrence; mOS, median overall survival. (B) Patient flow from random assignment *excluded according to ITT analysis-based criteria. Of the enrolled patients, 23 did not meet inclusion criteria and thus were excluded from random assignment. In addition, 12 more patients were excluded from the efficacy analysis because they were found to violate inclusion criteria after randomization.
There were no significant differences in baseline characteristics between the two study groups (Table 1). Sixty-two patients (43%) were treated with surgical resection, 61 (42.4%) with RFA, 10 (6.9%) with TACE, and 3 (1.4%) with PEI.
Table 1.
Baseline demographics and disease characteristics.
| Variable | Immunotherapy(n = 69) | Control group(n = 75) | p value |
|---|---|---|---|
| Male sex, n (%) | 48 (69.6) | 61 (81.3) | 0.10d |
| Age years, mean (SD) | 57.1 ± 9.5 | 58.1 ± 10.8 | 0.58e |
| Cause of liver disease, n (%) | 0.33 | ||
| HBV infection only | 51 (73.9) | 53 (70.7) | |
| HCV infection only | 8 (11.6) | 8 (10.7) | |
| HBV and HCV co-infection | 2 (2.9) | 0 (0) | |
| Others | 8 (11.6) | 14 (18.6) | |
| Treatment modality, n (%) | 1.00f | ||
| Surgical resection | 30 (43.5) | 32 (42.7) | |
| RFA | 29 (42.0) | 32 (42.7) | |
| TACE | 5 (7.3) | 5 (6.7) | |
| PEI | 1 (1.5) | 2 (2.7) | |
| Combination a | 4 (5.8) | 4 (5.3) | |
| HCC stage, n (%)b | 0.83d | ||
| Stage I | 20 (29.0) | 24 (32.0) | |
| Stage II or III | 49 (71.0) | 51 (68.0) | |
| Histological confirmation, n (%) | 0.62d | ||
| No | 15 (21.7) | 19 (25.3) | |
| Yes | 54 (78.3) | 56 (74.7) | |
| ECOG performance status, n (%)c | 0.85d | ||
| 0 | 45 (65.2) | 50 (66.7) | |
| 1 | 24 (34.8) | 25 (33.3) | |
| α-fetoprotein, ng/mL | 0.45e | ||
| Median | 4.5 | 4.5 | |
| Range | 0.9–242.4 | 0.4–731.5 | |
| PIVKA-II, mAU/mL | 0.85e | ||
| Median | 19.0 | 20.0 | |
| Range | 6.0–275.0 | 6.0–177.0 | |
| Aspartate aminotransferase, IU/L | 0.15e | ||
| Median | 32.0 | 31.0 | |
| Range | 15.0–362.0 | 13.0–127.0 | |
| Alanine aminotransferase, IU/L | 0.11e | ||
| Median | 23.0 | 24.0 | |
| Range | 5.0–512.0 | 6.0–104.0 | |
| Alkaline phosphatase, IU/L | 0.92e | ||
| Median | 93.0 | 91.0 | |
| Range | 47.0–629.0 | 48.0–362.0 | |
| Albumin, g/dL | 0.85e | ||
| Median | 4.0 | 4.0 | |
| Range | 2.8–5.1 | 2.9–4.8 | |
| Total bilirubin, mg/dL | 0.77e | ||
| Median | 0.7 | 0.8 | |
| Range | 0.2–2.4 | 0.3–4.5 | |
| Creatinine, mg/dL | 0.19e | ||
| Median | 0.8 | 0.8 | |
| Range | 0.5–1.2 | 0.4–1.4 | |
| Platelet, ×103/mm3 | 0.09e | ||
| Median | 133.0 | 148.0 | |
| Range | 54.0–314.0 | 45.0–500.0 | |
| Prothrombin time (INR) | 0.14e | ||
| Median | 1.1 | 1.1 | |
| Range | 1.0–1.4 | 0.9–1.7 | |
| Child-Pugh score | 0.83e | ||
| Mean(SD) | 5.3(0.5) | 5.3(0.6) | |
| Median | 5.0 | 5.0 | |
| Range | 5.0–7.0 | 5.0–7.0 |
Abbreviations: ECOG, Eastern Cooperative Oncology Group; HCC, hepatocellular carcinoma; PEI, percutaneous ethanol injection; RFA, radiofrequency ablation; TACE, transarterial chemoembolization..
Note. Data are expressed as n (%), mean ± SD, or median with range.
The combination therapy indicates 3 cases with TACE/RFA and 1 with TACE/surgical resection in the immunotherapy group and 3 with TACE/RFA and 1 with RFA/surgical resection in the control group.
HCC staging was done according to the modified UICC (mUICC) staging system.
ECOG performance status assesses the daily living abilities of the patient, on a scale ranging from 0 (fully active) to 5 (dead).
By Chi-square test.
By Student's t-test.
By Fisher's exact test.
Efficacy
Recurrence-free survival and subgroup analyses
Among the 144 patients in the efficacy population, 69 experienced tumor recurrence or death by the time of data cut-off: 33 (47.8%) in the immunotherapy group and 35 (46.7%) in the control group. Median RFS was not reached in either group. The difference in RFS between the two groups was not statistically significant (HR, 0.97; 95% confidence interval [CI], 0.60–1.56; p = 0.90 by log-rank; Fig. 2A).
Figure 2.
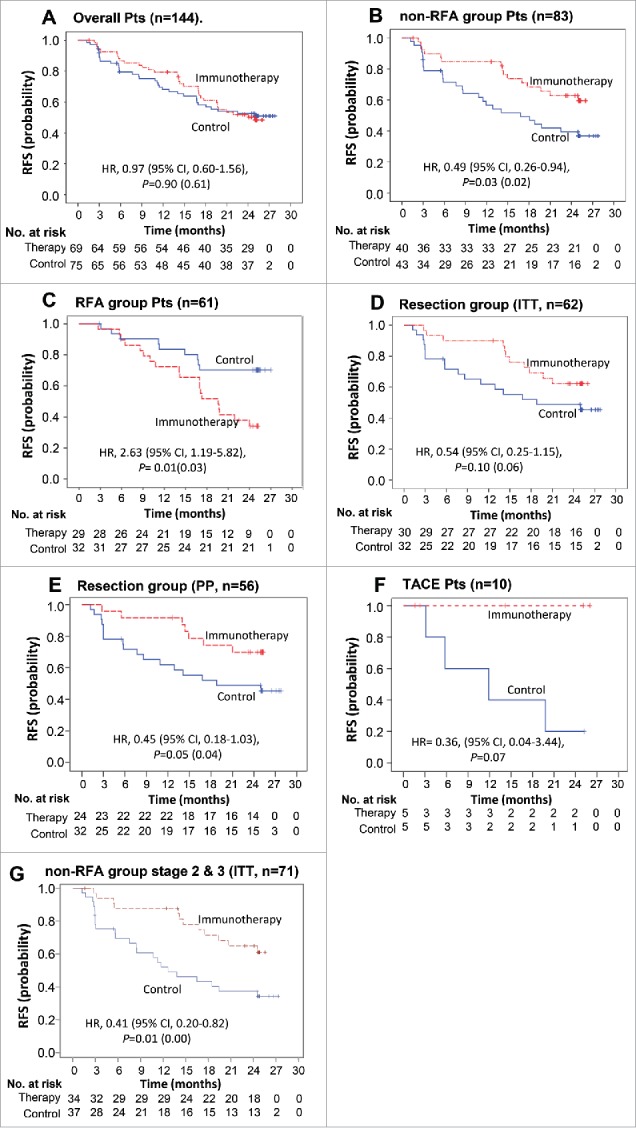
Kaplan–Meier estimates of recurrence-free survival (RFS) in (A) overall patients, (B) patients of non-RFA group, (C) patients of RFA group, (D) patients of surgical resection group and (E) patients of surgical resection group (PP analysis), (F) Kaplan–Meier estimates of RFS in patients of TACE group, (G) Kaplan-Meier estimates of RFS in non-RFA group patients with stage 2 and 3 diseases. RFS was computed on all patients included in the ITT population (except E). Patients who had not progressed or died were censored on data cut-off. p values calculated by log-rank test (and by Wilcoxon test). Note: HR, hazard ratio; CI, confidence interval.
In post-hoc subgroup analyses (Table 2), however, when RFA patients were excluded, DC immunotherapy significantly prolonged the RFS of non-RFA group (HR, 0.49; 95% CI, 0.26–0.94; p = 0.03); 26 of 43 in the control group and 14 of 40 in the immunotherapy group experienced tumor recurrence or death (Fig. 2B, Fig. S1A). On the contrary, in a subgroup of 61 RFA patients (29 immunotherapy and 32 control), DC immunotherapy significantly increased the risk of recurrence as compared with untreated controls (HR, 2.63; 95% CI, 1.19–5.82; p = 0.01) (Fig. 2C, Fig. S1B). There were no significant differences in baseline disease characteristics between the RFA group and non-RFA group except there were more patients with stage I in the RFA group, compared with the non-RFA group in which a higher proportion had stage II/IIII disease (p < 0.01). It means that the immunosuppressive tumor microenvironment of RFA group was likely to be less severe than that of non-RFA group, but the clinical outcome after DC immunotherapy was unexpectedly reversed. Also, patients in the non-RFA group had higher proportion of the histological diagnosis (non-RFA group>>RFA group, p ≤ 0.01) (Table S1). On the other hand, among the subgroup of 62 patients who received surgical resection (30 immunotherapy and 32 control), the immunotherapy patients showed a trend toward prolonged RFS (HR, 0.54; 95% CI, 0.25–1.15; p = 0.10) (Fig. 2D, Fig. S1C). When assessed among the 56 surgically resected patients who completed the full treatment as per-protocol (PP) analysis, the RFS was significantly improved by immunotherapy (HR, 0.45; 95% CI, 0.18–1.03; p = 0.04 by Wilcoxon test) (Fig. 2E, Fig. S2D). In a subgroup of 10 patients, who received TACE for HCC (5 immunotherapy and 5 control), none in immunotherapy group experienced tumor recurrence during the study period, compared with four of five controls, but the difference failed to reach statistical significance (HR, 0.36; 95% CI, 0.04–3.44; p = 0.07) due to the limited number of patients (Fig. 2F). Interestingly, DC immunotherapy significantly prolonged the RFS of the stage II/III patients (HR, 0.41; 95% CI, 0.20–0.82; p = 0.01) among the non-RFA group (34 immunotherapy and 37 control) stratified by stage I vs II/III (mUICC stage) according to the study protocol (Table 2 and Fig. 2G)
Table 2.
Subgroup analysis of recurrence rates between control and immunotherapy groups over 2 y.
| Treat. Mod | Sur. Resec. |
RFA |
TACE |
Combination |
PEIT |
Overalla |
Non-RFAb |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pt. Stage | DC therapy | Pt # | Rec. #(%) | Pt # | Rec. #(%) | Pt # | Rec. #(%) | Pt # | Rec. #(%) | Pt # | Rec. #(%) | Pt # | Rec. #(%) | Pt # | Rec. #(%) | |
| I | Control | 2 | 0 | 18 | 4 | 2 | 1 | 1 | 1 | 1 | 1 | 24 | 7 | 6 | 3 | |
| (0.0) | (22.2) | (50.0) | (100) | (100) | (29.2) | (50.0) | ||||||||||
| DC therapy | 3 | 1 | 14 | 7 | 2 | 0 | 0 | 0 | 1 | 1 | 20 | 9 | 6 | 2 | ||
| (33.3) | (46.7) | (0.0) | (NA) | (100) | (45.0) | (33.3) | ||||||||||
| Benefit (%)c | (−33.3) | (−24.5) | (+50.0) | (N/A) | (0.0) | (-15.8) | (+16.7) | |||||||||
| II + III | Control | 30 | 17 | 14 | 5 | 3 | 3 | 3 | 2 | 1 | 1 | 51 | 28 | 37 | 23 | |
| (55.7) | (35.7) | (100) | (66.7) | (100) | (54.9) | (62.2) | ||||||||||
| DC therapy | 27 | 10 | 15 | 12 | 3 | 0 | 4 | 2 | 0 | 0 | 49 | 24 | 34 | 12 | ||
| (37.0) | (80.0) | (0.0) | (50.0) | (0.0) | (49.0) | (29.4) | ||||||||||
| Benefit (%)c | (+18.7) | (-44.3) | (+100) | (+16.7) | (N/A) | (+5.9) | (+32.8) | |||||||||
| Total | Control | 32 | 17 | 32 | 9 | 5 | 4 | 4 | 3 | 2 | 2 | 75 | 35 | 43 | 26 | |
| (53.1) | (28.1) | (80.0) | (75.0) | (100) | (46.7) | (60.5) | ||||||||||
| DC therapy | 30 | 11 | 29 | 19 | 5 | 0 | 4 | 2 | 1 | 1 | 69 | 33 | 40 | 14 | ||
| (36.7) | (65.5) | (0.0) | (50.0) | (100) | (47.8) | (35.0) | ||||||||||
| Benefit (%)c | (+16.4) | (−37.4) | (+80.0) | (+25.0) | (0.0) | (-1.1) | (+25.5) | |||||||||
Abbreviation: N/A, not applicable; Pt #, patient number; Rec.# recurrent patient number; Treat Mod, treatment modality.
All subgroup analyses were assembled at the time of the data cut-off date.
Subgroups excluding patients who underwent RFA treatment.
% improvement of recurrence inhibition (control group recurrence rate – DC therapy group recurrence rate); (+) or (-) indicates improved or unimproved, respectively, by DC immunotherapy.
Overall survival (OS)
At the time of data cut-off, seven deaths had occurred in the efficacy population: two (2.9%) in the immunotherapy group and five (6.7%) in the control group. There was no significant difference in OS between the two groups (HR, 0.39; 95% CI, 0.08–2.01; log-rank p = 0.24; Fig. 3).
Figure 3.
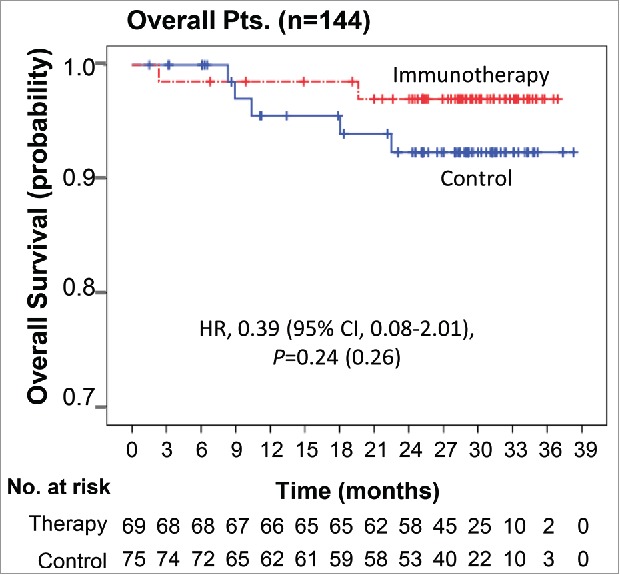
Overall survival Kaplan–Meier estimates of OS in overall patients. OS was computed on all patients included in the ITT population.
Immunologic response
DC immunotherapy augmented TAA-reactive T cell responses. In the lymphocyte proliferation assay, 60–85% of vaccinated patients showed the proliferation rate at least doubled to each TAA or TAA mixture as compared with baseline (Table S2). In the IFNγ ELISPOT assay, a positive response was detected in the immunotherapy group at a rate of 63.0% to AFP, 58.2% to GPC-3, 57.4% to MAGE-1, and 61.4% to the mixture of three TAAs (Table S3). IFNγ+ ELISPOT and TAA-specific T cell responses to the mixture of three TAAs were significantly enhanced after repeated vaccinations in the immunotherapy group when assessed by paired t-test (Fig. 4), and such patterns were replicated in the surgical resection subgroup within the immunotherapy group (Fig. S2). Among immunosuppressive factors, serum TGF-β level linearly decreased in the immunotherapy group over time (p < 0.001 by repeated measure ANOVA), while there were no significant changes in serum levels of either IL-10 or VEGF (Table S4). Nevertheless, neither IFNγ ELISPOT positivity (HR, 1.05; 95% CI, 0.49–2.22; p = 0.90), lymphocyte proliferation positivity (HR, 1.64; 95% CI, 0.22–12.11; p = 0.63), nor decrease of TGF-β (HR, 1.67; 95% CI, 0.79–3.55; p = 0.18) had a significant relationship with RFS in the immunotherapy group (data not shown). Lack of relationship between the immune responses and RFS might be due to the reversed clinical outcome of RFA group in which DC immunotherapy significantly increased the risk of recurrence.
Figure 4.

Immune responses after DC vaccination Changes in immune responses before and after DC vaccination against a tumor-associated antigen mixture (AFP, GPC-3, and MAGE-1). Data are represented after subtraction with the count of no Ag. (A) IFNγ ELISPOT assays were performed with PBMCs obtained from each patient at the indicated time points after the start of DC vaccination (0 w). Analysis was performed by paired t-test (GraphPad PRISM, ver 6.07) with the indicated number of patients. (B) Antigen-specific lymphocyte proliferation assays were performed after DC vaccinations using autologous PBMCs at each time point. Proliferation was determined by 3H-thymidine incorporation (DPM, disintegrations per minute) using a liquid scintillation counter and the results were analyzed using the same method as for the ELISPOT assay. **p < 0.01, ***p < 0.001, ****p < 0.0001.
Safety
Treatment-emergent adverse events (TEAEs) occurred in 108 patients (74.0%) in the safety population (n = 146) during the study period, and immunotherapy patients (69/70, 98.6%) experienced TEAEs more frequently than controls (39/76, 51.3%) (p < 0.001 by Chi-square test) (Table 3). However, the frequency of serious TEAEs between the two groups was comparable (14.2% immunotherapy vs. 17.1% control, p = 0.28 by Chi-square test) (Table 3). Sixty-five immunotherapy patients (92.9%) experienced a total of 503 TEAEs counted related to study drug; 502 (99.8%) were mild to moderate (grade 1 or 2) and 1 (0.2%) was grade 3 (injection site ulcer), and there was no drug-related deaths (Table S5).
Table 3.
Treatment-emergent adverse events (safety population).
| Immunotherapy (n = 70) |
Control (n = 76) | |||||||
|---|---|---|---|---|---|---|---|---|
| All TEAE |
Related TEAE |
All AE |
p value |
|||||
| Adverse event |
Any grade |
Grade 3 or 4 |
Any grade |
Grade 3 or 4 |
Any grade |
Grade 3 or 4 |
Any grade |
Grade 3 or 4 |
| Number of Pts | 69 (98.6%) | 10(14.2%) | 65(92.9%) | 1(1.4%) | 39 (51.3%) | 13 (17.1%) | <0.001a | 0.283a |
| General disorders and administration site conditions (# of Pts) | ||||||||
| Injection site pain | 28 (40.0%) | 0 | 28 (40.0%) | 0 | 0 | 0 | <.001a | NA |
| Injection site erythema | 24 (34.3%) | 0 | 24 (34.3%) | 0 | 0 | 0 | <.001a | NA |
| Injection site induration | 23 (32.9%) | 0 | 23 (32.9%) | 0 | 0 | 0 | <.001a | NA |
| Influenza like illness | 9 (12.9%) | 0 | 9 (12.9%) | 0 | 0 | 0 | 0.001b | NA |
| Injection site mass | 9 (12.9%) | 0 | 9 (12.9%) | 0 | 0 | 0 | 0.001b | NA |
| Injection site swelling | 9 (12.9%) | 0 | 9 (12.9%) | 0 | 0 | 0 | 0.001b | NA |
| Injection site ulcer | 1 (1,4%) | 1 (1.4%) | 1 (1.4%) | 1 (1.4%) | 0 | 0 | NA | |
| Pyrexia | 8 (11.4%) | 0 | 8 (11.4%) | 0 | 0 | 0 | 0.002b | NA |
| Fatigue | 5 (7.1%) | 0 | 1 (1.4%) | 0 | 2 (2.6%) | 0 | 0.260b | NA |
| Induration | 3 (4.3%) | 0 | 3 (4.3%) | 0 | 0 | 0 | 0.108b | NA |
| Chills | 3 (4.3%) | 0 | 3 (4.3%) | 0 | 0 | 0 | 0.108b | NA |
| Gastrointestinal disorders (#of Pts) | ||||||||
| Dyspepsia | 4 (5.7%) | 0 | 0 | 0 | 2 (2.6%) | 0 | 0.427b | NA |
| Epigastric discomfort | 3 (4.3%) | 0 | 0 | 0 | 3 (3.9%) | 0 | 1.000b | NA |
| Abdominal pain | 4 (5.7%) | 0 | 0 | 0 | 1 (1.3%) | 0 | 0.194b | NA |
| Abdominal pain upper | 3 (4.3%) | 0 | 0 | 0 | 1 (1.3%) | 0 | 0.350b | NA |
| Diarrhea | 4 (5.7%) | 0 | 0 | 0 | 0 | 0 | 0.050b | NA |
| Musculoskeletal and connective tissue disorders (# of Pts) | ||||||||
| Myalgia | 20 (28.6%) | 0 | 20 (28.6%) | 0 | 0 | 0 | <.001a | NA |
| Pain in extremity | 5 (7.1%) | 0 | 2 (2.9%) | 0 | 0 | 0 | 0.023b | NA |
| Muscular weakness | 3 (4.3%) | 0 | 0 | 0 | 1 (1.3%) | 0 | 0.350 b | NA |
| Investigations | ||||||||
| AST increased | 4 (5.7%) | 3 (4.3%) | 0 | 0 | 3 (3.9%) | 1 (1.3%) | 0.710b | 0.350b |
| ALT increased | 3 (4.3%) | 1 (1.4%) | 0 | 0 | 2 (2.6%) | 1 (1.3%) | 0.671b | 1.000b |
| Metabolism and nutrition disorders | ||||||||
| Hyperglycemia | 9 (12.9%) | 4 (5.7%) | 0 | 0 | 5 (6.6%) | 3 (3.9%) | 0.198a | 0.710b |
| Nervous system disorders | ||||||||
| Headache | 9 (12.9%) | 1 (1.4%) | 7 (10.0%) | 0 | 1 (1.3%) | 0 | 0.007b | 0.479b |
| Infections and infestations | ||||||||
| Upper resp. tract infect. | 6 (8.6%) | 0 | 1 (1.4%) | 0 | 2 (2.6%) | 0 | 0.153b | NA |
| Influenza | 3 (4.3%) | 0 | 0 | 0 | 2 (2.6%) | 1 (1.3%) | 0.671b | 1.000b |
| Respiratory, thoracic and mediastinal disorders | ||||||||
| Cough | 3 (4.3%) | 0 | 0 | 0 | 3 (3.9%) | 0 | 1.000b | NA |
| Oropharyngeal pain | 3 (4.3%) | 0 | 1 (1.4%) | 0 | 0 | 0 | 0.108b | NA |
| Skin and subcutaneous tissue disorders | ||||||||
| Pruritus | 3 (4.3%) | 0 | 0 | 0 | 2 (2.6%) | 0 | 0.671b | NA |
| Erythema | 5 (7.1%) | 0 | 4 (5.7%) | 0 | 0 | 0 | 0.023b | NA |
| Psychiatric disorders | ||||||||
| Insomnia | 3 (4.3%) | 0 | 0 | 0 | 0 | 0 | 0.108b | NA |
| Reproductive system and breast disorders | ||||||||
| BPH | 0 | 0 | 0 | 0 | 3 (3.9%) | 0 | 0.246b | NA |
Abbreviations: AE, adverse events in control group; ALT, alanine aminotransferase; AST, aspartate aminotransferase; BPH, benign prostate hyperplasia; TEAE, treatment-emergent adverse events..
Note. Listed are adverse events, as defined by the National Cancer Institute Common Terminology Criteria (version 3.0), that were considered drug-related or that occurred in at least three patients in either study group regardless of relationship to drug. Data are expressed as number of patients and (%).
TEAE incidences and detailed number of TEAEs are summarized in Table S5.
By chi-square test.
By Fisher's exact test.
Biomarker for DC-based HCC immunotherapy
Through a series of multiplex analysis, we could not find any cytokines/chemokines except IL-15, whose baseline concentrations showed a significant correlation with recurrence-free after DC immunotherapy (Table S6). The IL-15 level of healthy donors was 2.3 ± 0.4 pg/mL in the Luminex Multiplex Assay (sensitivity; minimum detectable concentration was 0.4 pg/mL at each protocol (n > 6 assays). Precision; intra-assay % coefficient of variance (CV) was 6.7 and extra-assay %CV was 9.5. Accuracy; 100.5 at 6 point spike recovery in serum matrix). On the other hand, the baseline IL-15 levels of recurrence-free patients after DC immunotherapy (2.2±1.1 pg/mL) were almost in the range of the healthy donors' while the patients who experienced recurrence even after immunotherapy showed baseline IL-15 mostly undetectable (Fig. 5A).
Figure 5.

Baseline IL-15 level in the sera of healthy donors (HD) and patients with (Recur) or without (R-free) tumor-recurrence at the indicated time points after DC vaccination in mixed group patients (A) and resected patients (B). IL-15 levels were assessed by Luminex Bead-based Milliplex#x08E8; Multiplex Assay system (Millipore, St Charles, MO, USA). Assay sensitivity of the system; minimum detectable concentration was 0.6 pg/mL at a short protocol (n = 4 assays). Precision; intra-assay % coefficient of variance (CV) was 6.7 and extra-assay %CV was 9.5. Accuracy; 100.5 at spike recovery in serum matrix (6 Point Spikes). Statistical analysis was performed by unpaired t-test with the number (n) of patients: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. (C) Baseline IL-15 level and RFS after DC immunotherapy. Twenty-eight patients from the DC vaccination group were assessed. IL-15(hi) and IL-15(lo) represent patients with baseline IL-15 >0.6 pg/mL (higher than minimal detectable concentration) and baseline IL-15<0.6 pg/mL (lower than minimal detectable concentration), respectively. HRs and p values were represented.
Patients who did not experience recurrence for a long time (at 15 and 24 mo) had significantly higher baseline IL-15 levels in overall immunotherapy group (1.59 ± 0.36 and 2.04 ± 0.41 pg/mL at 15 and 24 mo, respectively) and resected immunotherapy group (1.88 ± 0.62 and 2.15 ± 0.77 pg/mL at 15 and 24 mo, respectively) than those who experienced recurrence in the immunotherapy group at each time points (0.21 ± 0.21 and 0.43 ± 0.24 pg/mL) and in resected immunotherapy group (0.0 ± 0.0 and 0.39 ± 0.39 pg/mL), respectively, (all p < 0.05) (Fig. 5A and B). It is worth of note that the IL-15 level of each patient was not changed even after DC immunotherapy. Among patients in the immunotherapy group, high baseline serum IL-15 (> 0.6 pg/mL) was significantly correlated with longer RFS when including (HR, 0.16; 95% CI, 0.03–1.58; p = 0.001) or excluding RFA subgroup (HR, 0.11; 95% CI 0.02–2.47; p = 0.016) as well as in the surgical resection group (HR, 0.15; 95% CI, 0.03–1.36; p = 0.049) than low IL-15 group (Fig. 5C). Baseline serum IL-15 level shows potential as a biomarker for DC-based HCC immunotherapy.
Discussion
In this phase II trial, a triple TAA-pulsed DC therapy showed no significant improvement of RFS (primary end point) in overall patients with HCC that underwent standard treatment modalities. However, when patients treated with RFA were excluded, there was a significant prolongation of RFS in the immunotherapy group when compared with the untreated control group. In PP analysis, DC immunotherapy demonstrated 57% decrease of tumor recurrence or death among patients who underwent curative surgical resection as compared with each no adjuvant control group. TEAE was more frequent in the immunotherapy group, but the frequency of severe TEAE did not differ between the immunotherapy and control groups. We also found that IL-15 was significantly associated with RFS in patients treated with adjuvant DC immunotherapy.
DCs are crucial in activating T cell immunity, particularly for antigen-specific cytotoxic T cells (CTLs),9,10 but DC function is suppressed by tumor-mediated microenvironment.21-23 Thus, ex vivo-generated DC vaccine therapy is expected to stimulate antitumor immunity by enhancing specific CTLs.
We collectively applied several strategies to maximize DC vaccine potency. First, autologous DC vaccines were prepared by pulsing with the three most common HCC TAAs (AFP, MAGE-1, and GPC-3) to cover the heterogeneity of HCC. Second, instead of epitope peptides, recombinant TAA proteins were used to overcome the HLA-restriction. Third, cytoplasmic transduction peptide (CTP)24-fused TAAs facilitated antigen delivery into the cytoplasm of DCs, leading to better induction of TAA-specific CTL responses.25 Fourth, DC vaccines were injected subcutaneously near the inguinal lymph nodes, rather than intravenously.26,27 Last, a topical TLR-7 agonist, that facilitates DC migration to regional LNs, was applied around the injection site of DC vaccines.28
In the present study, TAA-pulsed DC immunotherapy unexpectedly accelerated HCC recurrence in RFA-treated patients, and the reversed clinical outcome was statistically significant (Fig. 2C), suggesting that DC immunotherapy is not compatible with RFA treatment of patients with HCC. This result is quite different from recent studies reporting that adjuvant cellular immunotherapy with natural killer cells, γδ T cells, or CIK cells in HCC patients following RFA treatment reduced the risk of HCC recurrence and improved progression-free survival compared with RFA alone.5,29 RFA has been reported to boost tumor-specific immune responses to thermally altered HCC antigens in the immune-tolerance environment.30 In our baseline analysis, RFA group showed significantly higher concentrations of IFNγ and IL-6 than non-RFA group among the pro-inflammatory cytokines and chemokines (Fig. S3), suggesting that DC vaccines generated from RFA group might be further exhausted as compared with those of non-RFA group. However, there was no clear difference in the surface phenotypes (MHC I/II and co-stimulatory molecules), migration and T cell proliferation capacities between RFA group and non-RFA group (Table S7). In addition, the RFA group showed significantly higher baseline levels of TAA-specific ELISPOTs when compared with the non-RFA group (Fig. S4A). However, we could not find any clear correlation between the level of baseline IL-6 and RFS. In addition, the ELISPOTs of the RFA group decreased from peak levels after 10 weeks and there was no further enhancement of T cell proliferation by repeated DC vaccination. In contrast, the non-RFA group showed significant enhancements in both TAA-specific ELISPOTs and T cell responses at week 22 by repeated DC vaccination (Fig. S4B). The TAA-pulsed DC vaccines generated from the non-RFA group patients were shown to express more IL-12 than those of the RFA group, but were not statistically significant (Fig. S5A), and there was no significant difference in the Th1 immunogenicity of DC vaccines between the RFA group and non-RFA group (Fig. S5B). These data suggested that differential immune responsiveness and/or T cell dysfunction/exhaustion through repetitive antigen encounter during DC immunotherapy might cause the opposing effects of immunotherapy in the RFA versus non-RFA subgroups. The detailed mechanism remains yet to be demonstrated by further investigation.
Although the efficacy of DC vaccine for patients with HCC has not been proven in phase III trial, this TAA-pulsed DC vaccine has several strengths over adjuvant CIK cell therapy of which efficacy is proven.5 First, subcutaneous administration of the DC vaccine is simpler than intravenous administration of CIK therapy. Second, DC vaccination required fewer injections than CIK therapy (6 times vs. 16 times). Third, unlike CIK therapy, the effect of the DC vaccine on antitumor immunity could be monitored by measuring antigen-specific T cell responses in vivo. Fourth, the level of baseline serum IL-15 was significantly correlated with RFS prolongation in the DC immunotherapy group (Fig. 5), suggesting that baseline IL-15 can be used as a novel biomarker for DC-based HCC immunotherapy. IL-15 is a well-established effector cytokine produced by monocytes/macrophages31 and DCs,32 which is essential for proliferation and activation of effecter CD8+ T cells and NK cells33-35 and effector T-cell survival and memory formation,36 thus has been used for adoptive T-cell immunotherapy.33,36-40 Considering the biologic functions of IL-15, higher baseline IL-15 might give synergistic effects on DC immunotherapy.
This phase II study is not without limitations. First, we included patients treated with TACE (n = 10), generally not regarded as a curative treatment in recommendations for the design of clinical trials from the National Cancer Institute.41 Second, patients with stage III HCC according to mUICC were included, which could include patients with vascular invasion of advanced HCC. In the following phase III trial, the inclusion criteria must be refined to involve only patients who underwent surgical resection and achieved complete remission without vascular invasion.
In conclusion, this study demonstrated that active adjuvant immunotherapy using a TAA-pulsed DC vaccine can prolong RFS in patients with HCC who have achieved complete remission by treatment modalities other than RFA. The DC vaccine was associated with a higher frequency of TEAE, but not with severe TEAE. Serum IL-15 was a candidate biomarker that could predict the treatment effect of adjuvant DC immunotherapy. Testing of the DC vaccine is warranted among patients who underwent curative surgical resection for HCC in a future phase III trial.
Patients and methods
Patients
Patients who achieved complete remission after one or two treatment(s), including surgical resection, radiofrequency ablation (RFA), transarterial chemoembolization (TACE), and percutaneous ethanol injection (PEI) for HCC of stage between I and III within 4 weeks before enrollment were eligible for this study. Appropriate antiviral treatment was done for patients with active hepatitis B among the HCC patients as reported previously.42 The diagnosis of HCC was made by histological examination or radiological imaging tests, mainly based on the guidelines from the American Association for the Study of Liver Diseases.6 Detailed eligibility and exclusion criteria are described in Supplementary Methods.
Study design
Study design is summarized in Fig. 1A. All patients provided written informed consent before enrollment. Study protocol and procedures were approved by the institutional review boards at the respective participating centers. All methods and procedures associated with this study were conducted in accordance with the good clinical practice guidelines and ethically with the principles of the Declaration of Helsinki and local law. The study was designed by the sponsor (JW CreaGene Inc.) in conjunction with the principal academic investigators. Data were managed in parallel by the Data and Safety Monitoring Board and the principal investigators. Clinical research information is available on the International Clinical Trials Registry Platform website provided by the World Health Organization (http://apps.who.int/trialsearch/Trial2.aspx?TrialID=KCT0000008). All authors had access to the study data and had reviewed and approved the final manuscript.
This phase II clinical study was a prospective, randomized, open-labeled trial, conducted at five academic hospitals in South Korea. All eligible participants were randomly assigned, at a 1:1 ratio, to receive adjuvant immunotherapy with TAA-pulsed DC vaccine (the vaccination group) or no adjuvant treatment (the control group). Randomization was performed through a web-based allocation system using a computer-generated permuted-block with a block size of 4 or 6 and stratified according to both HCC stage (according to the modified Union for International Cancer Control (mUICC) stage I vs. II/III) and study center.
DC vaccine generation and quality control
Autologous DCs were generated from peripheral blood mononuclear cells (PBMC) obtained from the HCC patients through leukapheresis at each center 2 weeks before the first vaccination. DC vaccine was generated and prepared as described previously,19,20 with minor modifications at a central Good Manufacturing Practice-compliant facility in JW CreaGene Inc. (Seongnam-si, Gyeonggi-do, Korea). In brief, monocytes were separated from the patient PBMC and cultured with supplements of granulocyte macrophage-colony stimulating factor (GM-CSF) and interleukin (IL)-4 for 5 d. On day 5, immature DCs were harvested and pulsed with CTP24-fused human AFP, GPC-3, and MAGE-1 recombinant proteins, and then matured in the presence of a cytokine cocktail consisting of IL-6, IL-1β, tumor necrosis factor-α, prostaglandin E2, interferon (IFN)γ, OK432, and poly I:C for 2 d. On day 7, the DCs were harvested, washed, and resuspended in cryopreserving solution. Finally, fully equipped DC vaccines were packed into a sterile glass vial, sealed with a snap-cap, and stored at an ultralow cryopreservation system.
Sterility, antigen uptake of imDC, cell viability, surface phenotype, and purity (lineage negativity) were evaluated (Table S8) as described20 with minor modifications (Supplementary Methods) for quality control of DC vaccine. DC migration, T-cell stimulation capacity, and Th1 cytokine in the culture supernatants were assessed (Table S9).
Treatment protocol
Patients who provided informed consent were screened within 4 weeks before the start of immunotherapy. Multiple TAA-pulsed DC vaccine (3 × 107 cells/injection) was injected subcutaneously into the thigh near the inguinal lymph nodes. Topical toll-like receptor-7 (TLR-7) agonist (Imiquimod: Mochida Pharmaceutical Co., Tokyo, Japan) was applied around the injection site for two consecutive days before injection. Patients received DC vaccine six times over 14 weeks (four times every 2 week and then two times every 4 week). Systemic review, blood tests, and urinalysis were performed at each vaccination. Vital signs were monitored during and after each vaccine injection. Response was evaluated 4 weeks after the fourth vaccination (10 weeks after the first vaccination) and 4 weeks after the sixth vaccination (18 weeks after the first vaccination).
Endpoints and assessments
The primary end point was RFS, which was measured from the date of randomization to the first recurrence or to death from any cause. Secondary endpoints included safety and induction of TAA-specific immune responses; changes in serum levels of immunosuppressive cytokines. TAA-specific cell-mediated immunity induced by DC vaccination was evaluated using interferon (IFN)γ enzyme-linked immunospot (ELISPOT), and lymphocyte proliferation assays following Minimal Information about T cell Assays (MIATA) guidelines43 with some modifications as described previously.20 TEAEs, classified and graded according to Common Terminology Criteria for Adverse Events version 3.0, were assessed from the time of written informed consent to the end of the study or drop-out, or until at least 30 d after the last dose of immunotherapy. Multiple occurrences of specific events were counted once per patient; the event with the greatest severity was summarized.
Tumor assessments were performed using dynamic computed tomography (CT) or magnetic resonance imaging (MRI) every 12 weeks from week 10 to week 104 after 1st DC vaccination.
Biomarker analysis
To find a pre-treatment biomarker that could predict treatment response, multiplex cytokine analyses were performed with baseline patients' sera via customized Luminex Bead-based Milliplex#x08E8; Multiplex Assay (Millipore, St Charles, MO, USA). IFNγ, interleukin (IL)-1β, IL-2, IL-4, IL-6, IL-8, IL-12, IL-15, IFNγ-induced protein 10 (IP-10, CXCL10), macrophage inflammatory protein-1β, soluble CD25, tumor necrosis factor-α, and chemokine (C-C motif) ligand-5 (CCL5) were examined.
Statistical analysis
Sample size was determined based on the primary end point of RFS. Assuming a one-sided type I error of 0.05, power of 80%, and randomization ratio of 1:1 between the two study groups, 41 recurrence events were needed to detect a 20% increase (from 60% to 80%) in 2-y RFS (hazard ratio (HR), 0.437). When the potential “loss to follow-up” rate was set at 15%, 156 patients were needed to record 41 recurrence events. Efficacy outcomes were basically assessed according to the ITT principle. Detailed statistical methods are provided in Supplementary Methods. All statistical tests were two-sided and statistical significance was set at p < 0.05. Statistical analysis was performed by statisticians at LSK Global Pharma Services Co., Ltd. (Seoul, Korea) using SAS software version 9.3 (SAS Institute Inc., Cary, NC).
Supplementary Material
Disclosure of potential conflicts of interest
Yoon Lee, Min-Kyu Heo, Jae-Sung Song, Hak-Yeop Kim, and Ki-Hwan Kim are the employees in JW CreaGene Inc. Yong-Soo Bae was involved in this DC vaccine study as a consultant and scientific advisor.
Acknowledgments
We thank the patients, physicians, nurses, trial coordinators, and pathologists who participated in this Phase II study of CreaVax-HCC at five academic hospitals. We acknowledge the members of the Data and Safety Monitoring Board (DSMB) for data monitoring, evaluation of interim analysis, and valuable criticism. We are grateful to personnel in the Medical Research Cooperating Center at Seoul National University Hospital (MRCC, http://mrcc.snuh.org) for their web-based randomization of enrolled patients according to the study design. We acknowledge Young-Jak Lee and his colleagues at LSK Global Pharma Services Co. Ltd for their study consulting, data monitoring and management, CRF development, statistical analysis, and CSR. We acknowledge Seung-Ho Hong, Jun-Eui Park, Jinah Jang, and research fellows at JW CreaGene Res. Institute for providing clinical grade CTP-attached HCC antigens, as well as Woo-Jeong Son and his colleagues who were involved in the production of DC vaccines and QC/QA at JW CreaGene Inc.
Funding
This trial was funded by a Bio New Drug grant (HI11C0049, A110054) from the Ministry of Health & Welfare, Republic of Korea, and supported by JW CreaGene Inc. (Seongnam-si, Gyeonggi-do, Korea).
Ethics approval
The Institutional Review Boards at five participating centers (Seoul National University Hospital, Samsung Medical Center, Catholic St. Marry Hospital, Kyoungbook National University Hospital, Pusan National University Hospital).
References
- 1.Bolondi L, Sofia S, Siringo S, Gaiani S, Casali A, Zironi G, Piscaglia F, Gramantieri L, Zanetti M, Sherman M. Surveillance programme of cirrhotic patients for early diagnosis and treatment of hepatocellular carcinoma: a cost effectiveness analysis. Gut 2001; 48:251-9; PMID:11156649; https://doi.org/ 10.1136/gut.48.2.251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yuen MF, Cheng CC, Lauder IJ, Lam SK, Ooi CG, Lai CL. Early detection of hepatocellular carcinoma increases the chance of treatment: Hong Kong experience. Hepatology 2000; 31:330-5; PMID:10655254; https://doi.org/ 10.1002/hep.510310211 [DOI] [PubMed] [Google Scholar]
- 3.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin 2011; 61:69-90; PMID:21296855; https://doi.org/ 10.3322/caac.20107 [DOI] [PubMed] [Google Scholar]
- 4.Lai EC, Fan ST, Lo CM, Chu KM, Liu CL, Wong J. Hepatic resection for hepatocellular carcinoma. An audit of 343 patients. Ann Surg 1995; 221:291-8; PMID:7717783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee JH, Lee JH, Lim YS, Yeon JE, Song TJ, Yu SJ, Gwak GY, Kim KM, Kim YJ, Lee JW et al.. Adjuvant immunotherapy with autologous cytokine-induced killer cells for hepatocellular carcinoma. Gastroenterology 2015; 148:1383-91 e6; PMID:25747273; https://doi.org/ 10.1053/j.gastro.2015.02.055 [DOI] [PubMed] [Google Scholar]
- 6.Bruix J, Sherman M, American Association for the Study of Liver Diseases . Management of hepatocellular carcinoma: an update. Hepatology 2011; 53:1020-2; PMID:21374666; https://doi.org/ 10.1002/hep.24199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Verslype C, Rosmorduc O, Rougier P, ESMO Guidelines Working Group . Hepatocellular carcinoma: ESMO-ESDO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol 2012; 23(Suppl 7):vii41-8; https://doi.org/ 10.1093/annonc/mds225 [DOI] [PubMed] [Google Scholar]
- 8.European Association for The Study of The Liver, European Organisation for Research Treatment of Cancer EASL-EORTC clinical practice guidelines: management of hepatocellular carcinoma. J Hepatol 2012; 56:908-43; PMID:22424438; https://doi.org/ 10.1016/j.jhep.2011.12.001 [DOI] [PubMed] [Google Scholar]
- 9.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature 1998; 392:245-52; PMID:9521319; https://doi.org/ 10.1038/32588 [DOI] [PubMed] [Google Scholar]
- 10.Vacchelli E, Ma Y, Baracco EE, Sistigu A, Enot DP, Pietrocola F, Yang H, Adjemian S, Chaba K, Semeraro M et al.. Chemotherapy-induced antitumor immunity requires formyl peptide receptor 1. Science 2015; 350:972-8; PMID:26516201; https://doi.org/ 10.1126/science.aad0779 [DOI] [PubMed] [Google Scholar]
- 11.Palucka AK, Coussens LM. The Basis of Oncoimmunology. Cell 2016; 164:1233-47; PMID:26967289; https://doi.org/ 10.1016/j.cell.2016.01.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Palucka K, Banchereau J. Dendritic-cell-based therapeutic cancer vaccines. Immunity 2013; 39:38-48; PMID:23890062; https://doi.org/ 10.1016/j.immuni.2013.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nestle FO, Alijagic S, Gilliet M, Sun Y, Grabbe S, Dummer R, Burg G, Schadendorf D. Vaccination of melanoma patients with peptide- or tumor lysate-pulsed dendritic cells. Nat Med 1998; 4:328-32; PMID:9500607; https://doi.org/ 10.1038/nm0398-328 [DOI] [PubMed] [Google Scholar]
- 14.Small EJ, Fratesi P, Reese DM, Strang G, Laus R, Peshwa MV, Valone FH. Immunotherapy of hormone-refractory prostate cancer with antigen-loaded dendritic cells. J Clin Oncol 2000; 18:3894-903; PMID:11099318; https://doi.org/ 10.1200/JCO.2000.18.23.3894 [DOI] [PubMed] [Google Scholar]
- 15.Holtl L, Rieser C, Papesh C, Ramoner R, Herold M, Klocker H, Radmayr C, Stenzl A, Bartsch G, Thurnher M. Cellular and humoral immune responses in patients with metastatic renal cell carcinoma after vaccination with antigen pulsed dendritic cells. J Urol 1999; 161:777-82; PMID:10022683; https://doi.org/ 10.1016/S0022-5347(01)61767-1 [DOI] [PubMed] [Google Scholar]
- 16.Lee WC, Wang HC, Hung CF, Huang PF, Lia CR, Chen MF. Vaccination of advanced hepatocellular carcinoma patients with tumor lysate-pulsed dendritic cells: a clinical trial. J Immunother 2005; 28:496-504; PMID:16113606; https://doi.org/ 10.1097/01.cji.0000171291.72039.e2 [DOI] [PubMed] [Google Scholar]
- 17.Butterfield LH, Ribas A, Dissette VB, Lee Y, Yang JQ, De la Rocha P, Duran SD, Hernandez J, Seja E, Potter DM et al.. A phase I/II trial testing immunization of hepatocellular carcinoma patients with dendritic cells pulsed with four alpha-fetoprotein peptides. Clin Cancer Res 2006; 12:2817-25; PMID:16675576; https://doi.org/ 10.1158/1078-0432.CCR-05-2856 [DOI] [PubMed] [Google Scholar]
- 18.Palmer DH, Midgley RS, Mirza N, Torr EE, Ahmed F, Steele JC, Steven NM, Kerr DJ, Young LS, Adams DH. A phase II study of adoptive immunotherapy using dendritic cells pulsed with tumor lysate in patients with hepatocellular carcinoma. Hepatology 2009; 49:124-32; PMID:18980227; https://doi.org/ 10.1002/hep.22626 [DOI] [PubMed] [Google Scholar]
- 19.Tada F, Abe M, Hirooka M, Ikeda Y, Hiasa Y, Lee Y, Jung NC, Lee WB, Lee HS, Bae YS et al.. Phase I/II study of immunotherapy using tumor antigen-pulsed dendritic cells in patients with hepatocellular carcinoma. Int J Oncol 2012; 41:1601-9; PMID:22971679; https://doi.org/ 10.3892/ijo.2012.1626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee JH, Lee Y, Lee M, Heo MK, Song JS, Kim KH, Lee H, Yi NJ, Lee KW, Suh KS et al.. A phase I/IIa study of adjuvant immunotherapy with tumour antigen-pulsed dendritic cells in patients with hepatocellular carcinoma. Br J Cancer 2015; 113:1666-76; PMID:26657650; https://doi.org/ 10.1038/bjc.2015.430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Apetoh L, Locher C, Ghiringhelli F, Kroemer G, Zitvogel L. Harnessing dendritic cells in cancer. Semin Immunol 2011; 23:42-9; PMID:21295491; https://doi.org/ 10.1016/j.smim.2011.01.003 [DOI] [PubMed] [Google Scholar]
- 22.Ma Y, Aymeric L, Locher C, Kroemer G, Zitvogel L. The dendritic cell-tumor cross-talk in cancer. Curr Opin Immunol 2011; 23:146-52; PMID:20970973; https://doi.org/ 10.1016/j.coi.2010.09.008 [DOI] [PubMed] [Google Scholar]
- 23.Chen YX, Man K, Ling GS, Chen Y, Sun BS, Cheng Q, Wong OH, Lo CK, Ng IO, Chan LC et al.. A crucial role for dendritic cell (DC) IL-10 in inhibiting successful DC-based immunotherapy: superior antitumor immunity against hepatocellular carcinoma evoked by DC devoid of IL-10. J Immunol 2007; 179:6009-15; PMID:17947674; https://doi.org/ 10.4049/jimmunol.179.9.6009 [DOI] [PubMed] [Google Scholar]
- 24.Kim D, Jeon C, Kim JH, Kim MS, Yoon CH, Choi IS, Kim SH, Bae YS. Cytoplasmic transduction peptide (CTP): new approach for the delivery of biomolecules into cytoplasm in vitro and in vivo. Exp Cell Res 2006; 312:1277-88; PMID:16466653; https://doi.org/ 10.1016/j.yexcr.2005.12.029 [DOI] [PubMed] [Google Scholar]
- 25.Aguilar M, Aisa D, Alpat B, Alvino A, Ambrosi G, Andeen K, Arruda L, Attig N, Azzarello P, Bachlechner A et al.. Precision measurement of the helium flux in primary cosmic rays of rigidities 1.9 GV to 3 TV with the alpha magnetic spectrometer on the international space station. Phys Rev Lett 2015; 115:211101; PMID:26636836; https://doi.org/ 10.1103/PhysRevLett.115.211101 [DOI] [PubMed] [Google Scholar]
- 26.Lappin MB, Weiss JM, Delattre V, Mai B, Dittmar H, Maier C, Manke K, Grabbe S, Martin S, Simon JC. Analysis of mouse dendritic cell migration in vivo upon subcutaneous and intravenous injection. Immunology 1999; 98:181-8; PMID:10540216; https://doi.org/ 10.1046/j.1365-2567.1999.00850.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Okada N, Tsujino M, Hagiwara Y, Tada A, Tamura Y, Mori K, Saito T, Nakagawa S, Mayumi T, Fujita T et al.. Administration route-dependent vaccine efficiency of murine dendritic cells pulsed with antigens. Br J Cancer 2001; 84:1564-70; PMID:11384109; https://doi.org/ 10.1054/bjoc.2001.1801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Prins RM, Craft N, Bruhn KW, Khan-Farooqi H, Koya RC, Stripecke R, Miller JF, Liau LM. The TLR-7 agonist, imiquimod, enhances dendritic cell survival and promotes tumor antigen-specific T cell priming: relation to central nervous system antitumor immunity. J Immunol 2006; 176:157-64; PMID:16365406; https://doi.org/ 10.4049/jimmunol.176.1.157 [DOI] [PubMed] [Google Scholar]
- 29.Cui J, Wang N, Zhao H, Jin H, Wang G, Niu C, Terunuma H, He H, Li W. Combination of radiofrequency ablation and sequential cellular immunotherapy improves progression-free survival for patients with hepatocellular carcinoma. Int J Cancer 2014; 134:342-51; PMID:23825037; https://doi.org/ 10.1002/ijc.28372 [DOI] [PubMed] [Google Scholar]
- 30.Zerbini A, Pilli M, Penna A, Pelosi G, Schianchi C, Molinari A, Schivazappa S, Zibera C, Fagnoni FF, Ferrari C et al.. Radiofrequency thermal ablation of hepatocellular carcinoma liver nodules can activate and enhance tumor-specific T-cell responses. Cancer Res 2006; 66:1139-46; PMID:16424051; https://doi.org/ 10.1158/0008-5472.CAN-05-2244 [DOI] [PubMed] [Google Scholar]
- 31.Badolato R, Ponzi AN, Millesimo M, Notarangelo LD, Musso T. Interleukin-15 (IL-15) induces IL-8 and monocyte chemotactic protein 1 production in human monocytes. Blood 1997; 90:2804-9; PMID:9326248 [PubMed] [Google Scholar]
- 32.Ohteki T, Tada H, Ishida K, Sato T, Maki C, Yamada T, Maki C, Yamada T, Hamuro J, Koyasu S. Essential roles of DC-derived IL-15 as a mediator of inflammatory responses in vivo. J Exp Med 2006; 203:2329-38; PMID:16966429; https://doi.org/ 10.1084/jem.20061297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mueller K, Schweier O, Pircher H. Efficacy of IL-2- versus IL-15-stimulated CD8 T cells in adoptive immunotherapy. Euro J Immunol 2008; 38:2874-85; PMID:18825743; https://doi.org/ 10.1002/eji.200838426 [DOI] [PubMed] [Google Scholar]
- 34.Perna SK, De Angelis B, Pagliara D, Hasan ST, Zhang L, Mahendravada A, Heslop HE, Brenner MK, Rooney CM, Dotti G et al.. Interleukin 15 provides relief to CTLs from regulatory T cell-mediated inhibition: implications for adoptive T cell-based therapies for lymphoma. Clin Cancer Res 2013; 19:106-17; PMID:23149818; https://doi.org/ 10.1158/1078-0432.CCR-12-2143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Conlon KC, Lugli E, Welles HC, Rosenberg SA, Fojo AT, Morris JC, Fleisher TA, Dubois SP, Perera LP, Stewart DM et al.. Redistribution, hyperproliferation, activation of natural killer cells and CD8 T cells, and cytokine production during first-in-human clinical trial of recombinant human interleukin-15 in patients with cancer. J Clin Oncol 2015; 33:74-82; PMID:25403209; https://doi.org/ 10.1200/JCO.2014.57.3329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu A, Bhanumathy KK, Wu J, Ye Z, Freywald A, Leary SC, Li R, Xiang J. IL-15 signaling promotes adoptive effector T-cell survival and memory formation in irradiation-induced lymphopenia. Cell Biosci 2016; 6:30; PMID:27158441; https://doi.org/ 10.1186/s13578-016-0098-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hasan AN, Selvakumar A, Shabrova E, Liu XR, Afridi F, Heller G, Riviere I, Sadelain M, Dupont B, O'Reilly RJ. Soluble and membrane bound IL-15 Ralpha/IL-15 complexes mediate proliferation of high avidity central memory CD8+ T-cells for adoptive immunotherapy of cancer and infections. Clin Exp Immunol 2016; 186(2):249-65; PMID:27227483; https://doi.org/ 10.1111/cei.12816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huarte E, Fisher J, Turk MJ, Mellinger D, Foster C, Wolf B, Meehan KR, Fadul CE, Ernstoff MS. Ex vivo expansion of tumor specific lymphocytes with IL-15 and IL-21 for adoptive immunotherapy in melanoma. Cancer Lett 2009; 285:80-8; PMID:19501956; https://doi.org/ 10.1016/j.canlet.2009.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dotti G, Gottschalk S, Savoldo B, Brenner MK. Design and development of therapies using chimeric antigen receptor-expressing T cells. Immunol Rev 2014; 257:107-26; PMID:24329793; https://doi.org/ 10.1111/imr.12131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pilipow K, Roberto A, Roederer M, Waldmann TA, Mavilio D, Lugli E. IL15 and T-cell stemness in T-cell-based cancer immunotherapy. Cancer Res 2015; 75:5187-93; PMID:26627006; https://doi.org/ 10.1158/0008-5472.CAN-15-1498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Llovet JM, Di Bisceglie AM, Bruix J, Kramer BS, Lencioni R, Zhu AX, Sherman M, Schwartz M, Lotze M, Talwalkar J et al.. Design and endpoints of clinical trials in hepatocellular carcinoma. J Natl Cancer Inst 2008; 100:698-711; PMID:18477802; https://doi.org/ 10.1093/jnci/djn134 [DOI] [PubMed] [Google Scholar]
- 42.Korean Association for the Study of the L KASL clinical practice guidelines: management of chronic hepatitis B. Clin Mol Hepatol 2012; 18:109-62; PMID:22893865; https://doi.org/ 10.3350/cmh.2012.18.2.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Britten CM, Janetzki S, Butterfield LH, Ferrari G, Gouttefangeas C, Huber C, Kalos M, Levitsky HI, Maecker HT, Melief CJ et al.. T cell assays and MIATA: the essential minimum for maximum impact. Immunity 2012; 37:1-2; PMID:22840835; https://doi.org/ 10.1016/j.immuni.2012.07.010 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.