

Abstract
The balance of histone acetylation and deacetylation is an epigenetic layer with a critical role in the regulation of gene expression. Histone acetylation induced by histone acetyl transferases (HATs) is associated with gene transcription, while histone hypoacetylation induced by histone deacetylase (HDAC) activity is associated with gene silencing. Altered expression and mutations of genes that encode HDACs have been linked to tumor development since they both induce the aberrant transcription of key genes regulating important cellular functions such as cell proliferation, cell‐cycle regulation and apoptosis. Thus, HDACs are among the most promising therapeutic targets for cancer treatment, and they have inspired researchers to study and develop HDAC inhibitors.
Keywords: Histone deacetylases, Cancer, Review
1. Introduction
For a long time, cancer has been considered to be the result of a wide variety of genetic and genomic alterations, such as amplifications, translocations, deletions, and point mutations. These have as their dramatic end‐point the activation of oncogenes and the inactivation of tumor‐suppressor genes. However, cancer development is not restricted to the genetic changes described above, but may also involve epigenetic changes. Epigenetics is concerned with the inheritance of information based on gene‐expression levels, as opposed to genetics, whose realm is that of information transmitted on the basis of gene sequence. The main epigenetic modifications in mammals, and particularly in humans, are DNA methylation and posttranslational histone modifications (acetylation, methylation, phosphorylation, etc.).
Until now, DNA methylation, and in particular silencing of tumor‐suppressor genes by promoter hypermethylation, has been the most widely studied epigenetic modification in human tumors (Esteller, 2002). However, in recent years there has been a significant growth in our knowledge about the involvement of aberrant patterns of histone modifications in cancer development. In particular, acetylation of lysine residues of histone 3 and histone 4 has become one of the best studied of this type of modifications. Acetylation levels are the result of the balance of the activities of histone acetyltransferase (HAT) and histone deacetylase (HDAC). The levels of histone acetylation play a crucial role in chromatin remodeling and in the regulation of gene transcription. The presence of acetylated lysine in histone tails is associated with a more relaxed chromatin state and gene‐transcription activation, while the deacetylation of lysine residues is associated with a more condensed chromatin state and transcriptional gene silencing (Johnstone, 2002; Iizuka and Smith, 2003). Histone deacetylation increases ionic interactions between the positively charged histones and negatively charged DNA, which yields a more compact chromatin structure and represses gene transcription by limiting the accessibility of the transcription machinery. In addition, histone acetylation has been associated with other genome functions such as chromatin assembly, DNA repair, and recombination (Polo and Almouzni, 2005; Vidanes et al., 2005).
Furthermore, HDACs regulate gene expression in other ways. For example, HDACs form corepressor complexes with the nuclear receptor in the absence of a ligand. Other studies indicate that HDACs can regulate the expression of a large number of genes by direct interaction with transcription factors such as E2f, Stat3, p53, the retinoblastoma protein, NF‐κB, TFIIE, etc. (Lin et al., 2006). Moreover, HDACs are involved in the deacetylation not only of chromatin proteins, which can lead to altered gene‐transcription regulation, but also of non‐histone proteins, which regulate important functions that, in turn, regulate cellular homeostasis (cell‐cycle progression, differentiation, and apoptosis) (Minucci and Pelicci, 2006).
Eighteen mammalian histone deacetylase enzymes have been identified so far, which can be subdivided into different families according to their homology with yeast HDACs. Class I, which is homologous to Rpd3 in yeast, includes HDACs 1, 2, 3, and 8, has a nuclear localization. It is ubiquitously expressed in human cell lines and tissues. Class II is homologous to yeast Hda1 and can be subdivided into two subclasses: IIa (HDAC 4, 7, and 9) and IIb (HDAC 6 and 10). Class II exhibits tissue‐specific expression and can shuttle between the nucleus and cytoplasm, which suggests that this class of HDACs may be involved in the acetylation of non‐histone proteins. The recently discovered HDAC11 is the only member of the class 4 HDACs and is homologous with both class I and class II. The class III HDACs, or sirtuins (SIRT1‐7), includes a group of proteins that are homologous with the yeast Sir2 family of proteins. The subcellular distribution and pattern of tissue‐specific expression of this class are unknown. Class I and class II HDACs are sensitive to the classical HDAC inhibitor trichostatin A (TSA), whereas those of class III are insensitive to this inhibitor and require the coenzyme NAD+ as a cofactor.
2. Biological functions of histone deacetylases
Many HDACs exist as components of multiprotein complexes, such as the transcriptional corepressors mSin3, N‐CoR, and SMRT (Glass and Rosenfeld, 2000). These are then targeted to specific genomic regions by interactions with DNA binding factors that include transcription factors, nuclear receptors, and other epigenetic modifier genes, such as methyl‐binding proteins (MBDs), DNA methyl transferases (DNMTs) and histone methyl transferases (HMTs) (Figure 1). The best‐characterized interaction is the recruitment of HDACs to methylated DNA via methyl‐binding proteins. The most thoroughly studied example of interaction between HDACs and MBDs is that of MeCP2. This methyl‐binding protein recruits HDAC‐containing complexes to methylated gene promoters as a mechanism for gene‐transcription repression (Jones et al., 1998; Nan et al., 1998). HDACs also interact with DNA methyl transferases. For example, in DNMT1 knockout cancer cells there is an increase in the amount of acetylated forms of histone H3 and a decrease in that of the methylated forms of histone H3. These changes are associated with the loss of interaction of HDACs and the heterochromatin protein HP1 with histone H3. These data strongly indicate that histone hyperacetylation is not always the result of a loss of HDAC activity, but that it could be due to a loss of HDAC targeted to specific DNA sequences. One possible explanation is that changes in DNA methylation also cause histone modification due to direct interactions between the enzymes regulating different epigenetic modifications (Espada et al., 2004). HDAC2 is also involved in the regulation of neuronal differentiation through a direct interaction with the N‐terminal domain of DNMT3b. Treatment of the pheochromocytoma cell line PC12 with HDAC inhibitors prevents the nerve growth factor‐induced differentiation of this cell line, while the overexpression of the N‐terminal domain of DNMT3b facilitates differentiation (Bai et al., 2005).
Figure 1.
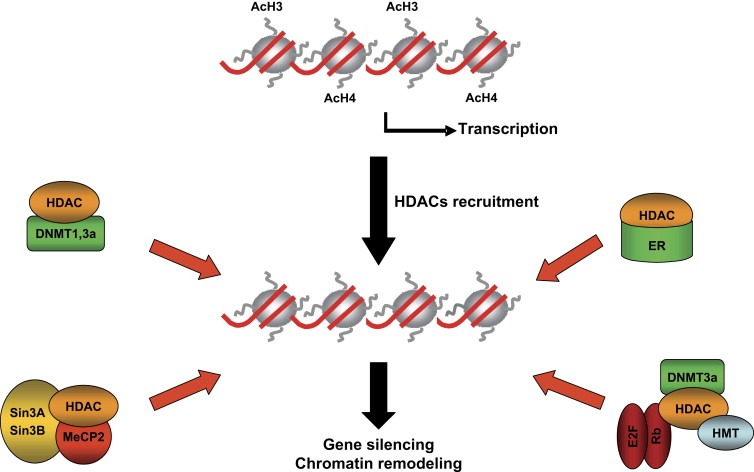
Different ways by which HDACs are recruited to gene promoters. An array of nucleosomes is shown. Histone octamers are represented by circles and the DNA is shown in red. Histone deacetylation induced by recruitment of HDAC to gene promoters by different factors including, DNA methyl transferases (DNMT), the methyl binding protein MeCP2, Estrogen receptor (ER) and transcription factors (E2F, Rb).
As mentioned above, HDACs are key enzymes regulating important cell processes such as cell‐cycle progression and apoptosis. Another way by which HDACs are recruited to DNA independently of DNA methylation involves the interaction with transcription factors and nuclear receptors. Focusing on the interaction with transcription factors, HDAC1 and HDAC2 are involved in transcriptional repression regulated by the retinoblastoma protein Rb (Robertson et al., 2000). E2F is a family of transcription factors involved in cell‐cycle control. E2F‐containing promoters are repressed by members of the Rb family that are recruited by a physical interaction with the E2F protein. One possibility is that the repression of E2f‐regulated promoters by Rb implies the recruitment of HDACs to the E2F‐containing promoters (Brehm et al., 1998; Ferreira et al., 1998; Robertson et al., 2000). Treatment with TSA, a classical HDAC inhibitor, prevents the Rb‐mediated repression of gene transcription (Siddiqui et al., 2003).
HDACs also participate in gene expression regulation mediated by nuclear receptors. Estrogen receptors (ERs) belong to a large superfamily of nuclear receptors that modulate the expression of genes regulating critical breast and ovary functions. HDAC1 interacts with ER‐α and suppresses its transcriptional activity. The interaction of HDAC1 with ER‐α is mediated by the activation function‐2 (AF‐2) domain and DNA‐binding domain of ER‐α, and this interaction is weakened in the presence of estrogens (Kawai et al., 2003). Furthermore, another study indicates that the ER‐gene transcription is regulated by a multiprotein complex that includes HDACs, DNMTs, and retinoblastoma protein Rb (Macaluso et al., 2003).
3. Role of HDACs in cancer
A typical characteristic of human cancer is the deregulation of DNA methylation and posttranslational histone modifications, in particular histone acetylation, which has the fatal consequence of gene transcription‐deregulation. The data from our studies of a panel of normal tissues, primary tumors, and human cancer cell lines indicate that a loss of acetylated Lys16 (K16‐H4) and trimethylated Lys20 (K20‐H4) of histone H4 is a common event in human cancer (Fraga et al., 2005) that is associated with the hypomethylation of repetitive sequences. Moreover, these changes occur early in tumorogenesis, as shown by the data from a mouse model of multistage skin cancer tumorogenesis, which indicates that the global loss of monoacetylated and trimethylated forms of histone H4 is a crucial event in cancer development (Fraga et al., 2005). Another study of gastrointestinal tumors concluded that the decrease in histone acetylation is not only involved in tumorogenesis but also in tumor invasion and metastasis (Yasui et al., 2003).
We are largely ignorant about all the mechanisms involved in histone hypoacetylation. These changes can be explained by a decrease in HAT activity due to the mutations or chromosomal translocations characteristic of leukemias, or to changes that result in the increased activity of HDACs. Focusing on the role of HDACs in cancer, the available data indicate that there is more than one mechanism by which HDACs function in cancer development. To date, most studies have focused on the role of the aberrant recruitment of HDACs to specific promoters through the interaction with fusion proteins that result from chromosomal translocations typical of hematological malignancies. An archetypal example that serves as a model for several other hematological malignancies is acute promyelocytic leukemia (APL). A genetic characteristic of this disease is the chromosomal translocation that produces the fusion proteins containing RAR‐PML and RAR‐PLZF. These fusion proteins bind to retinoic acid‐responsive elements (RAREs) and recruit the HDAC repressor complex with a high affinity, preventing the binding of retinoic acid, and repressing the expression of genes that regulate normal differentiation and proliferation of myeloid cells (Lin et al., 2001). Thus, in this case, HDACs are key elements in APL development. A similar mechanism underlies the action of the fusion protein AML1‐ETO (Wang et al., 1998).
However, the changes in gene expression described above are not related to specific alterations in HDAC expression. Although there are no conclusive data about the pattern of HDAC expression in human cancer, there are a number of studies showing altered expression of individual HDACs in tumor samples. For example, there is an increase in HDAC1 expression in gastric (Choi et al., 2001), prostate (Halkidou et al., 2004), colon (Wilson et al., 2006), and breast (Zhang et al., 2005) carcinomas. Overexpression of HDAC2 has been found in cervical (Huang et al., 2005), and gastric (Song et al., 2005) cancers, and in colorectal carcinoma with loss of APC expression (Zhu et al., 2004). Other studies have reported high levels of HDAC3 and HDAC6 expression in colon and breast cancer specimens, respectively (Zhang et al., 2004; Wilson et al., 2006).
The findings described above suggest that the transcriptional repression of tumor‐suppressor genes by overexpression and aberrant recruitment of HDACs to their promoter region could be a common phenomenon in tumor onset and progression. A typical example is the cyclin‐dependent kinase inhibitor p21WAF1, which inhibits cell‐cycle progression and whose expression is lost in many different tumors. In some tumors, p21WAF1 is epigenetically inactivated by hypoacetylation of the promoter, and treatment with HDAC inhibitors leads to the inhibition of tumor‐cell growth and an increase in both the acetylation of the promoter and gene expression (Gui et al., 2004). The transcription factor Snail recruits HDAC1, HDAC2, and the corepressor complex mSin3A to the E‐cadherin promoter to repress its expression (Peinado et al., 2004). Downregulation or loss of function of E‐cadherin has been implicated in the acquisition of invasive potential by carcinomas (Christofori and Semb, 1999; Hajra and Fearon, 2002), and so the aberrant recruitment of HDACs to this promoter may have a crucial role in tumor invasion and metastasis.
The role of HDACs in cancer is not restricted to their contribution to histone deacetylation, but also to their role in deacetylation of non‐histone proteins. For example, HDAC1 interacts with the tumor suppressor p53 and deacetylates it in vivo and in vitro (Juan et al., 2000; Luo et al., 2000). p53 is phosphorylated and acetylated under stress conditions. Since lysine residues acetylated in p53 overlap with those that are ubiquitinated, p53 acetylation serves to promote protein stability and activation, inducing checkpoints in the cell‐division cycle, permanent cell‐division arrest, and cell death.
The results from our laboratory are particularly interesting. We have recently found a mutation of HDAC2 in sporadic tumors with microsatellite instability and in tumors arising in individuals with hereditary non‐polyposis colorectal carcinoma. This mutation leads to the loss of HDAC2 expression and activity. Furthermore, the ectopic expression of HDAC2 in cancer cell lines harboring the truncated form of HDAC2 strongly indicates that this gene shares similar features to those of tumor‐suppressor genes, such as reduced colony formation, and the inhibition of tumor growth in xenograft nude mice (Ropero et al., 2006). Although we are largely ignorant of the molecular mechanisms underlying the role of HDAC2 loss in cancer development, we can speculate that the loss of HDAC2 function could induce oncogene expression. In fact, data exist suggesting that mutations or alterations that induce loss of function of class I HDACs may contribute to cancer development. The tumor‐suppressor gene Rb requires the recruitment of class I HDACs to repress gene transcription (Frolov and Dyson, 2004) Thus, the loss of class I HDAC activity could induce the expression of genes regulated by Rb, thereby suppressing their protective role in tumor development (Figure 2).
Figure 2.
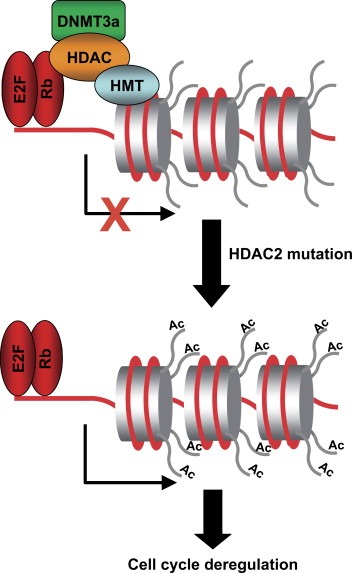
A model showing a possible effect of HDAC2 mutation in cancer development. class I HDACs are involved in gene transcription‐repression mediated by retinoblastoma protein. The lost of HDAC2 function could induce the hyperacetylation and reexpression of genes regulated by retinoblastoma protein Rb, and with crucial functions in cell cycle regulation.
In the last years, the sirtuins or class III HDACs has received much attention. These HDACs play important roles in the regulation of gene expression, apoptosis, stress responses, DNA repair, cell cycle, genomic stability, etc, indicating that this group of HDACs are key regulators of normal cell growth and proliferation. In particular, the most prominent family member, SIRT1, regulates histone acetylation levels (mainly K16‐H4 and K9‐H3 positions) (Vaquero et al., 2004; Pruitt et al., 2006), and the acetylation of transcription factors such as p53 (Vaziri et al., 2001), p300 histone acethyltransferase (Bouras et al., 2005), E2F1 (Wang et al., 2006), the DNA repair ku70 (Cohen et al., 2004), NF‐KB (Yeung et al., 2004), and the androgen receptor (Fu et al., 2006). Considering these together, it is clear that the deregulation of sirtuins has relevance in cancer development. There are several reports showing either up‐ or downregulation of SIRTs genes in cancer. For instance, SIRT1 is upregualted in human lung cancer (Yeung et al., 2004), prostate cancer (Kuzmichev et al., 2005) and leukemia (Bradbury et al., 2005) and has been found downregulated in colon tumors (Ozdag et al., 2006). Furthermore, the acetylation levels of K16‐H4 and K9‐H3, the histone substrates of SIRT1, have been found altered in different tumor types. As we mentioned above, the data from our studies showed the loss of monoacetylated K16‐H4 is a common event of human cancer and that this alteration is an early event in cancer development (Fraga et al., 2005) Treatment with SIRT1 inhibitors induces the reexpression of tumor suppressor genes together with an increase in the acetylation levels of K16‐H4 and K9‐H3 in breast and colon cancer cell lines (Pruitt et al., 2006). K16‐H4 is also the substrate of SIRT2, but the alteration of this HDAC in cancer it is not clear because SIRT2 is frequently downregulated in human gliomas (Hiratsuka et al., 2003).
The upregulation of SIRT1 expression in human cancer can also induce the deregulation of key proteins regulating important cellular functions. For example, the increase of SIRT1 expression in cancer cells produces the deacetylation and inactivation of p53, and inhibiting the tumor suppressor functions of this protein (Chen et al., 2005). Ku70 is other stress response‐related protein regulated by SIRT1. SIRT1 inhibits stress induced cell death through deacetylation of the DNA repair factor Ku70, allowing the long‐term survival of irreparable cancer cells (Cohen et al., 2004). The interaction of SIRT1 with the transcription factor E2F1 decreases its transcriptional activity and apoptotic functions (Wang et al., 2006). Taken together all these data indicate that SIRT1 might be involved in tumorogenesis.
4. Therapeutic implications
The range of proteins and processes regulated by HDACs described above demonstrates that these proteins are key elements in the regulation of gene expression, differentiation and development, and the maintenance of cellular homeostasis. Thus, altered expression of HDACs could play an active role in tumor onset and progression, and make them attractive candidate targets for anticancer drugs and therapies. To date, a wide range of natural and synthetic compounds have been identified that are able to inhibit the activity of class I, II, and IV HDACs. Given their chemical nature and mechanism of inhibition, the HDAC inhibitors can be classified as hydroxamic acids, carboxylic acids, benzamides, epoxides, and cyclic peptides (Villar‐Garea and Esteller, 2004). All these compounds inhibit many of the known mammalian HDACs, although a few exceptions are known. For example, MS‐275 is more active against HDAC1 than against HDAC3 (Hu et al., 2003). Depsipeptide is more strongly active against HDAC1 and HDAC2 than against HDAC4 and HDAC6 (Furumai et al., 2002). The knowledge of the specificity of different HDAC inhibitors against specific HDACs should be very useful in clinical management, but first we have to define the role of different HDACs in cancer. For example, we know that the loss of function of HDAC2 in colon cancer cells with microsatellite instability induces resistance of these cell lines to treatment with the archetypal HDAC inhibitor TSA (Ropero et al., 2006). Since this mutation is present in about 20% of primary tumors with microsatellite instability, this finding may be of significance for the pharmacogenetic selection of patients treated with HDAC inhibitors.
As we have mentioned above, the acetylation state of histones H3 and H4 is associated with active chromatin and gene expression, so we would expect the treatment with HDAC inhibitors to induce a general increase in the gene‐expression profile. However, the available data indicate that HDAC inhibitors cause transcriptional changes of a limited set of genes (Gray et al., 2004; Marks et al., 2004), and that the number of upregulated and downregulated genes is very similar, indicating that chromatin structure and transcription are closely regulated by several mechanisms that involve other histone modifications, DNA methylation, and the binding of a multiprotein complex. Many of the genes affected by HDAC inhibitors are key regulators of crucial biological processes in tumor progression. For example, treatment with HDAC inhibitors induces the expression of the cyclin‐dependent inhibitor p21WAF1. In addition, HDAC inhibitors exhibit antiangiogenic activities by downregulating proangiogenic genes, such as vascular endothelial growth factor (VEGF), basic fibroblast growth factor (bFGF), and hypoxia‐inducible factor 1‐α (Sasakawa et al., 2003; Qian et al., 2006).
As discussed above, histone acetylation is involved in nuclear functions other than gene transcription, including DNA repair and replication. Another effect of HDAC inhibitors is the inhibition of DNA repair, so they can also sensitize cancer cells to chemotherapy and radiotherapy by increasing the effects on DNA damage induced by these treatments (Kim et al., 2003; Munshi et al., 2005).
The knowledge of the implications of sirtuins in cancer has inspired to develop and study sirtuins inhibitors. In addition of nicotinamide, that was the first sirtuin inhibitor identified, in the last years a growing number of sirtuins inhitors have been identified, including sirtinol, cambinol, dihydrocoumarin and indoles. All these compounds have a common activity: inhibition of cell proliferation by inducing a increase in the acetylated levels of K16‐H4 and the reexpression of silenced tumor suppressor genes in cancer (Pruitt et al., 2006). The eventual translation of these compounds into the clinic may represent a new direction in cancer treatment.
Taken together, the results and the mechanism described indicate that HDACs are excellent targets for cancer treatment. The efficacy of HDAC inhibitors such as TSA, SAHA, and MS‐275 as anticancer agents has been demonstrated in a wide range of hematological and solid tumor cell lines, and in experimental animal models. These inhibitors showed potent antitumoral activity with no apparent toxicity in an animal model (Saito et al., 1999). Once the preclinical studies have given satisfactory results, the next step has been to test their efficacy in clinical trials. Currently, phase I‐II clinical trials are evaluating the efficacy of several HDAC inhibitors for the treatment of hematological and solid tumors as monotherapy, or in combination with other therapeutic agents (Kelly and Marks, 2005; Lin et al., 2006); and SAHA has already reached the bedside of cancer patients with its approval by the U.S. Food and Drug Administration for the treatment of cutaneous T‐cell lymphoma, a form of non‐Hodgkin's lymphoma.
Ropero Santiago, Esteller Manel, (2007), The role of histone deacetylases (HDACs) in human cancer, Molecular Oncology, 1, doi:10.1016/j.molonc.2007.01.001.
References
- Bai, S. , Ghoshal, K. , Datta, J. , Majumder, S. , Yoon, S.O. , Jacob, S.T. , 2005. DNA methyltransferase 3b regulates nerve growth factor-induced differentiation of PC12 cells by recruiting histone deacetylase 2. Mol. Cell Biol.. 25, 751–766. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Bouras, T. , Fu, M. , Sauve, A.A. , Wang, F. , Quong, A.A. , Perkins, N.D. , Hay, R.T. , Gu, W. , Pestell, R.G. , 2005. SIRT1 deacetylation and repression of p300 involves lysine residues 1020/1024 within the cell cycle regulatory domain 1. J. Biol. Chem.. 280, 10264–10276. [DOI] [PubMed] [Google Scholar]
- Bradbury, C.A. , Khanim, F.L. , Hayden, R. , Bunce, C.M. , White, D.A. , Drayson, M.T. , Craddock, C. , Turner, B.M. , 2005. Histone deacetylases in acute myeloid leukaemia show a distinctive pattern of expression that changes selectively in response to deacetylase inhibitors. Leukemia. 19, 1751–1759. [DOI] [PubMed] [Google Scholar]
- Brehm, A. , Miska, E.A. , McCance, D.J. , Reid, J.L. , Bannister, A.J. , Kouzarides, T. , 1998. Retinoblastoma protein recruits histone deacetylase to repress transcription. Nature. 391, 597–601. [DOI] [PubMed] [Google Scholar]
- Chen, W.Y. , Wang, D.H. , Yen, R.C. , Luo, J. , Gu, W. , Baylin, S.B. , 2005. Tumor suppressor HIC1 directly regulates SIRT1 to modulate p53-dependent DNA-damage responses. Cell. 123, 437–448. [DOI] [PubMed] [Google Scholar]
- Choi, J.H. , Kwon, H.J. , Yoon, B.I. , Kim, J.H. , Han, S.U. , Joo, H.J. , Kim, D.Y. , 2001. Expression profile of histone deacetylase 1 in gastric cancer tissues. Jpn. J. Cancer Res.. 92, 1300–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christofori, G. , Semb, H. , 1999. The role of the cell-adhesion molecule E-cadherin as a tumour-suppressor gene. Trends Biochem. Sci.. 24, 73–76. [DOI] [PubMed] [Google Scholar]
- Cohen, H.Y. , Miller, C. , Bitterman, K.J. , Wall, N.R. , Hekking, B. , Kessler, B. , Howitz, K.T. , Gorospe, M. , de Cabo, R. , Sinclair, D.A. , 2004. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science. 305, 390–392. [DOI] [PubMed] [Google Scholar]
- Espada, J. , Ballestar, E. , Fraga, M.F. , Villar-Garea, A. , Juarranz, A. , Stockert, J.C. , Robertson, K.D. , Fuks, F. , Esteller, M. , 2004. Human DNA methyltransferase 1 is required for maintenance of the histone H3 modification pattern. J. Biol. Chem.. 279, 37175–37184. [DOI] [PubMed] [Google Scholar]
- Esteller, M. , 2002. CpG island hypermethylation and tumor suppressor genes: a booming present, a brighter future. Oncogene. 21, 5427–5440. [DOI] [PubMed] [Google Scholar]
- Ferreira, R. , Magnaghi-Jaulin, L. , Robin, P. , Harel-Bellan, A. , Trouche, D. , 1998. The three members of the pocket proteins family share the ability to repress E2F activity through recruitment of a histone deacetylase. Proc. Natl. Acad. Sci. USA. 95, 10493–10498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraga, M.F. , Ballestar, E. , Villar-Garea, A. , Boix-Chornet, M. , Espada, J. , Schotta, G. , Bonaldi, T. , Haydon, C. , Ropero, S. , Petrie, K. , Iyer, N.G. , Perez-Rosado, A. , Calvo, E. , Lopez, J.A. , Cano, A. , Calasanz, M.J. , Colomer, D. , Piris, M.A. , Ahn, N. , Imhof, A. , Caldas, C. , Jenuwein, T. , Esteller, M. , 2005. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat. Genet.. 37, 391–400. [DOI] [PubMed] [Google Scholar]
- Frolov, M.V. , Dyson, N.J. , 2004. Molecular mechanisms of E2F-dependent activation and pRB-mediated repression. J. Cell Sci.. 117, 2173–2181. [DOI] [PubMed] [Google Scholar]
- Fu, M. , Liu, M. , Sauve, A.A. , Jiao, X. , Zhang, X. , Wu, X. , Powell, M.J. , Yang, T. , Gu, W. , Avantaggiati, M.L. , Pattabiraman, N. , Pestell, T.G. , Wang, F. , Quong, A.A. , Wang, C. , Pestell, R.G. , 2006. Hormonal control of androgen receptor function through SIRT1. Mol. Cell Biol.. 26, 8122–8135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furumai, R. , Matsuyama, A. , Kobashi, N. , Lee, K.H. , Nishiyama, M. , Nakajima, H. , Tanaka, A. , Komatsu, Y. , Nishino, N. , Yoshida, M. , Horinouchi, S. , 2002. FK228 (depsipeptide) as a natural prodrug that inhibits class I histone deacetylases. Cancer Res.. 62, 4916–4921. [PubMed] [Google Scholar]
- Glass, C.K. , Rosenfeld, M.G. , 2000. The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev.. 14, 121–141. [PubMed] [Google Scholar]
- Gray, S.G. , Qian, C.N. , Furge, K. , Guo, X. , Teh, B.T. , 2004. Microarray profiling of the effects of histone deacetylase inhibitors on gene expression in cancer cell lines. Int. J. Oncol.. 24, 773–795. [DOI] [PubMed] [Google Scholar]
- Gui, C.Y. , Ngo, L. , Xu, W.S. , Richon, V.M. , Marks, P.A. , 2004. Histone deacetylase (HDAC) inhibitor activation of p21WAF1 involves changes in promoter-associated proteins, including HDAC1. Proc. Natl. Acad. Sci. USA. 101, 1241–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajra, K.M. , Fearon, E.R. , 2002. Cadherin and catenin alterations in human cancer. Genes Chromosomes Cancer. 34, 255–268. [DOI] [PubMed] [Google Scholar]
- Halkidou, K. , Gaughan, L. , Cook, S. , Leung, H.Y. , Neal, D.E. , Robson, C.N. , Halkidou, K. , 2004. Upregulation and nuclear recruitment of HDAC1 in hormone refractory prostate cancer. Prostate. 59, 177–189. [DOI] [PubMed] [Google Scholar]
- Hiratsuka, M. , Inoue, T. , Toda, T. , Kimura, N. , Shirayoshi, Y. , Kamitani, H. , Watanabe, T. , Ohama, E. , Tahimic, C.G. , Kurimasa, A. , Oshimura, M. , 2003. Proteomics-based identification of differentially expressed genes in human gliomas: down-regulation of SIRT2 gene. Biochem. Biophys. Res. Commun.. 309, 558–566. [DOI] [PubMed] [Google Scholar]
- Hu, E. , Dul, E. , Sung, C.M. , Chen, Z. , Kirkpatrick, R. , Zhang, G.F. , Johanson, K. , Liu, R. , Lago, A. , Hofmann, G. , Macarron, R. , de los Frailes, M. , Perez, P. , Krawiec, J. , Winkler, J. , Jaye, M. , 2003. Identification of novel isoform-selective inhibitors within class I histone deacetylases. J. Pharmacol. Exp. Ther.. 307, 720–728. [DOI] [PubMed] [Google Scholar]
- Huang, B.H. , Laban, M. , Leung, C.H. , Lee, L. , Lee, C.K. , Salto-Tellez, M. , Raju, G.C. , Hooi, S.C. , 2005. Inhibition of histone deacetylase 2 increases apoptosis and p21Cip1/WAF1 expression, independent of histone deacetylase 1. Cell Death Differ.. 12, 395–404. [DOI] [PubMed] [Google Scholar]
- Iizuka, M. , Smith, M.M. , 2003. Functional consequences of histone modifications. Curr. Opin. Genet. Dev.. 13, 154–160. [DOI] [PubMed] [Google Scholar]
- Johnstone, R.W. , 2002. Histone-deacetylase inhibitors: novel drugs for the treatment of cancer. Nat. Rev. Drug Discov.. 1, 287–299. [DOI] [PubMed] [Google Scholar]
- Jones, P.L. , Veenstra, G.J. , Wade, P.A. , Vermaak, D. , Kass, S.U. , Landsberger, N. , Strouboulis, J. , Wolffe, A.P. , 1998. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat. Genet.. 19, 187–191. [DOI] [PubMed] [Google Scholar]
- Juan, L.J. , Shia, W.J. , Chen, M.H. , Yang, W.M. , Seto, E. , Lin, Y.S. , Wu, C.W. , 2000. Histone deacetylases specifically down-regulate p53-dependent gene activation. J. Biol. Chem.. 275, 20436–20443. [DOI] [PubMed] [Google Scholar]
- Kawai, H. , Li, H. , Avraham, S. , Jiang, S. , Avraham, H.K. , 2003. Overexpression of histone deacetylase HDAC1 modulates breast cancer progression by negative regulation of estrogen receptor alpha. Int. J. Cancer. 107, 353–358. [DOI] [PubMed] [Google Scholar]
- Kelly, W.K. , Marks, P.A. , 2005. Drug insight: Histone deacetylase inhibitors–development of the new targeted anticancer agent suberoylanilide hydroxamic acid. Nat. Clin. Pract. Oncol.. 2, 150–157. [DOI] [PubMed] [Google Scholar]
- Kim, M.S. , Blake, M. , Baek, J.H. , Kohlhagen, G. , Pommier, Y. , Carrier, F. , 2003. Inhibition of histone deacetylase increase cytotoxicity to anticancer drugs targeting DNA. Cancer Res.. 63, 7291–7300. [PubMed] [Google Scholar]
- Kuzmichev, A. , Margueron, R. , Vaquero, A. , Preissner, T.S. , Scher, M. , Kirmizis, A. , Ouyang, X. , Brockdorff, N. , Abate-Shen, C. , Farnham, P. , Reinbe, D. , 2005. Composition and histone substrates of polycomb repressive group complexes change during cellular differentiation. Proc. Natl. Acad. Sci. USA. 102, 1859–1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, H.Y. , Chen, C.S. , Lin, S.P. , Weng, J.R. , Chen, C.S. , 2006. Targeting histone deacetylase in cancer therapy. Med. Res. Rev.. 26, 397–413. [DOI] [PubMed] [Google Scholar]
- Lin, R.J. , Sternsdorf, T. , Tini, M. , Evans, R.M. , 2001. Transcriptional regulation in acute promyelocytic leukaemia. Oncogene. 20, 7204–7216. [DOI] [PubMed] [Google Scholar]
- Luo, J. , Su, F. , Chen, D. , Shiloh, A. , Gu, W. , 2000. Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature. 408, 377–381. [DOI] [PubMed] [Google Scholar]
- Macaluso, M. , Cinti, C. , Russo, G. , Russo, A. , Giordano, A. , 2003. pRb2/p130-E2F4/5-HDAC1-SUV39H1-p300 and pRb2/p130-E2F4/5-HDAC1-SUV39H1-DNMT1 multimolecular complexes mediate the transcription of estrogen receptor-alpha in breast cancer. Oncogene. 22, 3511–3517. [DOI] [PubMed] [Google Scholar]
- Marks, P.A. , Richon, V.M. , Miller, T. , Kelly, W.K. , 2004. Histone deacetylase inhibitors. Adv. Cancer Res.. 91, 137–168. [DOI] [PubMed] [Google Scholar]
- Minucci, S. , Pelicci, P.G. , 2006. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat. Rev. Cancer. 6, 38–51. [DOI] [PubMed] [Google Scholar]
- Munshi, A. , Kurland, J.F. , Nishikawa, T. , Tanaka, T. , Hobbs, M.L. , Tucker, S.L. , Ismail, S. , Stevens, C. , Meyn, R.E. , 2005. Histone deacetylase inhibitors radiosensitize human melanoma cells by suppressing DNA repair activity. Clin. Cancer Res.. 11, 4912–4922. [DOI] [PubMed] [Google Scholar]
- Nan, X. , Ng, H.H. , Johnson, C.A. , Laherty, C.D. , Turner, B.M. , Eisenman, R.N. , Bird, A. , 1998. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 393, 386–389. [DOI] [PubMed] [Google Scholar]
- Ozdag, H. , Teschendorff, A.E. , Ahmed, A.A. , Hyland, S.J. , Blenkiron, C. , Bobrow, L. , Veerakumarasivam, A. , Burtt, G. , Subkhankulova, T. , Arends, M.J. , Collins, V.P. , Bowtell, D. , Kouzarides, T. , Brenton, J.D. , Caldas, C. , 2006. Differential expression of selected histone modifier genes in human solid cancers. BMC Genomics. 7, 90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peinado, H. , Ballestar, E. , Esteller, M. , Cano, A. , 2004. Snail mediates E-cadherin repression by the recruitment of the Sin3A/histone deacetylase 1 (HDAC1)/HDAC2 complex. Mol. Cell Biol.. 24, 306–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polo, S.E. , Almouzni, G. , 2005. Histone metabolic pathways and chromatin assembly factors as proliferation markers. Cancer Lett.. 220, 1–9. [DOI] [PubMed] [Google Scholar]
- Pruitt, K. , Zinn, R.L. , Ohm, J.E. , McGarvey, K.M. , Kang, S.H. , Watkins, D.N. , Herman, J.G. , Baylin, S.B. , 2006. Inhibition of SIRT1 reactivates silenced cancer genes without loss of promoter DNA hypermethylation. PLoS Genet.. 2, e40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian, D.Z. , Kato, Y. , Shabbeer, S. , Wei, Y. , Verheul, H.M. , Salumbides, B. , Sanni, T. , Atadja, P. , Pili, R. , 2006. Targeting tumor angiogenesis with histone deacetylase inhibitors: the hydroxamic acid derivative LBH589. Clin. Cancer Res.. 12, 634–642. [DOI] [PubMed] [Google Scholar]
- Robertson, K.D. , Ait-Si-Ali, S. , Yokochi, T. , Wade, P.A. , Jones, P.L. , Wolffe, A.P. , 2000. DNMT1 forms a complex with Rb, E2F1 and HDAC1 and represses transcription from E2F-responsive promoters. Nat. Genet.. 25, 338–342. [DOI] [PubMed] [Google Scholar]
- Ropero, S. , Fraga, M.F. , Ballestar, E. , Hamelin, R. , Yamamoto, H. , Boix-Chornet, M. , Caballero, R. , Alaminos, M. , Setien, F. , Paz, M.F. , Herranz, M. , Palacios, J. , Arango, D. , Orntoft, T.F. , Aaltonen, L.A. , Schwartz, S. , Esteller, M. , 2006. A truncating mutation of HDAC2 in human cancers confers resistance to histone deacetylase inhibition. Nat. Genet.. 38, 566–569. [DOI] [PubMed] [Google Scholar]
- Saito, A. , Yamashita, T. , Mariko, Y. , Nosaka, Y. , Tsuchiya, K. , Ando, T. , Suzuki, T. , Tsuruo, T. , Nakanishi, O. , 1999. A synthetic inhibitor of histone deacetylase, MS-27-275, with marked in vivo antitumor activity against human tumors. Proc. Natl. Acad. Sci. USA. 96, 4592–4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasakawa, Y. , Naoe, Y. , Noto, T. , Inoue, T. , Sasakawa, T. , Matsuo, M. , Manda, T. , Mutoh, S. , 2003. Antitumor efficacy of FK228, a novel histone deacetylase inhibitor, depends on the effect on expression of angiogenesis factors. Biochem. Pharmacol.. 66, (6) 897–906. [DOI] [PubMed] [Google Scholar]
- Siddiqui, H. , Solomon, D.A. , Gunawardena, R.W. , Wang, Y. , Knudsen, E.S. , 2003. Histone deacetylation of RB-responsive promoters: requisite for specific gene repression but dispensable for cell cycle inhibition. Mol. Cell Biol.. 23, 7719–7731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, J. , Noh, J.H. , Lee, J.H. , Eun, J.W. , Ahn, Y.M. , Kim, S.Y. , Lee, S.H. , Park, W.S. , Yoo, N.J. , Lee, J.Y. , Nam, S.W. , 2005. Increased expression of histone deacetylase 2 is found in human gastric cancer. APMIS. 113, 264–268. [DOI] [PubMed] [Google Scholar]
- Vaquero, A. , Scher, M. , Lee, D. , Erdjument-Bromage, H. , Tempst, P. , Reinberg, D. , 2004. Human SirT1 interacts with histone H1 and promotes formation of facultative heterochromatin. Mol. Cell. 16, 93–105. [DOI] [PubMed] [Google Scholar]
- Vaziri, H. , Dessain, S.K. , Ng Eaton, E. , Imai, S.I. , Frye, R.A. , Pandita, T.K. , Guarente, L. , Weinberg, R.A. , 2001. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 107, 149–159. [DOI] [PubMed] [Google Scholar]
- Vidanes, G.M. , Bonilla, C.Y. , Toczyski, D.P. , 2005. Complicated tails: histone modifications and the DNA damage response. Cell. 121, 973–976. [DOI] [PubMed] [Google Scholar]
- Villar-Garea, A. , Esteller, M. , 2004. Histone deacetylase inhibitors: understanding a new wave of anticancer agents. Int. J. Cancer. 112, 171–178. [DOI] [PubMed] [Google Scholar]
- Wang, C. , Chen, L. , Hou, X. , Li, Z. , Kabra, N. , Ma, Y. , Nemoto, S. , Finkel, T. , Gu, W. , Cress, W.D. , Chen, J. , 2006. Interactions between E2F1 and SirT1 regulate apoptotic response to DNA damage. Nat. Cell Biol.. 8, 1025–1031. [DOI] [PubMed] [Google Scholar]
- Wang, J. , Hoshino, T. , Redner, R.L. , Kajigaya, S. , Liu, J.M. , 1998. ETO, fusion partner in t(8;21) acute myeloid leukemia, represses transcription by interaction with the human N-CoR/mSin3/HDAC1 complex. Proc. Natl. Acad. Sci. USA. 95, 10860–10865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson, A.J. , Byun, D.S. , Popova, N. , Murray, L.B. , L'Italien, K. , Sowa, Y. , Arango, D. , Velcich, A. , Augenlicht, L.H. , Mariadason, J.M. , 2006. Histone deacetylase 3 (HDAC3) and other class I HDACs regulate colon cell maturation and p21 expression and are deregulated in human colon cancer. J. Biol. Chem.. 281, 13548–13558. [DOI] [PubMed] [Google Scholar]
- Yasui, W. , Oue, N. , Ono, S. , Mitani, Y. , Ito, R. , Nakayama, H. , 2003. Histone acetylation and gastrointestinal carcinogenesis. Ann. N. Y. Acad. Sci.. 983, 220–231. [DOI] [PubMed] [Google Scholar]
- Yeung, F. , Hoberg, J.E. , Ramsey, C.S. , Keller, M.D. , Jones, D.R. , Frye, R.A. , Mayo, M.W. , 2004. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J.. 23, 2369–2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Z. , Yamashita, H. , Toyama, T. , Sugiura, H. , Omoto, Y. , Ando, Y. , Mita, K. , Hamaguchi, M. , Hayashi, S. , Iwase, H. , 2004. HDAC6 expression is correlated with better survival in breast cancer. Clin. Cancer Res.. 10, 6962–6968. [DOI] [PubMed] [Google Scholar]
- Zhang, Z. , 2005. Quantitation of HDAC1 mRNA expression in invasive carcinoma of the breast. Breast Cancer Res. Treat.. 94, 11–16. [DOI] [PubMed] [Google Scholar]
- Zhu, P. , Martin, E. , Mengwasser, J. , Schlag, P. , Janssen, K.-P. , Göttlicher, M. , 2004. Induction of HDAc2 expression upon loss of APC in colorectal tumorogenesis. Cancer Cell. 5, 455–463. [DOI] [PubMed] [Google Scholar]