

Abstract
The development of cancer is driven by the accumulation of scores of alterations affecting the structure and function of the genome. Equally important in this process are genetic alterations and epigenetic changes. Whereas the former disrupt normal patterns of gene expression, sometimes leading to the expression of abnormal, constitutively active proteins, the latter deregulate the mechanisms such as transcriptional control leading to the inappropriate silencing or activation of cancer‐associated genes. Both types of changes are inheritable at the cellular level, thus contributing to the clonal expansion of cancer cells. In this review, we summarize current knowledge on how genetic alterations in oncogenes or tumour suppressor genes, as well as epigenetic changes, can be exploited in the clinics as biomarkers for cancer detection, diagnosis and prognosis. We propose a rationale for identifying alterations that may have a functional impact within a background of “passenger” alterations that may occur solely as the consequence of deregulated genetic and epigenetic stability. Such functional alterations may represent candidates for targeted therapeutic approaches.
Keywords: Genetic changes, Epigenetic changes, Cancer biomarkers, Molecular signature, Prognosis
1. Introduction
Carcinogenesis proceeds through the accumulation of genetic and epigenetic changes that allow cells to break free from the tight network of controls that regulate the homeostatic balance between cell proliferation and cell death (Hanahan and Weinberg, 2000). The experimental work of Weinberg and his collaborators has established that transforming a primary cell into a malignant one in vitro requires alterations in the functionality of a handful of mechanisms by which cells regulate their growth, division, position, differentiation and life span (Elenbaas et al., 2001). In the early 90s, studies on cancer tissues have popularized the view that stepwise acquisition of genetic alterations could determine the morphological changes that accompany cancer progression (Fearon and Vogelstein, 1990). More recently, however, this “sequential” concept has been challenged by the observation that individual tumours show a great heterogeneity in their patterns of genetic alterations, epigenetic changes and gene expression, even within homogenous histological groups (Feinberg et al., 2006). In addition, the fact that malignant phenotypes can be maintained solely by a tiny sub‐population of cell with stem cell properties (Kim et al., 2005; Pardal et al., 2003; Singh et al., 2004), argue that tumour heterogeneity is not simply a consequence of mutation acquisition, leading to a clonal expansion of mutated cells. Moreover, the notion that early‐stage cancers are systematically less aggressive than late‐stage cancers is being challenged by the identification of early‐stage cancers with similar gene expression profiles to fully metastatic cancers (Schedin and Elias, 2004). Thus, it is now clear that there are multiple mechanisms by which cells can progress into malignancy, and that it is the concerted accumulation and functional cooperation between genetic and epigenetic changes, rather than their order of occurrence, that drives carcinogenesis.
The elucidation of the human genome sequence has made it possible to identify genetic alterations in cancers in unprecedented detail. In a recent study, the sequence of 13,023 well‐annotated human genes has been analysed in 11 breast and 11 colorectal cancer specimens (Sjoblom et al., 2006). Based on this analysis, it appears that individual tumours may accumulate an average of approximately 90 mutant genes but that only a subset of these are likely to contribute to the neoplastic process. Using stringent criteria to delineate this subset, the authors identified that tumours contained, on average, 11 genes that were mutated at significant frequency, including well‐known oncogenes and tumour suppressor genes, as well as many new genes predicted to affect a wide range of cellular functions, such as transcription, cell adhesion, and invasion.
In addition to genetic changes, epigenetic events have emerged as key mechanisms in the development of human cancer. The research on epigenetics has become increasingly visible due to the remarkable progress in our understanding of the critical role of epigenetic mechanisms in normal cellular processes and the abnormal events that lead to diseases, most notably to cancer. Thus, genetic and epigenetic events represent two complementary mechanisms that are involved at every step of carcinogenesis, from responses to carcinogen exposures to progression into malignancy (Figure 1). In the present review, we summarize current concepts on genetic and epigenetic changes associated with cancer, and we discuss their potential relevance as biomarkers for cancer detection, diagnosis and prognosis.
Figure 1.
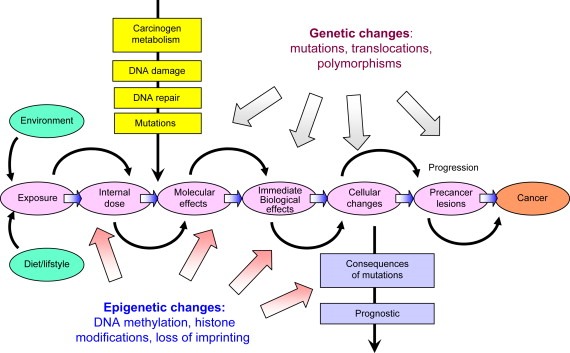
Cancers are the consequence of combined genetic and epigenetic changes induced by environmental/dietary factors that trigger inappropriate activation or inactivation of specific genes leading to neoplastic transformation.
2. Genetic alterations
In cancer cells, somatic mutations occur and accumulate at a rate significantly higher than in normal cells, a property referred to as “Mutator Phenotype”. This ability of cancer cells to accumulate mutations is critical for the development of cancer as well as for the rapid development of resistance to cytotoxic cancer treatments (Bielas et al., 2006). The Mutator Phenotype can be caused by a number of mechanisms, such as defects in cell‐cycle regulation, apoptosis, specific DNA repair pathways, or error‐prone DNA polymerase, and it can have its source in inherited genetic defects that make subjects prone to specific cancers. For example, patients with HNPCC (hereditary non‐polyposis colorectal cancer) exhibit microsatellite instability in relation with mutations in genes involved in the DNA mismatch repair system (Abdel‐Rahman et al., 2006; Fishel and Wilson, 1997). However, an important subset of colorectal cancers exhibiting microsatelitte instability did not contain mutations in mismatch repair genes (Thibodeau et al., 1996), but instead these genes were found silenced by promoter hypermethylation (Esteller et al., 1998; Herman et al., 1998; Kane et al., 1997).
Mutations in cancer cells cover a wide range of structural alterations in DNA, including changes in chromosomes copy numbers or chromosomal alterations encompassing millions of base‐pairs such as translocations, deletions or amplifications, as well as smaller changes in nucleotide sequences such as point mutations affecting a single nucleotide at a critical position of a cancer‐related gene (Sugimura et al., 1992). These different kinds of alterations often co‐exist within a single tumour.
Tumours as well as precursor lesions harbor heterogeneous cell populations, including normal cells such as stromal or inflammatory cells. When analysing such lesions, the presence of these non‐tumour cells may mask the detection of genetic alterations in cancer cell populations. The use of laser‐guided microdissection allows to selectively isolate groups of cells corresponding to specific population. This approach, coupled to refined, sensitive PCR‐based detection methods, provides the possibility of performing high resolution, molecular profiling of selected cell groups (Garnis et al., 2004). In addition, sensitive PCR methods make it possible to detect small amounts of DNA containing somatic genetic alterations in biological fluids such as saliva or plasma, as well as in exfoliated cells from diverse origins, thus providing opportunities for detecting cancer of precancerous lesions based on non‐invasive genetic screening for somatic mutations (Gormally et al., in press).
Several databases are available that compile mutations in cancer genes reported in the literature. The most extensive of these databases is the COSMIC database maintained at the Sanger Institute, Hinxton, UK (//www.sanger.ac.uk/genetics/CGP/cosmic/) (Forbes et al., 2006). This database contains the description of over 40,000 individual mutations occurring in a set of 291 genes that have been identified as mutated and causally implicated in cancer development (Futreal et al., 2004). The list of these genes is available at http://www.sanger.ac.uk/genetics/CGP/Census/. The most common mutation class is chromosomal translocation that creates a chimeric gene or apposes a gene to the regulatory elements of another gene, a genetic event that is common in cancers of the haematopoietic system. From a functional point of view, the most common domain that is encoded by cancer genes is the protein kinase domain. An example of a cancer gene commonly mutated in such a domain is EGFR, encoding the epidermal growth factor (EGF) receptor, which is frequently mutated in adenocarcinoma of the lung in never smokers (Shigematsu and Gazdar, 2006). Other commonly mutated functional domains are involved in DNA binding and transcriptional regulation. A typical example of such a cancer gene is the tumour suppressor TP53, which is altered by mutation and/or loss of alleles in about half of human cancer (Hainaut and Hollstein, 2000).
Despite intensive efforts for describing and cataloguing mutations, their significance as biomarkers in the clinics largely remains to be determined. So far, most studies have consisted of the analysis of retrospective clinical series that lack the design and power to assess the value of mutation detection for cancer diagnosis or prognosis. In this section, we briefly summarize current knowledge on the significance as biomarkers of mutations on TP53 and EGFR, as examples of clinically relevant mutations in human cancers.
2.1. TP53 mutations
TP53 encodes a 393‐residue transcription factor that regulates the expression of multiple genes involved in anti‐proliferative responses under certain stress conditions, such as in particular DNA damage. This gene thus functions as an important safeguard against untimely cell proliferation in genotoxic conditions. The database maintained at the International Agency for Research on Cancer (IARC TP53 database, http://www‐p53.iarc.fr/) compiles about 24,000 somatic TP53 mutations detected in almost every type of human cancers (Olivier et al., 2002). The majority of these mutations are missense substitutions (74%) resulting from single nucleotide substitutions that cluster within exons 5–8, altering the conformation and/or biochemical activity of the protein domain which is involved in specific DNA binding (Figure 2A). In the early 90s, it became apparent that TP53 mutations were occurring in a non‐random manner, and that there were significant differences in mutations patterns, in particular between cancers that are strongly associated with exposure to environmental mutagens. Thus, one of the main applications of TP53 mutations as biomarker is in molecular epidemiology, as potential reporter of specific mutagenic exposures. Indeed, there is evidence that mutation patterns in common cancers show significant differences in relation with geographic variations in incidence, perhaps indicative of differences in exposure to specific environmental carcinogens (Hainaut and Hollstein, 2000).
Figure 2.
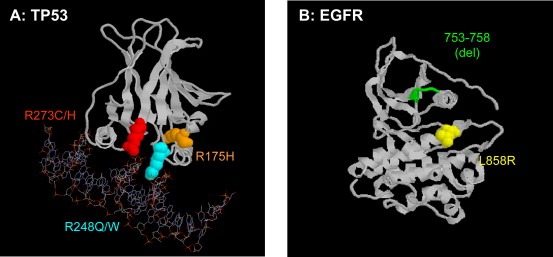
Most common mutations in TP53 (A) and EGFR (B). “Hotspot” mutations are colour‐coded. The region encompassing residues 753–758 in EGFR is often targeted by small, in‐frame deletions.
Several studies have addressed the significance of TP53 mutation detection in body fluids for early cancer detection. For example, mutations are sometimes detectable in DNA from sputum or from exfoliated bronchial cells in subjects with chronic obstructive pulmonary disease (COPD) (Wang et al., 2006). Potentially the most interesting source of DNA for early cancer detection is plasma, which contains small amounts of free DNA fragments shed by apoptotic or necrotic normal and cancer cells. TP53 mutations in plasma DNA have been reported in patients with cancers of the colon, pancreas, lung, and liver. For example, a specific, aflatoxin‐induced TP53 mutation (at codon 249) has been detected in the plasma of non‐cancer subjects who were chronic carriers of Hepatitis B virus, up to 5 years ahead of the development of liver cancer (Jackson et al., 2003). In a large prospective study, we have shown that the presence of TP53 and/or KRAS mutations in plasma DNA of healthy subjects was predictive of the risk of bladder cancer (Gormally et al., 2006). It is, however, unclear whether mutant TP53 plasma DNA originates from clinically undetected cancer or precancerous lesions, or from normal cells undergoing exposure to mutagens.
A large number of studies have investigated the predictive value of TP53 mutation status for tumor response to treatment and patient outcome in various cancers. In most cancers, presence of a mutation is correlated with decreased survival or poor response to treatment (for detailed see http://www‐p53.iarc.fr/Somatic.html). For example, mutations within the DNA‐binding domain have been repeatedly associated with bad prognosis in several types of cancer.
Based on data compiled from different European case series, we have recently performed a detailed assessment of the prognostic significance of TP53 mutations in 1794 patients with breast cancers (Olivier et al., 2006). In this series, the overall TP53 mutation prevalence in exons 5–8 was 21%. Results from this large cohort showed that TP53 mutation was a predictor of poor overall survival independently of the currently available prognostic factors such as tumour size, node status and estrogen and progesterone receptor contents. Missense mutations located within the DNA‐binding motifs and non‐missense (truncating) mutations were associated with the worse outcome. Recent studies on gene expression profiling using micro‐arrays have shown that TP53 mutation is associated with specific expression profiles. While tumour classification based on these profiles was a stronger predictor of outcome than any of the classical clinicopathological markers, TP53 mutation was strongly correlated with the profiles associated with bad outcome.
2.2. EGFR mutations
The epidermal growth factor receptor (EGFR) family corresponds to subclass I of the receptor Tyrosine Kinase (TK) superfamily. This subclass consists in four genes encoding monomeric transmembrane TK receptors, EGFR (ErbB1), HER2 (ErbB2), EGFR3 (ErbB3), and EGFR4 (ErbB4). Upon ligand binding, these receptors undergo homo‐ or heterodimerization and activation of the conserved intracellular kinase domain, resulting in activation of multiple downstream pathways mediating proliferative and anti‐apoptotic responses. EGFR and HER2 are often altered in diverse human cancers, by amplification, point mutation, or both. Amplifications of EGFR have been detected in brain cancers and in a small proportion of a number of epithelial cancers such as squamous oral or esophageal cancer. Amplification and overexpression of HER2 are a frequent event in breast and ovarian cancer (Harari and Yarden, 2000).
In recent years, mutations in the TK domain of EGFR have attracted considerable interest due to their potential clinical significance in predicting the response of lung cancer patients to small molecule Tyrosine Kinase Inhibitors (TKIs) such as erlotinib or gefinitib (Paez et al., 2004, 2004, 2005, 2006, 2005). A database of EGFR mutations in non‐small cell lung carcinomas is maintained at the City of Hope Hospital (http://www.cityofhope.org/cmdl/egfr_db/index.html). Activating mutations in the TK domain of EGFR occur predominantly in lung adenocarcinomas of never smokers, and are mutually exclusive with mutation in KRAS, which encodes a protein involved in signal transduction downstream of EGFR (Shigematsu and Gazdar, 2006). Thus, mutation in EGFR may represent interesting biomarker of a lung carcinogenic process different from the one initiated by exposure to tobacco smoke.
EGFR mutations concentrate in the first four exons of the TK domain (exons 18–21) and include point mutations, deletions, and insertions. The main types of mutations are deletions in exon 19 and a single point mutation in exon 21, L858R, which together account for over 80% of all mutations (Figure 2B). Occasional point mutations occur at several other sites. These activating mutations alter the structure of the region of the TK domain involved in ATP binding, thus conferring ligand independence and selective activation of downstream Akt and STAT pathways, which promote cell survival and induce a dependence on EGFR signals. Switching off this dependency by TKI may thus represent a powerful therapeutic approach. Initial clinical studies have shown occasional spectacular tumour regression in patients treated with TKI. However, recent clinical trials have shown that EGFR mutation per se does not fully predict the patient's response to TKI. Increased EGFR gene copy number and co‐expression of other members of the EGFR family may be important in determining the sensitivity and clinical response to TKI (Takano et al., 2005). Furthermore, treatment with TKI might also be beneficial in some patients without characterized molecular alteration in EGFR family members. Recently, a classification based on gene expression patterns has been shown to capture the majority of tumours with high levels of EGFR activation independently of the molecular mechanism of activation, making it possible to develop predictive models of EGFR TKI sensitivity that is not achieved with single biomarkers or clinical characteristics (Balko et al., 2006).
3. Epigenetic alterations
The term ‘epigenetic’ defines all heritable changes in gene expression and chromatin structure that are not coded in the DNA sequence itself. With minor exceptions (T‐ and B‐cells of the immune system), all differentiation processes are triggered and maintained through epigenetic mechanisms. Epigenetic inheritance includes DNA methylation, histone modifications and RNA‐mediated silencing (Table 1), all of which are essential mechanisms that allow the stable propagation of gene activity states from one generation of cells to the next (Feinberg et al., 2006; Jaenisch and Bird, 2003). Disruption of any of these three distinct and mutually reinforcing epigenetic mechanisms leads to inappropriate gene expression, resulting in cancer development and other ‘epigenetic diseases’ (Egger et al., 2004; Feinberg et al., 2006; Feinberg and Tycko, 2004; Jones and Baylin, 2002). Despite the great deal of uncertainty about the precise underlying mechanisms, in recent years we have witnessed a tremendous pace of the research on epigenetics and this field holds a great promise to advance our understanding of tumorigenesis and help in the development of strategies for cancer treatment and prevention (Table 2).
Table 1.
Epigenetic changes and possible mechanisms by which they promote tumorigenesis
| Epigenetic change | Putative mechanism | Biological consequence |
|---|---|---|
| DNA hypomethylation | Activation of cellular oncogenes | Increased proliferation, growth advantage |
| Activation of transposable element | Genomic instability, transcriptional noise | |
| DNA hypermethylation | De novo hypermethylation of CpG islands within gene promoters leading to silencing of tumour suppressors and cancer‐associated genes | Genomic and chromosomal instability, increased proliferation, growth advantage |
| Loss of imprinting (LOI) | Reactivation of silent alleles, biallelic expression of imprinted genes | Expansion of precursor cell population |
| Relaxation of X‐chromosome inactivation | Mechanisms is unknown but it appears to be age‐related | Altered gene dosage, growth advantage |
| Histone acetylation | Gain‐of‐function | Activation of tumour promoting genes |
| Loss‐of‐function | Defects in DNA repair and checkpoints | |
| Histone deacetylation | Silencing of tumour suppressor genes | Genomic instability, increased proliferation |
| Histone methylation | Loss of heritable patterns of gene expression (‘cellular memory’) | Genomic instability, growth advantage |
Table 2.
Epigenetic changes/patterns as potential biomarkers in clinics
| Epigenetic marker | Clinical association | Reference |
|---|---|---|
| Histone modifications | ||
| Histone acetylation and dimethylation of five residues in histones H3 and H4 | Predictors of outcome independently of tumour stage, preoperative prostate‐specific antigen levels, and invasion | Seligson et al. (2005) |
| DNA methylation | ||
| Analysis of methylation patterns of 45 DNA fragments using methylation‐sensitive arbitrarily primed‐PCR (MS‐AP‐PCR) screening | Methylation patterns varied between different tumours suggesting variable penetrance of the methylation phenotype (colon, prostate and bladder cancers) | Liang et al. (1998) |
| Analysis of 1,184 unselected CpG islands in 98 primary human tumours using restriction landmark genomic scanning (RLGS) | CpG island methylation patterns were shared within a given cancer type. Some tumours appears to have relatively lower levels of methylation compared to others | Costello et al. (2000) |
| Methylation analysis of 1104 CpG islands using CpG island arrays (differential methylation hybridization, DMH) | CpG island methylation patterns correlated with histological grade of breast cancer (poorly differentiated tumours exhibited more hypermethylated CpG islands) hypermethylation was associated with less high grade breast cancers | Yan et al. (2000) |
| Methylation of CpG island of 9 genes identified by MS‐RDA | Aberrant methylation of 9 genes is associated with diffuse‐type and intestinal‐type histology of gastric cancers | Kaneda et al. (2002) |
| Methylation of MGMT | Response of gliomas and lymphomas to chemotherapy using alkylating agents | Esteller et al. (2002) and Esteller and Herman (2004) |
| Methylation of DAPK | Recurrence of bladder cancers | Tada et al. (2002) |
| Methylation of RASSF1 and/or APC in serum DNA | Association with poor outcome of breast cancer cases | Muller et al. (2003) |
| Methylation analysis (MSP) of 12 cancer‐associated genes | In samples from 50 consecutive T‐ALL leukaemia patients, the methylation profile appears to be a biomarker of risk prediction | Roman‐Gomez et al. (2005) |
| Methylation analysis (MSP) of 8 genes in 105 specimens of NSCLC representing all stages of cancer | Three genes (APC, ATM, and RASSF1A) emerged as determinants of prognostic groups | Safar et al. (2005) |
| Methylation analysis of 9 genes (by QM‐MSP) on ductal lavage cells from women undergoing mastectomy | Methylation analysis was found to be more sensitive than cytology. Methylation marker could improve sensitivity and specificity of detection of breast cancer | Fackler et al. (2006) |
| Methylation of 4 GATA genes | Methylation of GATA‐4,‐5 was correlated with clinical parameters and age of patients | Guo et al. (2004) |
| Methylation of 7 fragments derived from screening of methylated CpGs (by MS‐RDA) | Strong association between methylation (methylator phenotype) and poor prognosis of neuroblastomas | Abe et al. (2005) |
3.1. DNA methylation
The best‐studied epigenetic mechanism is DNA methylation. The methylation of DNA refers to the covalent addition of a methyl group to the 5‐carbon (C5) position of cytosine bases that are located 5′ to a guanosine base in a CpG dinucleotide (Figure 3A). DNA methylation plays an important role in different cellular processes including gene expression, silencing of transposable elements, and defense against viral sequences (Bird, 2002; Jaenisch and Bird, 2003; Jones and Baylin, 2002). Importantly, aberrant DNA methylation is tightly connected to a wide variety of human cancer (see below). DNA methylation is mediated by enzymes DNA methyltransferases (DNMTs) among which DNMT1 is the principal enzyme in mammals responsible for post‐replicative methylation (known as maintenance of DNA methylation), and DNMT3A and 3B are responsible for methylation of new CpG sites (de novo methylation). The levels and patterns of DNA methylation undergo dramatic changes during embryonic development, starting with a wave of a profound demethylation during the cleavage stage and followed by a widespread de novo methylation after embryo implantation stage (Jaenisch, 1997). Interestingly, in contrast to the maternal genome, which is only partially demethylated after fertilization; demethylation is a remarkably active and rapid process in the male genome resulting in an almost complete removal of methyl groups (within hours) after fertilization. Recent studies indicated that during early embryonic development the promoter methylation is accompanied by specific histone modifications that are typical of heterochromatin, and that abnormal epigenetic premarking during early embryonic development may predispose certain genes to cancer promoting DNA methylation events (Schlesinger et al., 2006; Widschwendter et al., 2006).
Figure 3.
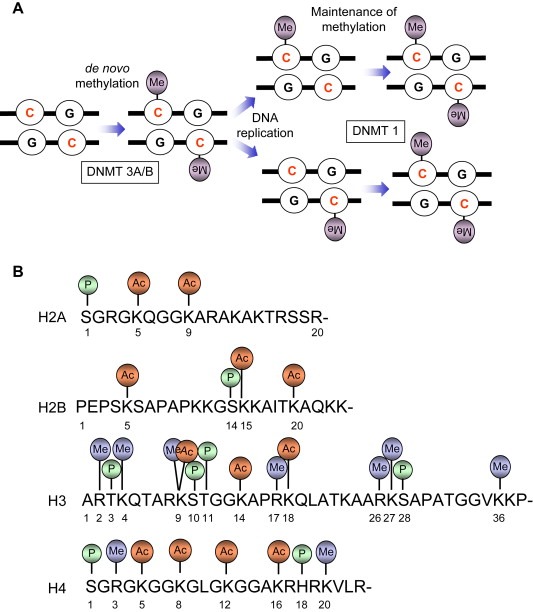
(A) DNA methylation, the covalent addition of a methyl group to the cytosine base in DNA, may be set up de novo (by DNA methyltransferase DNMT3A and DNMT3B) and maintained (by DNMT1) after DNA replication. (B) N‐terminal “tails” of core histones are subject of different covalent modifications, the combinations of which are proposed to constitute the “histone code” that extends and modulates genetic (DNA) code.
3.2. Histone modifications
Post‐translational marking of chromatin proteins (histones) is a major epigenetic mechanism of fundamental importance in the regulation of cellular processes that utilize genomic DNA as a template (Jenuwein and Allis, 2001; Loizou et al., 2006; Peterson and Cote, 2004). Acetylation, methylation, phosphorylation and ubiquitination are major histone modifications (Figure 3B), combination of which may constitute the ‘histone code’ that extends and modulates the genetic code (Jenuwein and Allis, 2001; Strahl and Allis, 2000). Histone modifications play multifaceted roles for several cellular processes including gene transcription, DNA repair, recombination and DNA replication, and their deregulation is implicated in human malignancies (Cairns, 2001; Feinberg et al., 2006; Rowley, 1998; Wolffe, 2001; Yang, 2004). Several lines of evidence implicated histone acetylation and protein complexes (HATs) responsible for this chromatin modifications in human malignancies. HATs have been shown to be involved in chromosomal translocations in which resulting fusion protein exhibits a “gain‐of‐function” by altered HAT activity on specific histones. This is well exemplified by p300 and CBP genes that are found translocated on specific chromosomes in certain leukaemia. In addition, a number of HAT proteins have been found among leukemic translocations, underscoring the importance of a tight control of HATs and histone acetylation for tissue homeostasis. Furthermore, screening of human cancers identified mutations in HAT genes. For example, mutations in p300 and CBP have been found (Gayther et al., 2000; Santos‐Rosa and Caldas, 2005; Shigeno et al., 2004). These results thus suggest that HATs may act as tumour suppressor genes. Similarly, histone deacetylases (HDACs), enzymes responsible for the removal of acetyl groups from histone tails, participate in a dynamic equilibrium of histone acetylation. By analogy, deregulation of this process is believed to be implicated in human cancer, and several tumour suppressor genes such as RB (retinoblastoma), APC (adenomatosis polyposis coli), and p53 may require HDAC activity for their function (Frolov and Dyson, 2004; Sengupta et al., 2005; Zhu et al., 2004).
Histone methylation occurs on lysine and arginine residues and is carried out by a family of enzymes called histone methyltransferases (HMTs). Lysine residues can be mono‐, di‐, or tri‐methylated, whereas, arginine can be either mono‐ or di‐methylated. Interestingly, histone methylation may be associated with either transcriptional activation or repression, depending on lysine/arginine modified by methylation and other histone modifications in the surrounding residues or even different histone tails. For example, tri‐methylation of histone H3 lysine‐4 is associated with transcriptional activation and creates a binding site for the chromodomain containing proteins which recruits HATs (Pray‐Grant et al., 2005). Similarly, methylation of lysine‐36 and lysine‐79 of histone H3 appears to be associated with active chromatin and transcriptional activation. In contrast, tri‐methylation of histone H3 lysine‐9 contributes to transcriptional silencing by recruiting heterochromatin protein (HP1) and triggering formation of heterochromatin (Jenuwein and Allis, 2001). Similarly, methylation of histone H3 lysine 27 is associated with transcriptional repression and maintenance of silent chromatin through recruitment of the Polycomb complex (PRC1) (Cao et al., 2002). Consistent with their important role in transcriptional control many HMTs are essential for cell proliferation and their deregulation is implicated in human diseases including cancer (Santos‐Rosa and Caldas, 2005; Schneider et al., 2002; Sparmann and van Lohuizen, 2006; Varambally et al., 2002).
3.3. RNA‐associated gene silencing
This type of epigenetic inheritance involves RNA molecule (i.e. RNA interference or noncoding RNA) and is found to play an important role in the maintenance of the gene transcription state through multiple cell divisions (Egger et al., 2004). Accumulating evidence indicates that deregulation in microRNAs (miRNAs) are linked to several steps of cancer initiation and progression. miRNAs are relatively small noncoding RNAs (usually 20–22 nucleotides long) that are excised from longer (60–110 nucleotides) RNA precursor (Bartel, 2004; Calin and Croce, 2006). miRNAs play essential roles in normal biological processes including development, proliferation, differentiation and cell death (Bartel, 2004; Pasquinelli et al., 2005). Interestingly, miRNA appears to be able to act as either tumour suppressors or oncogenes by affecting distinct genes of gene families involved in critical biological processes such as proliferation and differentiation. Many miRNA genes are located in the genomic loci known as fragile sites and are therefore susceptible to either loss of amplification (Zhang et al., 2006a).
Several recent studies indicated that miRNA profiles differ significantly between cancer and normal tissues and also between different tumours (Lu et al., 2005; Calin and Croce, 2006; Volinia et al., 2006). Interestingly, miRNA profiling revealed distinct patterns that may classify cancers according to the developmental lineage and differentiation status, lending miRNAs useful tools in cancer diagnostics and prognosis. Although we are just beginning to understand complexity of mechanisms regulated by miRNAs, future studies in this field are likely to provide important information to the overall knowledge in cancer biology.
3.4. Aberrant DNA methylation in cancer
Two forms of aberrant DNA methylation is found in human cancer: the overall loss of 5‐methyl‐cytosine (global hypomethylation) and gene promoter‐associated (CpG island‐specific) hypermethylation (Feinberg and Tycko, 2004; Jones and Baylin, 2002). Both global hypomethylation and CpG island hypermethylation are found in virtually all types of cancer. While the precise consequences of genome‐wide hypomethylation are still debated (activation of cellular proto‐oncogenes, induction of chromosome instability), hypermethylation of gene promoters is in turn associated with gene inactivation (Feinberg and Tycko, 2004; Jones and Baylin, 2002). Therefore, DNA methylation can act as a double‐edged sword, promoting the neoplastic process by local hypermethylation resulting in silencing of tumour suppressor genes and in parallel by global hypomethylation triggering reactivation of cellular proto‐oncogenes (Table 1). A plethora of studies reported the silencing of tumour suppressor genes and other cancer‐related genes, central to the development of many human cancers, may occur through hypermethylation in the absence of obvious genetic change.
3.5. p16INK4a (CDKN2A) promoter hypermethylation in cancer
Significant efforts have been made to discover the epigenetic target gene(s) suitable for early diagnosis, risk assessment and cancer prevention. Such gene would be the target of DNA hypermethylation early in the tumour development, in a high percentage of cases, and specific to cancer type. To the large extent, the p16INK4a (CDKN2A) gene appears to fulfill these criteria. p16INK4, a gene encoding a tumour suppressor, is among the most frequently silenced (by de novo hypermethylation) cancer‐associated genes in human cancer.
The p16INK4a gene resides on a complex locus on human chromosome 9p, which contains an alternative promoter and alternative reading frame encoding unrelated tumour suppressor p19ARF, known as positive regulator of p53 tumour suppressor. Interestingly, the third tumour suppressor (p15), another CDK inhibitor, is expressed from the genomic sequence adjacent to the p16INK4a/p19ARF locus. Curiously, all three tumour suppressors are silenced through epigenetic mechanism, although this does not seem to happen to the same extent and in the same tumour types. The p16INK4a gene is one of the most extensively studied genes with respect to DNA methylation in human cancers. The protein product of p16INK4a gene has long been recognized as a tumour suppressor and mediator of cell senescence (Gil and Peters, 2006). p16INK4a protein binds and inhibits CDK4/CDK6‐cyclin‐D kinase activity thereby maintaining pRB (the protein product of the retinoblastoma tumour suppressor gene) in its unphosphorylated and growth‐suppressive state, resulting in cell‐cycle arrest at G1 phase (Sherr and Roberts, 1999). De novo methylation of the p16INK4a promoter is one of the most frequent epigenetic alterations detected in a wide range of human cancer. In addition, silencing of p16INK4a by promoter hypermethylation is highly tumour specific (Shaw et al., 2006) and appears to be the earliest event in some cancer types (Belinsky et al., 2002), making this gene an attractive target for preventive strategies.
Examination of biopsies from different stages of human squamous‐cell lung carcinoma (SCC) revealed a progressive increase in p16INK4a promoter methylation (Belinsky, 2004; Belinsky et al., 1998; Nuovo et al., 1999). For example, the frequency of p16INK4a promoter methylation increased during lung cancer progression from basal cell hyperplasia (17%) to squamous‐cell metaplasia (24%) to carcinoma in situ (50%) to SSC (60%) (Belinsky, 2004; Belinsky et al., 1998). Even higher frequency (94%) of p16INK4a promoter methylation was observed in lung tumours of rats exposed to 4‐methylnitrosamino‐1‐(3‐pyridyl)‐1‐butanone, a carcinogen found in tobacco smoke (Belinsky et al., 1998). The study of O(6)‐methylguanine‐DNA methyltransferase (MGMT), another cancer‐related gene, also reveled a high prevalence of methylation (51%) which increased with tumour stage, however, in contrast to p16INK4a, it appears to be a late event in lung adenocarcinoma and does not correlate with tobacco smoking (Pulling et al., 2004). Thus, the progressive increase and high incidence of p16INK4a methylation as well as its concordance with the morphological changes defined for the development and progression of SCC makes this epigenetic change ideally suited for early diagnostic and risk assessment.
3.6. Epigenetic changes as biomarkers in the clinic and risk assessment
For many types of human tumours, symptoms are often not presented until the primary tumours have invaded surrounding tissue and/or metastasized, therefore the late presentation of neoplastic process prevents the timely detection of cancer, resulting in high mortality. The discovery of epigenetic biomarkers thus may prove to be extremely useful for early detection and prevention of cancer. Cancer patients have been shown to have increased amounts of cell free DNA in their plasma or serum. In addition to genetic changes, epigenetic alterations are increasingly characterized in circulating DNA from different types of tumours, and such changes can be used for sensitive and specific detection of tumour‐derived nucleic acids in the circulation. To date, nearly all tumour‐associated nucleic acids have been detected in the plasma or serum of cancer patients and successful detection of epigenetic marks in circulating DNA has opened up new possibilities in cancer detection and risk assessment. A number of cancer‐associated genes have been found methylated in plasma/serum DNA. Among these, p16INKa, p15INK4b, RASSF1A, MLH1, GSTP1, CDH1, APC, and DAPK1 are the genes found most frequently methylated in circulating DNA (Laird, 2003). Furthermore, efficient detection of methylated tumour‐associated genes in circulating DNA was reported for a wide range of human cancers including head and neck cancer (Sanchez‐Cespedes et al., 2000, 2002, 2003, 2004); oesophageal cancer (Kawakami et al., 2000), lung cancer (An et al., 2002; Belinsky et al., 2005; Esteller et al., 1999; Kurakawa et al., 2001; Usadel et al., 2002; Bearzatto et al., 2002), liver cancer (Wong et al., 1999, 2000, 2003, 2006), gastric cancer (Lee et al., 2002), bladder cancer (Dominguez et al., 2002; Goessl et al., 2002; Valenzuela et al., 2002); prostate cancer (Goessl et al., 2000, 2002, 2004, 2006), and colorectal cancer (Bazan et al., 2006; Grady et al., 2001; Lecomte et al., 2002; Nakayama et al., 2002; Taback et al., 2006; Zou et al., 2002). In addition to plasma/serum DNA, epigenetic changes may be detected in other bodily fluids, such as urine, sputum, and breast ductal lavage (Laird, 2003).
Importantly, several studies on methylation in bodily fluids (such as plasma and urine) from cancer patients reported a diagnostic coverage of 100% using panels of as few as four genes (Chan et al., 2002; Dulaimi et al., 2004). Therefore, since concomitant methylation of multiple gene promoters and bodily fluids were strongly associated with human cancer, clinical sensitivity of the methods for detection of methylation biomarkers in bodily fluids may be increased by using multiple epigenetic markers.
While cancer epigenetics have focused primarily on DNA methylation changes as biomarker (Laird, 2003), cancer‐specific histone modifications as potential biomarkers remain largely unexplored. Two recent studies have given us a glimpse of what could be the utility of histone modification patterns in the clinic and risk assessment (Fraga et al., 2005; Seligson et al., 2005). Future studies are likely to provide detailed information on the possible use of histone marks in the clinic, especially in the area of molecular diagnostics, early detection and prognosis. As we gain insight into the functional significance of chromatin alterations and as new tools for specific and efficient detection of histone marks become available, there will be enormous benefit to use histone modifications in clinics and risk assessment.
3.7. The CpG island methylator phenotype (CIMP)
The studies on DNA methylation involving multiple genes revealed that some cancer types exhibit concurrent methylation of groups of cancer‐associated genes. To define this phenomenon, Jean‐Pierre Issa coined the term the CpG island methylator (CIMP) phenotype (Issa, 2004; Toyota et al., 1999), which was most apparent in colorectal cancer where it was found that cancers with microsatellite instability exhibit a high prevalence of methylation of multiple gene promoters including those of p16INK4a and MLH1, the mismatch repair gene (Ahuja et al., 1997). A puzzling evidence emerged from another study showing that tumours with microsatellite instability show hypermethylation and silencing of MLH1 gene (Kane et al., 1997). This lead to the proposal that methylation‐mediated silencing of MLH1 causes microsatellite instability. This notion was subsequently supported by a study demonstrating that the reversal of hypermethylation with 5‐aza‐2′‐deoxycytidine treatment resulted in reexpression of the silenced MLH1 gene and restoration of the DNA mismatch repair capacity (Herman et al., 1998). A more recent study has shown that the CIMP phenotype may also be associated with mutations in other genes such as BRAF (Weisenberger et al., 2006).
In addition, several studies revealed other genes (including p16INK4a) that are silenced through hypermethylation in sporadic cancers with microsatellite instability (Shen et al., 2003; Shibata et al., 2002). It is important to note that these genes are not found preferentially methylated in inherited cancers with microsatellite instability associated with germline mutations in DNA mismatch repair genes, ruling out the possibility that the reduced capacity of mismatch repair itself may promote hypermethylation (Yamamoto et al., 2002). While the CIMP phenotype has been extensively studied in colorectal cancer (Ahuja et al., 1997; Goel et al., 2006; Issa, 2004; Shibata et al., 2002; Toyota et al., 1999; Weisenberger et al., 2006), several other studies provided evidence that the CIMP phenotype may also be present in other cancer types including hepatocellular carcinoma, gastric cancer, pancreatic cancer, glioblastomas, oral cancer, leukaemias, and solid tumours (An et al., 2005; Issa, 2004; Marsit et al., 2006; Shaw et al., 2006; and reference therein). However, the existence of CIMP has been challenged by some studies where no evidence of concordant hypermethylation of multiple genes was found (Eads et al., 1999, 2001, 2007). Therefore, it is likely that the CIMP‐positive tumours represent a subset of cancers with distinct epigenotype.
Despite wealth of studies providing strong evidence for existence of CIMP in a variety of human cancers, the causes and underlying mechanism of this phenomenon remain unclear (Issa, 2004). It is possible that the CIMP is a consequence of inactivation (possibly through mutations) of the genes involved in the process of DNA methylation, although such a possibility awaits an experimental proof. Alternatively, specific agents (epimutagens), of combination thereof, in the environmental, diet, or lifestyle may promote, and/or relieve resistance against, aberrant methylation, leading to altered gene expression and oncogenic process. The large population‐based cohorts and case‐control studies may offer excellent opportunities to test the contribution of repeated and chronic exposure to epimutagens in the environment and nutrition to CIMP in specific cancers.
Regardless of the origin of CIMP, molecular distinction of CIMP‐positive cancers seems to be reflected in their clinical, histopathological and epidemiological attributes (Issa, 2004; Samowitz et al., 2005). For example, CIMP‐positive cancers exhibit a low rate of p53 mutations and strikingly high frequency of KRAS and BRAF (Kambara et al., 2004; Toyota et al., 2000; Weisenberger et al., 2006). Importantly, CIMP‐positive cases seem to be associated with poor prognosis (Issa, 2003; Ward et al., 2003). Therefore, CIMP phenotype could be exploited in the clinic for risk assessment and diagnostic/prognostic purposes.
3.8. Loss of imprinting (LOI) and cancer
Genomic imprinting refers to the conditioning of parental genomes mediated by epigenetic mechanism during gametogenesis ensuring that a specific locus is exclusively expressed from either maternal or paternal genome in the offspring (John and Surani, 2000; Surani et al., 1984; Oakey and Beechey, 2002). Around 80 genes have so far been found imprinted in humans and mice, although a recent estimation suggested that as many as 600 genes are potentially imprinted (Luedi et al., 2005). Interestingly, imprinted genes are not equally distributed throughout the genome, but are clustered into a distinct imprinted domains in both humans and mice (e.g. about half of all imprinted mouse genes are clustered on chromosome 7) (Beechey C.V., Cattanach B.M., Blake A., Peters J. (2004) Harwell (United Kingdom) http://www.mgu.har.mrc.ac.uk/research/imprinting).
Imprinted genes play critical roles in developmental and cellular processes, therefore loss of imprinting (LOI) due to epigenetic alterations leads to abnormal biallelic expression resulting in several human syndromes. Importantly, pathological biallelic expression of several genes caused by LOI is associated with human cancer (Feinberg et al., 2006; Feinberg and Tycko, 2004).
In general, the potential significance of epigenetic dysfunction in human malignancies is illustrated by the fact that the LOI and loss of X‐inactivation occur at much higher frequency compared to genetic mutations (King et al., 1994).
One of the best‐studied example of genomic imprinting and its implication in human malignancies is the IGF2/H19 locus. The H19 gene, which encodes a nontranslated RNA, is monoallelically expressed as the paternal allele is normally silenced through its promoter hypermethylation. Since H19 gene lies at 100kb downstream from the IGF2 gene, the maternal‐specific expression of H19 induces silencing of IGF2 in cis, resulting in monoallelic expression of IGF2 from opposite (paternal) copy. Therefore, the reciprocal expression of these two genes is a tightly regulated mechanism, and when the H19 promoter is abnormally methylated on both alleles, pathological biparental expression (hyperexpression) of IGF2 occurs.
The IGF2/H19 locus has been intensively studied in childhood malignancies such as Wilms' tumours as well as overgrowth syndromes such as the Beckwith‐Wiedemann syndrome (Feinberg and Tycko, 2004). Early works from the Feinberg's, Reeve's and Tycko's laboratories revealed that embryonal neoplasms are in turn associated with hypermethylation and consequent silencing of H19 gene resulting in reciprocal increase in IGF2 expression (Moulton et al., 1994; Steenman et al., 1994). This lead to the notion of a gatekeeper role for LOI in tumours (Feinberg and Tycko, 2004). The strong support for a gatekeeper role for LOI of IGF2 in Wilms' tumours has come from the studies showing that Beckwith‐Wiedemann syndrome, a prenatal overgrowth disorder, predisposes to various embryonal tumours including Wilms' tumours.
It is estimated that LOI of IGF2 accounts for a significant subset (∼50%) of Wilms' tumours in children (Ravenel et al., 2001). Consistent with an important role of LOI of IGF2 in cancers of adults, recent studies demonstrated that epimutation of IGF2/H19 locus is a common epigenetic event in adults and is associated with fivefold increased incidence of colorectal cancer (Cui et al., 2003; Sakatani et al., 2005).
Interestingly, disrupted imprinting of IGF2/H19 locus is found relatively early in the development of Wilms' tumour and often in the adjacent normal tissue or precancerous lesions in kidney of Wilms' tumour patients (Moulton et al., 1994). Several studies showed that LOI of IGF2 frequently occurs in histologically normal colon tissues of patients with colon cancer associated with LOI (Cui et al., 1998, 2003, 2004). A more direct evidence for the role of LOI in tumorigenesis is provided by studies from Feinberg laboratory demonstrating specific changes and increased susceptibility to colon cancers in both humans and mice with biallelic expression of IGF2 (Cui et al., 2003; Sakatani et al., 2005). An important observation emerged from these studies, that is, LOI of IGF2 led to a shift toward a less differentiated normal intestinal epithelium in both humans and mice (Sakatani et al., 2005), a synonym for an altered maturation of non‐neoplastic tissue. This argues that LOI may be a frequent mechanism by which epigenetic alterations predisposes to the development of cancer. These observations, together with other advances in cancer epigenetics, led to the new concept of cancer development known as the epigenetic progenitor model (Feinberg et al., 2006), which challenges the widely accepted clonal genetic model of cancer.
4. Conclusion and perspectives
In recent years, the development of genome‐wide analytic methods has opened the possibility of identifying simultaneously multiple changes in gene expression as well as in genetic or epigenetic alterations affecting the genome of cancer cells. The main question raised by such studies is to determine which alterations, or combinations thereof, can be interpreted as reliable biomarkers for providing information about the carcinogenesis process. This assessment should be done from the viewpoint of the suspected, primary role of such alterations in the initial steps of tumorigenesis. Indeed, the molecular events that occur in early stage of cancers or in precursor lesions are more likely to have a direct influence on cancer occurrence and progression than those that accumulate at later stage of cancer development. Among the latter, many alterations may be considered as “passengers” that represent sequels of the highly disturbed genetic and genomic instability that accompanies the progression of many cancers.
Similar to genetic changes, epigenetic events are also shown to influence virtually each steps in tumour development, therefore, understanding epigenetic changes associated with cancer onset, progression and metastasis are fundamental to improving our abilities to successfully diagnose, treat and prevent cancer. Epigenetic changes (e.g. promoter‐specific hypermethylation) occur early and at high frequency in different human malignancies, this feature combined with high sensitivity and specificity of detection may be exploited in the area of molecular diagnostics and cancer risk assessment. As a result, the research on epigenetic changes as potential biomarkers is in full swing. To date, sensitive detection of cancer using epigenetic biomarkers in DNA extracted from plasma/serum and other bodily fluids has been demonstrated for a large number of human cancers. It is hoped that this type of universally applicable markers would be made available in a clinical diagnostic setting and population‐based screening in the near future.
A distinguishing feature of epigenetic changes in comparison with genetic changes is that they are reversible; therefore aberrant DNA methylation, histone acetylation and methylation are attractive targets for the epigenetic therapy. Indeed, a number of drugs that are capable of altering levels or patterns of DNA methylation or histone modifications have been discovered, and many of these drugs are now in clinical trials (Egger et al., 2004). The intrinsic reversibility of epigenetic alterations also represents an exciting opportunity for the development of novel strategies for cancer prevention.
Figure 4 summarizes how it may be possible to design tailored treatment strategies by taking into account the functional contribution of genetic and epigenetic changes to the disruption of the regulatory mechanisms that constitute the “hallmarks of cancer”. From this viewpoint, the molecular “signature” of each cancer would consist the description of a specific set of alterations by which a particular tumour escapes these regulatory mechanisms. Based on this knowledge, it may become feasible to select appropriate combinations of therapeutic agents to revert or block the functional consequences of these alterations in tumours. By targeting specifically and simultaneously multiple pathways based on molecular signatures, such approaches may confer a greater therapeutic efficacy, while having less side‐effects than conventional cytotoxic therapies.
Figure 4.
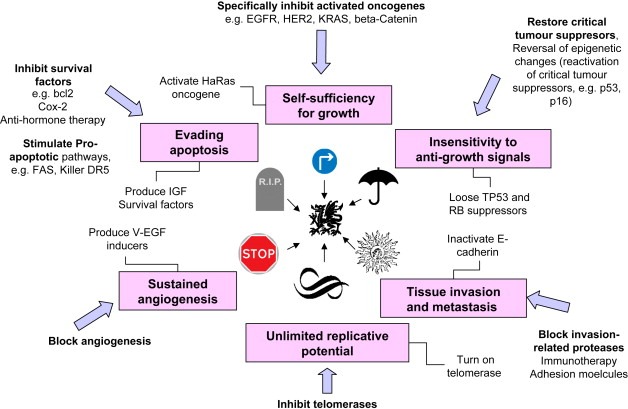
Rational treatment design. Six major mechanisms that are systematically deregulated in cancer are depicted (boxes), as well as the molecular mechanism that are responsible for their alterations. Blue arrows indicate possible therapeutic approaches specifically targetting each of these mechanisms.
Herceg Zdenko, Hainaut Pierre, (2007), Genetic and epigenetic alterations as biomarkers for cancer detection, diagnosis and prognosis, Molecular Oncology, 1, doi:10.1016/j.molonc.2007.01.004.
References
- Abdel-Rahman, W.M. , Mecklin, J.P. , Peltomaki, P. , 2006. The genetics of HNPCC: application to diagnosis and screening. Critical Reviews in Oncology–Hematology. 58, 208–220. [DOI] [PubMed] [Google Scholar]
- Abe, M. , Ohira, M. , Kaneda, A. , Yagi, Y. , Yamamoto, S. , Kitano, Y. , Takato, T. , Nakagawara, A. , Ushijima, T. , 2005. CpG island methylator phenotype is a strong determinant of poor prognosis in neuroblastomas. Cancer Research. 65, 828–834. [PubMed] [Google Scholar]
- Ahuja, N. , Mohan, A.L. , Li, Q. , Stolker, J.M. , Herman, J.G. , Hamilton, S.R. , Baylin, S.B. , Issa, J.P. , 1997. Association between CpG island methylation and microsatellite instability in colorectal cancer. Cancer Research. 57, 3370–3374. [PubMed] [Google Scholar]
- An, Q. , Liu, Y. , Gao, Y. , Huang, J. , Fong, X. , Li, L. , Zhang, D. , Cheng, S. , 2002. Detection of p16 hypermethylation in circulating plasma DNA of non-small cell lung cancer patients. Cancer Letters. 188, 109–114. [DOI] [PubMed] [Google Scholar]
- An, C. , Choi, I.S. , Yao, J.C. , Worah, S. , Xie, K. , Mansfield, P.F. , Ajani, J.A. , Rashid, A. , Hamilton, S.R. , Wu, T.T. , 2005. Prognostic significance of CpG island methylator phenotype and microsatellite instability in gastric carcinoma. Clinical Cancer Research. 11, 656–663. [PubMed] [Google Scholar]
- Balko, J.M. , Potti, A. , Saunders, C. , Stromberg, A. , Haura, E.B. , Black, E.P. , 2006. Gene expression patterns that predict sensitivity to epidermal growth factor receptor tyrosine kinase inhibitors in lung cancer cell lines and human lung tumors. BMC Genomics. 7, 289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel, D.P. , 2004. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 116, 281–297. [DOI] [PubMed] [Google Scholar]
- Bazan, V. , Bruno, L. , Augello, C. , Agnese, V. , Calo, V. , Corsale, S. , Gargano, G. , Terrasi, M. , Schiro, V. , Di Fede, G. , Adamo, V. , Intrivici, C. , Crosta, A. , Rinaldi, G. , Latteri, F. , Dardanoni, G. , Grassi, N. , Valerio, M.R. , Colucci, G. , Macaluso, M. , Russo, A. , 2006. Molecular detection of TP53, Ki-Ras and p16INK4A promoter methylation in plasma of patients with colorectal cancer and its association with prognosis. Results of a 3-year GOIM (Gruppo Oncologico dell'Italia Meridionale) prospective study. Annals of Oncology. 17, (Suppl. 7) vii84–vii90. [DOI] [PubMed] [Google Scholar]
- Bearzatto, A. , Conte, D. , Frattini, M. , Zaffaroni, N. , Andriani, F. , Balestra, D. , Tavecchio, L. , Daidone, M.G. , Sozzi, G. , 2002. p16(INK4A) Hypermethylation detected by fluorescent methylation-specific PCR in plasmas from non-small cell lung cancer. Clinical Cancer Research. 8, 3782–3787. [PubMed] [Google Scholar]
- Belinsky, S.A. , 2004. Gene-promoter hypermethylation as a biomarker in lung cancer. Nature Reviews Cancer. 4, 707–717. [DOI] [PubMed] [Google Scholar]
- Belinsky, S.A. , Nikula, K.J. , Palmisano, W.A. , Michels, R. , Saccomanno, G. , Gabrielson, E. , Baylin, S.B. , Herman, J.G. , 1998. Aberrant methylation of p16(INK4a) is an early event in lung cancer and a potential biomarker for early diagnosis. Proceedings of the National Academy of Sciences of the United States of America. 95, 11891–11896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belinsky, S.A. , Palmisano, W.A. , Gilliland, F.D. , Crooks, L.A. , Divine, K.K. , Winters, S.A. , Grimes, M.J. , Harms, H.J. , Tellez, C.S. , Smith, T.M. , Moots, P.P. , Lechner, J.F. , Stidley, C.A. , Crowell, R.E. , 2002. Aberrant promoter methylation in bronchial epithelium and sputum from current and former smokers. Cancer Research. 62, 2370–2377. [PubMed] [Google Scholar]
- Belinsky, S.A. , Klinge, D.M. , Dekker, J.D. , Smith, M.W. , Bocklage, T.J. , Gilliland, F.D. , Crowell, R.E. , Karp, D.D. , Stidley, C.A. , Picchi, M.A. , 2005. Gene promoter methylation in plasma and sputum increases with lung cancer risk. Clinical Cancer Research. 11, 6505–6511. [DOI] [PubMed] [Google Scholar]
- Bielas, J.H. , Loeb, K.R. , Rubin, B.P. , True, L.D. , Loeb, L.A. , 2006. Human cancers express a mutator phenotype. Proceedings of the National Academy of Sciences of the United States of America. 103, 18238–18242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird, A. , 2002. DNA methylation patterns and epigenetic memory. Genes and Development. 16, 6–21. [DOI] [PubMed] [Google Scholar]
- Cairns, B.R. , 2001. Emerging roles for chromatin remodeling in cancer biology. Trends in Cell Biology. 11, S15–S21. [DOI] [PubMed] [Google Scholar]
- Calin, G.A. , Croce, C.M. , 2006. MicroRNA signatures in human cancers. Nature Reviews Cancer. 6, 857–866. [DOI] [PubMed] [Google Scholar]
- Cao, R. , Wang, L. , Wang, H. , Xia, L. , Erdjument-Bromage, H. , Tempst, P. , Jones, R.S. , Zhang, Y. , 2002. Role of histone H3 lysine 27 methylation in polycomb-group silencing. Science. 298, 1039–1043. [DOI] [PubMed] [Google Scholar]
- Chan, M.W. , Chan, L.W. , Tang, N.L. , Tong, J.H. , Lo, K.W. , Lee, T.L. , Cheung, H.Y. , Wong, W.S. , Chan, P.S. , Lai, F.M. , To, K.F. , 2002. Hypermethylation of multiple genes in tumor tissues and voided urine in urinary bladder cancer patients. Clinical Cancer Research. 8, 464–470. [PubMed] [Google Scholar]
- Costello, J.F. , Fruhwald, M.C. , Smiraglia, D.J. , Rush, L.J. , Robertson, G.P. , Gao, X. , Wright, F.A. , Feramisco, J.D. , Peltomaki, P. , Lang, J.C. , Schuller, D.E. , Yu, L. , Bloomfield, C.D. , Caligiuri, M.A. , Yates, A. , Nishikawa, R. , Su Huang, H. , Petrelli, N.J. , Zhang, X. , O'Dorisio, M.S. , Held, W.A. , Cavenee, W.K. , Plass, C. , 2000. Aberrant CpG-island methylation has non-random and tumour-type-specific patterns. Nature Genetics. 24, 132–138. [DOI] [PubMed] [Google Scholar]
- Cui, H. , Horon, I.L. , Ohlsson, R. , Hamilton, S.R. , Feinberg, A.P. , 1998. Loss of imprinting in normal tissue of colorectal cancer patients with microsatellite instability. Nature Medicine. 4, 1276–1280. [DOI] [PubMed] [Google Scholar]
- Cui, H. , Cruz-Correa, M. , Giardiello, F.M. , Hutcheon, D.F. , Kafonek, D.R. , Brandenburg, S. , Wu, Y. , He, X. , Powe, N.R. , Feinberg, A.P. , 2003. Loss of IGF2 imprinting: a potential marker of colorectal cancer risk. Science. 299, 1753–1755. [DOI] [PubMed] [Google Scholar]
- Dominguez, G. , Carballido, J. , Silva, J. , Silva, J.M. , Garcia, J.M. , Menendez, J. , Provencio, M. , Espana, P. , Bonilla, F. , 2002. p14ARF promoter hypermethylation in plasma DNA as an indicator of disease recurrence in bladder cancer patients. Clinical Cancer Research. 8, 980–985. [PubMed] [Google Scholar]
- Dulaimi, E. , Uzzo, R.G. , Greenberg, R.E. , Al-Saleem, T. , Cairns, P. , 2004. Detection of bladder cancer in urine by a tumor suppressor gene hypermethylation panel. Clinical Cancer Research. 10, 1887–1893. [DOI] [PubMed] [Google Scholar]
- Eads, C.A. , Danenberg, K.D. , Kawakami, K. , Saltz, L.B. , Danenberg, P.V. , Laird, P.W. , 1999. CpG island hypermethylation in human colorectal tumors is not associated with DNA methyltransferase overexpression. Cancer Research. 59, 2302–2306. [PubMed] [Google Scholar]
- Eads, C.A. , Lord, R.V. , Wickramasinghe, K. , Long, T.I. , Kurumboor, S.K. , Bernstein, L. , Peters, J.H. , DeMeester, S.R. , DeMeester, T.R. , Skinner, K.A. , Laird, P.W. , 2001. Epigenetic patterns in the progression of esophageal adenocarcinoma. Cancer Research. 61, 3410–3418. [PubMed] [Google Scholar]
- Egger, G. , Liang, G. , Aparicio, A. , Jones, P.A. , 2004. Epigenetics in human disease and prospects for epigenetic therapy. Nature. 429, 457–463. [DOI] [PubMed] [Google Scholar]
- Elenbaas, B. , Spirio, L. , Koerner, F. , Fleming, M.D. , Zimonjic, D.B. , Donaher, J.L. , Popescu, N.C. , Hahn, W.C. , Weinberg, R.A. , 2001. Human breast cancer cells generated by oncogenic transformation of primary mammary epithelial cells. Genes and Development. 15, 50–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteller, M. , Herman, J.G. , 2004. Generating mutations but providing chemosensitivity: the role of O6-methylguanine DNA methyltransferase in human cancer. Oncogene. 23, 1–8. [DOI] [PubMed] [Google Scholar]
- Esteller, M. , Levine, R. , Baylin, S.B. , Ellenson, L.H. , Herman, J.G. , 1998. MLH1 promoter hypermethylation is associated with the microsatellite instability phenotype in sporadic endometrial carcinomas. Oncogene. 17, 2413–2417. [DOI] [PubMed] [Google Scholar]
- Esteller, M. , Sanchez-Cespedes, M. , Rosell, R. , Sidransky, D. , Baylin, S.B. , Herman, J.G. , 1999. Detection of aberrant promoter hypermethylation of tumor suppressor genes in serum DNA from non-small cell lung cancer patients. Cancer Research. 59, 67–70. [PubMed] [Google Scholar]
- Esteller, M. , Gaidano, G. , Goodman, S.N. , Zagonel, V. , Capello, D. , Botto, B. , Rossi, D. , Gloghini, A. , Vitolo, U. , Carbone, A. , Baylin, S.B. , Herman, J.G. , 2002. Hypermethylation of the DNA repair gene O(6)-methylguanine DNA methyltransferase and survival of patients with diffuse large B-cell lymphoma. Journal of the National Cancer Institute. 94, 26–32. [DOI] [PubMed] [Google Scholar]
- Fackler, M.J. , Malone, K. , Zhang, Z. , Schilling, E. , Garrett-Mayer, E. , Swift-Scanlan, T. , Lange, J. , Nayar, R. , Davidson, N.E. , Khan, S.A. , Sukumar, S. , 2006. Quantitative multiplex methylation-specific PCR analysis doubles detection of tumor cells in breast ductal fluid. Clinical Cancer Research. 12, 3306–3310. [DOI] [PubMed] [Google Scholar]
- Fearon, E.R. , Vogelstein, B. , 1990. A genetic model for colorectal tumorigenesis. Cell. 61, 759–767. [DOI] [PubMed] [Google Scholar]
- Feinberg, A.P. , Tycko, B. , 2004. The history of cancer epigenetics. Nature Reviews in Cancer. 4, 143–153. [DOI] [PubMed] [Google Scholar]
- Feinberg, A.P. , Ohlsson, R. , Henikoff, S. , 2006. The epigenetic progenitor origin of human cancer. Nature Reviews. 7, 21–33. [DOI] [PubMed] [Google Scholar]
- Fishel, R. , Wilson, T. , 1997. MutS homologs in mammalian cells. Current Opinion in Genetics and Development. 7, 105–113. [DOI] [PubMed] [Google Scholar]
- Forbes, S. , Clements, J. , Dawson, E. , Bamford, S. , Webb, T. , Dogan, A. , Flanagan, A. , Teague, J. , Wooster, R. , Futreal, P.A. , Stratton, M.R. , 2006. Cosmic 2005. British Journal of Cancer. 94, 318–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraga, M.F. , Ballestar, E. , Villar-Garea, A. , Boix-Chornet, M. , Espada, J. , Schotta, G. , Bonaldi, T. , Haydon, C. , Ropero, S. , Petrie, K. , Iyer, N.G. , Perez-Rosado, A. , Calvo, E. , Lopez, J.A. , Cano, A. , Calasanz, M.J. , Colomer, D. , Piris, M.A. , Ahn, N. , Imhof, A. , Caldas, C. , Jenuwein, T. , Esteller, M. , 2005. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nature Genetics. 37, 391–400. [DOI] [PubMed] [Google Scholar]
- Frolov, M.V. , Dyson, N.J. , 2004. Molecular mechanisms of E2F-dependent activation and pRB-mediated repression. Journal of Cell Science. 117, 2173–2181. [DOI] [PubMed] [Google Scholar]
- Futreal, P.A. , Coin, L. , Marshall, M. , Down, T. , Hubbard, T. , Wooster, R. , Rahman, N. , Stratton, M.R. , 2004. A census of human cancer genes. Nature Reviews in Cancer. 4, 177–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnis, C. , Buys, T.P. , Lam, W.L. , 2004. Genetic alteration and gene expression modulation during cancer progression. Molecular Cancer. 3, 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gayther, S.A. , Batley, S.J. , Linger, L. , Bannister, A. , Thorpe, K. , Chin, S.F. , Daigo, Y. , Russell, P. , Wilson, A. , Sowter, H.M. , Delhanty, J.D. , Ponder, B.A. , Kouzarides, T. , Caldas, C. , 2000. Mutations truncating the EP300 acetylase in human cancers. Nature Genetics. 24, 300–303. [DOI] [PubMed] [Google Scholar]
- Gil, J. , Peters, G. , 2006. Regulation of the INK4b-ARF-INK4a tumour suppressor locus: all for one or one for all. Nature Reviews in Molecular and Cellular Biology. 7, 667–677. [DOI] [PubMed] [Google Scholar]
- Goel, A. , Nagasaka, T. , Arnold, C.N. , Inoue, T. , Hamilton, C. , Niedzwiecki, D. , Compton, C. , Mayer, R.J. , Goldberg, R. , Bertagnolli, M.M. , Boland, C.R. , 2006. The CpG Island Methylator Phenotype and Chromosomal Instability Are Inversely Correlated in Sporadic Colorectal Cancer. Gastroenterology. [DOI] [PubMed] [Google Scholar]
- Goessl, C. , Sauter, T. , Michael, T. , Berge, B. , Staehler, M. , Miller, K. , 2000. Efficacy and tolerability of tolterodine in children with detrusor hyperreflexia. Urology. 55, 414–418. [DOI] [PubMed] [Google Scholar]
- Goessl, C. , Muller, M. , Straub, B. , Miller, K. , 2002. DNA alterations in body fluids as molecular tumor markers for urological malignancies. European Urology. 41, 668–676. [DOI] [PubMed] [Google Scholar]
- Gormally, E., Caboux, E., Vineis, P., Hainaut, P. Circulating free DNA in plasma or serum as biomarker of carcinogenesis: practical aspects and biological significance. Mutation Research, in press. (Epub ahead of print). doi:10.1016/j.mrrev.2006.11.002. [DOI] [PubMed]
- Gormally, E. , Vineis, P. , Matullo, G. , Veglia, F. , Caboux, E. , Le Roux, E. , Peluso, M. , Garte, S. , Guarrera, S. , Munnia, A. , Airoldi, L. , Autrup, H. , Malaveille, C. , Dunning, A. , Overvad, K. , Tjonneland, A. , Lund, E. , Clavel-Chapelon, F. , Boeing, H. , Trichopoulou, A. , Palli, D. , Krogh, V. , Tumino, R. , Panico, S. , Bueno-de-Mesquita, H.B. , Peeters, P.H. , Pera, G. , Martinez, C. , Dorronsoro, M. , Barricarte, A. , Navarro, C. , Quiros, J.R. , Hallmans, G. , Day, N.E. , Key, T.J. , Saracci, R. , Kaaks, R. , Riboli, E. , Hainaut, P. , 2006. TP53 and KRAS2 mutations in plasma DNA of healthy subjects and subsequent cancer occurrence: a prospective study. Cancer Research. 66, 6871–6876. [DOI] [PubMed] [Google Scholar]
- Grady, W.M. , Rajput, A. , Lutterbaugh, J.D. , Markowitz, S.D. , 2001. Detection of aberrantly methylated hMLH1 promoter DNA in the serum of patients with microsatellite unstable colon cancer. Cancer Research. 61, 900–902. [PubMed] [Google Scholar]
- Guo, M. , Akiyama, Y. , House, M.G. , Hooker, C.M. , Heath, E. , Gabrielson, E. , Yang, S.C. , Han, Y. , Baylin, S.B. , Herman, J.G. , Brock, M.V. , 2004. Hypermethylation of the GATA genes in lung cancer. Clinical Cancer Research. 10, 7917–7924. [DOI] [PubMed] [Google Scholar]
- Hainaut, P. , Hollstein, M. , 2000. p53 and human cancer: the first ten thousand mutations. Advances in Cancer Research. 77, 81–137. [DOI] [PubMed] [Google Scholar]
- Hanahan, D. , Weinberg, R.A. , 2000. The hallmarks of cancer. Cell. 100, 57–70. [DOI] [PubMed] [Google Scholar]
- Harari, D. , Yarden, Y. , 2000. Molecular mechanisms underlying ErbB2/HER2 action in breast cancer. Oncogene. 19, 6102–6114. [DOI] [PubMed] [Google Scholar]
- Herman, J.G. , Umar, A. , Polyak, K. , Graff, J.R. , Ahuja, N. , Issa, J.P. , Markowitz, S. , Willson, J.K. , Hamilton, S.R. , Kinzler, K.W. , Kane, M.F. , Kolodner, R.D. , Vogelstein, B. , Kunkel, T.A. , Baylin, S.B. , 1998. Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proceedings of the National Academy of Sciences of the United States of America. 95, 6870–6875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issa, J.P. , 2003. Methylation and prognosis: of molecular clocks and hypermethylator phenotypes. Clinical Cancer Research. 9, 2879–2881. [PubMed] [Google Scholar]
- Issa, J.P. , 2004. CpG island methylator phenotype in cancer. Nature Reviews in Cancer. 4, 988–993. [DOI] [PubMed] [Google Scholar]
- Jackson, P.E. , Kuang, S.Y. , Wang, J.B. , Strickland, P.T. , Munoz, A. , Kensler, T.W. , Qian, G.S. , Groopman, J.D. , 2003. Prospective detection of codon 249 mutations in plasma of hepatocellular carcinoma patients. Carcinogenesis. 24, 1657–1663. [DOI] [PubMed] [Google Scholar]
- Jaenisch, R. , 1997. DNA methylation and imprinting: why bother?. Trends in Genetics. 13, 323–329. [DOI] [PubMed] [Google Scholar]
- Jaenisch, R. , Bird, A. , 2003. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nature Genetics. 33, (Suppl.) 245–254. [DOI] [PubMed] [Google Scholar]
- Jenuwein, T. , Allis, C.D. , 2001. Translating the histone code. Science. 293, 1074–1080. [DOI] [PubMed] [Google Scholar]
- Jeronimo, C. , Usadel, H. , Henrique, R. , Silva, C. , Oliveira, J. , Lopes, C. , Sidransky, D. , 2002. Quantitative GSTP1 hypermethylation in bodily fluids of patients with prostate cancer. Urology. 60, 1131–1135. [DOI] [PubMed] [Google Scholar]
- John, R.M. , Surani, M.A. , 2000. Genomic imprinting, mammalian evolution, and the mystery of egg-laying mammals. Cell. 101, 585–588. [DOI] [PubMed] [Google Scholar]
- Jones, P.A. , Baylin, S.B. , 2002. The fundamental role of epigenetic events in cancer. Nature Reviews. 3, 415–428. [DOI] [PubMed] [Google Scholar]
- Kambara, T. , Simms, L.A. , Whitehall, V.L. , Spring, K.J. , Wynter, C.V. , Walsh, M.D. , Barker, M.A. , Arnold, S. , McGivern, A. , Matsubara, N. , Tanaka, N. , Higuchi, T. , Young, J. , Jass, J.R. , Leggett, B.A. , 2004. BRAF mutation is associated with DNA methylation in serrated polyps and cancers of the colorectum. Gut. 53, 1137–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane, M.F. , Loda, M. , Gaida, G.M. , Lipman, J. , Mishra, R. , Goldman, H. , Jessup, J.M. , Kolodner, R. , 1997. Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Research. 57, 808–811. [PubMed] [Google Scholar]
- Kaneda, A. , Kaminishi, M. , Yanagihara, K. , Sugimura, T. , Ushijima, T. , 2002. Identification of silencing of nine genes in human gastric cancers. Cancer Research. 62, 6645–6650. [PubMed] [Google Scholar]
- Kawakami, K. , Brabender, J. , Lord, R.V. , Groshen, S. , Greenwald, B.D. , Krasna, M.J. , Yin, J. , Fleisher, A.S. , Abraham, J.M. , Beer, D.G. , Sidransky, D. , Huss, H.T. , Demeester, T.R. , Eads, C. , Laird, P.W. , Ilson, D.H. , Kelsen, D.P. , Harpole, D. , Moore, M.B. , Danenberg, K.D. , Danenberg, P.V. , Meltzer, S.J. , 2000. Hypermethylated APC DNA in plasma and prognosis of patients with esophageal adenocarcinoma. Journal of the National Cancer Institute. 92, 1805–1811. [DOI] [PubMed] [Google Scholar]
- Kim, C.F. , Jackson, E.L. , Woolfenden, A.E. , Lawrence, S. , Babar, I. , Vogel, S. , Crowley, D. , Bronson, R.T. , Jacks, T. , 2005. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell. 121, 823–835. [DOI] [PubMed] [Google Scholar]
- King, C.M. , Gillespie, E.S. , McKenna, P.G. , Barnett, Y.A. , 1994. An investigation of mutation as a function of age in humans. Mutation Research. 316, 79–90. [DOI] [PubMed] [Google Scholar]
- Kurakawa, E. , Shimamoto, T. , Utsumi, K. , Hirano, T. , Kato, H. , Ohyashiki, K. , 2001. Hypermethylation of p16(INK4a) and p15(INK4b) genes in non-small cell lung cancer. International Journal of Oncology. 19, 277–281. [PubMed] [Google Scholar]
- Laird, P.W. , 2003. The power and the promise of DNA methylation markers. Nature Reviews in Cancer. 3, 253–266. [DOI] [PubMed] [Google Scholar]
- Lecomte, T. , Berger, A. , Zinzindohoue, F. , Micard, S. , Landi, B. , Blons, H. , Beaune, P. , Cugnenc, P.H. , Laurent-Puig, P. , 2002. Detection of free-circulating tumor-associated DNA in plasma of colorectal cancer patients and its association with prognosis. International Journal of Cancer. 100, 542–548. [DOI] [PubMed] [Google Scholar]
- Lee, T.L. , Leung, W.K. , Chan, M.W. , Ng, E.K. , Tong, J.H. , Lo, K.W. , Chung, S.C. , Sung, J.J. , To, K.F. , 2002. Detection of gene promoter hypermethylation in the tumor and serum of patients with gastric carcinoma. Clinical Cancer Research. 8, 1761–1766. [PubMed] [Google Scholar]
- Liang, G. , Salem, C.E. , Yu, M.C. , Nguyen, H.D. , Gonzales, F.A. , Nguyen, T.T. , Nichols, P.W. , Jones, P.A. , 1998. DNA methylation differences associated with tumor tissues identified by genome scanning analysis. Genomics. 53, 260–268. [DOI] [PubMed] [Google Scholar]
- Loizou, J.I. , Murr, R. , Finkbeiner, M.G. , Sawan, C. , Wang, Z.Q. , Herceg, Z. , 2006. Epigenetic information in chromatin: the code of entry for DNA repair. Cell Cycle. 5, 696–701. [DOI] [PubMed] [Google Scholar]
- Lu, J. , Getz, G. , Miska, E.A. , Alvarez-Saavedra, E. , Lamb, J. , Peck, D. , Sweet-Cordero, A. , Ebert, B.L. , Mak, R.H. , Ferrando, A.A. , Downing, J.R. , Jacks, T. , Horvitz, H.R. , Golub, T.R. , 2005. MicroRNA expression profiles classify human cancers. Nature. 435, 834–838. [DOI] [PubMed] [Google Scholar]
- Luedi, P.P. , Hartemink, A.J. , Jirtle, R.L. , 2005. Genome-wide prediction of imprinted murine genes. Genome Research. 15, 875–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsit, C.J. , McClean, M.D. , Furniss, C.S. , Kelsey, K.T. , 2006. Epigenetic inactivation of the SFRP genes is associated with drinking, smoking and HPV in head and neck squamous cell carcinoma. International Journal of Cancer. 119, 1761–1766. [DOI] [PubMed] [Google Scholar]
- Moulton, T. , Crenshaw, T. , Hao, Y. , Moosikasuwan, J. , Lin, N. , Dembitzer, F. , Hensle, T. , Weiss, L. , McMorrow, L. , Loew, T. , 1994. Epigenetic lesions at the H19 locus in Wilms' tumour patients. Nature Genetics. 7, 440–447. [DOI] [PubMed] [Google Scholar]
- Muller, H.M. , Widschwendter, A. , Fiegl, H. , Ivarsson, L. , Goebel, G. , Perkmann, E. , Marth, C. , Widschwendter, M. , 2003. DNA methylation in serum of breast cancer patients: an independent prognostic marker. Cancer Research. 63, 7641–7645. [PubMed] [Google Scholar]
- Nakayama, H. , Hibi, K. , Taguchi, M. , Takase, T. , Yamazaki, T. , Kasai, Y. , Ito, K. , Akiyama, S. , Nakao, A. , 2002. Molecular detection of p16 promoter methylation in the serum of colorectal cancer patients. Cancer Letters. 188, 115–119. [DOI] [PubMed] [Google Scholar]
- Nuovo, G.J. , Plaia, T.W. , Belinsky, S.A. , Baylin, S.B. , Herman, J.G. , 1999. In situ detection of the hypermethylation-induced inactivation of the p16 gene as an early event in oncogenesis. Proceedings of the National Academy of Sciences of the United States of America. 96, 12754–12759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakey, R.J. , Beechey, C.V. , 2002. Imprinted genes: identification by chromosome rearrangements and post-genomic strategies. Trends in Genetics. 18, 359–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivier, M. , Eeles, R. , Hollstein, M. , Khan, M.A. , Harris, C.C. , Hainaut, P. , 2002. The IARC TP53 database: new online mutation analysis and recommendations to users. Human Mutation. 19, 607–614. [DOI] [PubMed] [Google Scholar]
- Olivier, M. , Langerod, A. , Carrieri, P. , Bergh, J. , Klaar, S. , Eyfjord, J. , Theillet, C. , Rodriguez, C. , Lidereau, R. , Bieche, I. , Varley, J. , Bignon, Y. , Uhrhammer, N. , Winqvist, R. , Jukkola-Vuorinen, A. , Niederacher, D. , Kato, S. , Ishioka, C. , Hainaut, P. , Borresen-Dale, A.L. , 2006. The clinical value of somatic TP53 gene mutations in 1,794 patients with breast cancer. Clinical Cancer Research. 12, 1157–1167. [DOI] [PubMed] [Google Scholar]
- Paez, J.G. , Janne, P.A. , Lee, J.C. , Tracy, S. , Greulich, H. , Gabriel, S. , Herman, P. , Kaye, F.J. , Lindeman, N. , Boggon, T.J. , Naoki, K. , Sasaki, H. , Fujii, Y. , Eck, M.J. , Sellers, W.R. , Johnson, B.E. , Meyerson, M. , 2004. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 304, 1497–1500. [DOI] [PubMed] [Google Scholar]
- Pao, W. , Miller, V. , Zakowski, M. , Doherty, J. , Politi, K. , Sarkaria, I. , Singh, B. , Heelan, R. , Rusch, V. , Fulton, L. , Mardis, E. , Kupfer, D. , Wilson, R. , Kris, M. , Varmus, H. , 2004. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proceedings of the National Academy of Sciences of the United States of America. 101, 13306–13311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pao, W. , Miller, V.A. , Politi, K.A. , Riely, G.J. , Somwar, R. , Zakowski, M.F. , Kris, M.G. , Varmus, H. , 2005. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Medicine. 2, e73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulou, E. , Davilas, E. , Sotiriou, V. , Koliopanos, A. , Aggelakis, F. , Dardoufas, K. , Agnanti, N.J. , Karydas, I. , Nasioulas, G. , 2004. Cell-free DNA and RNA in plasma as a new molecular marker for prostate cancer. Oncology Research. 14, 439–445. [DOI] [PubMed] [Google Scholar]
- Papadopoulou, E. , Davilas, E. , Sotiriou, V. , Georgakopoulos, E. , Georgakopoulou, S. , Koliopanos, A. , Aggelakis, F. , Dardoufas, K. , Agnanti, N.J. , Karydas, I. , Nasioulas, G. , 2006. Cell-free DNA and RNA in plasma as a new molecular marker for prostate and breast cancer. Annals of the New York Academy of Sciences. 1075, 235–243. [DOI] [PubMed] [Google Scholar]
- Pardal, R. , Clarke, M.F. , Morrison, S.J. , 2003. Applying the principles of stem-cell biology to cancer. Nature Reviews in Cancer. 3, 895–902. [DOI] [PubMed] [Google Scholar]
- Pasquinelli, A.E. , Hunter, S. , Bracht, J. , 2005. MicroRNAs: a developing story. Current Opinion in Genetics and Development. 15, 200–205. [DOI] [PubMed] [Google Scholar]
- Peterson, C.L. , Cote, J. , 2004. Cellular machineries for chromosomal DNA repair. Genes and Development. 18, 602–616. [DOI] [PubMed] [Google Scholar]
- Pray-Grant, M.G. , Daniel, J.A. , Schieltz, D. , Yates, J.R. , Grant, P.A. , 2005. Chd1 chromodomain links histone H3 methylation with SAGA- and SLIK-dependent acetylation. Nature. 433, 434–438. [DOI] [PubMed] [Google Scholar]
- Pulling, L.C. , Vuillemenot, B.R. , Hutt, J.A. , Devereux, T.R. , Belinsky, S.A. , 2004. Aberrant promoter hypermethylation of the death-associated protein kinase gene is early and frequent in murine lung tumors induced by cigarette smoke and tobacco carcinogens. Cancer Research. 64, 3844–3848. [DOI] [PubMed] [Google Scholar]
- Ravenel, J.D. , Broman, K.W. , Perlman, E.J. , Niemitz, E.L. , Jayawardena, T.M. , Bell, D.W. , Haber, D.A. , Uejima, H. , Feinberg, A.P. , 2001. Loss of imprinting of insulin-like growth factor-II (IGF2) gene in distinguishing specific biologic subtypes of Wilms tumor. Journal of the National Cancer Institute. 93, 1698–1703. [DOI] [PubMed] [Google Scholar]
- Roman-Gomez, J. , Jimenez-Velasco, A. , Agirre, X. , Prosper, F. , Heiniger, A. , Torres, A. , 2005. Lack of CpG island methylator phenotype defines a clinical subtype of T-cell acute lymphoblastic leukemia associated with good prognosis. Journal of Clinical Oncology. 23, 7043–7049. [DOI] [PubMed] [Google Scholar]
- Rowley, J.D. , 1998. The critical role of chromosome translocations in human leukemias. Annual Review of Genetics. 32, 495–519. [DOI] [PubMed] [Google Scholar]
- Safar, A.M. , Spencer, H. , Su, X. , Coffey, M. , Cooney, C.A. , Ratnasinghe, L.D. , Hutchins, L.F. , Fan, C.Y. , 2005. Methylation profiling of archived non-small cell lung cancer: a promising prognostic system. Clinical Cancer Research. 11, 4400–4405. [DOI] [PubMed] [Google Scholar]
- Sakatani, T. , Kaneda, A. , Iacobuzio-Donahue, C.A. , Carter, M.G. , de Boom Witzel, S. , Okano, H. , Ko, M.S. , Ohlsson, R. , Longo, D.L. , Feinberg, A.P. , 2005. Loss of imprinting of Igf2 alters intestinal maturation and tumorigenesis in mice. Science. 307, 1976–1978. [DOI] [PubMed] [Google Scholar]
- Samowitz, W.S. , Albertsen, H. , Herrick, J. , Levin, T.R. , Sweeney, C. , Murtaugh, M.A. , Wolff, R.K. , Slattery, M.L. , 2005. Evaluation of a large, population-based sample supports a CpG island methylator phenotype in colon cancer. Gastroenterology. 129, 837–845. [DOI] [PubMed] [Google Scholar]
- Sanchez-Cespedes, M. , Esteller, M. , Wu, L. , Nawroz-Danish, H. , Yoo, G.H. , Koch, W.M. , Jen, J. , Herman, J.G. , Sidransky, D. , 2000. Gene promoter hypermethylation in tumors and serum of head and neck cancer patients. Cancer research. 60, 892–895. [PubMed] [Google Scholar]
- Santos-Rosa, H. , Caldas, C. , 2005. Chromatin modifier enzymes, the histone code and cancer. European Journal of Cancer. 41, 2381–2402. [DOI] [PubMed] [Google Scholar]
- Schedin, P. , Elias, A. , 2004. Multistep tumorigenesis and the microenvironment. Breast Cancer Research. 6, 93–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlesinger, Y. , Straussman, R. , Keshet, I. , Farkash, S. , Hecht, M. , Zimmerman, J. , Eden, E. , Yakhini, Z. , Ben-Shushan, E. , Reubinoff, B.E. , Bergman, Y. , Simon, I. , Cedar, H. , 2006. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nature Genetics. [DOI] [PubMed] [Google Scholar]
- Schneider, R. , Bannister, A.J. , Kouzarides, T. , 2002. Unsafe SETs: histone lysine methyltransferases and cancer. Trends in Biochemical Sciences. 27, 396–402. [DOI] [PubMed] [Google Scholar]
- Seligson, D.B. , Horvath, S. , Shi, T. , Yu, H. , Tze, S. , Grunstein, M. , Kurdistani, S.K. , 2005. Global histone modification patterns predict risk of prostate cancer recurrence. Nature. 435, 1262–1266. [DOI] [PubMed] [Google Scholar]
- Sengupta, S. , Shimamoto, A. , Koshiji, M. , Pedeux, R. , Rusin, M. , Spillare, E.A. , Shen, J.C. , Huang, L.E. , Lindor, N.M. , Furuichi, Y. , Harris, C.C. , 2005. Tumor suppressor p53 represses transcription of RECQ4 helicase. Oncogene. 24, 1738–1748. [DOI] [PubMed] [Google Scholar]
- Shaw, R.J. , Akufo-Tetteh, E.K. , Risk, J.M. , Field, J.K. , Liloglou, T. , 2006. Methylation enrichment pyrosequencing: combining the specificity of MSP with validation by pyrosequencing. Nucleic Acids Research. 34, e78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, L. , Kondo, Y. , Hamilton, S.R. , Rashid, A. , Issa, J.P. , 2003. P14 methylation in human colon cancer is associated with microsatellite instability and wild-type p53. Gastroenterology. 124, 626–633. [DOI] [PubMed] [Google Scholar]
- Sherr, C.J. , Roberts, J.M. , 1999. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes and Development. 13, 1501–1512. [DOI] [PubMed] [Google Scholar]
- Shibata, D.M. , Sato, F. , Mori, Y. , Perry, K. , Yin, J. , Wang, S. , Xu, Y. , Olaru, A. , Selaru, F. , Spring, K. , Young, J. , Abraham, J.M. , Meltzer, S.J. , 2002. Hypermethylation of HPP1 is associated with hMLH1 hypermethylation in gastric adenocarcinomas. Cancer Research. 62, 5637–5640. [PubMed] [Google Scholar]
- Shigematsu, H. , Gazdar, A.F. , 2006. Somatic mutations of epidermal growth factor receptor signaling pathway in lung cancers. International Journal of Cancer. 118, 257–262. [DOI] [PubMed] [Google Scholar]
- Shigeno, K. , Yoshida, H. , Pan, L. , Luo, J.M. , Fujisawa, S. , Naito, K. , Nakamura, S. , Shinjo, K. , Takeshita, A. , Ohno, R. , Ohnishi, K. , 2004. Disease-related potential of mutations in transcriptional cofactors CREB-binding protein and p300 in leukemias. Cancer Letters. 213, 11–20. [DOI] [PubMed] [Google Scholar]
- Singh, S.K. , Hawkins, C. , Clarke, I.D. , Squire, J.A. , Bayani, J. , Hide, T. , Henkelman, R.M. , Cusimano, M.D. , Dirks, P.B. , 2004. Identification of human brain tumour initiating cells. Nature. 432, 396–401. [DOI] [PubMed] [Google Scholar]
- Sjoblom, T. , Jones, S. , Wood, L.D. , Parsons, D.W. , Lin, J. , Barber, T.D. , Mandelker, D. , Leary, R.J. , Ptak, J. , Silliman, N. , Szabo, S. , Buckhaults, P. , Farrell, C. , Meeh, P. , Markowitz, S.D. , Willis, J. , Dawson, D. , Willson, J.K. , Gazdar, A.F. , Hartigan, J. , Wu, L. , Liu, C. , Parmigiani, G. , Park, B.H. , Bachman, K.E. , Papadopoulos, N. , Vogelstein, B. , Kinzler, K.W. , Velculescu, V.E. , 2006. The consensus coding sequences of human breast and colorectal cancers. Science. 314, 268–274. [DOI] [PubMed] [Google Scholar]
- Slattery, M.L. , Curtin, K. , Sweeney, C. , Levin, T.R. , Potter, J. , Wolff, R.K. , Albertsen, H. , Samowitz, W.S. , 2007. Diet and lifestyle factor associations with CpG island methylator phenotype and BRAF mutations in colon cancer. International Journal of Cancer. 120, 656–663. [DOI] [PubMed] [Google Scholar]
- Sparmann, A. , van Lohuizen, M. , 2006. Polycomb silencers control cell fate, development and cancer. Nature Reviews in Cancer. 6, 846–856. [DOI] [PubMed] [Google Scholar]
- Steenman, M.J. , Rainier, S. , Dobry, C.J. , Grundy, P. , Horon, I.L. , Feinberg, A.P. , 1994. Loss of imprinting of IGF2 is linked to reduced expression and abnormal methylation of H19 in Wilms' tumour. Nature Genetics. 7, 433–439. [DOI] [PubMed] [Google Scholar]
- Strahl, B.D. , Allis, C.D. , 2000. The language of covalent histone modifications. Nature. 403, 41–45. [DOI] [PubMed] [Google Scholar]
- Sugimura, T. , Terada, M. , Yokota, J. , Hirohashi, S. , Wakabayashi, K. , 1992. Multiple genetic alterations in human carcinogenesis. Environmental Health Perspectives. 98, 5–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surani, M.A. , Barton, S.C. , Norris, M.L. , 1984. Development of reconstituted mouse eggs suggests imprinting of the genome during gametogenesis. Nature. 308, 548–550. [DOI] [PubMed] [Google Scholar]
- Taback, B. , Saha, S. , Hoon, D.S. , 2006. Comparative analysis of mesenteric and peripheral blood circulating tumor DNA in colorectal cancer patients. Annals of the New York Academy of Sciences. 1075, 197–203. [DOI] [PubMed] [Google Scholar]
- Tada, Y. , Wada, M. , Taguchi, K. , Mochida, Y. , Kinugawa, N. , Tsuneyoshi, M. , Naito, S. , Kuwano, M. , 2002. The association of death-associated protein kinase hypermethylation with early recurrence in superficial bladder cancers. Cancer Research. 62, 4048–4053. [PubMed] [Google Scholar]
- Takano, T. , Ohe, Y. , Sakamoto, H. , Tsuta, K. , Matsuno, Y. , Tateishi, U. , Yamamoto, S. , Nokihara, H. , Yamamoto, N. , Sekine, I. , Kunitoh, H. , Shibata, T. , Sakiyama, T. , Yoshida, T. , Tamura, T. , 2005. Epidermal growth factor receptor gene mutations and increased copy numbers predict gefitinib sensitivity in patients with recurrent non-small-cell lung cancer. Journal of Clinical Oncology. 23, 6829–6837. [DOI] [PubMed] [Google Scholar]
- Thibodeau, S.N. , French, A.J. , Roche, P.C. , Cunningham, J.M. , Tester, D.J. , Lindor, N.M. , Moslein, G. , Baker, S.M. , Liskay, R.M. , Burgart, L.J. , Honchel, R. , Halling, K.C. , 1996. Altered expression of hMSH2 and hMLH1 in tumors with microsatellite instability and genetic alterations in mismatch repair genes. Cancer Research. 56, 4836–4840. [PubMed] [Google Scholar]
- Toyooka, S. , Kiura, K. , Mitsudomi, T. , 2005. EGFR mutation and response of lung cancer to gefitinib. The New England Journal of Medicine. 352, 2136 (author reply 2136) [DOI] [PubMed] [Google Scholar]
- Toyota, M. , Ahuja, N. , Ohe-Toyota, M. , Herman, J.G. , Baylin, S.B. , Issa, J.P. , 1999. CpG island methylator phenotype in colorectal cancer. Proceedings of the National Academy of Sciences of the United States of America. 96, 8681–8686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyota, M. , Ohe-Toyota, M. , Ahuja, N. , Issa, J.P. , 2000. Distinct genetic profiles in colorectal tumors with or without the CpG island methylator phenotype. Proceedings of the National Academy of Sciences of the United States of America. 97, 710–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usadel, H. , Brabender, J. , Danenberg, K.D. , Jeronimo, C. , Harden, S. , Engles, J. , Danenberg, P.V. , Yang, S. , Sidransky, D. , 2002. Quantitative adenomatous polyposis coli promoter methylation analysis in tumor tissue, serum, and plasma DNA of patients with lung cancer. Cancer Research. 62, 371–375. [PubMed] [Google Scholar]
- Valenzuela, M.T. , Galisteo, R. , Zuluaga, A. , Villalobos, M. , Nunez, M.I. , Oliver, F.J. , Ruiz de Almodovar, J.M. , 2002. Assessing the use of p16(INK4a) promoter gene methylation in serum for detection of bladder cancer. European Urology. 42, 622–628. discussion 628–630 [DOI] [PubMed] [Google Scholar]
- Varambally, S. , Dhanasekaran, S.M. , Zhou, M. , Barrette, T.R. , Kumar-Sinha, C. , Sanda, M.G. , Ghosh, D. , Pienta, K.J. , Sewalt, R.G. , Otte, A.P. , Rubin, M.A. , Chinnaiyan, A.M. , 2002. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. 419, 624–629. [DOI] [PubMed] [Google Scholar]
- Volinia, S. , Calin, G.A. , Liu, C.G. , Ambs, S. , Cimmino, A. , Petrocca, F. , Visone, R. , Iorio, M. , Roldo, C. , Ferracin, M. , Prueitt, R.L. , Yanaihara, N. , Lanza, G. , Scarpa, A. , Vecchione, A. , Negrini, M. , Harris, C.C. , Croce, C.M. , 2006. A microRNA expression signature of human solid tumors defines cancer gene targets. Proceedings of the National Academy of Sciences of the United States of America. 103, 2257–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y.C. , Hsu, H.S. , Chen, T.P. , Chen, J.T. , 2006. Molecular diagnostic markers for lung cancer in sputum and plasma. Annals of the New York Academy of Sciences. 1075, 179–184. [DOI] [PubMed] [Google Scholar]
- Ward, R.L. , Cheong, K. , Ku, S.L. , Meagher, A. , O'Connor, T. , Hawkins, N.J. , 2003. Adverse prognostic effect of methylation in colorectal cancer is reversed by microsatellite instability. Journal of Clinical Oncology. 21, 3729–3736. [DOI] [PubMed] [Google Scholar]
- Weisenberger, D.J. , Siegmund, K.D. , Campan, M. , Young, J. , Long, T.I. , Faasse, M.A. , Kang, G.H. , Widschwendter, M. , Weener, D. , Buchanan, D. , Koh, H. , Simms, L. , Barker, M. , Leggett, B. , Levine, J. , Kim, M. , French, A.J. , Thibodeau, S.N. , Jass, J. , Haile, R. , Laird, P.W. , 2006. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nature Genetics. 38, 787–793. [DOI] [PubMed] [Google Scholar]
- Widschwendter, M. , Fiegl, H. , Egle, D. , Mueller-Holzner, E. , Spizzo, G. , Marth, C. , Weisenberger, D.J. , Campan, M. , Young, J. , Jacobs, I. , Laird, P.W. , 2006. Epigenetic stem cell signature in cancer. Nature Genetics. [DOI] [PubMed] [Google Scholar]
- Wolffe, A.P. , 2001. Chromatin remodeling: why it is important in cancer. Oncogene. 20, 2988–2990. [DOI] [PubMed] [Google Scholar]
- Wong, I.H. , Lo, Y.M. , Zhang, J. , Liew, C.T. , Ng, M.H. , Wong, N. , Lai, P.B. , Lau, W.Y. , Hjelm, N.M. , Johnson, P.J. , 1999. Detection of aberrant p16 methylation in the plasma and serum of liver cancer patients. Cancer Research. 59, 71–73. [PubMed] [Google Scholar]
- Wong, I.H. , Lo, Y.M. , Yeo, W. , Lau, W.Y. , Johnson, P.J. , 2000. Frequent p15 promoter methylation in tumor and peripheral blood from hepatocellular carcinoma patients. Clinical Cancer Research. 6, 3516–3521. [PubMed] [Google Scholar]
- Wong, T.S. , Chang, H.W. , Tang, K.C. , Wei, W.I. , Kwong, D.L. , Sham, J.S. , Yuen, A.P. , Kwong, Y.L. , 2002. High frequency of promoter hypermethylation of the death-associated protein-kinase gene in nasopharyngeal carcinoma and its detection in the peripheral blood of patients. Clinical Cancer Research. 8, 433–437. [PubMed] [Google Scholar]
- Wong, T.S. , Man, M.W. , Lam, A.K. , Wei, W.I. , Kwong, Y.L. , Yuen, A.P. , 2003. The study of p16 and p15 gene methylation in head and neck squamous cell carcinoma and their quantitative evaluation in plasma by real-time PCR. European Journal of Cancer. 39, 1881–1887. [DOI] [PubMed] [Google Scholar]
- Wong, T.S. , Kwong, D.L. , Sham, J.S. , Wei, W.I. , Kwong, Y.L. , Yuen, A.P. , 2004. Quantitative plasma hypermethylated DNA markers of undifferentiated nasopharyngeal carcinoma. Clinical Cancer Research. 10, 2401–2406. [DOI] [PubMed] [Google Scholar]
- Woodson, K. , Flood, A. , Green, L. , Tangrea, J.A. , Hanson, J. , Cash, B. , Schatzkin, A. , Schoenfeld, P. , 2004. Loss of insulin-like growth factor-II imprinting and the presence of screen-detected colorectal adenomas in women. Journal of the National Cancer Institute. 96, 407–410. [DOI] [PubMed] [Google Scholar]
- Yamamoto, H. , Min, Y. , Itoh, F. , Imsumran, A. , Horiuchi, S. , Yoshida, M. , Iku, S. , Fukushima, H. , Imai, K. , 2002. Differential involvement of the hypermethylator phenotype in hereditary and sporadic colorectal cancers with high-frequency microsatellite instability. Genes, Chromosomes and Cancer. 33, 322–325. [DOI] [PubMed] [Google Scholar]
- Yan, P.S. , Perry, M.R. , Laux, D.E. , Asare, A.L. , Caldwell, C.W. , Huang, T.H. , 2000. CpG island arrays: an application toward deciphering epigenetic signatures of breast cancer. Clinical Cancer Research. 6, 1432–1438. [PubMed] [Google Scholar]
- Yang, X.J. , 2004. The diverse superfamily of lysine acetyltransferases and their roles in leukemia and other diseases. Nucleic acids research. 32, 959–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, L. , Huang, J. , Yang, N. , Greshock, J. , Megraw, M.S. , Giannakakis, A. , Liang, S. , Naylor, T.L. , Barchetti, A. , Ward, M.R. , Yao, G. , Medina, A. , O'Brien-Jenkins, A. , Katsaros, D. , Hatzigeorgiou, A. , Gimotty, P.A. , Weber, B.L. , Coukos, G. , 2006. microRNAs exhibit high frequency genomic alterations in human cancer. Proceedings of the National Academy of Sciences of the United States of America. 103, 9136–9141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y.J. , Rossner, P. , Chen, Y. , Agrawal, M. , Wang, Q. , Wang, L. , Ahsan, H. , Yu, M.W. , Lee, P.H. , Santella, R.M. , 2006. Aflatoxin B1 and polycyclic aromatic hydrocarbon adducts, p53 mutations and p16 methylation in liver tissue and plasma of hepatocellular carcinoma patients. International Journal of Cancer. 119, 985–991. [DOI] [PubMed] [Google Scholar]
- Zhu, P. , Martin, E. , Mengwasser, J. , Schlag, P. , Janssen, K.P. , Gottlicher, M. , 2004. Induction of HDAC2 expression upon loss of APC in colorectal tumorigenesis. Cancer Cell. 5, 455–463. [DOI] [PubMed] [Google Scholar]
- Zou, H.Z. , Yu, B.M. , Wang, Z.W. , Sun, J.Y. , Cang, H. , Gao, F. , Li, D.H. , Zhao, R. , Feng, G.G. , Yi, J. , 2002. Detection of aberrant p16 methylation in the serum of colorectal cancer patients. Clinical Cancer Research. 8, 188–191. [PubMed] [Google Scholar]