

Abstract
The human tissue kallikreins, 15 secreted serine proteases, may play diverse roles in pathophysiology. The National Center for Biotechnology Information's dbSNP was mined for polymorphisms located within the kallikrein (KLK) locus using custom‐designed “ParSNPs” and “LocusAnnotator” software tools. Using “ParSNPs”, a filterable catalogue of 1856 KLK polymorphisms (1023 validated) was generated. “LocusAnnotator” was used to annotate the KLK locus sequence with gene and polymorphism features. A second locus was examined to validate the use of both programs on a non‐kallikrein locus. This report may assist in the informed selection of KLK polymorphisms for future association and biochemical studies in relation to disease. Furthermore, “ParSNPs” and “LocusAnnotator” are available at no cost from our website (www.acdcLab.org/annotations) to examine other loci.
Keywords: Genetic polymorphism, Single nucleotide polymorphism, Kallikreins
1. Introduction
In the post‐genomic era, a growing proportion of human genetics research is turning its focus to the identification and characterization of genotypic variation and its contribution at the population level. This has precipitated an international initiative to develop and execute a Human Variome Project, to catalogue genetic variations associated with disease (Ring et al., 2006). The most common type of variation in the human genome is the single nucleotide polymorphism (SNP), representing approximately 90% of all sequence variation (Collins et al., 1998). SNPs are conventionally defined as common variations at a single nucleotide position in the genome such that the least common allele is present in at least 1% of a given population. However, some researchers distinguish between these ‘polymorphic SNPs’ and ‘common SNPs’ with a minor allele frequency of at least 10% in the population (Ladiges et al., 2004; Kruglyak and Nickerson, 2001; Brookes, 1999). Considering that all SNPs may not have yet been discovered, validated, or deposited in public databases, the overall SNP density in the human genome is now estimated to be approximately one SNP every 150–300 base pairs (bp) (Ladiges et al., 2004; Botstein and Risch, 2003; Kruglyak and Nickerson, 2001). It is likely that despite large‐scale SNP discovery efforts (Sachidanandam et al., 2001; Collins et al., 1998; Wang et al., 1998), population‐specific or less common SNPs may remain unidentified or unvalidated (Botstein and Risch, 2003). SNP density may therefore require further refinement as more data are accumulated.
Insertion and deletion polymorphisms (‘indels’) represent another source of genomic variation. Compared to the extensive discovery efforts for SNPs, genome‐wide studies of indels have only recently been performed. A number of recent studies have composed libraries of indels of various lengths (Conrad et al., 2006; Mills et al., 2006; Hinds et al., 2006; McCarroll et al., 2006). Notably, Hinds et al. (2006) have reported that many deletions lie in linkage disequilibrium with nearby SNPs, which implies that indels may be surveyed indirectly for association with disease through genome‐wide SNP association studies.
One gene family of interest when considering the association of polymorphisms with disease is the human tissue kallikrein (KLK) family (Yousef and Diamandis, 2001; Yousef et al., 2000). The kallikrein (KLK) locus is a region of approximately 300 kilobases (kb) on chromosome 19q13.4, containing 15 genes clustered in a tandem array. These genes encode 15 serine proteases (KLKs), secreted as propeptides which are processed into mature peptides with trypsin‐ or chymotrypsin‐like endopeptidase activities (Yousef and Diamandis, 2001). The kallikreins have 30–50% similarity at both the nucleotide and amino acid sequence levels, and subsets of the KLKs exhibit coexpression and coregulation patterns in various tissues (Borgono and Diamandis, 2004; Yousef and Diamandis, 2001; Yousef et al., 2000). At the physiological level, kallikreins have been implicated in diverse processes such as blood pressure regulation, tissue remodeling, cell growth regulation, seminal clot liquefaction, and the desquamation of skin cells, although many kallikreins have yet to be fully functionally characterized (Borgono and Diamandis, 2004; Borgono et al., 2004). Conversely, the deregulation of kallikreins may play a role in many pathological processes, most notably in adenocarcinomas and other cancers (Obiezu and Diamandis, 2005; Borgono and Diamandis, 2004; Borgono et al., 2004). These reports have led to proposals that several kallikreins may have clinical utility as cancer biomarkers.
Recent kallikrein research reflects the appeal of associating specific polymorphisms with disease, either to contribute to the condition's etiology or to indicate which individuals might be susceptible. We are aware of association studies that have been published for seven of the 15 KLK genes. Most of these investigations have focused on the association of SNPs in the proximal promoter region of KLK3 (PSA) with prostate and breast cancers (Chiang et al., 2004; Wang et al., 2003; Bharaj et al., 2000). Similarly, SNPs in KLK2 and KLK10 have been associated with prostate cancer (Nam et al., 2006; Chiang et al., 2005; Bharaj et al., 2002). Other studies have investigated the potential association of polymorphisms in KLK1 with hypertension and renal disease (Hua et al., 2005; Yu et al., 2002), in KLK7 with atopic dermatitis (Vasilopoulos et al., 2004), in KLK8 with intracranial aneurysms (Weinsheimer et al., 2007), and in KLK12 with a splicing defect (Shinmura et al., 2004). While many of these studies have observed significant associations of KLK polymorphisms with disease, parallel studies by independent research groups to date have obtained conflicting or non‐significant results (Nam et al., 2004, 2005, 2006, 2003). The KLK family, therefore, presents many research opportunities, both to reach a consensus regarding previously reported associations of polymorphisms with disease in specific populations and to investigate the association of polymorphisms in all KLK genes with various other conditions.
Arguably, the most popular public database of SNPs and other polymorphisms in the human genome is the National Center for Biotechnology Information's (NCBI) dbSNP (Sherry et al., 2001), containing over 11,000,000 non‐redundant records in Build 126 (2006). Here, we present a comprehensive catalogue of 1856 unique polymorphisms found in the human tissue kallikrein locus represented in dbSNP, of which 1023 are validated. We have also generated a graphical representation of KLK polymorphisms in their genomic context. Custom‐designed software tools “ParSNPs” and “LocusAnnotator” were used to parse the information contained in the dbSNP records and to graphically annotate polymorphisms within the KLK locus, respectively. To validate the use of our “ParSNPs” and “LocusAnnotator” programs for cataloguing polymorphisms in an additional locus, we also parsed information for polymorphisms in an approximately 400kb region around the Plasminogen activator, urokinase (PLAU) gene locus. PLAU is another serine protease that may be activated in a cascade by KLK2 or KLK4 (Borgono and Diamandis, 2004). This report provides the first consolidated library of SNPs, indels, and other polymorphisms with characteristics of interest found in the KLK locus, while also demonstrating the usefulness of “ParSNPs” and “LocusAnnotator” for examining dbSNP polymorphisms in other non‐KLK loci. To this end, these software tools can be obtained at no cost through our website (www.acdcLab.org/annotations), for research purposes. Furthermore, it is proposed that the polymorphism catalogue and annotated KLK locus will facilitate the informed selection of KLK polymorphisms for further studies including their effects on gene expression or protein function, and may also provide a general overview of polymorphisms that may be selected for future disease association studies.
2. Results
2.1. Polymorphisms in the human tissue kallikrein locus
A batch query of dbSNP was executed to obtain all polymorphisms identified in the human tissue kallikrein locus (Figure 1A) as of February 2007. Overall, 1856 records of polymorphisms mapping to unique locations within the KLK locus were identified. A custom software tool developed in‐house named “ParSNPs” was used to parse the information contained in the batch report output into a filterable spreadsheet format. For each SNP or polymorphism record, “ParSNPs” gathers and organizes the following information: dbSNP record identification number (rsID), chromosome and base position, alleles and their average frequency, heterozygosity, validation status, associated gene loci, functional class of the polymorphism, and additional information for synonymous and non‐synonymous coding SNPs.
Figure 1.
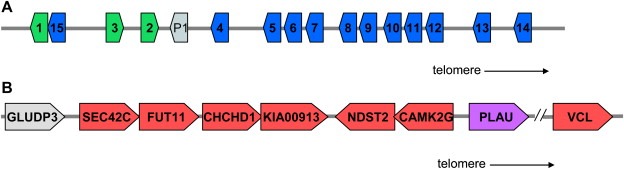
Loci examined for polymorphisms represented in dbSNP. (A) The human tissue kallikrein locus. Green arrowheads numbered 1–3 represent the three classical kallikrein genes KLK1, KLK2, and KLK3, while the blue arrowheads numbered 4–15 represent the KLK4–KLK15 genes. The grey arrowhead annotated as P1 represents the sole kallikrein pseudogene, KLKP1. (B) PLAU (purple arrowhead) and its associated 200kb flanking regions. Arrowheads are annotated with their respective gene names. GLUD3P (grey arrowhead) is a pseudogene. Relative lengths of genes and loci are not drawn to scale.
When creating “ParSNPs”, our main interest was the functional class annotation of each polymorphism (Table 1) (Kitts and Sherry, 2006). Given the double‐stranded nature of genomic DNA and the observation of alternative mRNA splicing in all KLK transcripts except possibly KLK14 (Kurlender et al., 2005), one SNP or polymorphism can be annotated with more than one functional class, given its context. For example, a SNP in a given gene could be classified as intronic in the context of one alternative mRNA transcript, but as a non‐synonymous coding SNP in the context of another. To accommodate these context‐dependent functional classes, “ParSNPs” creates a row of data for each functional class associated with a given polymorphism. Thus, the list of polymorphisms in the “ParSNPs” output was redundant and was examined manually to obtain non‐redundant counts of polymorphism records.
Table 1.
Functional classes associated with polymorphisms in dbSNPa
| Functional class | Definition |
|---|---|
| Null (inter‐gene or unspecified) | No functional annotation assigned in dbSNP |
| Gene locus region | Within 2kb 5′ or 500bp 3′ of a gene, but not in a transcribed region |
| Untranslated | In a transcribed region of the gene, but not in a coding region |
| Intronic | In an intron, excluding the first or last two intron bases |
| Splice junction | In the first or last two bases of an intron |
| Coding synonymous | Variant allele results in no amino acid change upon translation compared to reference contig allele |
| Coding non‐synonymous | Variant allele results in amino acid change upon translation compared to reference contig allele |
Adapted from Kitts and Sherry (2006).
An examination of the non‐redundant “ParSNPs” data revealed that, of the polymorphisms in the KLK locus, 1541 (83.0%) were SNPs and 304 (16.4%) were indels (Figure 2A). The remaining 11 (0.6%) records represented seven “mixed” polymorphism records whose alleles belong to more than one class of polymorphism (e.g. an insertion and a SNP), three multi‐nucleotide polymorphisms (e.g., CA/TG), and one named large deletion element. “ParSNPs” identified 1023 polymorphisms (55.1%) as validated, according to dbSNP. By the standards of dbSNP, a polymorphism is deemed validated if confirmation experiments have directly been performed, if genotype or allele frequency information is available, if the alleles have been observed in at least two homologous chromosomes sequenced, or if other independent submissions replicate the initial polymorphism submission (Kitts and Sherry, 2006). The vast majority of validated polymorphisms in the KLK locus found in dbSNP are SNPs (1012/1023; 98.9%, Figure 2B). In addition, a small number of validated insertions or deletions have been observed (6/1023; 0.6%). The remaining records retrieved (5/1023; 0.5%) represent “mixed” polymorphism records. Further analysis was limited to these validated polymorphisms in an attempt to avoid analysis of false positive records in dbSNP that may have arisen due to sequencing artifacts. Over the 285kb of the KLK locus, this corresponded to an average of one validated SNP per 282bp and one validated indel per 47.5kb.
Figure 2.
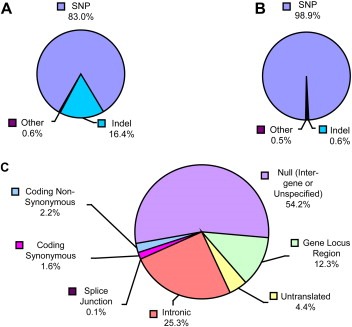
Polymorphisms in the kallikrein locus represented in dbSNP. Total (A) and validated (B) dbSNP polymorphisms identified in the KLK locus, separated by variation class. (C) Functional class annotations associated with validated KLK polymorphisms in dbSNP.
The SNPs observed in the KLK locus may be classified according to the nature of their alleles. SNPs begin as single base substitutions in an individual that are subsequently established in the population over time. They may therefore be classified as transitions (C↔T or G↔A) or as transversions (C↔A or G↔T, C↔G, T↔A). Of the validated SNPs in the KLK locus, 716 (70.8%) were C↔T transitions (or G↔A on the opposite strand). Of the transversion SNPs, 161 (15.9%) were C↔A (G↔T on the opposite strand), while 89 (8.8%) and 45 (4.5%) were C↔G and T↔A transversions, respectively. The record for the rs7256917 SNP states that the alleles at this position can be either C/G or C/T, so this record was not classified in the groups above.
The full “ParSNPs” catalogue of KLK polymorphisms is available on our website (www.acdcLab.org/annotations).
2.2. Functional class annotations of kallikrein polymorphisms
By examining the “ParSNPs” output, the proportion of validated dbSNP polymorphisms in the KLK locus for each of the polymorphism functional classes was determined (Figure 2C). The most prevalent functional annotations were those corresponding to the non‐coding intronic, gene locus region, or UTR polymorphisms (443/1057; 41.9%), whereas a smaller proportion of polymorphisms were associated with coding regions in the KLK locus (40/1057; 3.8%). In addition, a single splice‐site SNP was identified in KLK12. The remaining polymorphism records for which no functional class was specified by dbSNP (573/1057; 54.2%) may refer to either intergenic or unannotated polymorphisms. The number of polymorphisms associated with each KLK gene is stratified by functional class in Table 2. KLK1 had the most polymorphisms identified and deposited in dbSNP of any of the KLK genes (55/484; 11.4%), whereas KLK11 had the fewest (14/484; 2.9%). An examination of the 23 validated non‐synonymous coding SNPs identified in the KLK locus (Table 3) revealed that, based on the SNPs currently identified and validated in dbSNP, KLK1 also contains the most non‐synonymous SNPs, whereas none have yet been identified and validated in KLK6, KLK7, KLK9, KLK12 or KLK13.
Table 2.
Functional class annotation for validated KLK polymorphisms in dbSNP
| Gene | Total annotations | Gene locus region | Untranslated | Intronic | Splice junction | Coding synonymous | Coding non‐synonymous |
|---|---|---|---|---|---|---|---|
| KLK1 | 55 | 18 | 0 | 31 | 0 | 2 | 4 |
| KLK2 | 21 | 11 | 3 | 5 | 0 | 1 | 1 |
| KLK3 | 42 | 11 | 7 | 18 | 0 | 3 | 3 |
| KLK4 | 28 | 6 | 3 | 15 | 0 | 2 | 2 |
| KLK5 | 44 | 5 | 4 | 32 | 0 | 0 | 3 |
| KLK6 | 23 | 8 | 4 | 10 | 0 | 1 | 0 |
| KLK7 | 28 | 6 | 5 | 15 | 0 | 2 | 0 |
| KLK8 | 37 | 11 | 0 | 24 | 0 | 1 | 1 |
| KLK9 | 22 | 5 | 4 | 12 | 0 | 1 | 0 |
| KLK10 | 39 | 9 | 6 | 19 | 0 | 3 | 2 |
| KLK11 | 14 | 3 | 2 | 6 | 0 | 0 | 3 |
| KLK12 | 16 | 8 | 1 | 6 | 1 | 0 | 0 |
| KLK13 | 30 | 5 | 0 | 25 | 0 | 0 | 0 |
| KLK14 | 29 | 5 | 2 | 21 | 0 | 0 | 1 |
| KLK15 | 49 | 12 | 5 | 28 | 0 | 1 | 3 |
| KLKP1 | 7 | 7 | 0 | 0 | 0 | 0 | 0 |
| Total | 484 | 130 | 46 | 267 | 1 | 17 | 23 |
Table 3.
Validated non‐synonymous KLK SNPs in dbSNP
| Gene | dbSNP ID | SNP | Position in codon | Amino acid changea | Exon | Predicted peptide locationb |
|---|---|---|---|---|---|---|
| KLK1 | rs5515 | G/A | 2 | Arg77His | 3 | Mature |
| rs5516 | G/C | 1 | Glu145Gln | 3 | Mature | |
| rs5517 | A/G | 1 | Lys186Glu | 4 | Mature | |
| rs5518 | T/A | 2 | Val193Glu | 4 | Mature | |
| KLK2 | rs198977 | C/T | 1 | Arg250Trp | 5 | Mature |
| KLK3 | rs2271092 | G/A | 1 | Glu32Lys | 2 | Mature |
| rs2003783 | C/A | 1 | Leu132Ile | 3 | Mature | |
| rs17632542 | T/C | 2 | Ile179Thr | 4 | Mature | |
| KLK4 | rs1654551 | T/G | 1 | Ser22Ala | 2 | Signal peptide |
| rs2569527 | C/A | 3 | His197Gln | 4 | Mature | |
| KLK5 | rs182854 | A/G | 1 | Asn153Asp | 3 | Mature |
| rs2232534 | A/G | 1 | Ile172Val | 3 | Mature | |
| rs2232535 | G/C | 2 | Ser210Thr | 4 | Mature | |
| KLK8 | rs16988799 | G/A | 1 | Val154Ile | 3 | Mature |
| KLK10 | rs3745535 | T/G | 1 | Ser50Ala | 2 | Mature |
| rs2075690 | T/C | 2 | Leu149Pro | 3 | Mature | |
| KLK11 | rs2288892 | G/A | 1 | Ala32Thrc | 1 | Signal peptide |
| rs3745539 | G/A | 2 | Gly17Glu | 2 | Signal peptide | |
| rs1048328 | C/T | 1 | Arg134Cys | 3 | Mature | |
| KLK14 | rs2569491 | C/T | 1 | His29Tyr | 3 | Mature |
| KLK15 | rs7247190 | C/T | 1 | Pro128Serd | 3 | Mature |
| rs3212805 | C/T | 2 | Pro134Leud | 3 | Mature | |
| rs10403407 | C/T | 1 | Leu163Phed | 4 | Mature | |
Amino acid numbering refers to the sequences for isoform 1 or A of each KLK pre‐pro‐protein, unless otherwise noted.
Predictions based on Yousef and Diamandis (2001).
Amino acid numbering refers to KLK11 isoform 2 (GenBank accession NP_659196). In isoform 1, this SNP is in an untranslated region.
Amino acid numbering refers to KLK15 isoform 4 (GenBank accession NP_059979).
2.3. Graphical annotation of the kallikrein locus
We developed an additional custom software tool named “LocusAnnotator” that uses the “ParSNPs” output, in addition to genomic and mRNA sequences, to generate web pages of the genomic KLK locus annotated with polymorphism information. In addition to annotating the SNPs and other polymorphisms identified in the locus, “LocusAnnotator” annotates the start and stop codons, introns, exons, splice donors and acceptors, untranslated exon bases, poly‐A tails, and TATA boxes of the KLK genes. For the sake of clarity, one reference mRNA sequence for each gene with multiple transcript variants was used in the annotation process (see Section 4 for details). However, “LocusAnnotator” can accept the mRNA sequence of any splice variant as input. Each polymorphism annotation is accompanied by a pop‐up “tool tip text” information balloon that appears when the user hovers the mouse pointer over it, and by a hypertext link to the original full dbSNP record at NCBI. The KLK locus annotated by “LocusAnnotator” is also available online (www.acdcLab.org/annotations), allowing the KLK polymorphisms from dbSNP to be viewed graphically in their genomic context.
2.4. Examination of the PLAU locus and surrounding region using “ParSNPs” and “LocusAnnotator”
To demonstrate that a larger genomic region than the kallikrein locus with different characteristics can also easily be examined with “ParSNPs” and “LocusAnnotator”, we chose to study the Plasminogen activator, urokinase gene and 200kb of genomic sequence on either side of the gene locus (Figure 1B). This region also includes the calcium‐/calmodulin‐dependent protein kinase II gamma (CAMK2G), coiled‐coil‐helix‐coiled‐coil‐helix domain containing 1 (CHCHD1), fucosyltransferase 11 (alpha (1,3) fucosyltransferase) (FUT11), KIAA0913 (KIA00913), N‐deacetylase/N‐sulfotransferase (heparan glucosaminyl) 2 (NDST2), SEC24 related gene family, member C (Saccharomyces cerevisiae) (SEC24C), and vinculin (VCL) genes. After manually merging nine duplicate polymorphism records (see Section 4 for details), 1197 unique polymorphism records were parsed in “ParSNPs.” Of these, 918 (76.7%) were SNPs and 272 (22.7%) were indels (Figure 3A). The remaining seven records (0.06%) represented one microsatellite, two large deletions of named elements, and four polymorphisms of mixed classes. In total, 578 polymorphisms (48.3%) were classified as validated by dbSNP (Figure 3B). While 572 (99.0%) of these validated polymorphisms were true SNPs, only two (0.3%) indels were validated. The remaining four (0.7%) polymorphisms represent three mixed class polymorphisms and one microsatellite. Over the 406kb examined for polymorphisms, dbSNP therefore contained, on average, one validated SNP per 442bp and one validated indel per 203kb.
Figure 3.
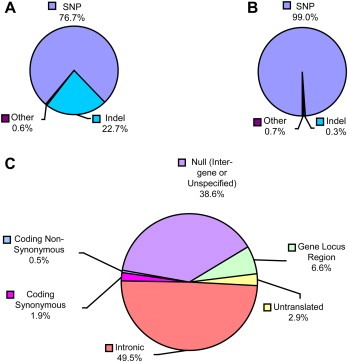
Polymorphisms in the PLAU region represented in dbSNP. Total (A) and validated (B) dbSNP polymorphisms identified either in the PLAU gene or within 200kb of either ends of this gene, separated by variation class. (C) Functional class annotations associated with validated polymorphisms in the PLAU region found in dbSNP.
Similarly to the KLK locus, in PLAU and the surrounding region studied, 372 (65.0%) validated SNPs were C↔T transitions (or G↔A on the opposite strand). Ninety‐three (16.3%) SNPs were C↔A (G↔T on the opposite strand) transversions, 58 (10.1%) were C↔G transversions, and 49 (8.6%) were T↔A transversions.
Examining the “ParSNPs” output for the validated polymorphisms revealed that, like the KLK locus, polymorphism annotations in PLAU and the surrounding region were mostly associated with non‐coding regions of gene loci (348/590; 59.0%) (Figure 3C). The proportion of non‐coding polymorphisms associated with genes compared to intergenic or unannotated polymorphisms was greater in the PLAU region compared to the KLK locus. In addition, whereas the coding SNPs in the KLK data were divided approximately equally between synonymous and non‐synonymous SNPs, more synonymous SNPs were identified in the PLAU region (11/590; 1.9%). Only three validated non‐synonymous SNPs were identified in dbSNP region, two of which were found in the PLAU gene. The first PLAU SNP was a T to C transition substituting a proline for the reference leucine residue at amino acid position 141, while the second SNP was an A to C transversion substituting a glutamine for the reference lysine at position 231. The third non‐synonymous SNP was found in the KIAA0913 gene, and was a G to A transition substituting a lysine for a glutamic acid at amino acid position 1075. The functional class annotations were also stratified by gene, as shown in Table 4. Finally, the PLAU locus region was annotated using “LocusAnnotator” as described for the KLK locus.
Table 4.
Functional class annotations for validated dbSNP polymorphisms in the PLAU region
| Gene | Total annotations | Gene locus region | Untranslated | Intronic | Splice junction | Coding synonymous | Coding non‐synonymous |
|---|---|---|---|---|---|---|---|
| PLAU | 26 | 4 | 3 | 15 | 0 | 2 | 2 |
| CAMK2G | 79 | 3 | 5 | 70 | 0 | 1 | 0 |
| CHCHD1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| FUT11 | 4 | 1 | 0 | 3 | 0 | 0 | 0 |
| GLUDP3 | 12 | 12 | 0 | 0 | 0 | 0 | 0 |
| KIAA0913 | 12 | 5 | 1 | 3 | 0 | 2 | 1 |
| NDST2 | 19 | 8 | 6 | 5 | 0 | 0 | 0 |
| SEC24C | 35 | 3 | 2 | 27 | 0 | 3 | 0 |
| VCL | 175 | 3 | 0 | 169 | 0 | 3 | 0 |
| Total | 362 | 39 | 17 | 292 | 0 | 11 | 3 |
The full “ParSNPs” catalogue of polymorphisms in the PLAU region is available online (www.acdcLab.org/annotations). At our website, one can also examine the genomic sequence of this region, annotated by “LocusAnnotator”.
3. Discussion
As of February 2007, the kallikrein locus was found to contain 1856 non‐redundant polymorphisms in dbSNP, of which 1023 had been validated. Examining the validated dbSNP polymorphism records with the “ParSNPs” and “LocusAnnotator” programs developed by our group allowed these genomic variants to be classified by functional class and to be visualized by genomic context. The information contained in “ParSNPs” generated a comprehensive catalogue of polymorphisms in the KLK locus and their characteristics, while “LocusAnnotator” created an annotated KLK locus.
As expected, most of the polymorphisms identified in the KLK locus were true SNPs (or candidate SNPs in the case of unvalidated SNP records). While indels represented the second most common class of polymorphism, very few of these indels have been validated. This may be explained with reference to two observations. First, polymorphic indels with minor allele frequencies less than 5% may be difficult to identify in genotyping studies. Of the six validated indels, all but one were flagged as validated by dbSNP due to the presence of genotype or allele frequency data, yet only one of these indels had an average minor allele frequency of less than 5%. Second, a larger problem may be that although the methods currently used to detect structural variation such as indel polymorphisms or copy number variation (CNV) are rapidly evolving, current efforts at constructing indel libraries have still resulted in data sets with little overlap between studies, even those using the same initial input data (Abecasis et al., 2007; Eichler, 2006). This would suggest that fewer indels could be validated by clustering of multiple submissions, supported by the fact that only one of the validated indels was validated by this method. It is therefore likely that as SNP and indel detection technology progresses, the number of validated polymorphisms in the KLK locus represented in dbSNP will increase.
The average SNP density observed in the KLK locus agrees with the estimates of one SNP per 150–300bp proposed for the entire genome (Ladiges et al., 2004; Botstein and Risch, 2003; Kruglyak and Nickerson, 2001). This does not necessarily imply that nearly all polymorphisms in the KLK locus have already been discovered, since SNP density varies along the genome, and many of the unvalidated candidate SNPs may represent true SNPs. Unvalidated SNPs in the KLK locus should be further examined in future genotyping studies to assess their status as true SNPs. The expected genome‐wide indel density is much more difficult to define given that different discovery efforts have yielded different indel libraries (Abecasis et al., 2007; Eichler, 2006). Currently, approximately 14,900 non‐redundant validated indels have been submitted to dbSNP, corresponding to an approximate average of one indel per 201kb in the human genome. The density of validated indels observed in the KLK locus and deposited in dbSNP is much higher than this crude average for the whole genome. In addition, the proportion of SNPs representing transitions versus transversions agrees with previous observations that two‐thirds of all SNPs are due to C↔T transitions (or G↔A on the opposite strand) (Brookes, 1999). This is proposed to be accounted for by the role of spontaneous deamination of 5‐methyl‐cytosines to thymines in CpG islands (Holliday and Grigg, 1993).
The distribution of functional class annotations associated with the validated KLK polymorphisms reflects two factors. First, to an extent, the distribution reflects the degree of conservation throughout the genome. Coding SNPs, and in particular non‐synonymous coding SNPs, were observed at a relatively low frequency, reflecting the high conservation of coding regions throughout the genome (Waterston et al., 2002; Cargill et al., 1999). To complement this observation, polymorphisms in non‐coding regions of the KLK locus have been observed at a higher frequency. However, it is important to note that many elements of non‐coding DNA in the human genome also show a high degree of conservation, referred to as conserved non‐genic sequences (CNGs). These CNGs account for approximately two‐thirds of sequence elements conserved between mice and humans (Dermitzakis et al., 2005; Waterston et al., 2002). Nevertheless, the proportions of validated polymorphisms associated with each functional class may not completely mirror the effect of selection pressure and sequence conservation in the different regions. This is because the distribution is biased by a second factor, the tendency of polymorphism discovery efforts to concentrate on particular genomic areas. For instance, many discovery efforts have focused on genic regions or on identifying non‐synonymous coding SNPs due to their potential for having direct effects on the protein product (Bhangale et al., 2005; Stephens et al., 2001; Cargill et al., 1999). The distribution of functional classes associated with KLK polymorphisms likely reflects a balance of both of these factors.
The catalogue of polymorphisms generated for the PLAU region by “ParSNPs” and the associated annotated sequence generated by “LocusAnnotator” validates the use of these two programs for loci other than the kallikrein locus, for which they were originally developed. While the observed density of validated dbSNP SNPs in this region was slightly less than the predicted genome‐wide average, this is not surprising since SNP density should vary throughout the genome. Conversely, the observed validated indel density in the PLAU region was quite consistent with the rough prediction of average genome‐wide indel density as described above. Overall, the proportion of transitions and transversions and functional classes associated with the validated polymorphism records was similar to those observed in the human tissue kallikrein locus. These results demonstrate that although “ParSNPs” and “LocusAnnotator” were first designed to analyze the kallikrein locus, a locus of different composition can just as easily be examined.
The “ParSNPs” and “LocusAnnotator” catalogues of KLK polymorphisms create a “one‐stop” resource for interested researchers to select polymorphisms associated with certain functional classes for further study. While not all non‐synonymous SNPs are predicted to lead to changes at the protein level that can be associated with disease (Rudd et al., 2005), those identified in the KLK locus still represent a group of SNPs that may be mined for causal association with various conditions. For example, the effects of non‐synonymous polymorphisms in other serine proteases such as factor VII‐activating protease (FASP) and Omi/Htr2 on proteolytic activity and its regulation have been shown to play roles in cardiovascular disease and Parkinson's disease, respectively (Sedding et al., 2006; Strauss et al., 2005). In addition, polymorphisms in non‐coding regulatory regions of the KLKs may have effects at the transcriptional level due to altered promoter activity (Buckland et al., 2005; Cramer et al., 2003). Moreover, splice‐site mutations such as that identified in KLK12 can lead to splicing defects resulting in non‐functional proteins (Shinmura et al., 2004).
The main advantage of using the “ParSNPs” and “LocusAnnotator” catalogues to identify polymorphisms of interest, in comparison with simply searching dbSNP, is that they allow the user to examine polymorphisms with a given set of multiple characteristics more rapidly and efficiently than going through many records by hand. “LocusAnnotator” also allows loci of theoretically any length to be visualized at the genomic sequence level, with various useful annotations. However, the catalogues derived by these programs using dbSNP data are inherently limited by the quality of data that is initially submitted to dbSNP. Polymorphisms that have been identified in the KLK locus but have not been submitted to dbSNP, such as a unique minisatellite and other repeat sequences (Yousef et al., 2001), will be absent from the catalogue. Similarly, any errors or ambiguities in submitted data will be propagated, for example in a few cases of indels with ambiguous stated lengths. However, by restricting analysis to validated polymorphisms, the impact of these issues should be small.
In response to the increasing volume of publicly available polymorphism data, numerous other tools have been developed to organize and visualize polymorphisms and their characteristics in a high‐throughput fashion. Among these tools are SNPper (Riva and Kohane, 2004), SNPHunter (Wang et al., 2005), and SNPselector (Xu et al., 2005). SNPper is a web‐based resource developed to provide a local database of dbSNP polymorphisms mapping uniquely to the human genome, to allow parsing and filtering of data pertaining to these polymorphisms' characteristics, and to present the polymorphisms graphically in their genomic context. While “ParSNPs” and “LocusAnnotator” share SNPper's ability to organize a large amount of polymorphism data into an intuitive catalogue, to filter this data for characteristics of interest, and to show the location of polymorphisms within genes of interest, our programs have some unique features as well. For instance, since all filtering by “ParSNPs” is executed in Microsoft Excel, the users may rapidly sort, filter, and analyze the polymorphism data according to complex criteria independent of a web connection, and these filtered polymorphism sets can be saved and exported in various common formats for downstream use. In addition, whereas SNPper will visually represent polymorphisms in the sequence context of genes as defined by the UCSC Genome Browser (Kent et al., 2002), “LocusAnnotator” uses user‐supplied sequence data and will display any polymorphisms in large intergenic regions as well. SNPper possesses unique features as well, such as the ability to search for polymorphisms associated with particular Gene Ontology annotations.
SNPHunter and SNPselector are resources that focus on appropriate selection of polymorphisms specifically for association studies, and as such their main features are centred on selecting polymorphisms in and near query genes, chosen to be evenly spaced within the genomic region. In contrast, “ParSNPs” and “LocusAnnotator” were designed to give the user a general idea of interesting polymorphisms in any user‐defined genomic region, at a glance. These programs are thus more appropriate for selecting a few specific polymorphisms of interest for functional studies or association studies on a smaller scale. While SNPHunter offers a schematic graphical representation of the location of polymorphisms within queried genes, neither SNPHunter nor SNPselector generate displays of the location of each polymorphism in the context of a gene's nucleotide sequence or its location within the sequence of an entire defined genomic region, as does “LocusAnnotator”.
Finally, while SNPHunter is designed to act as a portal between the users and the information contained in dbSNP, SNPper, and SNPselector query their own databases of polymorphisms derived from dbSNP or the UCSC Genome Browser database, respectively. “ParSNPs” and “LocusAnnotator” do not maintain local databases; rather they are tools to examine the output from a direct batch query of dbSNP. For this reason, the user has no requirement to maintain a database and constantly update the programs with each dbSNP release, barring any drastic changes to dbSNP's batch report output format. In addition, by querying dbSNP directly and later analyzing the results with “ParSNPs” and “LocusAnnotator”, the user can take advantage of the complex queries available in dbSNP. For example, whereas SNPper's database is limited to polymorphisms mapping uniquely to the human genome, certain users may be interested in other ambiguously mapped polymorphisms as well.
Ultimately, the catalogue of polymorphisms in the kallikrein locus reported here promises to be a valuable resource for rational selection of KLK polymorphisms for further study. While coding non‐synonymous SNPs lend themselves to biochemical studies of variation in KLK proteolytic activity, others may be more appropriate for disease association or linkage studies. By making the “ParSNPs” and “LocusAnnotator” programs freely available to the research community, it is our hope that consolidated polymorphism catalogues for other gene families will be generated.
4. Experimental procedures
4.1. Retrieval of SNP and other polymorphism records
All non‐redundant human tissue kallikrein polymorphisms in dbSNP Build 126 (2006) were retrieved as of February 2007 using the Entrez SNP index (www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=snp). Polymorphisms retrieved were limited to those mapping only once to the human reference genome (Build 36.1) and found in the kallikrein locus, defined as base position 56,004,216–56,289,314 on chromosome 19 (Figure 1A). A batch report of these records was retrieved in both “flat file” and “genotype report” formats. This included information for 1856 non‐redundant dbSNP records.
Polymorphisms within the PLAU gene or within 200kb flanking either side of the gene (Figure 1B) had been similarly retrieved in November 2006, limiting results to polymorphisms mapping only once to the human reference genome (Build 36.1) and between base positions 75,140,896–75,547,260 on chromosome 10. A total of 1206 polymorphism records were retrieved from dbSNP. Although no records were flagged by dbSNP as duplicates during the batch report query, nine records were manually classified as duplicates based on reporting identical alleles at identical chromosome base positions. If any of these duplicates was a validated record, it was kept for analysis. If neither duplicate was validated, the record containing the most information was kept (i.e. merged records rs33913415, rs34978424, rs34236710, rs35084254, rs33971368, rs34958445, rs34985773, rs34063648, and rs9416032 into rs6143983, rs57866121, rs10577271, rs5786126, rs11339212, rs11356021, rs5786132, rs10709887, and rs2461867, respectively). This yielded information for 1197 non‐redundant dbSNP records.
4.2. “ParSNPs” analysis of polymorphism characteristics
“ParSNPs”, a macro written in‐house for use in Microsoft Excel, was used to parse the information contained in the dbSNP flat files and genotype reports for the KLK and PLAU loci, generating spreadsheets with filterable columns. The following information columns were included (if known): dbSNP reference SNP record identification number (rsID), chromosome, chromosome position, alleles, average allele frequency, heterozygosity, validation status, associated gene loci, polymorphism functional class, and additional information for coding SNPs. Since “ParSNPs” generates a new row in the spreadsheet for each functional class associated with each polymorphism, the “ParSNPs” output was filtered and examined manually to obtain a non‐redundant set of polymorphisms for Figures 2 and 3. Data pertaining to the annotation of validated polymorphism functional classes were extracted from the complete output file. This allowed each polymorphism to be associated with annotations of more than one functional class. Functional classes were defined as in Table 1.
4.3. “LocusAnnotator” annotation of genomic loci
“LocusAnnotator”, a second software tool developed in‐house, was used to annotate the genomic sequence of the KLK locus, and also of PLAU and its flanking regions. For the annotation of the KLK locus, the input for this program was the KLK “ParSNPs” output, along with the genomic sequence of the locus (GenBank accession NT_011109.15, bases 23,580,594–23,865,692) and a reference mRNA sequence for each KLK gene. For KLKs with alternative splice variants, transcript variant 1 (or transcript variant A in the case of KLK6) was chosen for use as the reference mRNA in “LocusAnnotator” (GenBank accessions for KLK1: NM_002257.2, KLK2: NM_005551.3, KLK3: NM_001648.2, KLK4: NM_004917.3, KLK5: NM_012427.4, KLK6: NM_002774.3, KLK7: NM_005046.2, KLK8: NM_007196.2, KLK9: NM_012315.1, KLK10: NM_002776.4, KLK11: NM_006853.2, KLK12: NM_019598.2, KLK13: NM_015596.1, and KLK14: NM_022046.4). The exception to this was KLK15, for which no reference sequence for transcript variant 1 is available. For KLK15, transcript variant 4 (GenBank accession NM_017509.2) was chosen as the reference mRNA sequence, as in Kurlender et al. (2005). For the annotation of PLAU and the 200kb flanking regions, the input for “LocusAnnotator” was the PLAU “ParSNPs” output, the genomic sequence of the region (GenBank accession NT_008583.16, bases 24,022,045–24,428,409) and the mRNA sequences of PLAU (GenBank accession NM_002658.2), CHCHD1 (GenBank accession NM_203298.1), CAMK2G (GenBank accession NM_172171.1), FUT11 (GenBank accession NM_173540.1), KIAA0913 (GenBank accession NM_015037.2), NDST2 (GenBank accession NM_003635.2), SEC24C (GenBank accession NM_004922.2), and VCL (GenBank accession NM_014000.2). For transcripts with alternative splice variants, transcript variant 1 was chosen as the reference form for “LocusAnnotator”.
“LocusAnnotator” uses this input to generate a spreadsheet containing the location of all annotations to be added on the locus genomic sequence. This encompasses not only the polymorphisms (both validated and unvalidated) and selected characteristics, but also the start and stop codons, intron and exon structures, splice donors and acceptors, untranslated exon bases, TATA boxes, and poly‐A tails for each gene. To determine the location of the intron/exon boundaries, “LocusAnnotator” uses NCBI's Spidey program (Wheelan et al., 2001) to align the mRNA transcripts to the genomic locus sequence. Finally, this information is used to generate a series of web pages depicting the KLK or PLAU locus sequences and all associated annotations, including hypertext links to the original online dbSNP records for each polymorphism.
Acknowledgements
We would like to thank Julie L.V. Shaw and Carla A. Borgono for helpful discussions and advice. This work was supported in part by an Undergraduate Student Research Award to C.A.G. from the Natural Sciences and Engineering Research Council of Canada. The authors declare that they have no competing financial interests. The programs “ParSNPs” and “LocusAnnotator” can be obtained at no cost for research purposes. Please see our website for instructions for obtaining these programs, system requirements, and other related information (www.acdcLab.org/annotations).
Goard Carolyn A., Bromberg Irvin L., Elliott Marc B., Diamandis Eleftherios P., (2007), A consolidated catalogue and graphical annotation of dbSNP polymorphisms in the human tissue kallikrein (KLK) locus, Molecular Oncology, 1, doi: 10.1002/mol2.2007.1.issue-3.
References
- Abecasis, G. , Tam, P.K. , Bustamante, C.D. , Ostrander, E.A. , Scherer, S.W. , Chanock, S.J. , Kwok, P.Y. , Brookes, A.J. , 2007. Human genome variation 2006: emerging views on structural variation and large-scale SNP analysis. Nat. Genet.. 39, 153–155. [DOI] [PubMed] [Google Scholar]
- Bhangale, T.R. , Rieder, M.J. , Livingston, R.J. , Nickerson, D.A. , 2005. Comprehensive identification and characterization of diallelic insertion–deletion polymorphisms in 330 human candidate genes. Hum. Mol. Genet.. 14, 59–69. [DOI] [PubMed] [Google Scholar]
- Bharaj, B. , Scorilas, A. , Diamandis, E.P. , Giai, M. , Levesque, M.A. , Sutherland, D.J. , Hoffman, B.R. , 2000. Breast cancer prognostic significance of a single nucleotide polymorphism in the proximal androgen response element of the prostate specific antigen gene promoter. Breast Cancer Res. Treat.. 61, 111–119. [DOI] [PubMed] [Google Scholar]
- Bharaj, B.B. , Luo, L.Y. , Jung, K. , Stephan, C. , Diamandis, E.P. , 2002. Identification of single nucleotide polymorphisms in the human kallikrein 10 (KLK10) gene and their association with prostate, breast, testicular, and ovarian cancers. Prostate. 51, 35–41. [DOI] [PubMed] [Google Scholar]
- Borgono, C.A. , Diamandis, E.P. , 2004. The emerging roles of human tissue kallikreins in cancer. Nat. Rev. Cancer. 4, 876–890. [DOI] [PubMed] [Google Scholar]
- Borgono, C.A. , Michael, I.P. , Diamandis, E.P. , 2004. Human tissue kallikreins: physiologic roles and applications in cancer. Mol. Cancer Res.. 2, 257–280. [PubMed] [Google Scholar]
- Botstein, D. , Risch, N. , 2003. Discovering genotypes underlying human phenotypes: past successes for Mendelian disease, future approaches for complex disease. Nat. Genet.. 33, (Suppl.) 228–237. [DOI] [PubMed] [Google Scholar]
- Brookes, A.J. , 1999. The essence of SNPs. Gene. 234, 177–186. [DOI] [PubMed] [Google Scholar]
- Buckland, P.R. , Hoogendoorn, B. , Coleman, S.L. , Guy, C.A. , Smith, S.K. , O'Donovan, M.C. , 2005. Strong bias in the location of functional promoter polymorphisms. Hum. Mutat.. 26, 214–223. [DOI] [PubMed] [Google Scholar]
- Cargill, M. , Altshuler, D. , Ireland, J. , Sklar, P. , Ardlie, K. , Patil, N. , Shaw, N. , Lane, C.R. , Lim, E.P. , Kalyanaraman, N. , 1999. Characterization of single-nucleotide polymorphisms in coding regions of human genes. Nat. Genet.. 22, 231–238. [DOI] [PubMed] [Google Scholar]
- Chiang, C.H. , Chen, K.K. , Chang, L.S. , Hong, C.J. , 2004. The impact of polymorphism on prostate specific antigen gene on the risk, tumor volume and pathological stage of prostate cancer. J. Urol.. 171, 1529–1532. [DOI] [PubMed] [Google Scholar]
- Chiang, C.H. , Hong, C.J. , Chang, Y.H. , Chang, L.S. , Chen, K.K. , 2005. Human kallikrein-2 gene polymorphism is associated with the occurrence of prostate cancer. J. Urol.. 173, 429–432. [DOI] [PubMed] [Google Scholar]
- Collins, F.S. , Brooks, L.D. , Chakravarti, A. , 1998. A DNA polymorphism discovery resource for research on human genetic variation. Genome Res.. 8, 1229–1231. [DOI] [PubMed] [Google Scholar]
- Conrad, D.F. , Andrews, T.D. , Carter, N.P. , Hurles, M.E. , Pritchard, J.K. , 2006. A high-resolution survey of deletion polymorphism in the human genome. Nat. Genet.. 38, 75–81. [DOI] [PubMed] [Google Scholar]
- Cramer, S.D. , Chang, B.L. , Rao, A. , Hawkins, G.A. , Zheng, S.L. , Wade, W.N. , Cooke, R.T. , Thomas, L.N. , Bleecker, E.R. , Catalona, W.J. , 2003. Association between genetic polymorphisms in the prostate-specific antigen gene promoter and serum prostate-specific antigen levels. J. Natl. Cancer Inst.. 95, 1044–1053. [DOI] [PubMed] [Google Scholar]
- Database of Single Nucleotide Polymorphisms (dbSNP), 2006. Bethesda (MD): National Center for Biotechnology Information, National Library of Medicine (dbSNP Build ID: {build 126}). Available from: <http://www.ncbi.nlm.nih.gov/SNP/> (accessed 2006 November 20 to February 28).
- Dermitzakis, E.T. , Reymond, A. , Antonarakis, S.E. , 2005. Conserved non-genic sequences – an unexpected feature of mammalian genomes. Nat. Rev. Genet.. 6, 151–157. [DOI] [PubMed] [Google Scholar]
- Eichler, E.E. , 2006. Widening the spectrum of human genetic variation. Nat. Genet.. 38, 9–11. [DOI] [PubMed] [Google Scholar]
- Hinds, D.A. , Kloek, A.P. , Jen, M. , Chen, X. , Frazer, K.A. , 2006. Common deletions and SNPs are in linkage disequilibrium in the human genome. Nat. Genet.. 38, 82–85. [DOI] [PubMed] [Google Scholar]
- Holliday, R. , Grigg, G.W. , 1993. DNA methylation and mutation. Mutat. Res.. 285, 61–67. [DOI] [PubMed] [Google Scholar]
- Hua, H. , Zhou, S. , Liu, Y. , Wang, Z. , Wan, C. , Li, H. , Chen, C. , Li, G. , Zeng, C. , Chen, L. , 2005. Relationship between the regulatory region polymorphism of human tissue kallikrein gene and essential hypertension. J. Hum. Hypertens.. 19, 715–721. [DOI] [PubMed] [Google Scholar]
- Kent, W.J. , Sugnet, C.W. , Furey, T.S. , Roskin, K.M. , Pringle, T.H. , Zahler, A.M. , Haussler, D. , 2002. The human genome browser at UCSC. Genome Res.. 12, 996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitts, A., Sherry, S., 2006. The Single Nucleotide Polymorphism Database (dbSNP) of Nucleotide Sequence Variation. In: The NCBI Handbook. The National Library of Medicine, Bethesda, MD. Available from: <http://www.ncbi.nlm.nih.gov/books/bv.fcgi?rid=handbook.chapter.ch5> (accessed 2006 May 20).
- Kruglyak, L. , Nickerson, D.A. , 2001. Variation is the spice of life. Nat. Genet.. 27, 234–236. [DOI] [PubMed] [Google Scholar]
- Kurlender, L. , Borgono, C. , Michael, I.P. , Obiezu, C. , Elliott, M.B. , Yousef, G.M. , Diamandis, E.P. , 2005. A survey of alternative transcripts of human tissue kallikrein genes. Biochim. Biophys. Acta. 1755, 1–14. [DOI] [PubMed] [Google Scholar]
- Ladiges, W. , Kemp, C. , Packenham, J. , Velazquez, J. , 2004. Human gene variation: from SNPs to phenotypes. Mutat. Res.. 545, 131–139. [DOI] [PubMed] [Google Scholar]
- McCarroll, S.A. , Hadnott, T.N. , Perry, G.H. , Sabeti, P.C. , Zody, M.C. , Barrett, J.C. , Dallaire, S. , Gabriel, S.B. , Lee, C. , Daly, M.J. , 2006. Common deletion polymorphisms in the human genome. Nat. Genet.. 38, 86–92. [DOI] [PubMed] [Google Scholar]
- Mills, R.E. , Luttig, C.T. , Larkins, C.E. , Beauchamp, A. , Tsui, C. , Pittard, W.S. , Devine, S.E. , 2006. An initial map of insertion and deletion (INDEL) variation in the human genome. Genome Res.. 16, 1182–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam, R.K. , Zhang, W.W. , Klotz, L.H. , Trachtenberg, J. , Jewett, M.A. , Sweet, J. , Toi, A. , Teahan, S. , Venkateswaran, V. , Sugar, L. , 2006. Variants of the hK2 protein gene (KLK2) are associated with serum hK2 levels and predict the presence of prostate cancer at biopsy. Clin. Cancer Res.. 12, 6452–6458. [DOI] [PubMed] [Google Scholar]
- Obiezu, C.V. , Diamandis, E.P. , 2005. Human tissue kallikrein gene family: applications in cancer. Cancer Lett.. 224, 1–22. [DOI] [PubMed] [Google Scholar]
- Ring, H.Z. , Kwok, P.Y. , Cotton, R.G. , 2006. Human Variome Project: an international collaboration to catalogue human genetic variation. Pharmacogenomics. 7, 969–972. [DOI] [PubMed] [Google Scholar]
- Riva, A. , Kohane, I.S. , 2004. A SNP-centric database for the investigation of the human genome. BMC Bioinformatics. 5, 33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudd, M.F. , Williams, R.D. , Webb, E.L. , Schmidt, S. , Sellick, G.S. , Houlston, R.S. , 2005. The predicted impact of coding single nucleotide polymorphisms database. Cancer Epidemiol. Biomark. Prev.. 14, 2598–2604. [DOI] [PubMed] [Google Scholar]
- Sachidanandam, R. , Weissman, D. , Schmidt, S.C. , Kakol, J.M. , Stein, L.D. , Marth, G. , Sherry, S. , Mullikin, J.C. , Mortimore, B.J. , Willey, D.L. , 2001. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature. 409, 928–933. [DOI] [PubMed] [Google Scholar]
- Sedding, D. , Daniel, J.M. , Muhl, L. , Hersemeyer, K. , Brunsch, H. , Kemkes-Matthes, B. , Braun-Dullaeus, R.C. , Tillmanns, H. , Weimer, T. , Preissner, K.T. , 2006. The G534E polymorphism of the gene encoding the factor VII-activating protease is associated with cardiovascular risk due to increased neointima formation. J. Exp. Med.. 203, 2801–2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherry, S.T. , Ward, M.H. , Kholodov, M. , Baker, J. , Phan, L. , Smigielski, E.M. , Sirotkin, K. , 2001. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res.. 29, 308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinmura, K. , Tao, H. , Yamada, H. , Kataoka, H. , Sanjar, R. , Wang, J. , Yoshimura, K. , Sugimura, H. , 2004. Splice-site genetic polymorphism of the human kallikrein 12 (KLK12) gene correlates with no substantial expression of KLK12 protein having serine protease activity. Hum. Mutat.. 24, 273–274. [DOI] [PubMed] [Google Scholar]
- Stephens, J.C. , Schneider, J.A. , Tanguay, D.A. , Choi, J. , Acharya, T. , Stanley, S.E. , Jiang, R. , Messer, C.J. , Chew, A. , Han, J.H. , 2001. Haplotype variation and linkage disequilibrium in 313 human genes. Science. 293, 489–493. [DOI] [PubMed] [Google Scholar]
- Strauss, K.M. , Martins, L.M. , Plun-Favreau, H. , Marx, F.P. , Kautzmann, S. , Berg, D. , Gasser, T. , Wszolek, Z. , Muller, T. , Bornemann, A. , 2005. Loss of function mutations in the gene encoding Omi/HtrA2 in Parkinson's disease. Hum. Mol. Genet.. 14, 2099–2111. [DOI] [PubMed] [Google Scholar]
- Vasilopoulos, Y. , Cork, M.J. , Murphy, R. , Williams, H.C. , Robinson, D.A. , Duff, G.W. , Ward, S.J. , Tazi-Ahnini, R. , 2004. Genetic association between an AACC insertion in the 3′UTR of the stratum corneum chymotryptic enzyme gene and atopic dermatitis. J. Invest Dermatol.. 123, 62–66. [DOI] [PubMed] [Google Scholar]
- Wang, D.G. , Fan, J.B. , Siao, C.J. , Berno, A. , Young, P. , Sapolsky, R. , Ghandour, G. , Perkins, N. , Winchester, E. , Spencer, J. , 1998. Large-scale identification, mapping, and genotyping of single-nucleotide polymorphisms in the human genome. Science. 280, 1077–1082. [DOI] [PubMed] [Google Scholar]
- Wang, L. , Liu, S. , Niu, T. , Xu, X. , 2005. SNPHunter: a bioinformatic software for single nucleotide polymorphism data acquisition and management. BMC Bioinformatics. 6, 60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, L.Z. , Sato, K. , Tsuchiya, N. , Yu, J.G. , Ohyama, C. , Satoh, S. , Habuchi, T. , Ogawa, O. , Kato, T. , 2003. Polymorphisms in prostate-specific antigen (PSA) gene, risk of prostate cancer, and serum PSA levels in Japanese population. Cancer Lett.. 202, 53–59. [DOI] [PubMed] [Google Scholar]
- Waterston, R.H. , Lindblad-Toh, K. , Birney, E. , Rogers, J. , Abril, J.F. , Agarwal, P. , Agarwala, R. , Ainscough, R. , Alexandersson, M. , An, P. , 2002. Initial sequencing and comparative analysis of the mouse genome. Nature. 420, 520–562. [DOI] [PubMed] [Google Scholar]
- Weinsheimer, S. , Goddard, K.A. , Parrado, A.R. , Lu, Q. , Sinha, M. , Lebedeva, E.R. , Ronkainen, A. , Niemela, M. , Khusnutdinova, E.K. , Khusainova, R.I. , 2007. Association of kallikrein gene polymorphisms with intracranial aneurysms. Stroke. PMID:17761919 [DOI] [PubMed] [Google Scholar]
- Wheelan, S.J. , Church, D.M. , Ostell, J.M. , 2001. Spidey: a tool for mRNA-to-genomic alignments. Genome Res.. 11, 1952–1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, H. , Gregory, S.G. , Hauser, E.R. , Stenger, J.E. , Pericak-Vance, M.A. , Vance, J.M. , Zuchner, S. , Hauser, M.A. , 2005. SNPselector: a web tool for selecting SNPs for genetic association studies. Bioinformatics. 21, 4181–4186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yousef, G.M. , Bharaj, B.S. , Yu, H. , Poulopoulos, J. , Diamandis, E.P. , 2001. Sequence analysis of the human kallikrein gene locus identifies a unique polymorphic minisatellite element. Biochem. Biophys. Res. Commun. 285, 1321–1329. [DOI] [PubMed] [Google Scholar]
- Yousef, G.M. , Chang, A. , Scorilas, A. , Diamandis, E.P. , 2000. Genomic organization of the human kallikrein gene family on chromosome 19q13.3–q13.4. Biochem. Biophys. Res. Commun. 276, 125–133. [DOI] [PubMed] [Google Scholar]
- Yousef, G.M. , Diamandis, E.P. , 2001. The new human tissue kallikrein gene family: structure, function, and association to disease. Endocr. Rev.. 22, 184–204. [DOI] [PubMed] [Google Scholar]
- Yu, H. , Song, Q. , Freedman, B.I. , Chao, J. , Chao, L. , Rich, S.S. , Bowden, D.W. , 2002. Association of the tissue kallikrein gene promoter with ESRD and hypertension. Kidney Int.. 61, 1030–1039. [DOI] [PubMed] [Google Scholar]