

Abstract
The estrogen receptor (ER) is a ligand inducible transcription factor that regulates a large number of target genes. These targets are particularly relevant in breast cancer, where the sensitivity of the tumor to estrogens determines whether the patients can be treated with endocrine therapy such as tamoxifen. Identifying genomic ER targets is a daunting task. Quantifying expression levels of suspected target genes after estradiol stimulation or, more recently, using expression microarrays to this effect will reveal which genes are regulated by estradiol, however, without discriminating between direct and indirect targets. The identification of the palindromic sequence that defines the estrogen responsive element (ERE) allows for the in silico discovery of putative ER targets in the genome. However the ER can also bind imperfect EREs and half sites, and can bind indirectly via other factors. Chromatin immunoprecipitation (ChIP) can yield all ER genomic target sites. Coupling of ChIP with genome‐wide tiling arrays allows for the genome‐wide unbiased identification of direct ER target sequences.
Keywords: Estrogen receptor, Chromatin immunoprecipitation, Breast cancer, Tiling array
1. Introduction
In 1896 Beatson observed that removing the ovaries could lead to remission of breast cancer (Beatson, 1896). Although at that time hormones were not yet discovered, his experiments were the first to connect estrogens with breast cancer. More than 60years later it was demonstrated that estrogens were retained in target tissues (Jensen and Jacobson, 1962), laying the foundation for the subsequent identification of steroid receptors. Indeed, in 1968 O'Malley described that changes in gene expression occurred after estrogen stimulation, indicating estrogen receptor (ER) functions as a transcription factor (O'Malley et al., 1968). Soon after, a protein that specifically bound estrogens was found in breast tumors, and its quantity could predict the response of these tumors to endocrine disruption (Jensen et al., 1971; McGuire, 1973). With the cloning of the ER in 1986 (Green et al., 1986; Greene et al., 1986) and subsequent identification of its functional domains the role for the ER as a ligand dependent transcription factor became apparent (Green et al., 1986; Greene et al., 1986; Kumar et al., 1987).
The ER is a member of the superfamily of nuclear receptors, which are structurally related ligand‐inducible transcription factors, including steroid receptors (SRs), thyroid/retinoids receptors (TR, RARs and RXRs), vitamin D receptors (VDR), LXR, PPARs, and orphan receptors for which no ligand has been yet identified. After binding of estrogen to the estrogen‐binding site on the ER, the receptor dimerizes, translocates to the nucleus of the target cell, and binds to specific regions on chromatin, the so‐called estrogen response elements (EREs). In addition, the ER can interact with other transcription factors, SP‐1, AP‐1 and NF‐κB bound to their regulatory regions. These interactions, and the specific genes that are regulated by the interaction of the ER with chromatin regions have been subject to research over the years. This review will focus on the methods and research done to identify ER targets genes.
2. Expression
Elucidating ER target genes has for long been done on a gene‐by‐gene basis, measuring the effect of estrogen stimulation on the expression of a single gene. By differentially screening cDNA libraries of induced and non‐induced cells several target genes have been discovered, e.g. pS2/TFF1 (Brown et al., 1984; Jakowlew et al., 1984). Recently, investigation of global expression changes upon estradiol induction by SAGE or microarray has identified many ER target genes (Charpentier et al., 2000; Cunliffe et al., 2003; Frasor et al., 2003; Inoue et al., 2002; Seth et al., 2002). Interestingly, these global expression experiments indicated that around half of ER target genes are down regulated upon induction with estrogen. Many of the down‐regulated genes are known to inhibit the cell cycle, are pro‐apoptotic or are cytokines and growth factors that inhibit proliferation. This is in agreement with the view that estrogen promotes cell survival by down regulating pro‐apoptotic genes. Although global expression profiling provides a wealth of information on estrogen induced gene expression, it cannot distinguish between direct and indirect ER targets, i.e. genes that are regulated by other genes that are directly regulated by the ER. For this, protein synthesis inhibitors cycloheximide or puromycin can be used, although unspecific effects of these drugs cannot be excluded. All targets that are regulated by estrogens in the presence of protein synthesis inhibitors should be bona fide direct ER targets. Soulez and Parker used this to confirm that the P450‐IIB enzyme was a direct target for the ER in ZR75‐1 cells (Soulez and Parker, 2001), and Wang et al. (2004) found EEIG1 to be a direct ER target in MCF7 cells. To the best of our knowledge, no genome‐wide expression profile has been performed in breast cancer cells in the presence of protein synthesis inhibitors.
3. ERE
Klein‐Hitpass et al. (1986) identified an estrogen response element in the Xenopus vittelogenin A2 gene in 1986. They showed that this element functions in humans and defined a palindromic sequence (5′‐GGTCACAGTGACC‐3′) as the core ERE (Klein‐Hitpass et al., 1986). With the sequencing of the human genome it became possible to search in silico for the presence of EREs. These computational approaches have been used to identify ER target genes with limited success. From an extensive study where 71,119 EREs were identified, only 3 were perfect ERE palindromes (Bourdeau et al., 2004). By narrowing down to promoter regions still 12,515 EREs were identified and by including conservation with mouse, 660 EREs remained of which several could be validated. Other authors have used similar approaches (Kamalakaran et al., 2005). As the ER can bind imperfect EREs and half sites, it is computationally very difficult to distinguish between real binding sites and noise. In addition, ER can bind indirectly via other factors, which cannot be assessed using this approach.
4. ChIP
Chromatin immunoprecipitation (ChIP) is a new and very powerful technique by which transcription factor/co‐factor occupancy of a given locus can be determined in its chromatin context in vivo. In brief, protein and DNA are cross‐linked in the living cell using formaldehyde, chromatin is fragmented, and the transcription (co)factor of interest is immunoprecipitated with specific antibodies. The relative amount of a particular DNA fragment cross‐linked to the transcription factor (and therefore present in the precipitate) can be determined by real‐time quantitative PCR, and is a measure of the occupancy of the factor at that particular position in the genome (Figure 1). Such ChIP approaches provide valuable information about the involvement and temporal order of transcription factor and co‐factor recruitment during activation or repression of a gene or locus. Furthermore, ChIP provides a means to accurately determine the epigenetic status of the locus.
Figure 1.
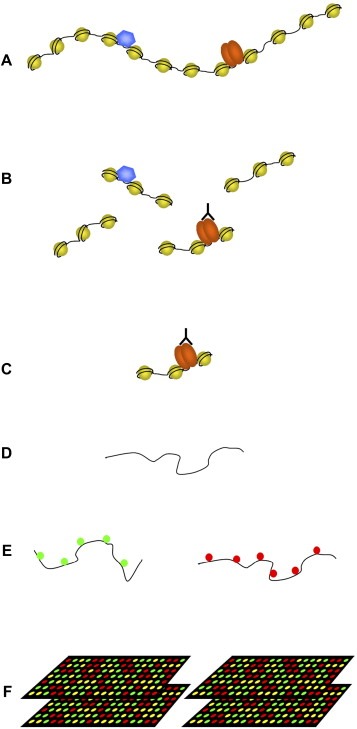
Overview of ChIP‐chip. (A) Using formaldehyde protein–protein and protein–DNA are cross‐linked in vivo. The cross‐linked chromatin is subsequently isolated. (B) The isolated chromatin is sheared in smaller fragments by sonication yielding fragments of 500–1000bp. The protein or histone modification of interest is precipitated using an antibody. The cross‐linked DNA is co‐precipitated. (C) The unbound chromatin is washed away. (D) The cross‐linking is reversed and the DNA is isolated. The DNA is amplified by either LM‐PCR or T7 linear amplification of DNA. (E) Total genomic DNA and ChIP DNA are differently labeled with Cy3 and Cy5. (F) Labeled DNA is hybridized on promoter arrays or arrays spanning the total non‐repetitive genome.
First ChIP experiments directed at the ER focused on a limited number of known binding sites and investigated binding of ER and cofactors. For example, Shang et al. (2000) found that the ER and a number of coactivators rapidly associate with chromatin at the c‐Myc, pS2 and CATD estrogen responsive promoters following estrogen treatment in a cyclic fashion. More recently, a high throughput ChIP approach, ChIP cloning, was described by Laganiere et al. (2005a). Here, the co‐precipitated DNA fragments were cloned and subsequently sequenced. This enabled the identification of unknown ER binding sites without any bias towards annotated genes and promoter regions. A disadvantage of this method is that large scale sequencing facility is needed and that it is difficult to discriminate between true binding sites and background DNA resulting in the necessity to do many work intensive validations.
Recently, ChIP has been coupled to microarray experiments, enabling the unbiased identification of ER binding sites on a genome wide scale (Figure 1). Initially, promoter arrays were used, specifically containing upstream regions from known genes as the size of the genome made it impossible to completely cover it sequence. These arrays are obviously biased and will miss a large number of binding sites as recent data shows that many transcription factors also bind in intergenic regions, introns and downstream of known genes. Despite this, this approach has been used with some success. Laganiere et al. identified 153 promoters bound by ER in the presence of estradiol (Laganiere et al., 2005b). Cheng used a promoter/CpG island array to identify ER binding, H3K9 acetylation and H3K9 dimethylation at different time points. Ninety‐two ER responsive promoters were identified, and a coregulatory role for c‐myc at a subset of promoters was demonstrated. In addition, the physical interaction between ER and c‐myc via the bridging factor TRRAP was shown (Cheng et al., 2006). Recently, a study using a ChIP‐promoter array variant, i.e. ChIP coupled to a DNA selection and ligation (DSL) strategy full genome promoter array, found 578 high confidence promoter ER gene targets in MCF‐7 cells (Kwon et al., 2007). In unsupervised clustering analyses, they found that the 54 genes that were regulated by estradiol and that were directly regulated by the estrogen receptor as discovered by ChIP‐DSL promoter array could identify a subset of patients with ER negative tumors, of higher grade and with a much poorer prognosis then the other patients.
By combining ChIP with genome wide tiling arrays (ChIP‐chip), ER binding can be investigated in a truly unbiased fashion. Tackling mammalian genomes is exceedingly more difficult and challenging due to its shear size, the presence of over 30,000 protein‐coding genes sprinkled into a bed of highly repetitive sequences. In mammals, the identification of transcription factor binding sites using bioinformatic tools is complicated because regulatory regions are much wider spread than in yeast and can be located as far as hundreds of kb upstream or downstream or within the transcribed region. Indeed, by using Affymetrix tiling arrays spanning chromosome 21 and 22, 57 ER binding sites were identified of which the majority was located distal from genes and not in promoter regions (Carroll et al., 2005). Comparing these data with a polymerase (pol) II ChIP‐chip showed the presence of pol II at the ER binding sites. Sequence analysis of the ER binding sites showed that these were enriched with a Forkhead motif. FoxA1/HNF3alpha expression correlates with the presence of ER in breast tumors and cell lines (van 't Veer et al., 2002; Lacroix and Leclercq, 2004). By ChIP‐qPCR Carroll showed that in the absence of estradiol FoxA1 was present for most targets and dissociated upon ligand induction (Carroll et al., 2005). They postulate a role for FoxA1 as a pioneering factor whereby FoxA1 is present on the chromatin and facilitates the interaction of the ER with its binding site.
Recently, Carroll published a genome wide ChIP‐chip for the estrogen receptor and RNA pol II, identifying 3665 high confidence ER binding sites and 3629 RNA pol II binding sites (Carroll et al., 2006). As described earlier for chromosome 21 and 22, mapping the location of the binding sites to nearby genes shows that only 4% is located within 1kb upstream of a gene. Analysis of ER binding sites for enriched DNA sequences identified ERE, Forkhead, AP‐1, Oct and C/EBP motifs. Pair wise analysis showed that the ERE and AP‐1 motif occur exclusively. The Oct, Forkhead, and C/EBP motifs have a positive correlation. Further investigation showed a role for the coregulator NRIP in ER induced repression of target genes. NRIP is a target of the ER and can interact with ER AP‐1 complexes to repress expression of target genes.
5. Amplification methods
For hybridizing ChIPed DNA micrograms of DNA are needed, while ChIP experiment yields are in the nanogram range. An amplification step is needed to obtain sufficient material. The majority of studies so far used LM‐PCR as their amplification method. Here, oligos are ligated to the ChIPed DNA, which are used as primer sites for a subsequent PCR reaction. This yields enough material for ChIP‐chip. The efficiency of LM amplification is unfortunately biased towards GC regions. This bias is partially compensated for by the fact that it is present both in the input as well as in the ChIP sample, so the ratio will be constant. Nevertheless this bias can cause false positives. A linear method of amplifying DNA is TLAD (T7 linear amplification of DNA) (Liu et al., 2003). In short, a T7 promoter is incorporated at the end of the ChIPed DNA fragments. For this the ChIPed DNA is tailed using a terminal transferase tailing step. A T7 promoter is then annealed to the DNA after which a second strand is synthesized to fill the DNA fragments. This is used as a template for an in vitro transcription reaction. The RNA produced is subsequently reverse transcribed to DNA. This linear amplification method has no bias towards CG content and preserves sample complexity.
6. Identifying ChIP sequences
There are several methods to determine the amount and sequence of ChIPed DNA fragments and their ChIP/input ratio. The most sensitive way is by qPCR, although this can only be used for a relative small number of sites and sequence information is necessary to develop primers, so no new sites can be identified. For qPCR, one needs to know the targets, only a few targets can be measured at the same time, but on the other hand, qPCR exhibits high sensitivity, there is no preamplification step required, and it is still the gold standard. By cloning the ChIP fragments (ChIP cloning) and sequencing all clones (Laganiere et al., 2005a), all information is unbiased, many targets are assessed at a time, the technique has a high sensitivity, but distinguishing real targets from background can be problematic. To identify a large number of binding sites microarrays (ChIP‐chip) can be used. These can contain up to 4.6 million probes and this number is increasing as technology develops further. Several types of arrays exist; promoter arrays contain promoter regions of known genes. All promoter regions can be spotted on one array, making it very affordable. Promoter arrays cover only a small portion of the whole genome, and recent studies show that the majority of ERs (Carroll et al., 2005, 2006) bind to non‐promoter regions. Promoter arrays hence show a limited view of transcription factor binding and will miss a large part of enhancers and non‐annotated transcripts. With the ChIP‐DSL (DNA selection and ligation) technique a promoter array is also used (Kwon et al., 2007). Each promoter is covered by one 40bp probe and for each probe there is a matching primer set of two 20bp primers. The ChIPed fragments are biotinylated and attached to a carrier. Unbound fragment are washed away and the primer pairs that contain the promoter specific sequence and a T7 or T3 sequence are hybridized to the fragments. Two matching primer pairs are then ligated allowing the amplification of the product by using T7 and T3 primers. With this method only fragments that are present on the array are amplified. The authors claim increased sensitivity and are able to use a small number of cells (Kwon et al., 2007). ChIP‐DSL has the same drawbacks as other promoter arrays, namely a very biased selection of binding sites. In addition, each promoter is covered with only one probe. The specific amplification of only fragments that are present on the array and the fact that the method can tolerate incomplete cross‐linking makes this a sensitive method. Using chromosome wide arrays, all binding site regions can be identified throughout a complete chromosome. An extension of chromosome wide array is the genome wide array set (currently 38 slides for Nimblegen or 14 slides for Affymetrix), which covers the whole genome at high resolution. Array synthesis and scanning is still developing fast, enabling more and more probes per array. Because of this development genome wide arrays will become cheaper and the use of promoter and chromosome wide arrays will decline.
7. Enhancer/promoter interactions
Recent data showed that ER binding sites could be located far from any gene (Carroll et al., 2005, 2006). They tested the hypothesis that these distal sites play a role during transcription of distal genes. To test this enhancer/promoter interaction chromosome conformation capture (3C) is used (Dekker et al., 2002; Horike et al., 2005). In short, proteins and DNA are cross‐linked using formaldehyde, sheared in small fragments and a chromatin immunoprecipitation is carried out using a specific antibody. The fragments are subsequently digested with a restriction enzyme and ligated. If two DNA fragments are in close vicinity because of promoter enhancer interaction they will be ligated to each other. By quantitative PCR using primers that span this ligation the enhancer promoter interaction can be determined. Recently this technique has been adapted for high throughput using microarrays, enabling the screening of promoter enhancer interactions on a large scale (Zhao et al., 2006). Identification of promoter enchancer interactions is an important step to obtain more biological information from ChIP‐chip data. The vast amount of data obtained by genome wide ChIP‐chip needs to be distilled into meaningful biological data. This is the next step and bottleneck of genome wide transcription factor profiling.
8. Clinical significance and future directions
What can we learn from knowledge of the profile of direct estrogen receptor gene targets? On a fundamental level, these experiments help expanding our understanding of how gene transcriptomes interact. Clinically, understanding of the functioning of the ER is still important in breast cancer, more than 110years after Beatson (1896). So far all prognostic expression array studies in breast cancer have found the ER status to be the most pronounced independent factor that dichotomizes patients after unsupervised clustering of expression profiles of their primary tumors (van 't Veer et al., 2002; Sorlie et al., 2001). Amplification of the ER gene has recently been described as a common early event in a large subset of breast cancers (Holst et al., 2007). ER status dependent expression profiles are being identified (Abba et al., 2005), and ER regulated gene expression profiles are used to predict survival in breast cancer patients (Oh et al., 2006). Thus, for the clinical evaluation of breast tumors, the ER is still crucial. The ER positive patients can be subdivided into so‐called luminal A and luminal B type tumors based on their gene expression profile (Sorlie et al., 2001), with the latter group having a relatively poor prognosis. Differences in functioning of the ER in these two patient subgroups will help elucidating the mechanisms involved in determining the prognosis of breast cancer patients. Furthermore, interest is now changing from defining patients with a poor prognosis (i.e. disease outcome irrespective of or in the absence of treatment) to identifying predictive gene profiles for, e.g. tamoxifen resistance (Jansen et al., 2005). Tamoxifen has been the gold standard for the endocrine treatment of patients with ER positive breast cancers for over 25years (Jordan, 2003). Differences in direct gene targets of the ER in the presence of agonists (estradiol) or antagonists (tamoxifen) might be relevant for identifying mechanisms involved in (in)sensitivity to tamoxifen or other ER antagonists (Jordan, 2004). With knowledge of the genome‐wide DNA binding sites of ER alpha, the effects of coregulators, ER beta or growth factors (Shou et al., 2004; Chang et al., 2006; Girault et al., 2006) on ER alpha interaction profiles can now be studied, which have relevance for revealing the mechanisms involved in tamoxifen resistance.
Welboren Willem-Jan, Stunnenberg Henk G., Sweep Fred C.G.J., Span Paul N., (2007), Identifying estrogen receptor target genes, Molecular Oncology, 1, doi:10.1016/j.molonc.2007.04.001.
References
- Abba, M.C. , Hu, Y. , Sun, H. , Drake, J.A. , Gaddis, S. , Baggerly, K. , Sahin, A. , Aldaz, C.M. , 2005. Gene expression signature of estrogen receptor alpha status in breast cancer. BMC Genomics. 6, 37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beatson, C.T. , 1896. On treatment of inoperable cases of carcinoma of the mamma: suggestions for a new method of treatment with illustrative cases. Lancet. ii, 104–107. [PMC free article] [PubMed] [Google Scholar]
- Bourdeau, V. , Deschenes, J. , Metivier, R. , Nagai, Y. , Nguyen, D. , Bretschneider, N. , Gannon, F. , White, J.H. , Mader, S. , 2004. Genome-wide identification of high-affinity estrogen response elements in human and mouse. Mol. Endocrinol.. 18, 1411–1427. [DOI] [PubMed] [Google Scholar]
- Brown, A.M. , Jeltsch, J.M. , Roberts, M. , Chambon, P. , 1984. Activation of pS2 gene transcription is a primary response to estrogen in the human breast cancer cell line MCF-7. Proc. Natl. Acad. Sci. USA. 81, 6344–6348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll, J.S. , Liu, X.S. , Brodsky, A.S. , Li, W. , Meyer, C.A. , Szary, A.J. , Eeckhoute, J. , Shao, W. , Hestermann, E.V. , Geistlinger, T.R. , Fox, E.A. , Silver, P.A. , Brown, M. , 2005. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell. 122, 33–43. [DOI] [PubMed] [Google Scholar]
- Carroll, J.S. , Meyer, C.A. , Song, J. , Li, W. , Geistlinger, T.R. , Eeckhoute, J. , Brodsky, A.S. , Keeton, E.K. , Fertuck, K.C. , Hall, G.F. , Wang, Q. , Bekiranov, S. , Sementchenko, V. , Fox, E.A. , Silver, P.A. , Gingeras, T.R. , Liu, X.S. , Brown, M. , 2006. Genome-wide analysis of estrogen receptor binding sites. Nat. Genet.. 38, 1289–1297. [DOI] [PubMed] [Google Scholar]
- Chang, E.C. , Frasor, J. , Komm, B. , Katzenellenbogen, B.S. , 2006. Impact of estrogen receptor beta on gene networks regulated by estrogen receptor alpha in breast cancer cells. Endocrinology. 147, 4831–4842. [DOI] [PubMed] [Google Scholar]
- Charpentier, A.H. , Bednarek, A.K. , Daniel, R.L. , Hawkins, K.A. , Laflin, K.J. , Gaddis, S. , MacLeod, M.C. , Aldaz, C.M. , 2000. Effects of estrogen on global gene expression: identification of novel targets of estrogen action. Cancer Res.. 60, 5977–5983. [PubMed] [Google Scholar]
- Cheng, A.S. , Jin, V.X. , Fan, M. , Smith, L.T. , Liyanarachchi, S. , Yan, P.S. , Leu, Y.W. , Chan, M.W. , Plass, C. , Nephew, K.P. , Davuluri, R.V. , Huang, T.H. , 2006. Combinatorial analysis of transcription factor partners reveals recruitment of c-MYC to estrogen receptor-alpha responsive promoters. Mol. Cell. 21, 393–404. [DOI] [PubMed] [Google Scholar]
- Cunliffe, H.E. , Ringner, M. , Bilke, S. , Walker, R.L. , Cheung, J.M. , Chen, Y. , Meltzer, P.S. , 2003. The gene expression response of breast cancer to growth regulators: patterns and correlation with tumor expression profiles. Cancer Res.. 63, 7158–7166. [PubMed] [Google Scholar]
- Dekker, J. , Rippe, K. , Dekker, M. , Kleckner, N. , 2002. Capturing chromosome conformation. Science. 295, 1306–1311. [DOI] [PubMed] [Google Scholar]
- Frasor, J. , Danes, J.M. , Komm, B. , Chang, K.C. , Lyttle, C.R. , Katzenellenbogen, B.S. , 2003. Profiling of estrogen up- and down-regulated gene expression in human breast cancer cells: insights into gene networks and pathways underlying estrogenic control of proliferation and cell phenotype. Endocrinology. 144, 4562–4574. [DOI] [PubMed] [Google Scholar]
- Girault, I. , Bieche, I. , Lidereau, R. , 2006. Role of estrogen receptor alpha transcriptional coregulators in tamoxifen resistance in breast cancer. Maturitas. 54, 342–351. [DOI] [PubMed] [Google Scholar]
- Green, S. , Walter, P. , Kumar, V. , Krust, A. , Bornert, J.M. , Argos, P. , Chambon, P. , 1986. Human oestrogen receptor cDNA: sequence, expression and homology to v-erb-A. Nature. 320, 134–139. [DOI] [PubMed] [Google Scholar]
- Greene, G.L. , Gilna, P. , Waterfield, M. , Baker, A. , Hort, Y. , Shine, J. , 1986. Sequence and expression of human estrogen receptor complementary DNA. Science. 231, 1150–1154. [DOI] [PubMed] [Google Scholar]
- Holst, F. , Stahl, P.R. , Ruiz, C. , Hellwinkel, O. , Jehan, Z. , Wendland, M. , Lebeau, A. , Terracciano, L. , Al-Kuraya, K. , Janicke, F. , Sauter, G. , Simon, R. , 2007. Estrogen receptor alpha (ESR1) gene amplification is frequent in breast cancer. Nat. Genet.. 39, 655–660. [DOI] [PubMed] [Google Scholar]
- Horike, S. , Cai, S. , Miyano, M. , Cheng, J.F. , Kohwi-Shigematsu, T. , 2005. Loss of silent-chromatin looping and impaired imprinting of DLX5 in Rett syndrome. Nat. Genet.. 37, 31–40. [DOI] [PubMed] [Google Scholar]
- Inoue, A. , Yoshida, N. , Omoto, Y. , Oguchi, S. , Yamori, T. , Kiyama, R. , Hayashi, S. , 2002. Development of cDNA microarray for expression profiling of estrogen-responsive genes. J. Mol. Endocrinol.. 29, 175–192. [DOI] [PubMed] [Google Scholar]
- Jakowlew, S.B. , Breathnach, R. , Jeltsch, J.M. , Masiakowski, P. , Chambon, P. , 1984. Sequence of the pS2 mRNA induced by estrogen in the human breast cancer cell line MCF-7. Nucleic Acids Res.. 26, 2861–2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen, M.P. , Foekens, J.A. , van Staveren, I.L. , Dirkzwager-Kiel, M.M. , Ritstier, K. , Look, M.P. , Meijer-van Gelder, M.E. , Sieuwerts, A.M. , Portengen, H. , Dorssers, L.C. , Klijn, J.G. , Berns, E.M. , 2005. Molecular classification of tamoxifen-resistant breast carcinomas by gene expression profiling. J. Clin. Oncol.. 23, 732–740. [DOI] [PubMed] [Google Scholar]
- Jensen, E.V. , Jacobson, H.I. , 1962. Basic guides to the mechanism of estrogen action. Recent Prog. Horm. Res.. 18, 387–414. [Google Scholar]
- Jensen, E.V. , Block, G.E. , Smith, S. , Kyser, K. , DeSombre, E.R. , 1971. Estrogen receptors and breast cancer response to adrenalectomy. Natl. Cancer Inst. Monogr.. 34, 55–70. [PubMed] [Google Scholar]
- Jordan, V.C. , 2003. Tamoxifen: a most unlikely pioneering medicine. Nat. Rev. Drug Discov.. 2, 205–213. [DOI] [PubMed] [Google Scholar]
- Jordan, V.C. , 2004. Selective estrogen receptor modulation: concept and consequences in cancer. Cancer Cell. 5, 207–213. [DOI] [PubMed] [Google Scholar]
- Klein-Hitpass, L. , Schorpp, M. , Wagner, U. , Ryffel, G.U. , 1986. An estrogen-responsive element derived from the 5′ flanking region of the Xenopus vitellogenin A2 gene functions in transfected human cells. Cell. 46, 1053–1061. [DOI] [PubMed] [Google Scholar]
- Kamalakaran, S. , Radhakrishnan, S.K. , Beck, W.T. , 2005. Identification of estrogen-responsive genes using a genome-wide analysis of promoter elements for transcription factor binding sites. J. Biol. Chem.. 280, 21491–21497. [DOI] [PubMed] [Google Scholar]
- Kumar, V. , Green, S. , Stack, G. , Berry, M. , Jin, J.R. , Chambon, P. , 1987. Functional domains of the human estrogen receptor. Cell. 51, 941–951. [DOI] [PubMed] [Google Scholar]
- Kwon, Y.S. , Garcia-Bassets, I. , Hutt, K.R. , Cheng, C.S. , Jin, M. , Liu, D. , Benner, C. , Wang, D. , Ye, Z. , Bibikova, M. , Fan, J.B. , Duan, L. , Glass, C.K. , Rosenfeld, M.G. , Fu, X.D. , 2007. Sensitive ChIP-DSL technology reveals an extensive estrogen receptor {alpha}-binding program on human gene promoters. Proc. Natl. Acad. Sci. USA. 104, 4852–4857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacroix, M. , Leclercq, G. , 2004. About GATA3, HNF3A, and XBP1, three genes co-expressed with the oestrogen receptor-alpha gene (ESR1) in breast cancer. Mol. Cell. Endocrinol.. 219, 1–7. [DOI] [PubMed] [Google Scholar]
- Laganiere, J. , Deblois, G. , Giguere, V. , 2005. Functional genomics identifies a mechanism for estrogen activation of the retinoic acid receptor alpha1 gene in breast cancer cells. Mol. Endocrinol.. 19, 1584–1592. [DOI] [PubMed] [Google Scholar]
- Laganiere, J. , Deblois, G. , Lefebvre, C. , Bataille, A.R. , Robert, F. , Giguere, V. , 2005. From the Cover: Location analysis of estrogen receptor alpha target promoters reveals that FOXA1 defines a domain of the estrogen response. Proc. Natl. Acad. Sci. USA. 102, 11651–11656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, C.L. , Schreiber, S.L. , Bernstein, B.E. , 2003. Development and validation of a T7 based linear amplification for genomic DNA. BMC Genomics. 4, 19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire, W.L. , 1973. Estrogen receptors in human breast cancer. J. Clin. Invest.. 52, 73–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh, D.S. , Troester, M.A. , Usary, J. , Hu, Z. , He, X. , Fan, C. , Wu, J. , Carey, L.A. , Perou, C.M. , 2006. Estrogen-regulated genes predict survival in hormone receptor-positive breast cancers. J. Clin. Oncol.. 24, 1656–1664. [DOI] [PubMed] [Google Scholar]
- O'Malley, B.W. , McGuire, W.L. , Middleton, P.A. , 1968. Altered gene expression during differentiation: population changes in hybridizable RNA after stimulation of the chick oviduct with oestrogen. Nature. 218, 1249–1251. [DOI] [PubMed] [Google Scholar]
- Seth, P. , Krop, I. , Porter, D. , Polyak, K. , 2002. Novel estrogen and tamoxifen induced genes identified by SAGE (Serial Analysis of Gene Expression). Oncogene. 21, 836–843. [DOI] [PubMed] [Google Scholar]
- Shang, Y. , Hu, X. , DiRenzo, J. , Lazar, M.A. , Brown, M. , 2000. Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell. 103, 843–852. [DOI] [PubMed] [Google Scholar]
- Shou, J. , Massarweh, S. , Osborne, C.K. , Wakeling, A.E. , Ali, S. , Weiss, H. , Schiff, R. , 2004. Mechanisms of tamoxifen resistance: increased estrogen receptor-HER2/neu cross-talk in ER/HER2-positive breast cancer. J. Natl. Cancer Inst.. 96, 926–935. [DOI] [PubMed] [Google Scholar]
- Sorlie, T. , Perou, C.M. , Tibshirani, R. , Aas, T. , Geisler, S. , Johnsen, H. , Hastie, T. , Eisen, M.B. , van de Rijn, M. , Jeffrey, S.S. , Thorsen, T. , Quist, H. , Matese, J.C. , Brown, P.O. , Botstein, D. , Eystein Lonning, P. , Borresen-Dale, A.L. , 2001. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA. 98, 10869–10874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soulez, M. , Parker, M.G. , 2001. Identification of novel oestrogen receptor target genes in human ZR75-1 breast cancer cells by expression profiling. J. Mol. Endocrinol.. 27, 259–274. [DOI] [PubMed] [Google Scholar]
- van 't Veer, L.J. , Dai, H. , van de Vijver, M.J. , He, Y.D. , Hart, A.A. , Mao, M. , Peterse, H.L. , van der Kooy, K. , Marton, M.J. , Witteveen, A.T. , Schreiber, G.J. , Kerkhoven, R.M. , Roberts, C. , Linsley, P.S. , Bernards, R. , Friend, S.H. , 2002. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 415, 530–536. [DOI] [PubMed] [Google Scholar]
- Wang, D.Y. , Fulthorpe, R. , Liss, S.N. , Edwards, E.A. , 2004. Identification of estrogen-responsive genes by complementary deoxyribonucleic acid microarray and characterization of a novel early estrogen-induced gene: EEIG1. Mol. Endocrinol.. 18, 402–411. [DOI] [PubMed] [Google Scholar]
- Zhao, Z. , Tavoosidana, G. , Sjolinder, M. , Gondor, A. , Mariano, P. , Wang, S. , Kanduri, C. , Lezcano, M. , Sandhu, K.S. , Singh, U. , Pant, V. , Tiwari, V. , Kurukuti, S. , Ohlsson, R. , 2006. Circular chromosome conformation capture (4C) uncovers extensive networks of epigenetically regulated intra- and interchromosomal interactions. Nat. Genet.. 38, 1341–1137 [DOI] [PubMed] [Google Scholar]