

Abstract
TIMP‐1 is a promising new candidate as a prognostic marker in colorectal and breast cancer. We now describe the discovery of two alternatively spliced variants of TIMP‐1 mRNA. The two variants lacking exon 2 (del‐2) and 5 (del‐5), respectively, were identified in human cancer cell lines by RT‐PCR. The del‐2 variant was, furthermore, detected in extracts from 12 colorectal cancer tissue samples. By western blotting additional bands of lower molecular mass than full‐length TIMP‐1 were identified in tumor tissue, but not in plasma samples obtained from cancer patients.
The two splice variants of TIMP‐1 may hold important clinical information, and either alone or in combination with measurement of full‐length TIMP‐1 they may improve the prognostic and/or predictive value of TIMP‐1 analyses.
Keywords: TIMP-1, Splice variants, Colon cancer, Cancer cell lines
Abbreviations
- CRC
colorectal cancer
- ELISA
enzyme-linked immunosorbent assay
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- HRP
horseradish peroxidase
- MMP
matrix metalloproteinase
- α-SMA
α-smooth muscle actin
- TIMP-1
tissue inhibitor of metalloproteinases-1
- TNP
trinitrophenyl hapten
1. Introduction
Despite an increasing knowledge on the molecular mechanisms underlying malignant transformation and dissemination of disease, the overall survival rates for the most common malignancies in the Western world have only improved slightly over the last decade. The obvious challenge in future management of cancer patients is therefore to improve existing therapy and/or to develop new therapy strategies. Another approach is to improve detection of early cancer disease, as patients diagnosed at the early stages of, e.g., colorectal cancer often will be cured by primary surgery alone (Ouyang et al., 2005). Furthermore, since stratification of cancer patients for therapy is based on prognostic evaluations, this calls for new and better prognostic markers. Tissue inhibitor of metalloproteinases‐1 (TIMP‐1) is a promising new marker for early detection and prognostic stratification of patients suffering from colorectal cancer (Holten‐Andersen et al., 2002b; Waas et al., 2005). In addition, TIMP‐1 also carries the potential as a predictive marker for response to chemotherapy (Davidsen et al., 2006; Schrohl et al., 2006).
TIMP‐1 is a naturally occurring inhibitor of matrix metalloproteinases (MMPs), a large family of proteases involved in many physiological and pathological processes like embryonic development, tissue morphogenesis, wound healing, inflammation and cancer invasion (for reviews see Egeblad and Werb, 2002; Lemaitre and D'Armiento, 2006). Four TIMPs (TIMP‐1 to ‐4) have been identified and they differ in tissue distribution and ability to inhibit different MMPs (Baker et al., 2002). Mature TIMP‐1 is a 28.5kDa glycoprotein, consisting of 184 amino acid residues. The unprocessed precursor contains a signal peptide of 23 residues, which is cleaved during the maturation of the protein (Brew et al., 2000). TIMP‐1 contains two sites of N‐glycan linkage and six disulfide bonds. The latter is folding the protein into a two‐domain structure. These disulfide bonds render TIMP‐1 very robust to changes in pH, temperature and denaturing conditions (Stricklin and Welgus, 1983). The MMP inhibitory activity has been located to the N‐terminal domain of TIMP‐1, which forms non‐covalent complexes with MMPs or proforms of these, in particular proMMP‐9 (Murphy et al., 1991).
Although now named for its ability to inhibit MMPs, TIMP‐1 was first identified for its growth stimulating activity of erythroid progenitors (Westbrook et al., 1984), and its mitogenic ability has since been demonstrated in many cell types (Bertaux et al., 1991; Hayakawa et al., 1992). TIMP‐1 has also been shown to inhibit apoptosis (Guedez et al., 1998; Liu et al., 2005) and to participate in regulation of angiogenesis (Akahane et al., 2004). It therefore seems that TIMP‐1 is a multifunctional protein demonstrating a range of activities that in relation to cancer can be both tumor promoting and suppressing. Some functions of TIMP‐1 can be ascribed to its MMP inhibitory ability, while others appear to be independent of binding to MMPs.
TIMP‐1 may exist in multiple molecular forms in the cancer environment and subsequently in the circulation, e.g. in complex with other proteins like MMPs (Holten‐Andersen et al., 2002a) or as different glycosylation variants (Thaysen‐Andersen et al., 2007). The clinical value of TIMP‐1 measurements may be improved by detection of specific forms of TIMP‐1, as shown in breast cancer, where measurement of unbound TIMP‐1 versus total TIMP‐1 adds to the prognostic evaluation of the patients (Holten‐Andersen et al., 2002b; Wurtz et al., 2005). We now describe the expression of two newly discovered splice variants of TIMP‐1 mRNA in cancer cell lines and in colon cancer tissue. They may hold important clinical information and could either alone or in combination with measurement of full‐length TIMP‐1 improve the already established clinical value of TIMP‐1 as a biological tumor marker.
2. Materials and methods
2.1. Cell culture
The following human breast cancer cell lines were used: MDA‐MB‐231‐BAG (Brunner et al., 1992), MDA‐MB‐435‐BAG (Brunner et al., 1992), CAMA‐1, EVSA‐T, ZR75.1, T47‐D, SKBR‐3 and MCF7‐S1 (kindly provided by Professor Marja Jäättela, Danish Cancer Society, Copenhagen, Denmark). Furthermore, three colon cancer cell lines DLD‐1, LoVo and LS 174T (all from ATCC, Manassas, USA) as well as the prostate cancer cell line PC‐3, the human acute promyelocytic leukemia cell line HL‐60 (kindly provided by Professor Hau C. Kwaan, Northwestern University, Chicago, USA) and the endothelial cell line EAHY‐926 were used. MCF7‐S1 cells were propagated in RPMI medium supplemented with 10% heat inactivated fetal calf serum (FCS), and HL‐60 cells were propagated in IMDM medium with 20% FCS. ZR75.1, T47‐D, SKBR‐3 and EAHY‐926 were propagated in HAMF/DME medium with 10% FCS, 0.15% Na2HCO3 and 45μg/ml Gentamycin. ZR75.1 cells were furthermore supplemented with 10nM estradiol and to EAHY‐926 cells 1× HAT medium was added. DLD‐1 cells were propagated in modified RPMI‐1640 medium (ATCC, Manassas, USA) supplemented with 10% FCS. LoVo cells were propagated in modified F‐12K medium (ATCC, Manassas, USA) supplemented with 10% FCS. LS 174T cells were propagated in modified EMEM (ATCC, Manassas, USA) supplemented with 10% FCS. All other cell lines were propagated in DMEM medium. MDA‐MB‐231‐BAG and MDA‐MB‐435‐BAG cells were supplemented with 10% FCS and CAMA‐1, EVSA‐T and PC‐3 cells with 5% FCS. All media (except for the colon cancer cell lines) used were purchased from Invitrogen, Taastrup, Denmark and were supplemented with penicillin and streptomycin. The cells were grown at 37°C in a humidified air atmosphere with 5% CO2.
2.2. Patient material
Tumor tissue and blood samples from 12 patients who underwent surgery for colorectal cancer (CRC) were obtained from Hvidovre Hospital, Denmark in accordance with an approval by the Scientific Ethical Committee for Copenhagen and Frederiksberg, Denmark (Journal no. KF 01‐078/93) and with informed consent from the patients. The tissue samples were taken from the peripheral area of the tumor, snap frozen in liquid nitrogen and stored at −80°C. At the time of surgical removal of the tumor, blood samples were collected and EDTA plasma samples prepared according to a previously described protocol (Holten‐Andersen et al., 1999). A pool of control EDTA plasma and platelets were obtained from the blood bank at Rigshospitalet, Copenhagen, Denmark.
Each tissue sample was divided into three parts. One part was used for RNA extraction and RT‐PCR and one part was used for protein extraction and ELISA. The middle part of each tissue was thawed and fixed in 4% neutral buffered paraformaldehyde overnight at 4°C and processed for paraffin embedding prior to histological analyses. The diagnosis of colon cancer was confirmed in H&E stained sections of the paraffin embedded material and the relative area of neoplastic cells was estimated (0–100%).
For western blotting analysis we obtained an additional six CRC tissue samples from the same source as the 12 CRC samples described above.
2.3. RNA extraction and reverse transcription
RNA was extracted from tissue samples or cells using a spin column kit (SV Total RNA isolation system, Promega, Madison, USA) according to the manufacturer's instructions. This procedure includes an on‐column DNase treatment, minimizing the risk of DNA contamination. Prior to extraction approx. 30mg of tissue sample was homogenized in 1ml lysis buffer. The cells were grown to a confluency of approx. 60% in 900ml flasks, washed twice in PBS and lysed in 0.5ml lysis buffer. RNA was extracted from 175μl lysate and the concentration of total RNA was measured spectrophotometrically.
One microgram of total RNA was transcribed into cDNA using the First Strand cDNA Synthesis Kit (Fermentas, Helsingborg, Sweden). The total volume of the reaction was 25μl consisting of 1× reaction buffer, 0.8mM dNTPs, 20U RiboLock RNase inhibitor, 0.5μg oligo(dT) primer, 0.2μg random hexamer primer and 40U M‐MuLV Reverse Transcriptase. Samples were incubated at 25°C for 10min, followed by 42°C for 1h. The reaction was terminated by incubating at 95°C for 5min followed by cooling on ice.
2.4. PCR
All primer sets used were intron‐spanning to avoid false positive results from contaminating genomic DNA. Prior to PCR with TIMP‐1 primers all cDNA samples were subjected to PCR with primers for GAPDH (Table 1) to ensure that RNA extraction and reverse transcription (RT) were carried out efficiently. PCR was performed in 25μl reactions containing 1× HotStarTaq Mastermix (Qiagen Nordic, West Sussex, UK), 1μM of each gene‐specific primer and 1μl cDNA. Reaction conditions were 95°C for 15min followed by 40 cycles of 94°C for 1min, 60°C for 30s and 72°C for 1min.
Table 1.
Primers used for RT‐PCR and qPCR analyses
| Gene | Specificity | Forward primer 5′→3′ | Reverse primer 5′→3′ | Product size (bp) |
|---|---|---|---|---|
| TIMP‐1 | All exon‐skipping variantsa | Forward exon 1 CCCTAGCGTGGACATTTATC | Reverse exon 6 AAGGTGACGGGACTGGAAG | 648 (full‐length) |
| TIMP‐1 | Full‐length+del‐2 | Forward exon 1 CCCTAGCGTGGACATTTATC | Reverse exon 3 GGTATAAGGTGGTCTGGTTG | 263+134 |
| TIMP‐1 | Full‐length+del‐5 | Forward exon 4 ACTTCCACAGGTCCCACAAC | Reverse exon 6 AAGGTGACGGGACTGGAAG | 252+127 |
| TIMP‐1 | Full‐lengthb | Forward exon 2 CTTCTGGCATCCTGTTGTTG | Reverse exon 3 GGTATAAGGTGGTCTGGTTG | 153 |
| TIMP‐1 | Del‐2b | Forward exon 1/3 CCCAGAGAGACACCAGAGTCA | Reverse exon 4 GTGGGACCTGTGGAAGTATC | 196 |
| GAPDH | Full‐length | CAATGACCCCTTCATTGACC | TTCACACCCATGACGAACAT | 309 |
Except variants lacking exon 1 or 6.
qPCR assay.
PCR products were separated on a 1.5% agarose gel (Fermentas, Sweden) stained with ethidium bromide and visualized by UV light.
2.5. Quantitative real‐time PCR (qPCR)
In order to estimate the expression level of the del‐2 TIMP‐1 variant compared with full‐length TIMP‐1, a qPCR assay determining relative expression levels was developed. Relative quantification was chosen in contrast to absolute quantification, since tumor samples contain various amounts of cells expressing TIMP‐1. The absolute amount of transcript will therefore depend on the composition of each sample. Relative quantification using either total TIMP‐1 or full‐length TIMP‐1 as reference gene will reflect only changes in transcription of the cells expressing TIMP‐1.
qPCR was carried out using SYBR Green I detection and the LightCycler System 2.0 (Roche Diagnostics, Hvidovre, Denmark) essentially as previously described (Offenberg and Thomsen, 2005). PCR conditions for each primer set were optimized by determining the MgCl2 concentration and annealing temperature at which only the specific product was seen. Reactions were carried out in 20μl volumes consisting of 1× FastStart Master SYBR Green Mix, 5mM MgCl2 and 0.5μM of the gene‐specific primer. The amplification program was as follows. Preincubation for Fast Start Polymerase activation at 95°C for 10min, followed by 45 amplification cycles (95°C for 5s (20°C/s), 65°C for 10s (20°C/s) and 72°C for 6–10s (20°C/s)). SYBR Green fluorescence was acquired at 72°C in each amplification cycle. After the end of the last cycle the melting curve was generated by starting the fluorescence acquisition at 65°C and taking measurements every 0.1s until 95°C was reached.
A standard curve for each primer set was generated using 10‐fold dilutions of a pool of tumor cDNA. The standard curve was used to correct differences in PCR efficiencies between primer sets. Dilutions of tumor cDNA samples were chosen for the generation of standard curves to ensure the same PCR efficiency in standards and samples. Relative quantification was done using the Relative Quantification software (LightCycler 2.0, Roche, Denmark).
2.6. Protein extraction for western blotting and ELISA
The frozen tissue samples used for ELISA were fragmented on dry ice using a pestle and mortar device (Arrow Fastener Co. Inc., Saddle Brook, N.J., USA). The powdered tissue was weighed and dissolved in Camiolo buffer (75mM CH3COOK, 0.3M NaCl, 0.1M l‐argenine, 10mM EDTA, 0.25% Triton X‐100, pH 4.2) in a 3:1 ratio of buffer to tissue. Samples were centrifuged for 1h at 20,000g, and the supernatant containing the proteins was used for further analysis. For western blotting, the tissue powder was dissolved in modified RIPA buffer (50mM Tris–HCl (pH 7.4), 1% NP‐40, 0.25% Na‐deoxycholate, 150mM NaCl, 1mM EDTA) supplemented with protease inhibitors (Pefabloc, Aprotinin, Leupeptin, Pepstatin: 1μg/ml of each).
The tumor cell lines were seeded in Petri dishes (1×106/dish). After 24h, the cells were harvested in ice‐cold PBS by scraping followed by centrifugation for 5min at 400g. Platelets were centrifuged for 5min at 300g. The tumor cells and platelets were resuspended in lysis buffer (25mM Hepes, 5mM MgCl2, 1mM EDTA, 0.5% Triton; pH 7.5) supplemented with protease inhibitors (see above), incubated for 30min on ice followed by centrifugation for 5min at 20,000g. Protein concentration in plasma, tissue samples and cell lysates was determined by protein BCA Protein assay kit (Pierce, Rockford, IL, USA).
2.7. Western blotting
Protein samples were mixed with 0.25× volume of 4× Laemmli sample buffer, boiled for 5min and equal amounts of protein (100 or 150μg) were separated on a 12% polyacrylamide gel and blotted on nitrocellulose membrane. The blot was blocked in PBS containing 0.1% Tween 20 and 5% dry milk powder for 1h, and then incubated overnight with monoclonal anti‐human TIMP‐1 (VT7; 1μg/ml) (Moller et al., 2005). Subsequently, the blot was washed 3×10min in PBS containing 0.1% Tween 20, incubated with horseradish peroxidase (HRP)‐conjugated goat anti‐mouse antibody diluted 1:1000 (DAKO, Glostrup, Denmark) in PBS with 0.1% Tween 20 and 1% dry milk powder for 1h followed by 3 times for 10min washing in PBS with 0.1% Tween 20. The blot was developed by the ECL+ detection system (Amersham, UK) according to the manufacturer's instructions. Recombinant human TIMP‐1 (rhTIMP‐1) and platelet lysate were used as positive controls, and as a control for the sensitivity of the assay we also included a pool of control EDTA plasma (see Section 2.2) with a known low level of TIMP‐1 (Table 2).
Table 2.
TIMP‐1 levels in plasma and tumor extracts measured by ELISA
| Sample number | EDTA plasma TIMP‐1 (ng/ml) | Tumor extracts TIMP‐1 (ng/mg protein) | Tumor area (%)a |
|---|---|---|---|
| 1 | 213 | 33.17 | 25 |
| 2 | 274.5 | 28.29 | 75 |
| 3 | 511.1 | 7 | 10 |
| 4 | 272.1 | 12.67 | 75 |
| 5 | 523.8 | 6.7 | 10 |
| 6 | 530 | 19.34 | 75 |
| 7 | 530 | 29.5 | 50 |
| 8 | 115.8 | 10.78 | 90 |
| 9 | 258.5 | 28.92 | 90 |
| 10 | 395.2 | 13.02 | 50 |
| 11 | 160.8 | 14.1 | 10 |
| 12 | 131.7 | 19.27 | 5 |
| Control plasma | 45.7 | ||
Relative area of neoplastic cells in tissue section.
2.8. ELISA
A well established sandwich ELISA was used to measure total levels of human TIMP‐1 in tissue and plasma samples (Holten‐Andersen et al., 1999). In brief, the immunoassay employs an affinity‐purified polyclonal sheep anti‐TIMP‐1 antibody as capture antibody and a monoclonal anti‐TIMP‐1 antibody for detection. The complex was detected with alkaline phosphatase‐conjugated rabbit anti‐mouse antibodies (Dako, Denmark) that enable a kinetic rate assay. The plate was read at 405nm, and the rate of the color development was collected automatically over a 1h period in a Power Wavex Microplate Reader (Biotek Instruments Inc., VT, USA) and the results were evaluated using the KC4 software (Biotek Instruments Inc., USA). A four‐parameter fitted standard curve was generated from which the TIMP‐1 concentration of each sample was calculated. TIMP‐1 concentrations in tissue samples were correlated for differences in protein concentrations.
2.9. Generation and sequencing of TIMP‐1 cDNA fragments
Full‐length human TIMP‐1 cDNA (Genbank accession no. NM_003254) was used as a template to generate two non‐overlapping cDNA fragments of TIMP‐1 by PCR named TIMP‐1 01 (56–378bp) and TIMP‐1 02 (398–680bp). The PCR reactions were carried out with upstream primers flanked by a T3 RNA polymerase site and downstream primers flanked by a T7 RNA polymerase site (polymerase sites in bold). The primers used for fragment 01 were: up 5′‐CAT TAA CCC TCA CTA AAG GGA GAA CCC ACC ATG GCC CCC TTG‐3′ and down 5′‐TAA TAC GAC TCA CTA TAG GGA GAC TCC TCG CTG CGG TTG TGG‐3′, and for fragment 02: up 5′‐CAT TAA CCC TCA CTA AAG GGA GAG CAG GAT GGA CTC TTG CAC A‐3′ and down 5′‐TAA TAC GAC TCA CTA TAG GGA GTA TCT GGG ACC GCA GGG ACT‐3′. The PCR was carried out in a 25μl reaction containing 1× Taq buffer, 0.2mM dNTP mix, 2mM MgCl2, 1μM of each primer, 1U Taq polymerase and 0.1–0.5μg cDNA template using standard reagents from Fermentas (Sweden). The PCR products were analysed by gel electrophoresis and purified using MinElute Gel Extraction kit from Qiagen Nordic (UK) according to the manufacturer's instructions.
For the two splice variants of TIMP‐1 lacking exon 2 (del‐2) or exon 5 (del‐5), RT‐PCR products from cell lines and colon cancer tissue samples were gel purified as described above and used as a template for a PCR reaction with the following T3 and T7 flanked primers: up 5′‐CAT TAA CCC TCA CTA AAG GGA GAA GTG GGT GGA TGA GTA ATG C‐3′ and down 5′‐TAA TAC GAC TCA CTA TAG GGA GTC TGG TTG ACT TCT GGT GTC C‐3′.
All TIMP‐1 fragments generated with T3 and T7 flanked primers were sequenced (AGOWA, Berlin, Germany) with both T3 and T7 primers and the sequences were confirmed by BLAST search.
2.10. Preparation of digoxigenin‐labeled RNA probes
Antisense and sense RNA probes were synthesized by in vitro transcription from TIMP‐1 fragment 01, 02 and del‐2 variant using T3 and T7 polymerases and Dig RNA labeling mix (Roche, Basel, Switzerland) according to the manufacturer's instructions. The labeled probes were purified using RNeasy MinElute Cleanup Kit (Qiagen, UK) and the yield of labeled probe was estimated by spot blot analysis. Antisense and corresponding sense probes were adjusted to the same concentration and diluted with de‐ionized formamide to a final concentration of 50%.
2.11. In situ hybridization
Paraffin sections were cut at 3μm, dried overnight, dewaxed and treated with 10–15μg/ml of proteinase K for 5min, dehydrated and air‐dried before 25μl of hybridization solution containing 5μl probe (50ng) was added to each section. Hybridization was carried out overnight at 42°C. After hybridization, the sections were washed with 2× SSC/0.1% SDS for 1h and with 0.5× SSC/0.1% SDS for 30min at 55°C. The sections were then treated with RNase A for 10min at 37°C to remove non‐hybridized probe followed by a final stringency wash with 0.1× SSC/0.1% SDS for 30min at 37°C. Prior to immunological detection of hybridized probe, the sections were blocked with 5% BSA in 0.1M Tris, 0.15M NaCl pH 7.5 containing 0.1% Triton X‐100 for 30min. Alkaline phosphatase‐labeled anti‐digoxigenin antibody diluted 1:500 was then applied to the sections for 2h, and the reaction was visualized by incubating the sections with NBT/BCIP (Roche, Switzerland) overnight in the dark. The sections were counter‐stained with Mayer's hematoxylin and mounted with Glycergel (Dako, Denmark). Sense probes were applied to each case as negative controls. For each in situ hybridization experiment, a positive control case (human colon carcinoma known to contain TIMP‐1 mRNA) was included.
2.12. Immunohistochemistry
Paraffin sections (3μm) were dewaxed in xylene and rehydrated through a graded series of ethanol. Antigen retrieval was carried out by boiling the sections for 10min in a conventional microwave oven in 10mM citrate buffer pH 6.00 followed by 30min in the hot buffer at room temperature. To block endogenous peroxidase activity, the sections were treated with 0.5% hydrogen peroxide in 99% ethanol for 10min. Sections were incubated at 4°C overnight with primary antibodies diluted to the following IgG concentrations: 0.25μg/ml monoclonal anti‐TIMP‐1 antibody, clone VT7 (Moller et al., 2005) and 3.75μg/ml monoclonal anti‐α‐smooth muscle actin (α‐SMA; Dako, Denmark). Anti‐TIMP‐1 antibody was detected with HRP‐labeled PowerVision Kit (ImmunoVision Technologies, Brisbane, CA, USA), and anti‐α‐SMA antibody was detected with HRP‐labeled rabbit Envision (Dako, Denmark). The reactions were in all cases visualized by incubating the sections with 0.1% diaminobenzidine containing 0.02% H2O2 for 5min. The sections were counter‐stained with Mayer's hematoxylin. For each tissue sample a negative control was performed on a serial section, where the primary antibody was substituted by an irrelevant monoclonal antibody against trinitrophenyl hapten (TNP) (IgG1 subtype) diluted to the same IgG concentration as the respective primary antibodies. For each immunohistochemical experiment, a positive control case (human colon carcinoma known to contain TIMP‐1) was included.
3. Results
3.1. RT‐PCR
The TIMP‐1 gene consists of six exons and the initial RT‐PCR screening of the cultured cell lines was carried out using a forward primer in exon 1 and a reverse primer in exon 6 (Table 1 and Figure 1A). After detection of more than one PCR product, indicating various splice variants, RT‐PCR was performed using combinations of primers located in two exons apart to identify any exon‐skipping variants. All cell lines except EVSA‐T and CAMA‐1 expressed more than one TIMP‐1 transcript (Figure 1A). These were identified as a variant lacking exon 2 (del‐2) (Figure 1B), and in MDA‐MB‐231‐BAG, MDA‐MB‐435‐BAG, PC‐3 and EAHY‐926 cells also a variant lacking exon 5 (del‐5) (Figure 1C). The identification of full‐length, del‐2 and del‐5 transcripts was confirmed by sequencing. No variants lacking exon 3 or 4 could be detected. MDA‐MB‐231‐BAG, MDA‐MB‐435‐BAG and PC‐3 cells also expressed additional variants, which could not be identified by the single exon‐skipping analysis and were therefore not analysed further.
Figure 1.
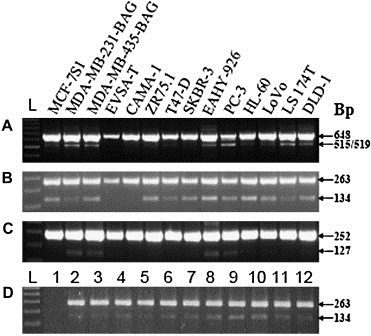
RT‐PCR on cultured cell lines (A–C) and colon cancer tissue samples (D). The following primer combinations were used. Panel A: forward exon 1 and reverse exon 6; panel B and D: forward exon 1 and reverse exon 3; panel C: forward exon 4 and reverse exon 6. Full‐length TIMP‐1 mRNA was detected in all cell lines (A, upper band at 648bp), and all cell lines except EVSA‐T and CAMA express more than one transcript, most abundantly a second transcript of 515/519bp (lower band in A). RT‐PCR with primers located in exon 1 and 3 shows that all cell lines except EVSA‐T and CAMA express del‐2 variant (B, lower band at 134bp), and RT‐PCR with primers located in exon 4 and 6 shows that MDA‐MB‐231‐BAG, MDA‐MD‐435‐BAG, EAHY‐926 and PC‐3 cell lines express del‐5 variant (C, lower band at 127bp). Both full‐length TIMP‐1 and del‐2 variant mRNA were detected in 11 of the 12 colon cancer tissue samples (D). L: 100bp DNA ladder.
Screening of the 12 colon cancer tissue samples revealed both full‐length and del‐2 variant in 11 samples (Figure 1D). In one of the samples no transcript could be detected when the primer located in exon 1 was used. However, amplification with both GAPDH primers and TIMP‐1 primers other than the one located in exon 1 revealed that there was TIMP‐1 expression in this sample (data not shown). qPCR in which the primer partly located in exon 1 (see below) was used, showed that in this sample the del‐2 variant was also expressed. The results indicate that in this sample exon 1 is either partially missing or the sequence located at the 5′ end is altered compared to the other samples. No del‐5 variant could be detected in any of the 12 tissue samples.
3.2. qPCR
To detect full‐length TIMP‐1 transcript, the forward primer located in exon 2 was combined with the reverse primer located in exon 3 (Table 1). To detect only the del‐2 transcript a forward primer designed to anneal to the last 18 bases of exon 1 and the first three bases of exon 3 (Table 1) was combined with a reverse primer in exon 4.
In the 12 CRC samples, the mean relative expression level of the del‐2 variant compared to the full‐length transcript was 0.00802 (±0.00206 SD). This means that the variant is transcribed at a level approx. 1/1000 of the full‐length transcript. The cancer cell lines generally displayed a higher level of del‐2 variant expression ranging from 0.0021 to 0.05.
3.3. Western blot analysis
Next, it was investigated if the splice variants detected by RT‐PCR and qPCR also generated a protein product detectable by western blotting analysis. To analyse if the presence of different transcripts results in different protein expression we chose to analyse one cell line that only express full‐length TIMP‐1 mRNA (EVSA‐T), two cell lines that express both full‐length TIMP‐1 mRNA and the del‐2 variant (MCF‐7S1 and HL‐60) and two cell lines that express both full‐length TIMP‐1 mRNA, the del‐2 variant and the del‐5 variant. We also included all three colon cancer cell lines to compare with the tumor tissue. As shown in Figure 2A, all the tumor cell lines, except for EVSA‐T, expressed full‐length TIMP‐1 protein, although at different levels of expression. The anti‐TIMP‐1 antibody recognized two bands around 24kDa in the positive controls and in the tumor cell lines, probably representing glycosylation variants of full‐length TIMP‐1. Below full‐length TIMP‐1 additional bands were detected, in particular, in PC‐3, DLD‐1, LoVo and LS 174T cell lines. When tumor tissue extracts from CRC patients were analysed for presence of TIMP‐1 protein, additional bands were detected below full‐length TIMP‐1 in tissue sample 1, 3 and 6 (Figure 2B). Both cells and tumor extracts were lysed in a buffer containing a cocktail of protease inhibitors minimizing the risk of break‐down products, and therefore the alternative products may represent splice variants. When plasma samples from CRC patients were analysed by western blotting no alternative TIMP‐1 products were detected (Figure 2C).
Figure 2.
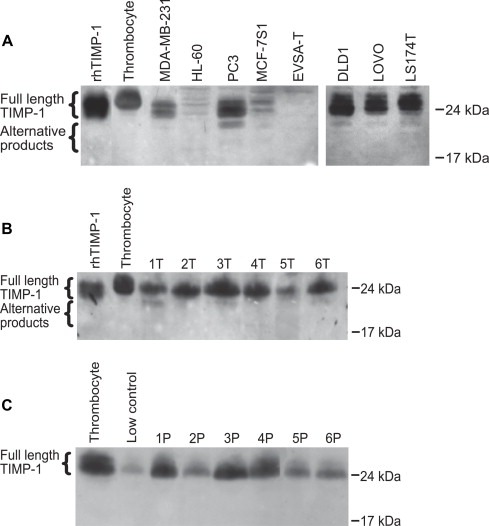
Detection of TIMP‐1 and alternative TIMP‐1 products in cancer cell lines (A), lysates of tumor samples from CRC patients (B) and in plasma from CRC patients (C). Equal amounts of protein were separated by SDS‐PAGE, and TIMP‐1 was subsequently detected with a monoclonal anti‐TIMP‐1 antibody (VT7). Low control, platelet lysate (contains high TIMP‐1 protein levels) and recombinant human (rh) TIMP‐1 serve as controls. The plasma samples are representative of a total of 12 samples analysed. The experiment was repeated once with similar results.
3.4. ELISA
ELISA detecting the amount of total TIMP‐1 protein was performed on both plasma samples and tumor tissue extracts. The results are displayed in Table 2. Compared with a control plasma pool, it is evident that all the plasma samples from CRC patients displayed an elevated level of TIMP‐1 protein (mean 326.4±163.4ng/ml), supporting our previous reports (Holten‐Andersen et al., 2002, 2000). In the tumor tissue samples, the mean concentration of TIMP‐1 was 18.6±9.3ng/mg of protein. In order to evaluate if the tumor extract levels were elevated we would need to compare with a matched sample of normal colon tissue, which unfortunately was not available for this study. There was no obvious correlation between TIMP‐1 levels in plasma samples and tumor extracts. As TIMP‐1 levels in the tumor tissue samples are likely to reflect the amount of tumor tissue/invasive front present in the sample, we evaluated histopathologically a section taken from the middle part of each biopsy. However, there was no apparent correlation between TIMP‐1 levels in the tumor extracts and the relative area of neoplastic cells in the corresponding tissue sections (Table 2).
3.5. Localization of TIMP‐1 mRNA and protein
TIMP‐1 full‐length mRNA expression was detected in nine of the 12 colon cancer samples by in situ hybridization. The positive signal was seen in single cells located adjacent to tumor cells. The cells were elongated and resembled in all cases fibroblasts (Figure 3A and C). No signal was observed in tumor cells in any of the samples. The specificity of the in situ hybridization signal was evaluated using probes transcribed from two non‐overlapping fragments of TIMP‐1 cDNA. In all nine positive samples, the two non‐overlapping antisense probes gave identical hybridization patterns (Figure 3A and C), while no specific signal was seen with the two corresponding sense probes (Figure 3D). The intensity of the hybridization signal was generally weaker than the signal observed in the positive control tissue, possibly due to differences in fixation procedure. The 12 colon cancer samples were also hybridized with a specific antisense probe against the TIMP‐1 del‐2 splice variant, and very weak signal was observed in two of the samples. However, a stronger signal was seen in the control tissue, where the positive cells co‐localized with a proportion of cells expressing full‐length TIMP‐1 mRNA (Figure 3B).
Figure 3.
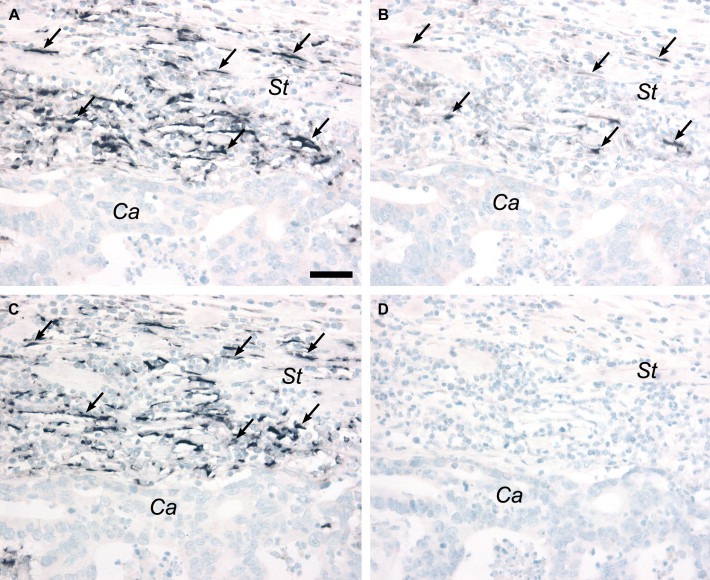
TIMP‐1 expressions in colon cancer. Serial sections from a colon adenocarcinoma were hybridized with antisense or sense probes for full‐length TIMP‐1 (A, C, D) or exon 2 splice variant of TIMP‐1 (B). Full‐length TIMP‐1 mRNA is seen in fibroblast‐like cells localized in the stroma (St) adjacent to the cancer cells (Ca) with similar hybridization patterns seen with two non‐overlapping antisense probes (transcribed from pTIMP‐1 01 and 02) (arrows in A and C). The exon 2 variant of TIMP‐1 mRNA is detected in some of the same fibroblast‐like cells seen in a parallel section (arrows in B). No signal is detected with a sense probe corresponding to the antisense probe shown in panel A (D). Scale bar: A–D=50μm.
TIMP‐1 protein was detected in all 12 colon cancer samples by immunohistochemistry using a monoclonal antibody against human TIMP‐1. This antibody recognizes a linear epitope located in the C‐terminal end of TIMP‐1 translated from part of exon 6 (Moller et al., 2005). Intense positive immunostaining was seen in fibroblast‐like cells in all samples (Figure 4B), and in addition tumor cells also showed weak staining in nine of the 12 samples investigated. No staining was observed in any of the samples when the primary antibody was replaced by an irrelevant control antibody against TNP (Figure 4D).
Figure 4.
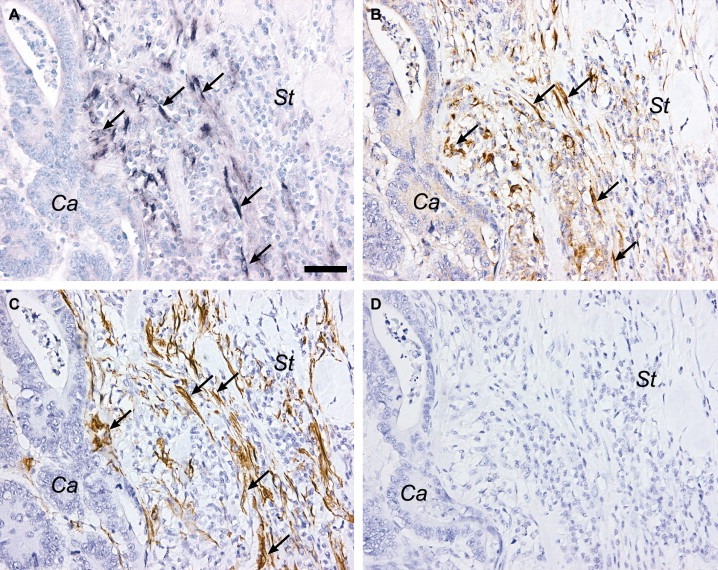
Identification of TIMP‐1 positive cells in colon cancer. Serial sections from a colon adenocarcinoma were processed for in situ hybridization with TIMP‐1 antisense probe 01 (A), or immunostained with monoclonal antibodies against TIMP‐1 (B) or α‐SMA (C). TIMP‐1 mRNA expressing cells (arrows in A) are seen the stroma (St) with no signal observed in the cancer cells (Ca), and immunoreactivity for TIMP‐1 is detected in the same cells (arrows in B). TIMP‐1 mRNA and protein co‐localize with a sub‐population of α‐SMA positive cells, as seen on an adjacent section (arrows in C). No immunoreactivity is seen when the primary antibody is substituted with an irrelevant antibody against TNP (D). Scale bar: A–D=50μm.
There was a good correlation between TIMP‐1 immunohistochemistry and in situ hybridization for full‐length TIMP‐1 mRNA performed on serial sections (Figure 4A and B). The positive cells were further characterized by immunostaining with a monoclonal antibody against α‐SMA. On serial sections, TIMP‐1 mRNA and protein positive cells co‐localized with a sub‐population of α‐SMA positive cells (Figure 4A–C), indicating that the TIMP‐1 expressing cells are smooth muscle cells or myofibroblasts.
4. Discussion
By screening tumor tissue from 12 patients suffering from colorectal cancer and a panel of human cancer cell lines, we have, in addition to full‐length TIMP‐1 mRNA, identified two exon‐skipping splice variants. In most cell lines and in all the cancerous tissue samples we detected a variant of TIMP‐1 lacking exon 2 (del‐2), and in a few of the cell lines a variant lacking exon 5 (del‐5). The observed splice variants were all sequenced, confirming that they represented TIMP‐1 transcripts.
In full‐length TIMP‐1 mRNA the translation is initiated from the first ATG site in exon 2. Sequence analysis of information on the del‐2 variant (Ensembl gene report OTTHUMG00000021447) revealed that the most likely initiation site containing part of the Kozak consensus sequence (Kozak, 1987) is located at the end of exon 3 (Figure 5). This does not change the reading frame, but results in a shorter protein (143 amino acids) lacking the signal peptide sequence, which directs full‐length TIMP‐1 to the secretory pathway. Thus, this variant, if translated, is probably a soluble, intracellular protein. In the del‐5 variant, any translation initiation is likely to start at the same ATG site in exon 2 as full‐length TIMP‐1. However, a theoretical translation (ExPASy translation tool) gives a shift in the reading frame of the exon 6 sequence with an introduction of a new stop codon, which would result in a protein consisting of 136 amino acids (Figure 5). Full‐length TIMP‐1 is N‐glycosylated at position 53 and 101 (Gomez et al., 1997), and although the second site is retained in the shorter del‐2 variant, it is unlikely to be exposed to the N‐glycosylation machinery because of the lack of signal peptide, and thus may not be glycosylated in vivo. Although residues throughout the TIMP‐1 molecule are involved in inhibition of MMPs, the main inhibitory activity has been assigned to the N‐terminal end of the molecule (Murphy et al., 1991), more specifically to a region surrounding the second disulfide knot (Bodden et al., 1994). Part of this region is missing in a putative del‐2 variant of TIMP‐1, which may therefore have compromised MMP inhibitory activity. The del‐5 variant of TIMP‐1 is more likely to retain its inhibitory activity.
Figure 5.
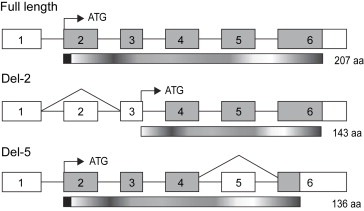
Schematic representation of full‐length and alternatively spliced TIMP‐1. Exons are numbered from 1 to 6 and translated regions are shown in grey. White boxes indicate untranslated regions. Translation initiation sites are marked by black arrow and ATG. The corresponding translated proteins are shown underneath the mRNAs and the theoretical size is marked with number of amino acids (aa). The signal peptide sequences are shown as black boxes at the N‐terminal end of full‐length TIMP‐1 and del‐5 variant. The translation initiation site in del‐2 TIMP‐1 is shifted from exon 2 to exon 3, which results in a shorter protein lacking the signal peptide. A theoretical translation of the del‐5 variant results in a protein consisting of 136 aa due a shift in reading frame of the exon 6 sequence.
By performing western blotting analyses on the tumor tissue samples, we could in addition to full‐length TIMP‐1 demonstrate additional TIMP‐1 bands of lower molecular mass. However, these extra products are absent in the plasma samples suggesting that they could represent the exon 2 variant, which if translated would not be secreted into the circulation because of the lacking signal peptide. Even though the del‐2 mRNA transcript was expressed in all the analysed cell lines (except EVSA‐T) we were only able to detect additional TIMP‐1 bands of lower molecular mass in the PC‐3, DLD‐1, LoVo and LS 174T cell lines. The western blot analysis shows different levels of TIMP‐1 in the cell lines and we could only detect the additional TIMP‐1 band in cells with high expression of the full‐length TIMP‐1 protein. The additional TIMP‐1 bands could therefore be present in the other cell lines as well, however, below detection level. This is further supported by the finding that the del‐2 variant mRNA is expressed at a much lower level than full‐length TIMP‐1 mRNA.
When we quantified the total level of TIMP‐1 protein by using a thoroughly validated ELISA assay, there was no correlation between tumor extracts and plasma either. This, however, could be explained by the fact that a tumor extract only represents a small part of the whole tumor, not necessarily from the invasive front, where we have previously demonstrated that the TIMP‐1 expressing fibroblast‐like cells are localized (Holten‐Andersen et al., 2005). These fibroblast‐like cells are in this study further characterized as myofibroblasts or smooth muscle cells and are now shown to also express the del‐2 variant, although at a much lower level than full‐length TIMP‐1, which is in agreement with our qPCR data.
The paradox of raised levels of inhibitors of tissue‐degrading enzymes in advanced disease is well‐described, but little is known about the biology of this phenomenon. High levels of TIMP‐1 in the circulation and in tumor extracts may simply reflect the raised levels of MMPs, but there is increasing data describing tumor‐promoting activities of TIMP‐1 independent of MMP inhibition, including inhibition of apoptosis. It is possible that different variant forms of TIMP‐1 are responsible for the diverse and contrasting functions of the protein, similarly to what have been found for other cancer‐related proteins (Venables, 2006). A further investigation of this requires expression and characterization of the variant forms of the protein.
The prognostic value of TIMP‐1 has been described at both mRNA and protein level. In breast cancer, however, the mRNA data are somewhat conflicting. One semi‐quantitative study has shown an inverse correlation between high level of TIMP‐1 expression and lymph node metastasis (Inoue et al., 2000), whereas other investigators have found high TIMP‐1 mRNA levels to be correlated with poor prognosis (Nakopoulou et al., 2002; Ree et al., 1997). However, Span et al. (2004) found no association between levels of full‐length TIMP‐1 mRNA and disease progression, when measured by a quantitative RT‐PCR assay. At the protein level, high TIMP‐1 has repeatedly proved to be a marker of poor prognosis in both colorectal and breast cancer (Holten‐Andersen et al., 2002, 2003, 2004, 2005, 2005), making measurements of TIMP‐1 protein more attractive in the clinical setting. The development of quantitative assays specific for full‐length and del‐2 variant of TIMP‐1 may improve the prognostic and predictive value and is useful in the management of cancer patients.
Acknowledgements
We wish to thank the surgeons and pathologists at University Hospital Hvidovre for collection of plasma and tumor tissue samples from colon cancer patients. We also thank Vibeke Jensen and Lise Larsen for excellent technical assistance. MCF7‐S1 cells were kindly provided by Professor Marja Jäättela, Danish Cancer Society, Copenhagen, Denmark, and PC‐3 and HL‐60 cells were kindly provided by Professor Hau C. Kwaan, Northwestern University, Chicago, USA. This work was supported by The Danish Cancer Society, The IMK Foundation, The Danish Medical Research Counsel, Beckett Foundation, The Danish Cancer Research Foundation, Eva og Henry Frænkels Foundation, Grosserer Valdemar Foersom og Hustru Thyra Foersoms Foundations, Ib Henriksens Foundation, Kathrine og Vigo Skovgaard Foundation, Knud og Dagny Gad Andresens Foundation, Redaktør Kaaresens Foundation, The Obel Family Foundation, The Kornerup Fund, The Aage and Johanne Louis‐Hansen Fund, The Aase and Einar Danielsens Fund, The Raimund and Dagmar Ringgaard‐Bohns Foundation, The A.P. Moeller Foundation for the Advancement of Medical Science and P.A. Messerschmidt og Hustrus Foundation. HJN is Danish Cancer Society professor of surgical oncology.
Usher P.A., Sieuwerts A.M., Bartels A., Lademann U., Nielsen H.J., Holten-Andersen L., Foekens J.A., Brünner N., Offenberg H., (2007), Identification of alternatively spliced TIMP‐1 mRNA in cancer cell lines and colon cancer tissue, Molecular Oncology, 1, doi:10.1002/mol2.2007.1.issue-2.
References
- Akahane, T. , Akahane, M. , Shah, A. , Connor, C.M. , Thorgeirsson, U.P. , 2004. TIMP-1 inhibits microvascular endothelial cell migration by MMP-dependent and MMP-independent mechanisms. Exp. Cell Res.. 301, 158–167. [DOI] [PubMed] [Google Scholar]
- Baker, A.H. , Edwards, D.R. , Murphy, G. , 2002. Metalloproteinase inhibitors: biological actions and therapeutic opportunities. J. Cell Sci.. 115, 3719–3727. [DOI] [PubMed] [Google Scholar]
- Bertaux, B. , Hornebeck, W. , Eisen, A.Z. , Dubertret, L. , 1991. Growth stimulation of human keratinocytes by tissue inhibitor of metalloproteinases. J. Investig. Dermatol.. 97, 679–685. [DOI] [PubMed] [Google Scholar]
- Bodden, M.K. , Harber, G.J. , Birkedal-Hansen, B. , Windsor, L.J. , Caterina, N.C. , Engler, J.A. , Birkedal-Hansen, H. , 1994. Functional domains of human TIMP-1 (tissue inhibitor of metalloproteinases). J. Biol. Chem.. 269, 18943–18952. [PubMed] [Google Scholar]
- Brew, K. , Dinakarpandian, D. , Nagase, H. , 2000. Tissue inhibitors of metalloproteinases: evolution, structure and function. Biochim. Biophys. Acta. 1477, 267–283. [DOI] [PubMed] [Google Scholar]
- Brunner, N. , Thompson, E.W. , Spang-Thomsen, M. , Rygaard, J. , Dano, K. , Zwiebel, J.A. , 1992. lacZ transduced human breast cancer xenografts as an in vivo model for the study of invasion and metastasis. Eur. J. Cancer. 28A, 1989–1995. [DOI] [PubMed] [Google Scholar]
- Davidsen, L.M. , Würtz, S.O. , Romer, M.U. , Moller, S.N. , Johansen, S.K. , Christensen, I.J. , Larsen, J.K. , Offenberg, H. , Brunner, N. , Lademann, U. , October 23, 2006. TIMP-1 gene deficiency increases tumor cell sensitivity to chemotherapy-induced apoptosis. Br. J. Cancer. 95, (8) 1114–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egeblad, M. , Werb, Z. , 2002. New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer. 2, 161–174. [DOI] [PubMed] [Google Scholar]
- Gomez, D.E. , Alonso, D.F. , Yoshiji, H. , Thorgeirsson, U.P. , 1997. Tissue inhibitors of metalloproteinases: structure, regulation and biological functions. Eur. J. Cell Biol.. 74, 111–122. [PubMed] [Google Scholar]
- Guedez, L. , Stetler-Stevenson, W.G. , Wolff, L. , Wang, J. , Fukushima, P. , Mansoor, A. , Stetler-Stevenson, M. , 1998. In vitro suppression of programmed cell death of B cells by tissue inhibitor of metalloproteinases-1. J. Clin. Investig.. 102, 2002–2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa, T. , Yamashita, K. , Tanzawa, K. , Uchijima, E. , Iwata, K. , 1992. Growth-promoting activity of tissue inhibitor of metalloproteinases-1 (TIMP-1) for a wide range of cells. A possible new growth factor in serum. FEBS Lett.. 298, 29–32. [DOI] [PubMed] [Google Scholar]
- Holten-Andersen, M.N. , Christensen, I.J. , Nielsen, H.J. , Lilja, H. , Murphy, G. , Jensen, V. , Brunner, N. , Piironen, T. , 2002. Measurement of the noncomplexed free fraction of tissue inhibitor of metalloproteinases 1 in plasma by immunoassay. Clin. Chem.. 48, 1305–1313. [PubMed] [Google Scholar]
- Holten-Andersen, M.N. , Christensen, I.J. , Nielsen, H.J. , Stephens, R.W. , Jensen, V. , Nielsen, O.H. , Sorensen, S. , Overgaard, J. , Lilja, H. , Harris, A. , Murphy, G. , Brunner, N. , 2002. Total levels of tissue inhibitor of metalloproteinases 1 in plasma yield high diagnostic sensitivity and specificity in patients with colon cancer. Clin. Cancer Res.. 8, 156–164. [PubMed] [Google Scholar]
- Holten-Andersen, M.N. , Hansen, U. , Brunner, N. , Nielsen, H.J. , Illemann, M. , Nielsen, B.S. , 2005. Localization of tissue inhibitor of metalloproteinases 1 (TIMP-1) in human colorectal adenoma and adenocarcinoma. Int. J. Cancer. 113, 198–206. [DOI] [PubMed] [Google Scholar]
- Holten-Andersen, M.N. , Murphy, G. , Nielsen, H.J. , Pedersen, A.N. , Christensen, I.J. , Hoyer-Hansen, G. , Brunner, N. , Stephens, R.W. , 1999. Quantitation of TIMP-1 in plasma of healthy blood donors and patients with advanced cancer. Br. J. Cancer. 80, 495–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holten-Andersen, M.N. , Stephens, R.W. , Nielsen, H.J. , Murphy, G. , Christensen, I.J. , Stetler-Stevenson, W. , Brunner, N. , 2000. High preoperative plasma tissue inhibitor of metalloproteinase-1 levels are associated with short survival of patients with colorectal cancer. Clin. Cancer Res.. 6, 4292–4299. [PubMed] [Google Scholar]
- Inoue, H. , Mimori, K. , Shiraishi, T. , Kataoka, A. , Sadanaga, N. , Ueo, H. , Barnard, G.F. , Mori, M. , 2000. Expression of tissue inhibitor of matrix metalloproteinase-1 in human breast carcinoma. Oncol. Rep.. 7, 871–874. [DOI] [PubMed] [Google Scholar]
- Kozak, M. , 1987. At least six nucleotides preceding the AUG initiator codon enhance translation in mammalian cells. J. Mol. Biol.. 196, 947–950. [DOI] [PubMed] [Google Scholar]
- Lemaitre, V. , D'Armiento, J. , 2006. Matrix metalloproteinases in development and disease. Birth Defects Res. C Embryo Today. 78, 1–10. [DOI] [PubMed] [Google Scholar]
- Liu, X.W. , Taube, M.E. , Jung, K.K. , Dong, Z. , Lee, Y.J. , Roshy, S. , Sloane, B.F. , Fridman, R. , Kim, H.R. , 2005. Tissue inhibitor of metalloproteinase-1 protects human breast epithelial cells from extrinsic cell death: a potential oncogenic activity of tissue inhibitor of metalloproteinase-1. Cancer Res.. 65, 898–906. [PubMed] [Google Scholar]
- Moller, S.N. , Dowell, B.L. , Stewart, K.D. , Jensen, V. , Larsen, L. , Lademann, U. , Murphy, G. , Nielsen, H.J. , Brunner, N. , Davis, G.J. , 2005. Establishment and characterization of 7 new monoclonal antibodies to tissue inhibitor of metalloproteinases-1. Tumor Biol.. 26, 71–80. [DOI] [PubMed] [Google Scholar]
- Murphy, G. , Houbrechts, A. , Cockett, M.I. , Williamson, R.A. , O'Shea, M. , Docherty, A.J. , 1991. The N-terminal domain of tissue inhibitor of metalloproteinases retains metalloproteinase inhibitory activity. Biochemistry. 30, 8097–8102. [DOI] [PubMed] [Google Scholar]
- Nakopoulou, L. , Giannopoulou, I. , Stefanaki, K. , Panayotopoulou, E. , Tsirmpa, I. , Alexandrou, P. , Mavrommatis, J. , Katsarou, S. , Davaris, P. , 2002. Enhanced mRNA expression of tissue inhibitor of metalloproteinase-1 (TIMP-1) in breast carcinomas is correlated with adverse prognosis. J. Pathol.. 197, 307–313. [DOI] [PubMed] [Google Scholar]
- Offenberg, H. , Thomsen, P.D. , 2005. Functional challenge affects aquaporin mRNA abundance in mouse blastocysts. Mol. Reprod. Dev.. 71, 422–430. [DOI] [PubMed] [Google Scholar]
- Ouyang, D.L. , Chen, J.J. , Getzenberg, R.H. , Schoen, R.E. , 2005. Noninvasive testing for colorectal cancer: a review. Am. J. Gastroenterol.. 100, 1393–1403. [DOI] [PubMed] [Google Scholar]
- Ree, A.H. , Florenes, V.A. , Berg, J.P. , Maelandsmo, G.M. , Nesland, J.M. , Fodstad, O. , 1997. High levels of messenger RNAs for tissue inhibitors of metalloproteinases (TIMP-1 and TIMP-2) in primary breast carcinomas are associated with development of distant metastases. Clin. Cancer Res.. 3, 1623–1628. [PubMed] [Google Scholar]
- Schrohl, A.S. , Christensen, I.J. , Pedersen, A.N. , Jensen, V. , Mouridsen, H. , Murphy, G. , Foekens, J.A. , Brunner, N. , Holten-Andersen, M.N. , 2003. Tumor tissue concentrations of the proteinase inhibitors tissue inhibitor of metalloproteinases-1 (TIMP-1) and plasminogen activator inhibitor type 1 (PAI-1) are complementary in determining prognosis in primary breast cancer. Mol. Cell Proteomics. 2, 164–172. [DOI] [PubMed] [Google Scholar]
- Schrohl, A.S. , Holten-Andersen, M.N. , Peters, H.A. , Look, M.P. , Meijer-van Gelder, M.E. , Klijn, J.G. , Brunner, N. , Foekens, J.A. , 2004. Tumor tissue levels of tissue inhibitor of metalloproteinase-1 as a prognostic marker in primary breast cancer. Clin. Cancer Res.. 10, 2289–2298. [DOI] [PubMed] [Google Scholar]
- Schrohl, A.S. , Meijer-van Gelder, M.E. , Holten-Andersen, M.N. , Christensen, I.J. , Look, M.P. , Mouridsen, H. , Brunner, N. , Foekens, J.A. , 2006. Primary tumour levels of tissue inhibitor of metalloproteinases-1 are predictive of response to chemotherapy in patients with metastatic breast cancer. Clin. Cancer Res.. 12, 7054–7058. [DOI] [PubMed] [Google Scholar]
- Span, P.N. , Lindberg, R.L. , Manders, P. , Tjan-Heijnen, V.C. , Heuvel, J.J. , Beex, L.V. , Sweep, C.G. , 2004. Tissue inhibitors of metalloproteinase expression in human breast cancer: TIMP-3 is associated with adjuvant endocrine therapy success. J. Pathol.. 202, 395–402. [DOI] [PubMed] [Google Scholar]
- Stricklin, G.P. , Welgus, H.G. , 1983. Human skin fibroblast collagenase inhibitor. Purification and biochemical characterization. J. Biol. Chem.. 258, 12252–12258. [PubMed] [Google Scholar]
- Thaysen-Andersen, M. , Thogersen, I.B. , Nielsen, H.J. , Lademann, U. , Brunner, N. , Enghild, J.J. , Hojrup, P. , January 6, 2007. Rapid and individual-specific glycoprofiling of a low-abundant N-glycosylated protein tissue inhibitor of metalloproteinases-1. Mol. Cell. Proteomics. [DOI] [PubMed] [Google Scholar]
- Venables, J.P. , 2006. Unbalanced alternative splicing and its significance in cancer. Bioessays. 28, 378–386. [DOI] [PubMed] [Google Scholar]
- Waas, E.T. , Hendriks, T. , Lomme, R.M. , Wobbes, T. , 2005. Plasma levels of matrix metalloproteinase-2 and tissue inhibitor of metalloproteinase-1 correlate with disease stage and survival in colorectal cancer patients. Dis. Colon Rectum. 48, 700–710. [DOI] [PubMed] [Google Scholar]
- Westbrook, C.A. , Gasson, J.C. , Gerber, S.E. , Selsted, M.E. , Golde, D.W. , 1984. Purification and characterization of human T-lymphocyte-derived erythroid-potentiating activity. J. Biol. Chem.. 259, 9992–9996. [PubMed] [Google Scholar]
- Wurtz, S.O. , Christensen, I.J. , Schrohl, A.S. , Mouridsen, H. , Lademann, U. , Jensen, V. , Brunner, N. , 2005. Measurement of the uncomplexed fraction of tissue inhibitor of metalloproteinases-1 in the prognostic evaluation of primary breast cancer patients. Mol. Cell. Proteomics. 4, 483–491. [DOI] [PubMed] [Google Scholar]