

Abstract
Gut-associated lymphoid tissues are enriched in CCR6+ Th17-polarized CD4+ T cells that contribute to HIV-1 persistence during antiretroviral therapy (ART). This raises the need for Th17-targeted immunotherapies. In an effort to identify mechanisms governing HIV-1 permissiveness/persistence in gut-homing Th17 cells, we analyzed the transcriptome of CCR6+ versus CCR6– T cells exposed to the gut-homing inducer retinoic acid (RA) and performed functional validations in colon biopsies of HIV-infected individuals receiving ART (HIV+ART). Although both CCR6+ and CCR6– T cells acquired gut-homing markers upon RA exposure, the modulation of unique sets of genes coincided with preferential HIV-1 replication in RA-treated CCR6+ T cells. This molecular signature included the upregulation of HIV-dependency factors acting at entry/postentry levels, such as the CCR5 and PI3K/Akt/mTORC1 signaling pathways. Of note, mTOR expression/phosphorylation was distinctively induced by RA in CCR6+ T cells. Consistently, mTOR inhibitors counteracted the effect of RA on HIV replication in vitro and viral reactivation in CD4+ T cells from HIV+ART individuals via postentry mechanisms independent of CCR5. Finally, CCR6+ versus CCR6– T cells infiltrating the colons of HIV+ART individuals expressed unique molecular signatures, including higher levels of CCR5, integrin β7, and mTOR phosphorylation. Together, our results identify mTOR as a druggable key regulator of HIV permissiveness in gut-homing CCR6+ T cells.
Keywords: AIDS/HIV, Immunology
mTOR inhibitors decrease HIV reservoirs and restore Th17-mediated immunity at the intestinal level during ART.
Introduction
HIV-1 causes the progressive depletion of CD4+ T cells, thus impairing the ability of the immune system to fight pathogens. Although current antiretroviral therapies (ART) efficiently control viral replication and restore CD4 counts, the HIV eradication has not been achieved (1–3). Major CD4+ T cell alterations occur in gut-associated lymphoid tissues (GALT) rapidly upon HIV transmission (4, 5), and these alterations persist despite viral suppressive ART (3, 6), even in patients treated during acute HIV infection (7). The GALT is considered an important site of HIV replication and viral reservoir persistence during ART (8). GALT-infiltrating CD4+ T cells represent optimal HIV targets, likely due to their high expression of CCR5 (4, 5, 8), a major coreceptor for HIV entry (9–11), and integrin α4β7, a gut-homing molecule (12) identified as an HIV-binding molecule (13). Very recently, integrin α4β7–blocking Abs proved efficacy in controlling viral replication upon ART interruption in an SIV infection model (14), indicative that interfering with HIV replication in gut-homing α4β7+CD4+ T cells represents a potential curative strategy.
Among GALT-infiltrating CD4+ T cells, Th17 cells play a beneficial role in maintaining epithelial cell integrity at barrier surfaces via the production of effector cytokines under the control of lineage-specific transcription factors (15). Studies by our group and others demonstrated that Th17-polarized CCR6+CD4+ T cells are highly permissive to infection (16–18) as a consequence of their expression of HIV dependency factors (HDFs) acting at entry and postentry levels (19, 20), combined with the lack of HIV restriction mechanisms (21–23). Most recently, CD4+ T cells expressing the Th17 markers CCR6 and RORγt were identified as the first targets of SIV during vaginal transmission (24); in addition, the detrimental role of viral permissive CCR6+ T cell infiltration into the gut was documented (25). The depletion of Th17 cells from the GALT upon HIV infection persists despite viral-suppressive ART (26–28), even when ART is intensified with integrase and CCR5 inhibitors (29). Other promising strategies tested for mucosal Th17 restoration include an ideal ART initiation at very early stages of infection (Fiebig I/II but not III) (7), the use of probiotics, and IL-21 supplementation (30–32). The nonrestoration of GALT Th17 cells leads to significant damages of the intestinal barrier, thus allowing microbial translocation, a cause for chronic immune activation and occurrence of non-AIDS comorbidities during ART (3, 6, 33). Despite their dramatic depletion, long-lived Th17 cells contribute to HIV reservoir persistence during ART (34, 35). Consistently, our group identified a subset of CCR6+CCR4–CXCR3– Th17 cells predominant in blood and lymph nodes and carrying replication-competent HIV reservoirs (36) and further demonstrated preferential HIV DNA persistence in colon-infiltrating CCR6+CD4+ T cells during ART (37). These advances orient our search for immunotherapeutic strategies to limit HIV infection/persistence in mucosal Th17 cells in the ART era.
The homing of CCR6+ Th17 cells into the GALT is mediated via the CCR6/CCL20 axis (28, 38) as well as the adhesion molecule integrin α4β7 (12) and chemokine receptor CCR9 (39). The imprinting for gut homing is induced by all-trans retinoic acid (ATRA) (40), a vitamin A metabolite produced by GALT dendritic cells (41). Our previous studies demonstrated that ATRA increases HIV-1 permissiveness preferentially in CCR6+ T cells compared with CCR6– T cells (42). This effect coincided with the preferential ability of ATRA to upregulate the expression of the HIV coreceptor CCR5 on CCR6+ T cells, indicative of a most efficient HIV entry. Nevertheless, exposure to single-round VSV-G–pseudotyped HIV entering cells by endocytosis provided evidence that ATRA also promotes HIV replication in gut-homing CCR6+ T cells via postentry mechanisms (42). These findings led to the current hypothesis that CCR6 is a marker for Th17-polarized CD4+ T cells transcriptionally programmed to become HIV targets in the gut, in which ATRA promotes the expression of unidentified HIV permissiveness factors, which may be manipulated to protect gut-homing Th17 cells from HIV.
In our search for new druggable molecular targets to prevent HIV replication/persistence in gut-homing Th17 cells, herein we performed a genome-wide transcriptional profiling to identify HDFs modulated by ATRA in memory CCR6+ T cells. Together, our studies (a) provide an in-depth characterization of molecular mechanisms contributing to HIV replication/persistence in ATRA-exposed CCR6+ Th17 cells; (b) identify mTOR as a druggable target modulated by ATRA in these cells; and (c) support the beneficial use of mTOR inhibitors in clinics to preserve mucosal Th17 cells from HIV infection/persistence during ART.
Results
Transmitted/founder HIV preferentially infects retinoic acid–treated CCR6+CD4+ T cells.
We previously reported that ATRA-mediated imprinting for gut-homing increases replication of the laboratory-adapted R5 NL4.3BaL HIV preferentially in CCR6+ versus CCR6– T cells (42). Transmitted/founder (T/F) HIV strains are isolated early upon seroconversion and are unique in their virulence/sensitivity to antiviral mechanisms (43). To determine whether T/F HIV also targets gut-homing CCR6+ T cells for preferential replication, FACS-sorted memory CCR6+ and CCR6– T cells stimulated via CD3/CD28 in the presence/absence of ATRA were exposed in parallel to T/F THRO (44) and NL4.3BaL HIV (Figure 1A). The relative frequency of CCR6+ and CCR6– T cells in peripheral blood mononuclear cells (PBMCs) before sort is depicted in Figure 1, B and C. The dose of ATRA used (10 nM) (42) is consistent with physiological plasma levels (40) and had no influence on cell viability (data not shown). ATRA robustly increases replication of both NL4.3BaL and T/F THRO HIV in CCR6+ T cells, while the effects on CCR6– T cells were only minor, as reflected by the quantification of early (RU5), late (Gag), and integrated (Alu/HIV) HIV reverse transcripts (Figure 1, D and E). Thus, T/F HIV, similar to NL4.3BaL HIV, also targets ATRA-treated CCR6+ T cells for preferential replication, suggesting a critical role played by gut-homing CCR6+ T cells during the early steps of mucosal HIV transmission.
Figure 1. Replication advantage of transmitted/founder HIV in CCR6+ versus CCR6– T cells upon exposure to ATRA.

Memory CCR6+ and CCR6– T cells were isolated by MACS (magnetic-activated cell sorting) and FACS from the PBMCs (peripheral blood mononuclear cells) of HIV-uninfected individuals and tested for their ability to replicate HIV in vitro. (A) The experimental flow chart. (B) The expression of CCR6 and CD45RA on total CD4+ T cells isolated using magnetic beads and on memory (CD45RA–) CCR6+ and CCR6– T cells isolated by FACS. (C) The frequency of CCR6– and CCR6+ T cells within memory CD4+ T cells in n = 10 donors. (D and E) Paired t test P values are indicated on the graphs. Sorted cells were stimulated via CD3/CD28 in presence/absence of ATRA (all-trans retinoic acid) (10 nM) for 4 days and exposed to (D) T/F (transmitted/founder) THRO HIV or (E) laboratory-adapted NL4.3BaL HIV (25 ng HIV-p24/106 cells). Early (RU5 primers) and late (Gag primers) HIV DNA reverse transcripts, as well as integrated HIV DNA levels (Alu/HIV-LTR primers), were quantified by nested real-time PCR in matched subsets isolated from n = 2 different donors at day 3 after infection (mean ± SD of triplicate wells). Each symbol represents one triplicate value. One-way ANOVA, with Tukey’s multiple comparisons test (*P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001).
CCR6+ versus CCR6– T cells exhibit higher proliferation, TNF-α production, and NF-κB DNA-binding activity in the presence/absence of ATRA.
In search of postentry permissiveness mechanisms, we first investigated whether ATRA modulated proliferation and IL-2 production. In the absence of ATRA, CCR6+ compared with CCR6– T cells exhibited higher proliferation upon TCR triggering but a markedly lower production of IL-2 (Supplemental Figure 1, A–C; supplemental material available online with this article; https://doi.org/10.1172/jci.insight.93230DS1), consistent with previous studies (16), likely as a consequence of RORγt-mediated IL-2 inhibition (45). ATRA further decreased IL-2 production in CCR6+ T cells but had no significant effect on cell proliferation (Supplemental Figure 1, A–C). Levels of CD25 (IL-2Rα) were similarly high on CCR6+ and CCR6– T cells exposed or not to ATRA (Supplemental Figure 1, D–G), indicating that differences in IL-2 production are not related to IL-2 autoconsumption. Thus, increased HIV replication in CCR6+ T cells upon exposure to ATRA cannot be explained by enhanced cell proliferation and/or IL-2 production.
HIV permissiveness in CCR6+Th17 versus CCR6–Th1 cells is associated with higher NF-κB nuclear translocation (20). The proinflammatory cytokine TNF-α enhances HIV replication in part by triggering the NF-κB pathway (46). This prompted us to investigate differences in TNF-α production and NF-κB-p65 activity. Upon TCR triggering, CCR6+ versus CCR6– T cells produced significantly higher levels of TNF-α in the absence of ATRA; although ATRA slightly decreased TNF-α production in CCR6+ T cells, levels remained significantly higher in ATRA-treated CCR6+ versus CCR6– T cells (Supplemental Figure 2A). The NF-κB-p65 DNA-binding activity was higher in CCR6+ versus CCR6– T cell nuclei when cells were cultured in the presence/absence of ATRA; exposure to ATRA slightly decreased NF-κB-p65 DNA-binding activity in CCR6+ T cells (Supplemental Figure 2B), consistent with previous reports (47). Nevertheless, NF-κB-p65 DNA-binding activity remained significantly higher in ATRA-treated CCR6+ versus CCR6– T cells (Supplemental Figure 2B). Together, these results demonstrate that TNF-α production and NF-κB-p65 nuclear translocation/DNA-binding activity are significantly higher in memory CCR6+ versus CCR6– T cells upon TCR triggering but that ATRA has no positive effect on these differences.
ATRA promotes a unique transcriptional program associated with HIV permissiveness in CCR6+ T cells.
To gain insights into mechanisms underlying the differential effects of ATRA on CCR6+ versus CCR6– T cells, the ability of these two cell subsets to integrate ATRA-mediated signals was investigated. CCR6+ and CCR6– T cells expressed similar levels of RA receptor α (RARα) mRNA ex vivo (Figure 2A), and ATRA upregulated the expression of RA-inducible gene I (RIG-I)/retinoic acid receptor responder protein 3 (RARRES3), a direct target of RA (48), in both cell subsets (Figure 2B). We hypothesized that CCR6+ and CCR6– T cell subsets respond to ATRA in distinct fashions, with the expression of HDFs being promoted preferentially in CCR6+ T cells. To test this hypothesis, genome-wide transcriptional profiling was performed in CCR6+ and CCR6– T cells stimulated via the TCR in the presence/absence of ATRA for 4 days; this time point coincided with the time of HIV exposure in Figure 1.
Figure 2. ATRA differentially modulates gene expression in CCR6– and CCR6+ T cells.

Memory CCR6+/CCR6– T cells were sorted and stimulated as described in Figure 1. (A and B) The SYBR Green real-time RT-PCR quantification of RARα (retinoic acid receptor α) mRNA ex vivo (A; n = 2, mean ± SD) and RIG-I/RARRES3 (retinoic acid–induced gene I) mRNA upon CD3/CD28 triggering in the presence/absence of ATRA (B; n = 4, mean ± SEM). Paired t test P values are indicated on the graphs. Each symbol represents (A) one duplicate value or (B) one different donor. (C and D) Total RNA from matched T cell subsets of n = 6 different HIV-uninfected donors was used for genome-wide transcriptional profiling. One-way ANOVA analysis identified differentially expressed genes based on P values or adjusted P values (P < 0.05) and/or fold-change (FC, cutoff 1.3). (C) Volcano plots for all probes in each linear model with the log2 FC on the x axis and the negative logarithm of the adjusted P values for false discovery rate (FDR) on the y axis. The red/green color code is based on the 5% FDR threshold. (D) The Venn diagram depicts the number of probe sets modulated by ATRA uniquely in CCR6+ (gray) and CCR6– T cells (white) (P < 0.05) as well as 466 probe sets commonly modulated.
Differentially expressed genes were identified based on P values or adjusted P values (adj. P) and fold change (FC) of gene expression. Results in Figure 2C and Supplemental Tables 1–4 illustrate transcriptional differences between CCR6+ and CCR6– T cells upon TCR triggering in the presence/absence of ATRA. In the absence of ATRA, upregulated transcripts (n = 348; adj. P < 0.05, FC cutoff 1.3) in CCR6+ versus CCR6– T cells included Th17 markers, such as CCL20, IL-22, IL-17F, CTSH, KLRB1/CD161, IL-8, RORA, CSF2/GM-CSF, IL-26, RORC, CCR6, SGK1, PPARG, IL-23R, ARNTL, and PTPN13 (Supplemental Table 1). Transcripts upregulated by ATRA in CCR6+ T cells (n = 95; adj. P < 0.05, FC cutoff 1.3) included RARRES3, CD38, DHRS3, UBD, LGMN, EVI5, LZTFL1, NAPSA, GBP4, HIC1, CCR9, CCR2, CXCR6, and ITGB7 (Supplemental Table 2). Transcripts upregulated by ATRA in CCR6– T cells (n = 119; adj. P < 0.05, FC cutoff 1.3) included NAPSA, CD38, EVI5, SAMD9, GALR2, DHRS3, ANKRD37, PTGER2, SLC26A4, RARRES3, ARG2, APOBEC3B, RIPK2, and NFIL3 (Supplemental Table 3). The analysis of the n = 319 transcripts overexpressed in ATRA-treated CCR6+ versus CCR6– T cells revealed the preservation of a Th17 signature (e.g., CCL20, IL-17F, CTSH, IL-22, KLRB1, CSF2, ANTXR2, GPR56, CXCR6, KLF2, RORC, RORA, S100A4, CCR6, MAP3K4, CD96, IL-26, FURIN, ABCB1, ARNTL, and PTPN13) (Supplemental Table 4). Finally, in the presence/absence of ATRA, CCR6+ versus CCR6– T cells show superior expression of the Th17 markers RORC, ARNTL, and CCR6 and inferior expression of the RORC nuclear repressor NFIL3 (49) (Supplemental Figure 3). Together, these results demonstrate that CCR6+ and CCR6– T cells respond to ATRA in a unique transcriptional fashion and that ATRA exposure does not modify the Th17-polarization profile of CCR6+ T cells.
An extended intelligent search within top modulated transcripts revealed that ATRA induced expression of gut-homing markers integrin α4, integrin β7, and CCR9 (12) in both CCR6+ and CCR6– T cells. Of note, CXCR6, a minor HIV coreceptor (50, 51), was significantly upregulated by ATRA in CCR6+ cells but not CCR6– T cells (Supplemental Tables 2–4 and Supplemental Figure 3). FACS analysis validated the microarray results for integrin β7 and CXCR6 expression (Supplemental Figure 4, A–C). Although CCR5 transcripts were not differentially expressed in microarrays (data not shown), we confirm (42) that ATRA significantly upregulated CCR5 preferentially on CCR6+ T cells (Supplemental Figure 4, A and D). This coincided with the upregulation of KLF2 (Supplemental Figure 3), a positive regulator of CCR5 transcription (52). The downregulation of CCR5 ligands CCL3 and CCL3L1 by ATRA was mainly observed in CCR6+ T cells, indicative of a more efficient CCR5-mediated HIV entry (53). Although the T cell activation marker CD38 was similarly upregulated by ATRA in both subsets (Supplemental Figure 3), transcripts for MAP3K4, a Th17-specific kinase (54) we previously identified as a HDF (20), were expressed at superior levels and upregulated by ATRA in CCR6+ versus CCR6– T cells (Supplemental Figure 3). Moreover, the transcripts for PPARγ, a transcriptional repressor of RORγt (55) and Th17 intrinsic negative regulator of HIV replication (19), were downregulated by ATRA specifically in CCR6+ T cells (Supplemental Figure 3). Furthermore, CCR6+ versus CCR6– T cells stimulated via the TCR in the presence/absence of ATRA expressed decreased levels of IFN-stimulated genes, such as IFITM1, IFITM2, IFITM3, and IRF8 (Supplemental Tables 1–4), consistent with the low Th17 ability to respond to IFN (23). Finally, although differences in the expression of SAMHD1, a key IFN-induced HIV restriction factor (22, 56, 57), were not observed in the microarrays, in the presence/absence of ATRA, levels of SAMHD1 mRNA measured by RT-PCR were significantly lower in CCR6+ versus CCR6– T cells (Supplemental Figure 5). This stresses the existence of false negatives in high-throughput transcriptional screenings. Together, these findings reveal multiple HDFs acting at entry and postentry levels to facilitate HIV replication in ATRA-treated CCR6+ T cells as well as a state of antiviral immunity acting at different levels in CCR6– T cells.
Among probe sets differentially modulated by ATRA in CCR6+ and CCR6– T cells (P < 0.05), only 466 transcripts were shared, while 1,285 and 1,538 transcripts were unique to CCR6+ and CCR6– T cells, respectively (Figure 2D). A meta-analysis using the NCBI HIV-1 interactions database allowed the identification of human genes previously described to enhance HIV-1 protein expression that were differentially regulated by ATRA in CCR6+ versus CCR6– T cells (Supplemental Figure 6). Among those transcripts, LGALS3, ADAM15, TRIM8, IL-1α, FOXO3, and ARNTL were upregulated by ATRA in CCR6+ versus CCR6– T cells (Supplemental Figure 3 and 6).
Finally, gene set variation analysis (GSVA) identified pathways modulated by ATRA in CCR6+ T cells, including the Akt and PI3K signaling pathways (Figure 3), critical for the Th17 polarization (58–60). A custom pathway was created using the genes from BIOCARTA_AKT_PATHWAY and PID_PI3K_PLC_TRK_PATHWAY, genes that were differentially modulated by ATRA in CCR6+ T cells (Figure 4A). Of note, the mammalian target of rapamycin (mTOR) appeared upregulated by ATRA in CCR6+ T cells in this gene interaction network representation (Figure 4A). Ingenuity pathway analysis was further used to illustrate genes associated with the mTORC1 pathway and regulated by ATRA in CCR6+ T cells (Figure 4B).
Figure 3. Canonical pathways modulated by ATRA in CCR6+ T cells.

Memory CCR6+/CCR6– T cells were sorted and stimulated as described in Figure 1. Total RNA from matched T cell subsets of n = 6 different HIV-uninfected donors was used for genome-wide transcriptional profiling, as described in Figure 2. One-way ANOVA analysis identified differentially expressed genes based on P values or adjusted P values (P < 0.05) and/or fold change (FC, cutoff 1.3). The heatmap depicts the top 34 pathways upmodulated (red) and downmodulated (blue) by ATRA in CCR6+ T cells, identified using gene-set variation analysis (GSVA; P < 0.01;enrichment Z score).
Figure 4. Components of the AKT/PI3K pathways modulated by ATRA in CCR6+ T cells.

Memory CCR6+/CCR6– T cells were sorted and stimulated as described in Figure 1. Total RNA from matched T cell subsets of n = 6 different HIV-uninfected donors was used for genome-wide transcriptional profiling, as described in Figure 2. One-way ANOVA analysis identified differentially expressed genes based on P values or adjusted P values (P < 0.05) and/or fold-change (FC, cutoff 1.3). (A) Combined BIOCARTA_AKT_PATHWAY and PID_PI3K_PLC_TRK_PATHWAY canonical pathways significantly modulated by ATRA in CCR6+ T cells, identified using Ingenuity Pathway Analysis. (B) Gene expression network generated using individual genes representing REACTOME_MTORC1_MEDIATED_SIGNALING significantly modulated by ATRA in CCR6+ T cells identified through GSVA. The color code is based on expression FC (red and green for upregulated and downregulated transcripts, respectively). Nodes representing individual genes are annotated with their corresponding P values and FC (*P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001).
Together, this genome-wide transcriptional analysis reveals unique molecular signatures associated with HIV permissiveness in Th17-polarized CCR6+ T cells upon exposure to ATRA and points to a critical role played by mTOR in this process.
ATRA induces mTOR expression and phosphorylation selectively in CCR6+ T cells.
To explore the potential contribution of mTOR to HIV permissiveness in ATRA-treated CCR6+ T cells, the expression of total and phosphorylated mTOR was first investigated by Western blotting in sorted CCR6+ and CCR6– T cells stimulated via the TCR in the presence/absence of ATRA. Levels of total/phosphorylated mTOR were low to undetectable in TCR-activated CCR6+ T cells but markedly increased by ATRA treatment. In contrast, TCR-activated CCR6– T cells expressed high levels of total/phosphorylated mTOR, and ATRA did not modify this expression (Figure 5). The molecular weight of the main mTOR band was 80 kDa, which corresponds to the β isoform of mTOR typically expressed in T cells (61). To further validate these results, we performed semiquantitative RT-PCR and visualized on gel the expression of mTOR mRNA corresponding to isoform β (2,118 Kb) using established primers (61). Levels of mTOR isoform β mRNA were significantly higher in CCR6+ T cells upon exposure to ATRA (Supplemental Figure 7). The findings that mTOR mRNA expression and mTOR phosphorylation are induced by ATRA preferentially in CCR6+ T cells point to the existence of mTOR-dependent HIV permissiveness mechanisms in gut-homing CCR6+ T cells.
Figure 5. ATRA induces mTOR expression and phosphorylation selectively in CCR6+ T cells.
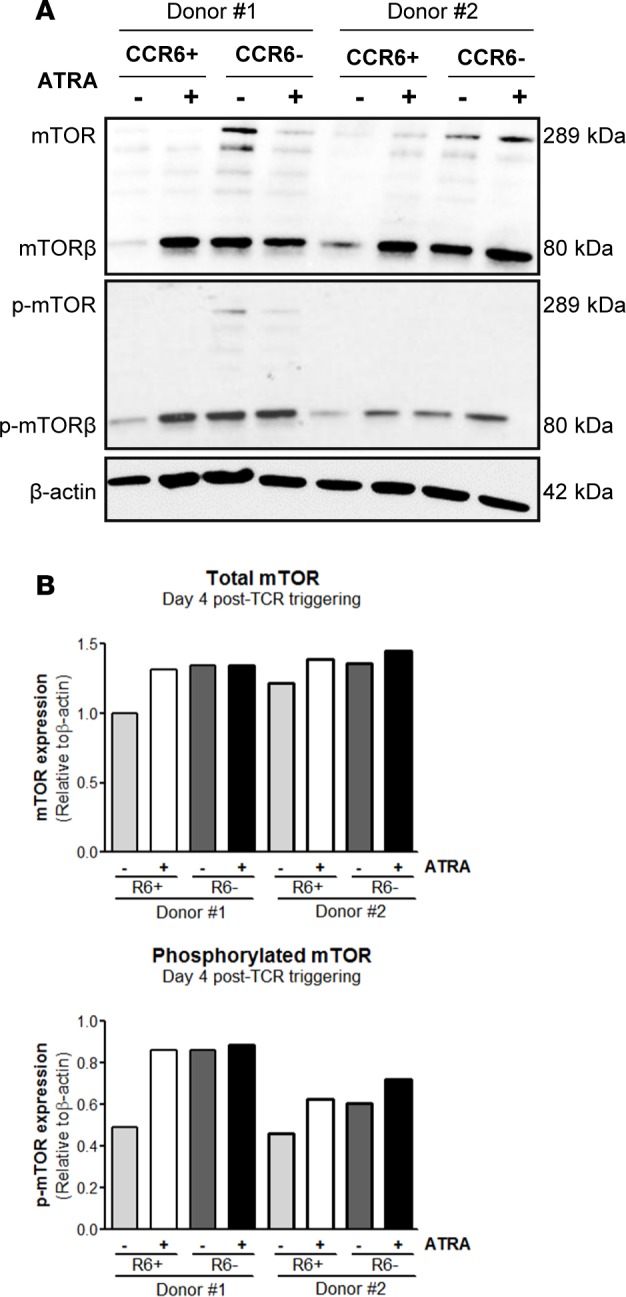
Memory CCR6+ and CCR6– T cell subsets were sorted and stimulated as described in Figure 1. (A) Cell lysates were used to visualize total and phosphorylated mTOR expression by Western blotting. (B) Levels of total and phosphorylated mTOR expression were quantified by densitometry and normalized relative to β-actin levels. Of note, the molecular weight of the predominant mTOR band was 80 kDa, corresponding to the β isoform of mTOR (1). Results obtained with cells from two different donors are shown.
mTORC1/2 inhibitors counteract the effect of ATRA on HIV replication.
To determine the functional role of ATRA-mediated mTOR activity, two commercially available mTOR inhibitors were tested for their ability to counteract the effect of ATRA (Sigma-Aldrich) on HIV permissiveness: rapamycin (Sigma-Aldrich), an inhibitor of mTORC1, and INK128 (Cayman Chemical Co.), an inhibitor of mTORC1/2 (62). Memory CD4+ T cells were stimulated via the TCR and further exposed to HIV in the presence/absence of ATRA, rapamycin, or INK128 (Figure 6A). Rapamycin and INK128 significantly reduced levels of early/late reverse transcripts and integrated HIV DNA in ATRA-treated T cells from five different donors, with INK128 having the most robust inhibitory effects, especially when added before and after infection (Figure 6, B–D). Similarly, rapamycin and, most significantly, INK128 had a robust inhibitory effect on HIV replication measured by HIV-p24 ELISA at days 3, 6, and 9 after infection (Figure 6E). Of note, the treatment with mTOR inhibitors in combination with ATRA significantly reduced the ability of cells to produce IL-17A but did not alter IFN-γ production, cell viability, and proliferation (Supplemental Figure 8, A–D). Similar experiments were further performed with FACS-sorted memory CCR6+/CCR6– T cells. INK128 significantly reduced HIV integration/replication in ATRA-treated CCR6+ T cells. Although levels of HIV integration/replication were lower in CCR6– compared with CCR6+ T cells, INK128 similarly acted on CCR6– T cells (Figure 7), consistent with the mTOR expression in these cells (Figure 5 and Supplemental Figure 7). Together, these results support the role of ATRA-induced mTOR signaling pathway as a positive regulator of HIV permissiveness in gut-homing CCR6+ T cells.
Figure 6. mTOR inhibitors counteract the effect of ATRA on HIV permissiveness in memory CD4+ T cells.

(A) The ability of mTOR inhibitors to counteract the effects of ATRA on HIV replication was tested in total memory CD4+ T cells. Briefly, total memory CD4+ T cells were stimulated via CD3/CD28 in presence/absence of ATRA and/or rapamycin (10 nM) or INK128 (50 nM) for 4 days. Then, cells were exposed to replication-competent T/F HIV THRO (25 ng HIV-p24/106 cells). Further, cells were cultured for 9 additional days in presence/absence of ATRA and/or rapamycin or INK128. (B–D) The relative RU5, Gag, and integrated HIV DNA levels quantified by real-time nested PCR at day 3 after infection (with the range of absolute HIV DNA copies/106 cells in ATRA-treated cells, considered 100%, being indicated in the figure) and (E) HIV-p24 levels quantified by ELISA in cell culture supernatants at days, 3, 6 and 9 after infection (n = 5, mean ± SEM) shown. Each symbol represents one different donor. Repeated-measures 1-way ANOVA (B–E), with Dunnett’s (relative to cells stimulated via the TCR in the presence of ATRA) (B–D) or Tukey’s (E) multiple comparisons test (*P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001).
Figure 7. mTOR inhibitors counteract the effect of ATRA on HIV permissiveness in sorted CCR6+ and CCR6– T cells.

Memory CCR6+ and CCR6– T cells were sorted as depicted in Figure 1 and stimulated via CD3/CD28 in presence of ATRA or ATRA and INK128 (50 nM) for 4 days. Then, cells were exposed to replication-competent T/F HIV THRO (25 ng HIV-p24/106 cells). Further, cells were cultured for 9 additional days in presence of ATRA or ATRA and INK128. (A) Absolute levels of integrated HIV DNA at day 3 after infection and (B) HIV-p24 levels in cell culture supernatants at day 6 after infection (n = 3, mean ± SD of triplicate wells). Each symbol represents one experimental triplicate value in individual donors. Ordinary 1-way ANOVA with Tukey’s multiple comparisons test (**P < 0.01; ***P < 0.001; ****P < 0.0001).
mTORC1/2 inhibitors limit HIV replication at the postentry level in ATRA-treated T cells.
Studies in humanized mice demonstrated the ability of rapamycin and INK128 to inhibit HIV replication via the regulation of CCR5 expression (62). To investigate this possibility, we tested the effects of rapamycin and INK128 on CCR5 expression in memory CD4+ T cells stimulated via TCR in the presence/absence of ATRA. Although rapamycin and INK128 substantially reduced CCR5 expression in the absence of ATRA, no significant changes were observed in the presence of ATRA (Figure 8, A and B). Similarly, rapamycin and INK128 had no significant effects on the expression of integrin β7 (Figure 8, C and D), a gut-homing molecule induced by ATRA (12) that was also identified as an HIV-gp120–binding molecule (13). To further distinguish between HIV entry and postentry mTOR-mediated mechanisms in the context of ATRA, we performed a single-round infection using replication-defective VSV-G–pseudotyped HIV and tested the effects of rapamycin or INK128 on HIV reverse transcription and integration. Both rapamycin and INK128 induced a partial decrease in levels of early (59% ± 3% and 65% ± 7% inhibition) and late (88% ± 2% and 90% ± 5% inhibition) HIV reverse transcripts and an almost complete abrogation of HIV DNA integration (98% ± 0.1% and 99% ± 0.1% inhibition, n = 3) (Figure 8, E–G). Together, these results demonstrate that mTORC1/2 inhibitors limit HIV replication in gut-homing CCR6+CD4+ T cells via postentry mechanisms, during reverse transcription and prior to integration, without effect on CCR5- and/or integrin β7-mediated viral entry or cell homing into the gut.
Figure 8. mTOR inhibitors limit HIV replication at postentry levels in ATRA-treated CD4+ T cells.

(A–D) Memory CD4+ T cells were first analyzed for the surface expression of CCR5 and integrin β7 at day 4 after TCR triggering in the presence/absence of ATRA (10 nM), rapamycin (10 nM), and/or INK128 (50 nM). (A) Levels of CCR5 and (C) integrin β7 expression in one representative donor, and (B) statistical analysis of CCR5 (n = 3) and (D) integrin β7 (n = 5) expression in different donors (mean ± SEM). (E–G) Then, stimulated cells were exposed to VSV-G–pseudotyped HIV (25 ng HIV-p24/106 cells) (single round infection) and cultured in presence/absence of ATRA and/or rapamycin or INK128 for 3 additional days. (E) Levels of RU5, (F) Gag, and (G) integrated HIV DNA were quantified by nested real-time PCR in cells harvested at day 3 after infection. Relative HIV DNA levels (mean ± SEM; n = 3); the range of absolute HIV DNA copies/106 cells in ATRA-treated CCR6+ T cells (considered 100%) is indicated on the figure. Each symbol represents one different donor. Repeated-measures 1-way ANOVA with Tukey’s (B–D) or Dunnett’s (relative to cells stimulated via the TCR the presence of ATRA) (E–G) multiple comparisons tests (***P < 0.001; ****P < 0.0001).
ATRA promotes HIV reactivation in CD4+ T cells of ART-treated individuals in an mTOR-dependent manner.
In a very recent study, we demonstrated that ATRA facilitates HIV reactivation from latency in CCR6+CD4+ T cells from ART-treated individuals (37). To determine whether HIV reservoir reactivation was dependent on mTOR activity, an optimized viral outgrowth assay (VOA) was performed in the presence/absence of ATRA and mTORC1/2 inhibitor INK128 (Figure 9A). Consistent with previous findings (37), ATRA significantly improved the efficacy of HIV reactivation in memory CD4+ T cells isolated from ART-treated individuals (Supplemental Table 5), as measured by intracellular HIV-p24 staining (Figure 9, B and C) and soluble HIV-p24 in cell culture supernatants at day 12 after reactivation (Figure 9D). Of particular importance, INK128 inhibited HIV reactivation mediated by TCR triggering in the presence of ATRA (Figure 9, B–D). Further, we tested the effects of INK128 on HIV reservoir reactivation in memory CCR6+ and CCR6– T cells isolated by FACS from ART-treated individuals (Supplemental Table 5). In the absence of ATRA, HIV reactivation was observed in CCR6+ but not CCR6– T cells in 1 of 3 donors (ART 3), while, in the presence of ATRA, HIV reactivation occurred in 3 of 3 donors preferentially (ART 3) or only (ART 4 and ART 6) in CCR6+ T cells. Of particular importance, INK128 dramatically reduced HIV reactivation in ATRA-treated CCR6+ T cells, as measured by intracellular HIV-p24 expression (Figure 9, E and F) and soluble HIV-p24 production in cell culture supernatants at day 12 after reactivation (data not shown). Together, these results reveal mTOR as a key player in ATRA-mediated HIV reactivation in CCR6+ T cells of ART-treated individuals.
Figure 9. Drug-mediated mTOR blockade counteracts the enhancing effects of ATRA on HIV reactivation in a viral outgrowth assay.

(A) HIV reactivation was measured in a viral outgrowth assay (VOA), as summarized in the flowchart. (B–D) Briefly, total memory CD4+ T cells (n = 5) and (E and F) FACS-sorted memory CCR6+ and CCR6– T cells (n = 3) were isolated from PBMCs of HIV+ART individuals (Supplemental Table 5; ART 1–6). Cells were activated via CD3/CD28 and cultured in the presence/absence of ATRA (10 nM) and/or INK128 (50 nM). (B and C) Intracellular HIV-p24 expression in total memory CD4+ T cells and (E and F) sorted memory CCR6+ and CCR6– T cells quantified by FACS as well as (D) HIV-p24 levels quantified by ELISA in cell culture supernatants of total memory CD4+ T cells harvested at day 12 after activation. Results are depicted in individual donors. Each point represents one experimental triplicate (mean ± SD of triplicate wells). Repeated-measures 1-way ANOVA with Dunnett’s multiple comparisons test (relative to cells stimulated via the TCR the presence of ATRA) (C and D) and ordinary 1-way ANOVA with Tukey’s multiple comparisons test (F) (****P < 0.0001).
Colon-infiltrating CCR6+CD4+ T cells express high levels of CCR5, integrin β7, and phosphorylated mTOR.
The GALT is enriched in DCs metabolizing vitamin A into ATRA (41). We further explored whether the molecular “signature” induced by ATRA in vitro, including the upregulation of CCR5, integrin β7, and mTOR phosphorylation on CCR6+ T cells, can be observed as well in the GALT. To test this possibility, we had access to matched blood and sigmoid biopsies from n = 10 HIV-infected individuals receiving ART (Supplemental Table 5; COL 1–10). Memory CD4+ T cells were identified using the gating strategy depicted in Figure 10A and further analyzed for the expression of CCR5, integrin β7, and phosphorylated mTOR in CCR6+ versus CCR6– T cells. The expression of CCR5 and integrin β7 was higher on cells from the colon versus the blood, with colon-infiltrating CCR6+ versus CCR6– T cells expressing significantly higher levels of CCR5 and integrin β7 (Figure 10, B–E). Similarly, mTOR phosphorylation was mainly observed in the colon, with colon-infiltrating CCR6+ versus CCR6– T cells expressing the highest levels of phosphorylated mTOR in all individuals tested (Figure 10, F and G). Thus, the activation of the mTOR pathway occurs almost exclusively in colon-infiltrating CCR6+ T cells and coincides with the highest levels of CCR5 and integrin β7 expression. These results provide a molecular explanation for the preferential HIV replication in colon CCR6+ T cells and point to mTOR as an antiviral drug target in these cells (Figure 11).
Figure 10. Memory CCR6+CD4+ T cells infiltrating the colons of HIV+ART individuals express high levels of CCR5, integrin β 7, and phosphorylated mTOR.

Matched blood and sigmoid biopsies were collected from HIV-infected individuals with undetectable plasma viral load (<40 HIV-RNA copies/ml) under ART (Supplemental Table 5; COL 1–10). Cells were extracted and stained with a cocktail of specific Abs. (A) The gating strategy for the flow cytometry identification of CD3+CD4+CD45RA– T cells. (B and C) The surface expression of CCR5 and (D and E) integrin β7 and (F and G) the intracellular expression of phosphorylated mTOR on CCR6+ and CCR6– subsets from sigmoid biopsies versus PBMCs in one representative donor (B, D, and F) and statistical analysis in all donors (C, E, and G). Paired t test values are indicated.
Figure 11. Mechanisms contributing to HIV replication in gut-homing CCR6+ T cells.

In this manuscript, we provide evidence that CCR6 is a marker for memory CD4+ T cells programmed to become HIV targets, especially in the intestinal environment rich in retinoic acid, due to their expression of a unique Th17-polarized transcriptional program and a mTOR-governed metabolic state. We propose the use of mTOR inhibitors together with ART to protect gut-homing Th17 cells from HIV infection/persistence.
Discussion
The role of ATRA in regulating tolerance and immunity via the modulation of regulatory and effector functions of CD4+ T cells, respectively, is well established (41). Our current results support the idea that, in the intestinal environment, HIV takes advantage of ATRA-mediated signaling pathways for replication/persistence in CCR6+CD4+ T cells. Briefly, we (a) identified mTOR as a major regulator of HIV permissiveness in CCR6+CD4+ T cells, with gut-homing tropism mimicked in vitro by exposure to ATRA; (b) demonstrated the ability of mTOR inhibitors to counteract the effects of ATRA on HIV replication in vitro and viral reservoir reactivation in T cells from ART-treated individuals; and (c) revealed that CCR6+CD4+ T cells infiltrating the colons of HIV-infected individuals on ART express a signature comprising high levels of CCR5, integrin β7, and phosphorylated mTOR, likely reflecting exposure to ATRA in vivo (Figure 11). Considering the strategic location of Th17 cells at portal sites of HIV/SIV entry (6, 24, 27, 28, 63), as well as their important role in maintaining mucosal immunity homeostasis in the context of a complex microbiota (6, 64, 65), our results point to the potential beneficial use of mTOR inhibitors in preventing HIV infection/persistence in gut-homing Th17 cells.
In this manuscript, we used Illumina bead array technology to unveil postentry HIV regulatory mechanisms modulated by ATRA in CCR6+ versus CCR6– T cells. Among genes commonly modulated by ATRA in CCR6+ and CCR6– T cells, the RIG-I is essential for sensing of viral nucleic acids and promoting antiviral immunity (66). HIV is known to subvert RIG-I–mediated antiviral mechanisms (67); this may explain robust HIV replication in RIG-I–expressing ATRA-treated CCR6+ T cells. Of particular importance, Li et al. recently demonstrated that acitretin, a RA derivative, increases HIV transcription in CD4+ T cells from ART-treated HIV-infected individuals and enhances RIG-I signaling, thus leading to an antiviral response and the apoptosis of infected cells (68). Similarly, in a previous report (37) and in this manuscript, we demonstrated that ATRA significantly increases HIV reactivation in a VOA without significant changes observed in T cell survival/proliferation. Differences between our results and those published by Li et al. (68) may be explained by differences in the experimental design. Nevertheless, these findings commonly support the fact that ATRA promotes HIV reservoir reactivation.
A meta-analysis using the NCBI HIV interaction database revealed HDFs preferentially expressed by ATRA-exposed CCR6+ T cells. These HDFs include LGALS3, promoting HIV-1 budding via association with Alix and Gag p6 (69); ADAM15, involved in the nuclear import of the HIV-1 preintegration complex (70); TRIM8, a regulator of NF-κB and STAT3-dependent signaling cascades (20, 71); and ARNTL (72), a component of the circadian clock machinery and regulator of Th17 development (49). All these HDFs contribute to robust HIV replication in gut-homing CCR6+ T cells.
GSVA of transcripts uniquely modulated by ATRA in CCR6+ T cells identified pathways, including Akt and PI3K. The network representation of Akt/PI3K pathways, previously linked to Th17 lineage polarization (58–60) and reported to be upregulated in human lamina propria T cells (73), pointed to the upregulation of mTOR, a metabolic sensor involved in the regulation of numerous cellular functions via the formation of two different signaling complexes, mTORC1 and mTORC2 (74–77). Considering the documented role of mTOR-mediated processes in the positive regulation of HIV replication (62, 78), we proceeded to the validation of mTOR expression at protein and mRNA levels as well as functional validations in cells from the blood and/or colons of HIV-infected individuals receiving ART and uninfected study participants.
The regulation of HIV permissiveness via mTOR-dependent mechanisms was demonstrated by the use of mTORC1 (rapamycin) and/or mTORC1/2 (INK128) inhibitors, which counteracted the effects of ATRA on HIV replication in CCR6+ T cells in vitro and inhibited HIV reservoir reactivation in an ATRA-based VOA performed with CCR6+ T cells from ART-treated individuals. Nevertheless, mTOR expression/phosphorylation is not always associated with HIV permissiveness. Indeed, CCR6– T cells are relatively resistant to HIV, despite high levels of mTOR expression, likely due to the existence of HIV restriction mechanisms acting at different levels, including entry (low/undetectable CCR5 expression), reverse transcription (high SAMHD1 expression), and/or transcription (limited TNF-α production and NF-κB activity). Thus, mTOR favors HIV replication/reactivation only within the unique transcriptional context of ATRA-exposed CCR6+ T cells.
Evidence exists in the literature that mTOR regulates HIV replication via multiple mechanisms (78, 79). One possible mechanism is via the regulation of CCR5 expression (62). However, CCR5 expression on ATRA-treated T cells was not affected by mTOR inhibitors. This suggests that ATRA induces CCR5 expression via mTOR-independent mechanisms and that mTOR regulation of HIV permissiveness in gut-homing CD4+ T cells likely occurs at postentry levels. Consistent with the evidence that mTOR regulates nucleotide biosynthesis essential for transcription (76, 80, 81), mTOR inhibitors may limit HIV reverse transcription by reducing the pool of nucleotides. Indeed, our single-round infection using VSV-G–pseudotyped HIV revealed restriction mechanisms acting during reverse transcription and prior to integration. Moreover, mTOR is a well-established regulator of autophagy (79, 82), a process that may be involved in the degradation of the incoming HIV upon fusion/uncoating (83, 84). By promoting the mTOR activity, ATRA may impair the autophagy process in CCR5+CCR6+ T cells at mucosal sites. Furthermore, considering the fact that ATRA-treated CCR6+ T cells maintain their Th17 features (42) and that mTORC1, via the induction of the kinase S6K2, is involved in the nuclear translocation of RORC (60, 85), one other possibility is that mTOR inhibitors interfere with the RORC-mediated transcriptional program in Th17 cells, which is favorable to HIV replication (20). Consistent with this hypothesis, we demonstrated that decreased HIV replication in the presence of rapamycin and INK128 coincided with a significant reduction in IL-17A production by ATRA-treated CD4+ T cells.
Of particular importance, we found that CCR6+ cells but not CCR6– T cells infiltrating the colons of HIV-infected individuals receiving ART express a unique molecular signature, including superior levels of CCR5, integrin β7, and phosphorylated mTOR expression, likely as a consequence of ATRA exposure in the intestinal environment (12, 41). In line with our recent report that HIV DNA persists in colon-infiltrating CCR6+ T cells during ART (37), it remains to be determined whether mTOR expression identifies a fraction of CCR6+ T cells enriched in HIV DNA and whether mTOR activation contributes to residual HIV replication in colon-infiltrating CCR6+ T cells during ART.
While this manuscript was in preparation for submission, Besnard et al. reported the results of a shRNA screen that revealed mTOR as a regulator of HIV latency (86) via mechanisms involving CDK9 and NF-κB activation that controls Tat-dependent HIV transcription (86). In addition, Martin et al. reported on the potential beneficial use of rapamycin in the context of HIV “shock-and-kill” strategies (87). These two reports (86, 87) further support the conclusions of our current study, which reveals the key role played by mTOR in regulating multiple postentry and postintegration HIV replication steps in gut-homing Th17-polarized CCR6+ T cells.
In conclusion, our findings point to CCR6 as a “zip code” molecule expressed on the surface of CD4+ T cells transcriptionally programmed to become HIV targets upon recruitment into the intestine and provide a detailed molecular explanation for preferential HIV/SIV replication and persistence in gut-homing CCR6+CD4+ T cells (24, 25, 37, 42). Most significantly, we reveal the role of the mTOR pathway in regulating effector functions as well as HIV permissiveness in gut-homing CCR6+ Th17 cells at multiple postentry levels. Additionally, we demonstrate the ability of mTOR inhibitors to counteract the effects of ATRA on HIV replication/reactivation in CCR6+ T cells. Our findings raise questions about the potential beneficial effects of mTOR inhibitors in decreasing HIV reservoirs and restoring the Th17-mediated immunity at the intestinal level during ART. This knowledge will open the way for further studies on the implementation of new therapeutic strategies aimed at HIV functional cure.
Methods
Study subjects.
Study subjects were recruited at the Montreal Chest Institute, McGill University Health Centre, and Centre Hospitalier de l’Université de Montréal–Hôpital Saint-Luc (Montreal, Québec, Canada). PBMCs were collected by leukapheresis (88) and cryopreserved until use. Sigmoid biopsies (≈32 biopsies/donor) were collected from HIV-infected individuals receiving ART (Supplemental Table 5) during colonoscopy and processed using Liberase DL (Roche Diagnostics), as previously described (37, 89, 90). Matched peripheral blood (20 ml/donor) was collected the same day from biopsy donors.
Flow cytometry staining.
Surface staining was performed with fluorochrome-conjugated Abs (Supplemental Table 6), as previously described (16). Cells extracted from sigmoid biopsies and matched PBMCs were stained on the surface with a cocktail of CD3-Alexa700, CD4-PE-Cy7, CD45RA-APC-eF780, CCR5-FITC, integrin β7-FITC, and CCR6-PE Abs as well as non–T cell lineage Abs: CD326-BV650, CD8-PerCP-Cy5.5, CD19-PerCP-Cy5.5, and CD66b-PerCP-Cy5.5 (37). Intracellular staining was subsequently performed with phosphorylated mTOR (S2448) eFluor-450 Abs (eBiosciences). The viability dye LIVE/DEAD Fixable Aqua Dead Cell Stain Kit (Invitrogen) was used to exclude dead cells. Cells were analyzed using a LSRII cytometer (Diva, version 6, BD Biosciences), and FlowJo (version 10.0.6, Tree Star Inc.). Flow cytometry gates were defined using the fluorescence minus one strategy (16, 91).
Magnetic-activated cell sorting and FACS.
Total or memory CD4+ T cells were enriched from PBMCs by negative selection using magnetic beads (magnetic-activated cell sorting [MACS], Miltenyi), with a purity of >95%, as previously described (42). Cells were then stained with CD45RA-APC-Cy7 and CCR6-PE Abs and a cocktail of FITC-conjugated Abs to exclude CD8+ T (CD8), NK (CD56), and B cells (CD19). The sorting gates were set on FITCneg memory (CD45RAneg) CCR6+/CCR6– T cells that were subsequently sorted by flow cytometry (BDAria III), with dead cells being excluded. Postsort quality control analysis demonstrated >97% CCR6+/CCR6– T cell purity, as previously reported (42).
HIV-1 infection in vitro.
The following HIV-1 molecular clones were used in this study: (a) replication-competent CCR5 using (R5) NL4.3BAL; (b) single-round VSVG-HIV-GFP, an env-deficient NL4.3 provirus pseudotyped with the VSV-G envelope and encoding for gfp in place of nef (42, 92); and (c) replication-competent T/F THRO. The full-length T/F THRO HIV-1 Infectious Molecular Clone (catalog 11919) was obtained from John Kappes through the NIH AIDS Reagent Program, Division of AIDS, National Institute of Allergy and Infectious Diseases, NIH (44). HIV stocks were produced, titrated, and used to infect cells (25–50 ng HIV-p24/106 cells), as previously described (16, 19, 20, 36, 42).
Quantification of integrated and Gag HIV DNA.
Levels of integrated and Gag HIV DNA were quantified in cell lysates by ultrasensitive nested real-time PCR (105 cells/test in triplicates; detection limit, 3 HIV DNA copies), as previously described (16, 19, 20, 36, 37, 42, 93).
Quantification of early HIV reverse transcripts.
Early HIV reverse transcripts (RU5) were quantified using a SYBR Green master mix (Qiagen). The external primers were forward AA55 5′-CGTCTAGAGATTTTCCACAC-3′ and reverse M667 5′-CTAACTAGGGAACCCACTG-3′. The internal primers were forward SK29 5′-ACTAGGGAACCCACTGCT-3′ and reverse SK30 5′-GGTCTGAGGGATCTCTAG-3′ (94). The HIV and CD3 DNA were amplified together in the first PCR and separately during the second PCR (LightCycler; Roche). The first PCR products (diluted 1:10) were used as template for the second PCR: 5 minutes at 94°C; 35 cycles of 1 minute at 94°C, 1 minute at 50°C, and 30 seconds at 72°C; and final elongation of 15 minutes at 72°C. The ACH2 cells carrying one copy of integrated HIV DNA per cell (NIH AIDS Reagent Program) were used as a standard curve, as previously described (16, 19, 20, 36, 37, 42, 93).
Genome-wide transcriptional profiling.
Genome-wide transcriptional profiling was performed in collaboration with Génome Québec (Montreal, Québec, Canada) using the Illumina bead array technology, as previously described (20). Briefly, total RNA was isolated using the RNeasy Kit (Qiagen) and quantified using the Pearl nanophotometer (Implen). The RNA quality was determined using the Agilent 2100 Bioanalyzer chip. High-quality RNA was reverse transcribed and hybridized on the Illumina HumanHT-12 v4 Expression BeadChip, providing coverage for more than 47,000 transcripts and known spliced variants across the human transcriptome. The expression of differentially expressed genes was analyzed as previously described (19, 20, 36). The entire microarray data set and the technical information requested by Minimum Information About a Microarray Experiment (MIAME) are available at the Gene Expression Omnibus database under accession GSE93660. Differentially expressed genes (cutoff 1.3-fold; P < 0.05) were classified through gene ontology using the NetAffx web-based application (Affymetrix), while differentially expressed pathways were identified using Ingenuity Pathway Analysis and GSVA. Corresponding heatmaps for biological function categories were generated using programming language R (19, 20, 36).
Real-time RT-PCR.
One-step SYBR Green real-time RT-PCR (Qiagen) was carried out in a LightCycler 480 II (Roche) according to the manufacturer’s recommendations, as we previously reported (16, 19, 20, 36, 42). Briefly, a standard curve was prepared with cDNA obtained upon reverse transcription of total RNA using specific QuantiTect Primers (Qiagen). The expression of each gene was normalized to the internal control 28S rRNA (forward 5′-CGAGATTCCTGTCCCCACTA-3′; reverse 5′-GGGGCCACCTCCTTATTCTA-3, Integrated DNA Technologies). Melting curve analysis performed after real-time amplification revealed the uniformity of thermal dissociation profile for each amplification product. Samples without template or without reverse transcriptase were used as negative controls. Each RT-PCR reaction was performed in triplicate.
mTOR protein and mRNA quantification.
Cells were lysed with RIPA buffer 1× (Cell Signaling) containing phosphatase inhibitors (PhosSTOP) and protease inhibitors (Complete Mini EDTA-free, Roche) for 10 minutes at 4°C and centrifuged at 14,000 g for 10 minutes. Proteins were quantified in supernatants by Bradford assay (Bio-Rad). Samples (15 μg protein/well) were loaded onto 7% acrylamide gels, and SDS-PAGE migration was performed (90 minutes, 100 V). Proteins were transferred to PVDF membranes and further blotted with phosphorylated (D9C2) or total mTOR (7C10) Abs (Cell signaling). Proteins were revealed with ECL prime Western blotting detection reagent (Amersham, GE Healthcare). In parallel, the expression of the mTOR mRNA was measured by semiquantitative RT-PCR performed using the Superscript III one-step RT-PCR system (Invitrogen) and specific primers (forward: S1, ATGCTTGGAACCGGACCTGCCG; reverse: AS1, TACCAGAAAGGGCACCAGCCAAT) (Integrated DNA Technologies) that could amplify both mTOR isoforms α (7.647Kb Kb) and β (2.118 Kb) (61) (Supplemental Figure 7A). However, the RT-PCR conditions were set for the optimal amplification of the short β mTOR isoform as follows: reverse transcription (30 minutes at 55°C); 40 cycles of PCR amplification (denaturation, 15 seconds at 94°C; annealing, 30 seconds at 63°C; elongation, 130 seconds at 68°C); and final elongation (15 minutes at 68°C). mTOR expression was normalized relative to 28S rRNA. Total RNA was extracted using the RNeasy Kit (Qiagen) and used for RT-PCR amplification of mTOR (250 ng/test) and 28S rRNA (5 ng/test).
Nuclear NF-κB quantification.
The NF-κB DNA-binding activity was quantified using an activity-based ELISA kit (Assay Designs & Stressgen) according to the manufacturer’s protocol, as described previously (20). Briefly, nuclear extracts were obtained using the BD transfactor extraction kit (Clonetech Laboratories). Nuclear protein content was quantified by Nanodrop (Thermo Scientific). The active form of NF-κB-p65 was quantified in nuclear extracts (1 μg protein/test). The assay used streptavidin-coated plates with bound NF-κB biotinylated-consensus sequence to capture only the active form of NF-κB. The captured NF-κB was then incubated with specific NF-κB-p65 Abs further detected using HRP-conjugated Abs. The assay was developed with a chemiluminescent substrate and the signal was detected using a luminometer. Results were expressed as light units. The specificity of the binding was demonstrated by the use of a wild-type and mutated NF-κB Competitor Duplex included in the kit.
ELISA quantification.
HIV-p24 levels were quantified in cell culture supernatants using a homemade ELISA, as described previously (16, 19, 20, 36, 37, 42). Cytokine levels in cell culture supernatants were quantified by ELISA assays specific for TNF-α and CCL20 (R&D Systems) and IL-17, IFN-γ, and IL-2 (eBiosciences) according to the manufacturers’ protocols.
VOA.
The VOA was designed to allow optimal HIV reservoir reactivation from small numbers of primary CD4+ T cells (36, 37). Briefly, memory CD4+ T cells (2 × 106 cells/ml/well) or FACS-sorted memory CCR6+/CCR6– T cells (106 cells/ml/well) were cultured in 48-well plates in the presence of immobilized CD3 and soluble CD28 Abs (1 μg/ml) for 3 days. Cells were further cultured in the presence of IL-2 (5 ng/ml) up to day 12. Every 3 days, cells were split and media were refreshed. The VOA was performed in the presence/absence of ATRA (10 nM) and/or INK128 (50 nM). HIV-p24 levels were quantified by ELISA in cell culture supernatants, while intracellular HIV-p24 expression was quantified by flow cytometry at day 12 after reactivation.
Statistics.
Statistical analyses were performed using GraphPad Prism 6. Repeated-measures 1-way ANOVA or ordinary ANOVA with Tukey’s or Dunnett’s post-test were used for multiple comparisons. Two-tailed paired t test was used for matched pairwise analyses. A P value of less than 0.05 was considered significant. For details, see figure legends.
Study approval.
This study used biological samples (blood and sigmoid biopsies) from HIV-infected and uninfected participants and was conducted in compliance with the principles included in the Declaration of Helsinki. This study received approval from the Institutional Review Boards of the McGill University Health Centre and the Centre Hospitalier de l’Université de Montréal Research Centre, Montreal, Québec, Canada. All human participants provided written informed consent for their participation in the study and agreed with the publication of the scientific results.
Author contributions
DP performed research, analyzed data, and wrote the manuscript. YZ and PM performed research and contributed to manuscript writing. JPG performed microarray data analysis and generated figures. AG performed research. AF contributed to research design and manuscript writing. NG and TJH provided protocols and reagents and contributed to research design. JPR provided access to clinical samples/information, set up clinical research protocols, and contributed to manuscript writing. PA designed research, analyzed data, and wrote the manuscript. All authors reviewed and accepted the manuscript.
Supplementary Material
Acknowledgments
The authors acknowledge the contribution of Dominique Gauchat (Flow Cytometry Core Facility, CHUM-Research Centre, Montreal, Québec, Canada), with expert technical support with flow cytometry analysis and sorting; Mario Legault for help with ethical approvals and informed consents; and Josée Girouard and Angie Massicotte for their critical contribution to sigmoid biopsy, blood, and leukapheresis collection from HIV-infected and uninfected donors. The authors thank Rosalie Ponte, Franck Dupuy, and Vikram Meraj for their contribution to colon biopsies access and processing and Andrew Mouland for the critical reading of the manuscript. The authors also thank Natalia Zamorano Cuervo and Sandra L. Cervantes-Ortiz for their technical assistance with Western blotting. Finally, the authors acknowledge study participants for their gift of biological samples essential for this study. This study was supported by grants from the Canadian Institutes of Health Research (CIHR) (MOP-82849; MOP-114957 to PA), the CIHR Canadian HIV Trials Network (CTN 247 to JPR), the Fonds de Recherche du Québec-Santé/AIDS and Infectious Diseases Network, Québec, Canada (to PA and JPR), and Canadian HIV Cure Enterprise Team Grant HIG-133050 from the CIHR, in partnership with the Canadian Foundation for AIDS Research and International AIDS Society (to PA and JPR). JPR holds a Louis Lowenstein Chair in Hematology and Oncology, McGill University. The funding institutions played no role in the design, collection, analysis, and interpretation of data.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information: JCI Insight. 2017;2(15):e93230. https://doi.org/10.1172/jci.insight.93230.
Contributor Information
Delphine Planas, Email: delphine.planas@gmail.com.
Yuwei Zhang, Email: zhangyw927@gmail.com.
Patricia Monteiro, Email: patriciamonteiro2003@yahoo.fr.
Jean-Philippe Goulet, Email: gouletjp@me.com.
Annie Gosselin, Email: annie.gosselin.chum@gmail.com.
Nathalie Grandvaux, Email: nathalie.grandvaux@umontreal.ca.
Ariberto Fassati, Email: a.fassati@ucl.ac.uk.
Jean-Pierre Routy, Email: jean-pierre.routy@mcgill.ca.
Petronela Ancuta, Email: petronela.ancuta@umontreal.ca.
References
- 1.Deeks SG, et al. International AIDS Society global scientific strategy: towards an HIV cure 2016. Nat Med. 2016;22(8):839–850. doi: 10.1038/nm.4108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Martin AR, Siliciano RF. Progress toward HIV eradication: case reports, current efforts, and the challenges associated with cure. Annu Rev Med. 2016;67:215–228. doi: 10.1146/annurev-med-011514-023043. [DOI] [PubMed] [Google Scholar]
- 3.Lederman MM, et al. A cure for HIV infection: “not in my lifetime” or “just around the corner”? Pathog Immun. 2016;1(1):154–164. doi: 10.20411/pai.v1i1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mehandru S, et al. Primary HIV-1 infection is associated with preferential depletion of CD4+ T lymphocytes from effector sites in the gastrointestinal tract. J Exp Med. 2004;200(6):761–770. doi: 10.1084/jem.20041196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brenchley JM, et al. CD4+ T cell depletion during all stages of HIV disease occurs predominantly in the gastrointestinal tract. J Exp Med. 2004;200(6):749–759. doi: 10.1084/jem.20040874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brenchley JM, Douek DC. Microbial translocation across the GI tract. Annu Rev Immunol. 2012;30:149–173. doi: 10.1146/annurev-immunol-020711-075001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schuetz A, et al. Initiation of ART during early acute HIV infection preserves mucosal Th17 function and reverses HIV-related immune activation. PLoS Pathog. 2014;10(12):e1004543. doi: 10.1371/journal.ppat.1004543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wong JK, Yukl SA. Tissue reservoirs of HIV. Curr Opin HIV AIDS. 2016;11(4):362–370. doi: 10.1097/COH.0000000000000293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu R, et al. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell. 1996;86(3):367–377. doi: 10.1016/S0092-8674(00)80110-5. [DOI] [PubMed] [Google Scholar]
- 10.Wilkin TJ, Gulick RM. CCR5 antagonism in HIV infection: current concepts and future opportunities. Annu Rev Med. 2012;63:81–93. doi: 10.1146/annurev-med-052010-145454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jessen H, Allen TM, Streeck H. How a single patient influenced HIV research--15-year follow-up. N Engl J Med. 2014;370(7):682–683. doi: 10.1056/NEJMc1308413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mora JR, von Andrian UH. Retinoic acid: an educational “vitamin elixir” for gut-seeking T cells. Immunity. 2004;21(4):458–460. doi: 10.1016/j.immuni.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 13.Arthos J, et al. HIV-1 envelope protein binds to and signals through integrin alpha4beta7, the gut mucosal homing receptor for peripheral T cells. Nat Immunol. 2008;9(3):301–309. doi: 10.1038/ni1566. [DOI] [PubMed] [Google Scholar]
- 14.Byrareddy SN, et al. Sustained virologic control in SIV+ macaques after antiretroviral and α4β7 antibody therapy. Science. 2016;354(6309):197–202. doi: 10.1126/science.aag1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gaffen SL, Jain R, Garg AV, Cua DJ. The IL-23-IL-17 immune axis: from mechanisms to therapeutic testing. Nat Rev Immunol. 2014;14(9):585–600. doi: 10.1038/nri3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gosselin A, et al. Peripheral blood CCR4+CCR6+ and CXCR3+CCR6+CD4+ T cells are highly permissive to HIV-1 infection. J Immunol. 2010;184(3):1604–1616. doi: 10.4049/jimmunol.0903058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.El Hed A, et al. Susceptibility of human Th17 cells to human immunodeficiency virus and their perturbation during infection. J Infect Dis. 2010;201(6):843–854. doi: 10.1086/651021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alvarez Y, et al. Preferential HIV infection of CCR6+ Th17 cells is associated with higher levels of virus receptor expression and lack of CCR5 ligands. J Virol. 2013;87(19):10843–10854. doi: 10.1128/JVI.01838-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bernier A, et al. Transcriptional profiling reveals molecular signatures associated with HIV permissiveness in Th1Th17 cells and identifies peroxisome proliferator-activated receptor gamma as an intrinsic negative regulator of viral replication. Retrovirology. 2013;10:160. doi: 10.1186/1742-4690-10-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cleret-Buhot A, et al. Identification of novel HIV-1 dependency factors in primary CCR4(+)CCR6(+)Th17 cells via a genome-wide transcriptional approach. Retrovirology. 2015;12:102. doi: 10.1186/s12977-015-0226-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Christensen-Quick A, Lafferty M, Sun L, Marchionni L, DeVico A, Garzino-Demo A. Human Th17 cells lack HIV-inhibitory RNases and are highly permissive to productive HIV infection. J Virol. 2016;90(17):7833–7847. doi: 10.1128/JVI.02869-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ruffin N, et al. Low SAMHD1 expression following T-cell activation and proliferation renders CD4+ T cells susceptible to HIV-1. AIDS. 2015;29(5):519–530. doi: 10.1097/QAD.0000000000000594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Touzot M, et al. Combinatorial flexibility of cytokine function during human T helper cell differentiation. Nat Commun. 2014;5:3987. doi: 10.1038/ncomms4987. [DOI] [PubMed] [Google Scholar]
- 24.Stieh DJ, et al. Th17 cells are preferentially infected very early after vaginal transmission of SIV in macaques. Cell Host Microbe. 2016;19(4):529–540. doi: 10.1016/j.chom.2016.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McGary CS, et al. The loss of CCR6(+) and CD161(+) CD4(+) T-cell homeostasis contributes to disease progression in SIV-infected rhesus macaques. Mucosal Immunol. 2017;10(4):1082–1096. doi: 10.1038/mi.2016.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Macal M, et al. Effective CD4+ T-cell restoration in gut-associated lymphoid tissue of HIV-infected patients is associated with enhanced Th17 cells and polyfunctional HIV-specific T-cell responses. Mucosal Immunol. 2008;1(6):475–488. doi: 10.1038/mi.2008.35. [DOI] [PubMed] [Google Scholar]
- 27.McKinnon LR, Kaul R. Quality and quantity: mucosal CD4+ T cells and HIV susceptibility. Curr Opin HIV AIDS. 2012;7(2):195–202. doi: 10.1097/COH.0b013e3283504941. [DOI] [PubMed] [Google Scholar]
- 28.Loiseau C, et al. CCR6(-) regulatory T cells blunt the restoration of gut Th17 cells along the CCR6-CCL20 axis in treated HIV-1-infected individuals. Mucosal Immunol. 2016;9(5):1137–1150. doi: 10.1038/mi.2016.7. [DOI] [PubMed] [Google Scholar]
- 29.Kim CJ, et al. Impact of intensified antiretroviral therapy during early HIV infection on gut immunology and inflammatory blood biomarkers. AIDS. 2017;31(11):1529–1534. doi: 10.1097/QAD.0000000000001515. [DOI] [PubMed] [Google Scholar]
- 30.Ortiz AM, et al. IL-21 and probiotic therapy improve Th17 frequencies, microbial translocation, and microbiome in ARV-treated, SIV-infected macaques. Mucosal Immunol. 2016;9(2):458–467. doi: 10.1038/mi.2015.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Micci L, et al. Interleukin-21 combined with ART reduces inflammation and viral reservoir in SIV-infected macaques. J Clin Invest. 2015;125(12):4497–4513. doi: 10.1172/JCI81400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.d’Ettorre G, et al. Probiotic supplementation promotes a reduction in T-cell activation, an increase in Th17 frequencies, a recovery of intestinal epithelium integrity mitochondrial morphology in ART-treated HIV-1-positive patients. Immun Inflamm Dis. doi: 10.1002/iid3. [published online ahead of print April 20, 2017]. https://doi.org/10.1002/iid3.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ancuta P, et al. Microbial translocation is associated with increased monocyte activation and dementia in AIDS patients. PLoS One. 2008;3(6):e2516. doi: 10.1371/journal.pone.0002516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sun H, et al. Th1/17 polarization of CD4 T cells supports HIV-1 persistence during antiretroviral therapy. J Virol. 2015;89(22):11284–11293. doi: 10.1128/JVI.01595-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Khoury G, et al. Persistence of integrated HIV DNA in CXCR3 + CCR6 + memory CD4+ T cells in HIV-infected individuals on antiretroviral therapy. AIDS. 2016;30(10):1511–1520. doi: 10.1097/QAD.0000000000001029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wacleche VS, et al. New insights into the heterogeneity of Th17 subsets contributing to HIV-1 persistence during antiretroviral therapy. Retrovirology. 2016;13(1):59. doi: 10.1186/s12977-016-0293-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gosselin A, et al. HIV persists in CCR6+CD4+ T cells from colon and blood during antiretroviral therapy. AIDS. 2017;31(1):35–48. doi: 10.1097/QAD.0000000000001309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang C, Kang SG, Lee J, Sun Z, Kim CH. The roles of CCR6 in migration of Th17 cells and regulation of effector T-cell balance in the gut. Mucosal Immunol. 2009;2(2):173–183. doi: 10.1038/mi.2008.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mavigner M, et al. Altered CD4+ T cell homing to the gut impairs mucosal immune reconstitution in treated HIV-infected individuals. J Clin Invest. 2012;122(1):62–69. doi: 10.1172/JCI59011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang C, Kang SG, HogenEsch H, Love PE, Kim CH. Retinoic acid determines the precise tissue tropism of inflammatory Th17 cells in the intestine. J Immunol. 2010;184(10):5519–5526. doi: 10.4049/jimmunol.0903942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hall JA, Grainger JR, Spencer SP, Belkaid Y. The role of retinoic acid in tolerance and immunity. Immunity. 2011;35(1):13–22. doi: 10.1016/j.immuni.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Monteiro P, et al. Memory CCR6+CD4+ T cells are preferential targets for productive HIV type 1 infection regardless of their expression of integrin β7. J Immunol. 2011;186(8):4618–4630. doi: 10.4049/jimmunol.1004151. [DOI] [PubMed] [Google Scholar]
- 43.Parrish NF, et al. Phenotypic properties of transmitted founder HIV-1. Proc Natl Acad Sci USA. 2013;110(17):6626–6633. doi: 10.1073/pnas.1304288110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ochsenbauer C, et al. Generation of transmitted/founder HIV-1 infectious molecular clones and characterization of their replication capacity in CD4 T lymphocytes and monocyte-derived macrophages. J Virol. 2012;86(5):2715–2728. doi: 10.1128/JVI.06157-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.He YW, Deftos ML, Ojala EW, Bevan MJ. RORgamma t, a novel isoform of an orphan receptor, negatively regulates Fas ligand expression and IL-2 production in T cells. Immunity. 1998;9(6):797–806. doi: 10.1016/S1074-7613(00)80645-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Goff SP. Knockdown screens to knockout HIV-1. Cell. 2008;135(3):417–420. doi: 10.1016/j.cell.2008.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kimura K, et al. Inhibition by all-trans retinoic acid of collagen degradation mediated by corneal fibroblasts. Clin Experiment Ophthalmol. 2016;44(6):502–508. doi: 10.1111/ceo.12709. [DOI] [PubMed] [Google Scholar]
- 48.Balmer JE, Blomhoff R. Gene expression regulation by retinoic acid. J Lipid Res. 2002;43(11):1773–1808. doi: 10.1194/jlr.R100015-JLR200. [DOI] [PubMed] [Google Scholar]
- 49.Yu X, et al. TH17 cell differentiation is regulated by the circadian clock. Science. 2013;342(6159):727–730. doi: 10.1126/science.1243884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Unutmaz D, Xiang W, Sunshine MJ, Campbell J, Butcher E, Littman DR. The primate lentiviral receptor Bonzo/STRL33 is coordinately regulated with CCR5 and its expression pattern is conserved between human and mouse. J Immunol. 2000;165(6):3284–3292. doi: 10.4049/jimmunol.165.6.3284. [DOI] [PubMed] [Google Scholar]
- 51.Limou S, et al. Multiple-cohort genetic association study reveals CXCR6 as a new chemokine receptor involved in long-term nonprogression to AIDS. J Infect Dis. 2010;202(6):908–915. doi: 10.1086/655782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Richardson MW, Jadlowsky J, Didigu CA, Doms RW, Riley JL. Kruppel-like factor 2 modulates CCR5 expression and susceptibility to HIV-1 infection. J Immunol. 2012;189(8):3815–3821. doi: 10.4049/jimmunol.1201431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Casazza JP, et al. Autocrine production of beta-chemokines protects CMV-Specific CD4 T cells from HIV infection. PLoS Pathog. 2009;5(10):e1000646. doi: 10.1371/journal.ppat.1000646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ramesh R, et al. Pro-inflammatory human Th17 cells selectively express P-glycoprotein and are refractory to glucocorticoids. J Exp Med. 2014;211(1):89–104. doi: 10.1084/jem.20130301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Klotz L, et al. The nuclear receptor PPAR gamma selectively inhibits Th17 differentiation in a T cell-intrinsic fashion and suppresses CNS autoimmunity. J Exp Med. 2009;206(10):2079–2089. doi: 10.1084/jem.20082771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lahouassa H, et al. SAMHD1 restricts the replication of human immunodeficiency virus type 1 by depleting the intracellular pool of deoxynucleoside triphosphates. Nat Immunol. 2012;13(3):223–228. doi: 10.1038/ni.2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ryoo J, et al. The ribonuclease activity of SAMHD1 is required for HIV-1 restriction. Nat Med. 2014;20(8):936–941. doi: 10.1038/nm.3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nagai S, Kurebayashi Y, Koyasu S. Role of PI3K/Akt and mTOR complexes in Th17 cell differentiation. Ann N Y Acad Sci. 2013;1280:30–34. doi: 10.1111/nyas.12059. [DOI] [PubMed] [Google Scholar]
- 59.Wan Q, et al. Cytokine signals through PI-3 kinase pathway modulate Th17 cytokine production by CCR6+ human memory T cells. J Exp Med. 2011;208(9):1875–1887. doi: 10.1084/jem.20102516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kurebayashi Y, et al. PI3K-Akt-mTORC1-S6K1/2 axis controls Th17 differentiation by regulating Gfi1 expression and nuclear translocation of RORγ. Cell Rep. 2012;1(4):360–373. doi: 10.1016/j.celrep.2012.02.007. [DOI] [PubMed] [Google Scholar]
- 61.Panasyuk G, et al. mTORbeta splicing isoform promotes cell proliferation and tumorigenesis. J Biol Chem. 2009;284(45):30807–30814. doi: 10.1074/jbc.M109.056085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Heredia A, et al. Targeting of mTOR catalytic site inhibits multiple steps of the HIV-1 lifecycle and suppresses HIV-1 viremia in humanized mice. Proc Natl Acad Sci USA. 2015;112(30):9412–9417. doi: 10.1073/pnas.1511144112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brandtzaeg P. Pillars article: mucosal and glandular distribution of immunoglobulin components: differential localization of free and bound SC in secretory epithelial cells. J Immunol. 1974;112:1553–1559. [PubMed] [Google Scholar]
- 64.Bolduc JF, Ouellet M, Hany L, Tremblay MJ. Toll-like receptor 2 ligation enhances HIV-1 replication in activated CCR6+ CD4+ T cells by increasing virus entry and establishing a more permissive environment to infection. J Virol. 2017;91(4):e01402–16. doi: 10.1128/JVI.01402-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dillon SM, et al. Enhancement of HIV-1 infection and intestinal CD4+ T cell depletion ex vivo by gut microbes altered during chronic HIV-1 infection. Retrovirology. 2016;13:5. doi: 10.1186/s12977-016-0237-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liu Y, Olagnier D, Lin R. Host and viral modulation of RIG-I-mediated antiviral immunity. Front Immunol. 2016;7:662. doi: 10.3389/fimmu.2016.00662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Solis M, et al. RIG-I-mediated antiviral signaling is inhibited in HIV-1 infection by a protease-mediated sequestration of RIG-I. J Virol. 2011;85(3):1224–1236. doi: 10.1128/JVI.01635-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li P, et al. Stimulating the RIG-I pathway to kill cells in the latent HIV reservoir following viral reactivation. Nat Med. 2016;22(7):807–811. doi: 10.1038/nm.4124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang SF, et al. Galectin-3 promotes HIV-1 budding via association with Alix and Gag p6. Glycobiology. 2014;24(11):1022–1035. doi: 10.1093/glycob/cwu064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Endsley MA, et al. Nuclear trafficking of the HIV-1 pre-integration complex depends on the ADAM10 intracellular domain. Virology. 2014;454–455:60–66. doi: 10.1016/j.virol.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.König R, et al. Global analysis of host-pathogen interactions that regulate early-stage HIV-1 replication. Cell. 2008;135(1):49–60. doi: 10.1016/j.cell.2008.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yeung ML, Houzet L, Yedavalli VS, Jeang KT. A genome-wide short hairpin RNA screening of jurkat T-cells for human proteins contributing to productive HIV-1 replication. J Biol Chem. 2009;284(29):19463–19473. doi: 10.1074/jbc.M109.010033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Braunstein J, et al. Up-regulation of the phosphoinositide 3-kinase pathway in human lamina propria T lymphocytes. Clin Exp Immunol. 2008;151(3):496–504. doi: 10.1111/j.1365-2249.2007.03562.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Delgoffe GM, et al. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat Immunol. 2011;12(4):295–303. doi: 10.1038/ni.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Maciolek JA, Pasternak JA, Wilson HL. Metabolism of activated T lymphocytes. Curr Opin Immunol. 2014;27:60–74. doi: 10.1016/j.coi.2014.01.006. [DOI] [PubMed] [Google Scholar]
- 76.Inoki K, Kim J, Guan KL. AMPK and mTOR in cellular energy homeostasis and drug targets. Annu Rev Pharmacol Toxicol. 2012;52:381–400. doi: 10.1146/annurev-pharmtox-010611-134537. [DOI] [PubMed] [Google Scholar]
- 77.Pollizzi KN, Powell JD. Regulation of T cells by mTOR: the known knowns and the known unknowns. Trends Immunol. 2015;36(1):13–20. doi: 10.1016/j.it.2014.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nicoletti F, Fagone P, Meroni P, McCubrey J, Bendtzen K. mTOR as a multifunctional therapeutic target in HIV infection. Drug Discov Today. 2011;16(15–16):715–721. doi: 10.1016/j.drudis.2011.05.008. [DOI] [PubMed] [Google Scholar]
- 79.Le Sage V, Cinti A, Amorim R, Mouland AJ. Adapting the stress response: viral subversion of the mTOR signaling pathway. Viruses. 2016;8(6):E152. doi: 10.3390/v8060152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Robitaille AM, et al. Quantitative phosphoproteomics reveal mTORC1 activates de novo pyrimidine synthesis. Science. 2013;339(6125):1320–1323. doi: 10.1126/science.1228771. [DOI] [PubMed] [Google Scholar]
- 81.Chen W, et al. Reciprocal regulation of autophagy and dNTP pools in human cancer cells. Autophagy. 2014;10(7):1272–1284. doi: 10.4161/auto.28954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Matsuzawa Y, et al. TNFAIP3 promotes survival of CD4 T cells by restricting MTOR and promoting autophagy. Autophagy. 2015;11(7):1052–1062. doi: 10.1080/15548627.2015.1055439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Borel S, Espert L, Biard-Piechaczyk M. Macroautophagy regulation during HIV-1 infection of CD4+ T cells and macrophages. Front Immunol. 2012;3:97. doi: 10.3389/fimmu.2012.00097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sagnier S, et al. Autophagy restricts HIV-1 infection by selectively degrading Tat in CD4+ T lymphocytes. J Virol. 2015;89(1):615–625. doi: 10.1128/JVI.02174-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.McGuire DJ, et al. CD5 enhances Th17-cell differentiation by regulating IFN-γ response and RORγt localization. Eur J Immunol. 2014;44(4):1137–1142. doi: 10.1002/eji.201343998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Besnard E, et al. The mTOR complex controls HIV latency. Cell Host Microbe. 2016;20(6):785–797. doi: 10.1016/j.chom.2016.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Martin AR, Pollack RA, Capoferri A, Ambinder RF, Durand CM, Siliciano RF. Rapamycin-mediated mTOR inhibition uncouples HIV-1 latency reversal from cytokine-associated toxicity. J Clin Invest. 2017;127(2):651–656. doi: 10.1172/JCI89552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Boulassel MR, et al. Changes in immunological and virological parameters in HIV-1 infected subjects following leukapheresis. J Clin Apher. 2003;18(2):55–60. doi: 10.1002/jca.10051. [DOI] [PubMed] [Google Scholar]
- 89.Shacklett BL, et al. Optimization of methods to assess human mucosal T-cell responses to HIV infection. J Immunol Methods. 2003;279(1–2):17–31. doi: 10.1016/s0022-1759(03)00255-2. [DOI] [PubMed] [Google Scholar]
- 90.Hayes TL, et al. Impact of highly active antiretroviral therapy initiation on CD4(+) T-cell repopulation in duodenal and rectal mucosa. AIDS. 2013;27(6):867–877. doi: 10.1097/QAD.0b013e32835d85b4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Roederer M. Compensation in flow cytometry. In: Robinson JP, ed. Current Protocols in Cytometry. New York, NY: Wiley; 2002: chapter 1: unit 1.14. [DOI] [PubMed] [Google Scholar]
- 92.Ancuta P, et al. CD16+ monocytes exposed to HIV promote highly efficient viral replication upon differentiation into macrophages and interaction with T cells. Virology. 2006;344(2):267–276. doi: 10.1016/j.virol.2005.10.027. [DOI] [PubMed] [Google Scholar]
- 93.Chomont N, et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat Med. 2009;15(8):893–900. doi: 10.1038/nm.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bakri Y, et al. The maturation of dendritic cells results in postintegration inhibition of HIV-1 replication. J Immunol. 2001;166(6):3780–3788. doi: 10.4049/jimmunol.166.6.3780. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.