

Archaic Csu pili mediate the formation of A. baumannii biofilms on abiotic surfaces. To determine the structural basis for biofilm formation, native and selenomethionine-substituted pilus tip adhesin subunit CsuE complexed with the chaperone CsuC was produced in E. coli, purified and crystallized. Methylation of lysine residues was essential for crystallization of the complex. Initial phases were derived from selenomethionine-based single-wavelength anomalous dispersion.
Keywords: chaperone–usher pathway, archaic pili, biofilm, Acinetobacter baumannii, adhesion, CsuC, CsuE
Abstract
Acinetobacter baumannii is one of the most difficult Gram-negative bacteria to control and treat. This pathogen forms biofilms on hospital surfaces and medical devices using Csu pili assembled via the archaic chaperone–usher pathway. To uncover the mechanism of bacterial attachment to abiotic surfaces, it was aimed to determine the crystal structure of the pilus tip adhesin CsuE. The CsuC–CsuE chaperone–subunit pre-assembly complex was purified from the periplasm of Escherichia coli overexpressing CsuC and CsuE. Despite the high purity of the complex, no crystals could be obtained. This challenge was solved by the methylation of lysine residues. The complex was crystallized in 0.1 M bis-tris pH 5.5, 17% PEG 3350 using the hanging-drop vapour-diffusion method. The crystals diffracted to a resolution of 2.31 Å and belonged to the triclinic space group P1, with unit-cell parameters a = 53.84, b = 63.85, c = 89.25 Å, α = 74.65, β = 79.65, γ = 69.07°. Initial phases were derived from a single anomalous diffraction experiment using a selenomethionine derivative.
1. Introduction
Multidrug-resistant Acinetobacter baumannii, which thrives in hospital environments, has quickly become one of the most difficult Gram-negative bacteria to control and treat (Maragakis & Perl, 2008 ▸). This pathogen has been shown to colonize various objects, including medical equipment and tools, hospital furniture and even the gowns and gloves of healthcare providers (Wilks et al., 2006 ▸; Morgan et al., 2010 ▸). The outstanding survival properties and antibiotic resistance of this pathogen are strongly associated with its ability to form biofilms (Ahmad et al., 2016 ▸). This process is mediated by Csu pili assembled via the chaperone–usher (CU) pathway (Tomaras et al., 2003 ▸). A CU pilus is a long linear polymer composed of 1–7 types of protein subunit (Zav’yalov et al., 2010 ▸; Zavialov et al., 2007 ▸). Biogenesis of the fibre requires a periplasmic chaperone and an outer membrane assembly platform termed the usher (Busch & Waksman, 2012 ▸). The periplasmic chaperone binds to pilin subunits shortly following their translocation to the periplasm. The chaperone–subunit complex represents a high-energy intermediate state (Zavialov et al., 2003 ▸, 2005 ▸) and serves as a substrate for subunit assembly (Yu, Dubnovitsky et al., 2012 ▸). A subunit-induced conformational change in the chaperone enables the binding of the chaperone–subunit complexes to the usher (Yu, Fooks et al., 2012 ▸), where subunits are released from the chaperone, polymerize by donor-strand exchange (DSE) and donor-strand complementation (DSC) mechanisms, and are secreted to the cell surface (Zav’yalov et al., 2010 ▸; Busch & Waksman, 2012 ▸; Phan et al., 2011 ▸).
The Csu system belongs to the archaic CU pathway, which forms the largest ‘nonclassical’ branch of the CU superfamily (Pakharukova, Garnett et al., 2015 ▸; Nuccio & Bäumler, 2007 ▸). Although archaic systems have a far wider phylogenetic distribution and are associated with a broader range of diseases than their classical equivalents, little is known regarding their precise assembly and adhesion mechanisms. The pilus is elaborated from four subunits, CsuA/B (16.1 kDa), CsuA (17.3 kDa), CsuB (16.9 kDa) and CsuE (33.5 kDa), using the CsuC/CsuD CU machinery (Tomaras et al., 2003 ▸). CsuA/B is capable of self-association and forms the pilus shaft (Pakharukova, Tuittila et al., 2015 ▸). The adhesin subunit CsuE is located at the tip of the pilus and is linked to the CsuA/B polymer via CsuA and CsuB (Pakharukova, Garnett et al., 2015 ▸). The CsuE and its closest homologues from Pseudomonas and Vibrio species share 29–34% identity at the amino-acid level (Tomaras et al., 2003 ▸). Our recent crystal structure of the CsuC–CsuA/B pre-assembly complex revealed that nonclassical chaperones, unlike their classical counterparts, maintain subunits in a substantially disordered conformational state (Pakharukova, Garnett et al., 2015 ▸). Furthermore, we demonstrated that the subunit lacks the classical pre-folded initiation site for DSE, suggesting that the assembly process is substantially different from the classical assembly pathway. To provide further insight into the assembly mechanism in archaic systems and to uncover the structural basis for the attachment of A. baumannii to abiotic surfaces, we aimed to determine the crystal structure of the CsuE adhesin. Here, we report the crystallization and SAD phasing of CsuE complexed with CsuC. The study highlights the importance of methylation of lysine residues for improving the crystallizability of proteins.
2. Materials and methods
2.1. Macromolecule production
Synthetic genes for CsuC and CsuE were ordered from GenScript. The CsuC coding sequence was extended to introduce a His6 tag at the C-terminus of the protein (Table 1 ▸). The gene coding for CsuC-His6 was flanked by an EcoRI restriction site at the 5′-end and NheI and SacI restriction sites at the 3′-end. The gene for CsuE was extended by a sequence coding for the donor strand (ds) from the CsuB subunit in order to produce donor-strand complemented CsuE (CsuEdsB) for another study. Each of the genes was delivered on plasmid pUC57 (Table 1 ▸). The DNA fragment coding for CsuC-His6 was inserted into the pET101 expression vector (Invitrogen) using EcoRI and NheI restriction-enzyme sites to produce the pET101-CsuC6H plasmid. The CsuE gene was obtained from pUC57-CsuEdsB by PCR using the primers 5′-AATGCTAGCGAAGGAATTCAGGAGCCC-3′ and 5′- TAAGAGCTCTTAACCTCCACCTCCACCAAAC-3′ and was cloned into the pET101-CsuC6H plasmid downstream of the CsuC6H open reading frame using the NheI and SacI restriction sites. During PCR, the sequence coding for the donor strand of CsuB was removed to allow CsuC–CsuE binding. The sequence was verified by sequencing at the Finnish Microarray and Sequencing Centre. The resulting pET101-Csu6H-CsuE plasmid was transformed into Escherichia coli strain BL21-AI (Invitrogen).
Table 1. Macromolecule-production information.
| Source organism | A. baumannii |
| DNA source | Oligonucleotide synthesis |
| Cloning vector | pUC57 |
| Expression vector | pET101-Csu6H-CsuE |
| Expression host | E. coli strains BL21-AI and BL21(DE3) (Invitrogen) |
| Sequence of CsuE† | MNIKTKKLLRHLCMFSGLMLTGNMAHAACSVSASGTSSISVPSIYLMENGENSSQFNSGLSCTGFSLALANMTYLKYRVEQMSNSFTNAQTGEKLNAIILDSNNEIISLGQEKDMSSFTLVNLFSGPDGNLPFYIRLPAGQSVSPGVYQADSPLKVKWFYSVPAVAIVGIGVFFESPGFRRGALGIGFNWGSGADSLGSLSITVLPDCRILAQDVNFGTAAFASKLEPVQSSMGIRCSVNTPYYVSLNNGLSPQNGNQRAMKSQTGNTFLKYDIFKNSSNDRWGSGNERWSSLNATINPGVHNGVTQQNYVFTTKIVDENADTIPAGTYQDTVTVQVEF |
| Sequence of CsuC† ‡ | MVICMNNSAFIKNGILKSFLFASTLSLVTPVMAQATFLIWPIYPKIEANEKATAVWLQNTGKTDAMVQIRVFKWNQDGLKDNYSEQSEIIPSPPVAKIKAGEKHMLRLTKSVNLPDGKEQSYRLIVDELPIRLSDGNEQDASKVSFQMRYSIPLFAYGKGIGSGLTEESQKLNAKNALAKPVLQWSVRNNQQGQSELYLKNNGQKFARLSALKTSKTGNDISLGKAAFGYVLSNSTVKFAIDQSTAHELAKTSKIYGVDSSGIKQELIEITKMEDPSGHHHHHH |
The secretion signal sequence is shown in italics.
The His6 tag is underlined.
The CsuC–CsuE complex was expressed and extracted from the E. coli periplasm using the protocol developed for the production of the CsuC–CsuA/B complex (Pakharukova, Tuittila et al., 2015 ▸). About 90–95% of the CsuC–CsuE was extracted using ice-cold 5 mM MgSO4 (periplasmic fraction) and only 5–10% of the CsuC–CsuE leaked into the 20% sucrose, 5 mM EDTA, 50 mM Tris–HCl pH 8.0 solution (sucrose fraction). The complex was purified from the periplasmic fraction by metal-ion affinity chromatography at 277 K using a 5 ml HiTrap Ni–IMAC column (GE Healthcare) equilibrated with 20 mM sodium phosphate, 0.5 M sodium chloride, 5 mM imidazole buffer pH 7.3. The target protein was eluted with an 8–500 mM gradient of imidazole. The protein-containing fractions were dialyzed overnight against 20 mM HEPES buffer pH 7.3 and further purified by cation-exchange chromatography at 277 K using a Mono S 5/50 GL column (GE Healthcare) equilibrated with 20 mM HEPES pH 7.3. A 0–300 mM gradient of NaCl was used to elute the protein complex. The final yield of the pure protein was 0.5–1 mg per litre of cell culture. Selenomethionine-incorporated CsuC–CsuE was expressed in E. coli BL21(DE3) cells as described in Pakharukova, Tuittila et al. (2015 ▸) and purified as described above.
2.2. Methylation and crystallization
To facilitate crystallization, both the native and the selenomethionine-incorporated CsuC–CsuE complexes were subjected to a lysine-methylation reaction. The methylation reaction was performed overnight in 50 mM HEPES, 250 mM NaCl pH 7.5 at a protein concentration of 0.5 mg ml−1. 20 µl of freshly prepared 1 M dimethylamine–borane complex (ABC; Sigma–Aldrich) and 40 µl 1 M formaldehyde (made from a 37% stock; J. T. Baker) were added per millilitre of protein solution. The reactions were gently mixed and incubated at 277 K for 2 h. A further 20 µl of ABC and 40 µl formaldehyde were added and incubation continued for 2 h. Following a final addition of 10 µl ABC per millilitre of initial protein volume, the reaction mixture was incubated overnight. The next morning, the reaction was quenched by the addition of 80 µl 1 M glycine (to a final concentration of 5 mg ml−1) and the mixture was incubated on ice for 2 h. The modified protein was dialyzed overnight against 20 mM HEPES pH 7.2. The methylation reaction resulted in a significant amount of precipitated protein (about 50%), which was removed by centrifugation. The soluble methylated protein was subjected to size-exclusion chromatography using a Superdex 75 10/300 GL column (GE Healthcare) equilibrated in 20 mM HEPES 150 mM NaCl pH 7.2. Appropriate peak fractions were pooled and concentrated to 12 mg ml−1 using a Vivaspin device (GE Healthcare) with a molecular-weight cutoff of 10 kDa. The protein concentration was determined by absorption measurements at 280 nm using a molar extinction coefficient of 72 560 cm−1 M −1.
Initial crystallization conditions were obtained by the sitting-drop vapour-diffusion method at 289 K using the commercial screening kits Index and Crystal Screen HT from Hampton Research. Aliquoting was performed with a Mosquito liquid dispenser (TTP Labtech) by mixing 75–150 nl protein solution (in 10 mM HEPES, 75 mM NaCl pH 7.2) and 100 nl reservoir solution in three different ratios (0.75:1, 1:1 and 1.5:1) in a 96-well plate and equilibrating against 80 µl reservoir solution. A PX Scanner device (Agilent Technologies) was used to examine the diffraction of crystals in the crystallization plates (Pakharukova et al., 2013 ▸). Optimization of the crystallization conditions was carried out manually by the hanging-drop vapour-diffusion method (Table 2 ▸).
Table 2. Crystallization.
| Method | Hanging-drop vapour-diffusion method |
| Plate type | 24-well hanging-drop plate (Hampton Research) |
| Temperature (K) | 289 |
| Protein concentration (mg ml−1) | 12 |
| Buffer composition of protein solution | 10 mM HEPES, 75 mM NaCl pH 7.2 |
| Composition of reservoir solution | 0.1 M bis-tris pH 5.5, 17% PEG 3350 |
| Volume and ratio of drop | 2 µl, 1:1 |
| Volume of reservoir (µl) | 1000 |
2.3. Data collection and processing
Crystals were soaked for 30–60 s in a cryoprotectant solution prepared by mixing two parts of precipitant solution with one part 50% PEG 400 and were then cooled by plunging them into liquid nitrogen. Diffraction data were collected under liquid-nitrogen cryoconditions at 100 K on beamline ID23-1 at the European Synchrotron Radiation Facility (ESRF), Grenoble, France. Data were processed using the Grenoble automatic data-processing system (GrenADeS) system at the ESRF (Monaco et al., 2013 ▸). Initial phases were determined by the single-wavelength anomalous diffraction (SAD) phasing method using the selenomethionine derivative and the initial model was constructed using the PHENIX software package (Adams et al., 2002 ▸).
3. Results and discussion
Co-expression of CsuC and CsuE resulted in a high level of accumulation of stable CsuC–CsuE complex in the periplasm (Fig. 1 ▸). A highly homogenous protein sample was obtained after consecutive purification of CsuC–CsuE using nickel-chelate affinity and ion-exchange chromatography (Fig. 1 ▸). However, initial crystallization trials failed to produce protein crystals. Variation of the protein concentration (10–50 mg ml−1) and temperature (277–298 K) did not improve the result. Since archaic chaperones maintain subunits in a substantially unfolded conformation (Pakharukova, Garnett et al., 2015 ▸), we assumed that the high structural flexibility in CsuE may hinder crystallization. Structural flexibility could be reduced by the methylation of lysine residues (Walter et al., 2006 ▸). In addition, methylation increases hydrophobicity, which could also be beneficial as CsuC–CsuE, like other proteins from the CU pathway (Berry et al., 2014 ▸; Roy et al., 2012 ▸; Pakharukova et al., 2016 ▸), is highly soluble, requiring large amounts of protein for crystallization experiments. Reductive methylation of lysine residues in CsuC–CsuE led to a significant loss of the complex (nearly 50%) owing to precipitation. The soluble methylated protein was additionally purified by size-exclusion chromatography. Initial crystallization experiments of the modified CsuC–CsuE resulted in the appearance of three-dimensional crystals in a broad range of conditions. The largest crystals (0.1 × 0.1 × 0.3 mm) were observed in 0.1 M bis-tris pH 5.5, 17% PEG 3350 at 289 K (Table 2 ▸, Fig. 2 ▸). The native crystal diffracted to a resolution of 2.31 Å, revealing a triclinic space group. Calculation of the Matthews coefficient V M (the crystal volume per unit of protein molecular weight) suggested the presence of two molecules of the CsuC–CsuE complex per crystallographic asymmetric unit (V M = 2.24 Å3 Da−1).
Figure 1.
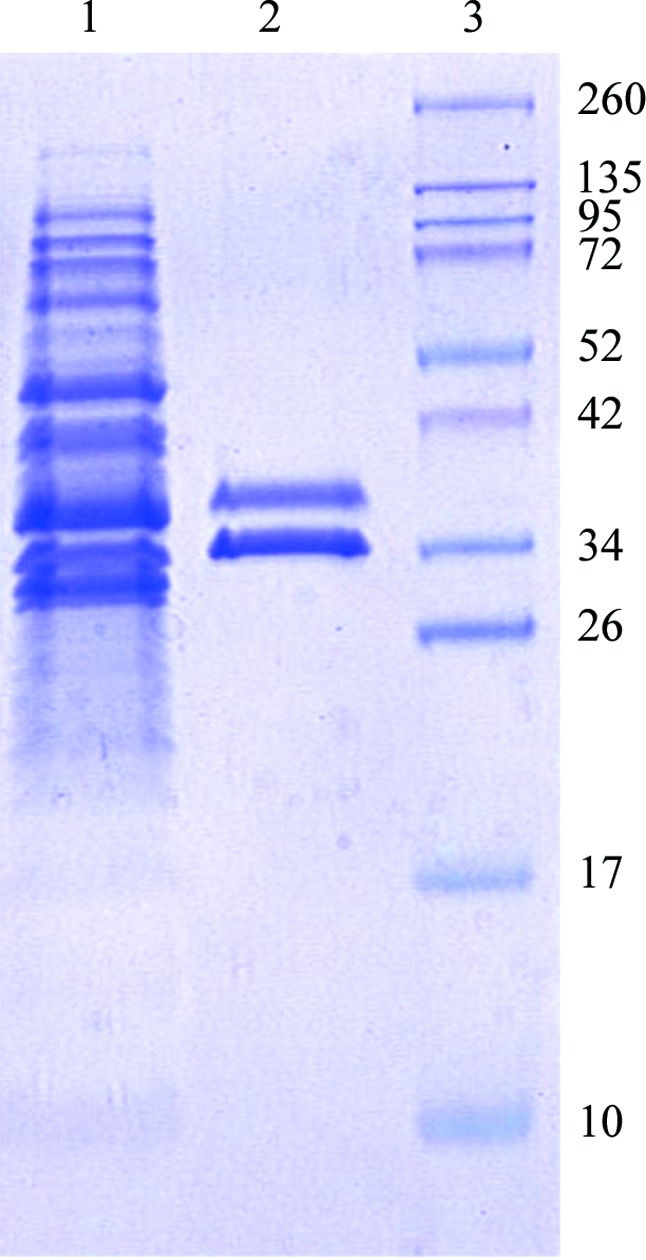
Coomassie Blue-stained SDS–polyacrylamide gel (12%) of the periplasmic extract from E. coli BL21-AI cells co-expressing CsuC and CsuE (lane 1), purified CsuC–CsuE complex (lane 2) and molecular-size marker proteins (lane 3; labelled in kDa).
Figure 2.

CsuC–CsuE crystal grown in 0.1 M bis-tris pH 5.5, 17% PEG 3350 at 289 K.
As the sequences of CsuA/B and the C-terminal domain of CsuE share 31% identity, the combined sequences of the CsuC–CsuE and CsuC–CsuA/B complexes share 54% identity. Therefore, we attempted to solve the structure by molecular replacement using CsuC–CsuA/B as a search model. The obtained phases enabled the CsuC chaperone and a few β-strands of the pilin domain of CsuE to be traced. However, missing or poor electron density in other regions of the protein prevented building of the complete model. The presence of ten methionine residues in the complex prompted us to apply the selenomethionine (SeMet) SAD method to determine the phases. An SeMet derivative of CsuC–CsuE was produced, purified and crystallized. The SeMet-derivative crystals diffracted to a resolution of 2.51 Å (Table 3 ▸ a). The anomalous signal in the SeMet-derivative data set extended to 4.0–4.4 Å resolution (Table 3 ▸ b). 22 Se atoms were found and refined using the PHENIX software package (Table 3 ▸ c). The sites were later assigned to ten SeMet residues in each molecule in the asymmetric unit (Table 3 ▸ c). Two Se atoms in the substructure clustered near SeMet20 in CsuE (chain B). In addition, one Se atom was placed near Cys210 in CsuE (chain B). The occupancies of the Se atoms generally exceeded 1.0, suggesting a slightly underestimated value for f′′. Phase improvement by density modification resulted in an experimental electron density that contained recognizable features of a β-structural protein (Fig. 3 ▸). About 90% of the asymmetric unit was chain-traced by AutoSol in the PHENIX software suite. The SAD phases were applied to the native data set and extended to a resolution of 2.31 Å. After a few cycles of refinement, we observed additional electron density around the NZ atom for several lysine residues, which can be interpreted as an N-linked methyl group. Structure refinement is in progress.
Table 3. Data statistics.
(a) Data collection and processing. Values in parentheses are for the outer shell. For the native data set, I/σ(I) in the outer shell falls below 2.0 at a resolution of 2.38 Å.
| Native data set | SeMet SAD data set | |
|---|---|---|
| Diffraction source | ID23-1, ESRF | ID23-1, ESRF |
| Wavelength (Å) | 0.98 | 0.98 |
| Temperature (K) | 100 | 100 |
| Detector | PILATUS 6M-F | PILATUS 6M-F |
| Crystal-to-detector distance (mm) | 337 | 497.7 |
| Rotation range per image (°) | 0.2 | 0.2 |
| Total rotation range (°) | 370 | 1080 |
| Exposure time per image (s) | 0.05 | 0.04 |
| Space group | P1 | P1 |
| a, b, c (Å) | 53.84, 63.85, 89.25 | 54.18, 63.85, 89.54 |
| α, β, γ (°) | 74.65, 79.65, 69.07 | 74.70, 79.56, 68.82 |
| Mosaicity (°) | 0.13 | 0.15 |
| Resolution range (Å) | 58.20–2.31 (2.43–2.31) | 45.34–2.51 (2.65–2.51) |
| Total No. of reflections | 166711 (22911) | 383562 (54037) |
| No. of unique reflections | 44881 (6462) | 36012 (5098) |
| Completeness (%) | 96 (94.3) | 97.9 (94.6) |
| Multiplicity | 3.7 (3.5) | 10.7 (10.6) |
| 〈I/σ(I)〉 | 13 (1.5) | 15.2 (2.0) |
| R meas | 0.033 (0.489) | 0.11 (1.160) |
| Overall B factor from Wilson plot (Å2) | 53.9 | 55.1 |
| 〈I〉 half-set correlation CC1/2 | 0.999 (0.665) | 0.999 (0.689) |
(b) Anomalous signal measurability as a function of resolution. The anomalous signal measurability is defined as the fraction of Bijvoet-related intensity differences for which |δI|/σ(δI) > 3.0 and [I(+)/σI(+), I(−)/σI(−)] > 3.0 hold. Measurability values were estimated using phenix.xtriage from the PHENIX software package.
| Resolution shells (Å) | 6.03 | 4.78 | 4.18 | 3.80 | 3.53 | 3.32 | 3.15 | 3.02 | 2.90 | 2.80 |
|---|---|---|---|---|---|---|---|---|---|---|
| Measurability | 0.35 | 0.18 | 0.08 | 0.03 | 0.02 | 0.01 | 0.006 | 0.005 | 0.004 | 0.002 |
(c) Substructure.
| Se-atom positions | ||||||
|---|---|---|---|---|---|---|
| Se atom | Assignment (residue No., chain†) | x | y | z | Occupancy | B (Å2) |
| Se1 | 206, B | −17.939 | −13.852 | −58.966 | 1.64 | 30.94 |
| Se2 | 234, D | −64.915 | −33.928 | −27.483 | 1.45 | 35.04 |
| Se3 | 88, B | −42.651 | −46.722 | −7.309 | 1.37 | 38.01 |
| Se4 | 71, C | 0.324 | 0.162 | −0.150 | 1.47 | 33.56 |
| Se5 | 206, D | −60.444 | −48.554 | −21.431 | 1.27 | 31.30 |
| Se6 | 71, A | −32.458 | −59.440 | −83.338 | 0.89 | 24.26 |
| Se7 | 239, A | −35.570 | −38.874 | −47.023 | 0.98 | 38.40 |
| Se8 | 32, A | −47.427 | −69.562 | −79.399 | 1.04 | 34.19 |
| Se9 | 45, D | −58.990 | −28.139 | −49.918 | 0.91 | 37.44 |
| Se10 | 55, B | −56.321 | −66.005 | −80.110 | 0.96 | 38.51 |
| Se11 | 45, B | −38.956 | −47.261 | −18.705 | 0.86 | 33.93 |
| Se12 | 114, A | −41.256 | −72.076 | −65.133 | 0.72 | 31.91 |
| Se13 | 88, D | −55.301 | −29.252 | −62.058 | 0.81 | 35.42 |
| Se14 | 32, C | −23.146 | −42.536 | −8.722 | 0.79 | 31.52 |
| Se15 | 20, B | −52.044 | −15.465 | −65.406 | 0.68 | 30.77 |
| Se16 | 55, D | −47.423 | −34.029 | −71.423 | 0.64 | 33.47 |
| Se17 | 114, C | −64.976 | −48.206 | −16.450 | 0.60 | 32.27 |
| Se18 | 234, B | −32.520 | −20.969 | −59.005 | 0.48 | 27.50 |
| Se19 | 239, C | −48.186 | −20.825 | −35.387 | 0.52 | 29.35 |
| Se20 | 20, B | −51.989 | −17.390 | −67.308 | 0.53 | 27.02 |
| Se21 | 20, D | −50.348 | −58.173 | −4.489 | 0.32 | 29.24 |
| Se22 | Cys210, B | −85.598 | −67.823 | −61.617 | 0.30 | 27.60 |
Chains A and B and chains C and D belong to CsuC and CsuE in the two molecules in the asymmetric unit, respectively.
Figure 3.

Experimental electron density computed using SAD phases. A fragment of the 2mF o − DF c (σA; Read, 1986 ▸) electron-density map contoured at 2σ is shown (produced using Coot; Emsley et al., 2010 ▸).
Acknowledgments
The authors would like to thank the staff at beamlines ID23-1 and BM14U, ESRF, Grenoble, France for their assistance in data collection.
References
- Adams, P. D., Grosse-Kunstleve, R. W., Hung, L.-W., Ioerger, T. R., McCoy, A. J., Moriarty, N. W., Read, R. J., Sacchettini, J. C., Sauter, N. K. & Terwilliger, T. C. (2002). Acta Cryst. D58, 1948–1954. [DOI] [PubMed]
- Ahmad, T., Tawfik, D., Sheweita, S., Haroun, M. & El-Sayed, L. (2016). Trials Vaccinol. 5, 53–60.
- Berry, A. A. et al. (2014). PLoS Pathog. 10, e1004404. [DOI] [PMC free article] [PubMed]
- Busch, A. & Waksman, G. (2012). Philos. Trans. R. Soc. Lond. B Biol. Sci. 367, 1112–1122. [DOI] [PMC free article] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Maragakis, L. L. & Perl, T. M. (2008). Clin. Infect. Dis. 46, 1254–1263. [DOI] [PubMed]
- Monaco, S., Gordon, E., Bowler, M. W., Delagenière, S., Guijarro, M., Spruce, D., Svensson, O., McSweeney, S. M., McCarthy, A. A., Leonard, G. & Nanao, M. H. (2013). J. Appl. Cryst. 46, 804–810. [DOI] [PMC free article] [PubMed]
- Morgan, D. J., Liang, S. Y., Smith, C. L., Johnson, J. K., Harris, A. D., Furuno, J. P., Thorn, K. A., Snyder, G. M., Day, H. R. & Perencevich, E. N. (2010). Infect. Control Hosp. Epidemiol. 31, 716–721. [DOI] [PMC free article] [PubMed]
- Nuccio, S. P. & Bäumler, A. J. (2007). Microbiol. Mol. Biol. Rev. 71, 551–575. [DOI] [PMC free article] [PubMed]
- Pakharukova, N., Garnett, J. A., Tuittila, M., Paavilainen, S., Diallo, M., Xu, Y., Matthews, S. J. & Zavialov, A. V. (2015). PLoS Pathog. 11, e1005269. [DOI] [PMC free article] [PubMed]
- Pakharukova, N., Roy, S., Tuittila, M., Rahman, M. M., Paavilainen, S., Ingars, A. K., Skaldin, M., Lamminmäki, U., Härd, T., Teneberg, S. & Zavialov, A. V. (2016). Mol. Microbiol. 102, 593–610. [DOI] [PubMed]
- Pakharukova, N., Tuittila, M., Paavilainen, S. & Zavialov, A. (2015). Acta Cryst. F71, 770–774. [DOI] [PMC free article] [PubMed]
- Pakharukova, N., Tuittila, M. & Zavialov, A. (2013). Acta Cryst. F69, 1389–1392. [DOI] [PMC free article] [PubMed]
- Phan, G. et al. (2011). Nature (London), 474, 49–53.
- Read, R. J. (1986). Acta Cryst. A42, 140–149.
- Roy, S. P., Rahman, M. M., Yu, X. D., Tuittila, M., Knight, S. D. & Zavialov, A. V. (2012). Mol. Microbiol. 86, 1100–1115. [DOI] [PubMed]
- Tomaras, A. P., Dorsey, C. W., Edelmann, R. E. & Actis, L. A. (2003). Microbiology, 149, 3473–3484. [DOI] [PubMed]
- Walter, T. S., Meier, C., Assenberg, R., Au, K. F., Ren, J., Verma, A., Nettleship, J. E., Owens, R. J., Stuart, D. I. & Grimes, J. M. (2006). Structure, 14, 1617–1622. [DOI] [PMC free article] [PubMed]
- Wilks, M., Wilson, A., Warwick, S., Price, E., Kennedy, D., Ely, A. & Millar, M. R. (2006). Infect. Control Hosp. Epidemiol. 27, 654–658. [DOI] [PubMed]
- Yu, X. D., Dubnovitsky, A., Pudney, A. F., Macintyre, S., Knight, S. D. & Zavialov, A. V. (2012). Structure, 20, 1861–1871. [DOI] [PubMed]
- Yu, X. D., Fooks, L. J., Moslehi-Mohebi, E., Tischenko, V. M., Askarieh, G., Knight, S. D., Macintyre, S. & Zavialov, A. V. (2012). J. Mol. Biol. 417, 294–308. [DOI] [PubMed]
- Zavialov, A. V., Berglund, J., Pudney, A. F., Fooks, L. J., Ibrahim, T. M., MacIntyre, S. & Knight, S. D. (2003). Cell, 113, 587–596. [DOI] [PubMed]
- Zavialov, A. V., Tischenko, V. M., Fooks, L. J., Brandsdal, B. O., Aqvist, J., Zav’yalov, V. P., Macintyre, S. & Knight, S. D. (2005). Biochem. J. 389, 685–694. [DOI] [PMC free article] [PubMed]
- Zavialov, A., Zav’yalova, G., Korpela, T. & Zav’yalov, V. (2007). FEMS Microbiol. Rev. 31, 478–514. [DOI] [PubMed]
- Zav’yalov, V., Zavialov, A., Zav’yalova, G. & Korpela, T. (2010). FEMS Microbiol. Rev. 34, 317–378. [DOI] [PubMed]