

Its haematophagic habits and urban habitat make the mosquito Aedes aegypti an important vector of a number of human viruses. Here, the high-resolution crystal structure of AaTI, a noncanonical Kazal inhibitor from the saliva of A. aegypti, is presented, providing a molecular explanation for its inhibitory profile.
Keywords: anticoagulants, thrombin, trypsin, salivary glands, protein–protein interactions, Kazal inhibitor, Aedes aegypti, Dengue fever, Yellow fever, Zika virus
Abstract
Blood-feeding exoparasites are rich sources of protease inhibitors, and the mosquito Aedes aegypti, which is a vector of Dengue virus, Yellow fever virus, Chikungunya virus and Zika virus, is no exception. AaTI is a single-domain, noncanonical Kazal-type serine proteinase inhibitor from A. aegypti that recognizes both digestive trypsin-like serine proteinases and the central protease in blood clotting, thrombin, albeit with an affinity that is three orders of magnitude lower. Here, the 1.4 Å resolution crystal structure of AaTI is reported from extremely tightly packed crystals (∼22% solvent content), revealing the structural determinants for the observed inhibitory profile of this molecule.
1. Introduction
Aedes aegypti is a well adapted urban mosquito that is capable of transmitting Dengue fever virus, Yellow fever virus and Chikungunya virus during blood feeding (Gubler, 2002 ▸; Vega-Rúa et al., 2014 ▸). Recently, A. aegypti has also been implicated in the transmission of Zika virus (Aliota et al., 2016 ▸; Chouin-Carneiro et al., 2016 ▸).
In haematophagous animals, proteases and their inhibitors play crucial roles by controlling coagulation (Corral-Rodríguez et al., 2009 ▸, 2010 ▸), platelet aggregation and vasoconstriction (Ribeiro, 1995 ▸) during blood acquisition (Takác et al., 2006 ▸) or storage (Araujo et al., 2007 ▸; Campos et al., 2002 ▸, 2004 ▸; Soares et al., 2012 ▸). Protease inhibitors are classified according to their primary structure and functional similarity among living organisms (Laskowski & Kato, 1980 ▸), and they are currently divided into 79 different families (Rawlings et al., 2016 ▸). In invertebrates, the Kazal, Kunitz and serpin inhibitor families contain the largest numbers of members.
Kazal-type inhibitors have characteristic structural features including two or three α-helices and a short three-stranded antiparallel β-sheet (Mühlhahn et al., 1994 ▸), six cysteine residues forming three disulfide bonds with 1–5/2–4/3–6 topology, and a canonical binding loop (Bode & Huber, 1992 ▸). The Kazal family members can be divided into classical and nonclassical inhibitors (Hemmi et al., 2005 ▸; Fink et al., 1986 ▸), and among the latter it is possible to find single-domain [for example leech-derived tryptase inhibitor (LDTI; Sommerhoff et al., 1994 ▸), bdellin B-3 (Fink et al., 1986 ▸), A. aegypti trypsin inhibitor (AaTI; Watanabe et al., 2010 ▸) and agaphelin (Waisberg et al., 2014 ▸)] and multi-domain [for example rhodniin (Friedrich et al., 1993 ▸) and infestin (Campos et al., 2002 ▸)] proteins. Moreover, the Kazal-type inhibitors present in the midgut of kissing bugs (Triatominae) display strong inhibitory properties against thrombin (Campos et al., 2002 ▸; Friedrich et al., 1993 ▸; Paim et al., 2011 ▸). In the particular case of infestins, a family of multi-domain Kazal-type molecules (Campos et al., 2002 ▸), an as yet unknown post-translational processing event generates one or two Kazal-domain inhibitors with distinct specificities from the parent protein (Lovato et al., 2006 ▸). Recently, AaTI, a single-domain Kazal-type inhibitor from the mosquito A. aegypti, was characterized as a trypsin and thrombin inhibitor (Watanabe et al., 2010 ▸). Expression of the gene encoding AaTI was detected in the mosquito salivary gland and midgut, suggesting a role in blood-coagulation control. Interestingly, the enzymatic activity of trypsin-like proteases from the midgut of A. aegypti females 24 h post-feeding was strongly inhibited by AaTI, but it only weakly affected enzymatic activity 3 h after feeding (Watanabe et al., 2010 ▸). This underscores the specificity of AaTI, since the content of trypsin-like enzymes in the mosquito midgut changes during blood digestion (Noriega & Wells, 1999 ▸). In addition, AaTI displays a negatively charged C-terminal sequence, which is reminiscent of that of hirudin (Rydel et al., 1990 ▸; Grütter et al., 1990 ▸; Fig. 1 ▸). Nevertheless, the mechanism of thrombin inhibition by AaTI seems to differ from that of hirudin, since there is no noticeable difference in the inhibition of α-thrombin and γ-thrombin by AaTI, and the removal of the C-terminal tail of the inhibitor does not significantly alter its ability to prolong the thrombin time, suggesting that, in contrast to hirudin, AaTI does not bind to exosite I of thrombin (Watanabe et al., 2011 ▸). In the present work, we report the crystallographic three-dimensional structure of AaTI and propose its inhibitory mechanism towards thrombin.
Figure 1.

Multiple amino-acid sequence alignment of insect-derived Kazal-type proteins. In the upper block AaTI from Aedes aegypti (UniProt entry Q1HRB8) is aligned with other putative Kazal inhibitors from A. albopictus (UniProt entry Q5MIW1), Psorophora albipes (UniProt entry T1D5N9), Culex pipiens pallens (UniProt entry I6XC79), C. quinquefasciatus (UniProt entry B0XEZ9), Ochlerotatus triseriatus (UniProt entry C6ZQX9), Anopheles sinensis (UniProt entry A0A084VPC8), A. gambiae (UniProt entry Q7QG67), A. aquasalis (UniProt entry T1DGP5) and A. darlingi (UniProt entry B6DDZ6). In the lower block, the structurally characterized infestin 1 (PDB entry 2f3c; Campos et al., 2012 ▸) and infestin 4 (PDB entry 2erw; Campos et al., 2012 ▸) from Triatoma infestans and the N-terminal portion of rhodniin (PDB entry 1tbq; van de Locht et al., 1995 ▸) from Rhodnius prolixus are also aligned with the upper sequences. The mature AaTI amino-acid numbering and secondary-structure elements are indicated above the alignment. A green-to-red colour gradient indicates increasing residue conservation. This figure was prepared with ALINE (Bond & Schüttelkopf, 2009 ▸).
2. Materials and methods
2.1. Macromolecule production
AaTI was expressed in a Pichia pastoris system and purified by affinity chromatography on trypsin-Sepharose as described previously (Watanabe et al., 2010 ▸).
2.2. Crystallization
The recombinant AaTI sample, concentrated to 20 mg ml−1 by ultrafiltration, was used for the screening and optimization of crystallization conditions at Laboratório Robotizado de Cristalização de Proteínas (Robolab-LNLS), Campinas, Brazil. Single crystals were grown at 18°C by sitting-drop vapour diffusion (Table 1 ▸). An iodide derivative was prepared by soaking a crystal briefly (∼15 s) in crystallization buffer supplemented with 0.5 M NaI. The crystals were directly transferred to the cryostream prior to data collection.
Table 1. Crystallization conditions.
| Method | Vapour diffusion, sitting-drop |
| Plate type | 96-well |
| Temperature (K) | 291 |
| Protein concentration (mg ml−1) | 20 |
| Buffer composition of protein solution | 10 mM Tris–HCl pH 8.0 |
| Composition of reservoir solution | 100 mM sodium acetate pH 5.5, 25%(w/v) PEG 3350, 5%(v/v) PEG 400, 3%(v/v) dioxane |
| Volume and ratio of drop | 1 µl (1:1) |
| Volume of reservoir (µl) | 80 |
2.3. Data collection and processing
Native (222 images with 1° oscillation and 20 s exposure) and derivative (960 images with 1° oscillation and 20 s exposure) X-ray diffraction data sets were collected from single monoclinic (space group P21) crystals using a MAR Mosaic 225 CCD detector (marXperts) on beamline MX2 of Laboratório Nacional de Luz Síncrotron (LNLS), Campinas, Brazil (Guimarães et al., 2009 ▸). Diffraction data were processed with XDS (Kabsch, 2010 ▸), scaled with XSCALE (Kabsch, 2010 ▸) and reduced with utilities from the CCP4 suite (Winn et al., 2011 ▸; Table 2 ▸).
Table 2. Data collection and processing.
Values in parentheses are for the outer shell.
| Native | Derivative | |
|---|---|---|
| Diffraction source | W01B-MX2, LNLS | W01B-MX2, LNLS |
| Wavelength (Å) | 1.000 | 1.459 |
| Temperature (K) | 100 | 100 |
| Detector | MAR Mosaic 225 CCD | MAR Mosaic 225 CCD |
| Crystal-to-detector distance (mm) | 89 | 99 |
| Rotation range per image (°) | 1.0 | 1.0 |
| Total rotation range (°) | 220 | 960 |
| Exposure time per image (s) | 20 | 20 |
| Space group | P21 | P21 |
| a, b, c (Å) | 26.1, 65.2, 27.7 | 26.4, 64.0, 27.7 |
| α, β, γ (°) | 90, 116.5, 90 | 90, 117.6, 90 |
| Mosaicity (°) | 0.18 | 0.57 |
| Resolution range (Å) | 65.2–1.40 (1.48–1.40) | 23.4–1.80 (1.89–1.80) |
| Total No. of reflections | 74704 (10645) | 144451 (20019) |
| No. of unique reflections | 16291 (2367) | 7491 (1058) |
| Completeness (%) | 99.5 (98.8) | 98.0 (94.8) |
| Multiplicity | 4.6 (4.5) | 19.3 (18.9) |
| 〈I/σ(I)〉 | 11.0 (1.2) | 25.7 (6.7) |
| R r.i.m. † | 0.080 (1.583) | 0.075 (0.371) |
| R p.i.m. ‡ | 0.037 (0.734) | 0.017 (0.084) |
| Half-set correlation CC1/2 | 0.998 (0.568) | 1.000 (0.985) |
| Anomalous half-set correlation CCanom | 0.853 (0.235) | |
| Overall B factor from Wilson plot (Å2) | 18.3 | 22.6 |
R
r.i.m. = 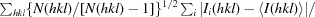
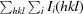 .
.
R
p.i.m. = 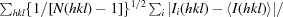 .
.
2.4. Structure solution and refinement
The three-dimensional structure of AaTI was solved by SAD using the anomalous signal of iodine with the SHELXC/D/E pipeline (Sheldrick, 2010 ▸) and the HKL2MAP GUI (Pape & Schneider, 2004 ▸). An initial atomic model was built with ARP/wARP (Langer et al., 2008 ▸) and was completed manually with Coot (Emsley et al., 2010 ▸), alternating with cycles of refinement with PHENIX (Adams et al., 2010 ▸; Table 3 ▸). The refined model coordinates and structure factors were deposited in the Protein Data Bank with accession code 5dae.
Table 3. Structure solution and refinement.
Values in parentheses are for the outer shell.
| Structure solution | |
| SHELXD | |
| No. of heavy atoms | 3 |
| SHELXE | |
| FOM | 0.570 |
| No. of residues built | 88 |
| Refinement | |
| Resolution range (Å) | 32.6–1.40 (1.49–1.40) |
| Completeness (%) | 99.3 |
| σ Cutoff | F > 1.35σ(F) |
| No. of reflections, working set | 16260 (2549) |
| No. of reflections, test set | 840 (142) |
| Final R cryst | 0.180 (0.320) |
| Final R free | 0.230 (0.363) |
| Cruickshank DPI | 0.0838 |
| No. of non-H atoms | |
| Protein | 838 |
| Water | 89 |
| Total | 927 |
| R.m.s. deviations | |
| Bonds (Å) | 0.011 |
| Angles (°) | 1.405 |
| Average B factors (Å2) | |
| Protein | 28.2 |
| Water | 33.9 |
| Ramachandran plot | |
| Most favoured (%) | 91.8 |
| Allowed (%) | 8.2 |
3. Results and discussion
3.1. Crystallization, data collection and structure solution
Recombinant AaTI crystallized readily at 18°C using a vapour-diffusion technique with a mixture of PEG 3350 and PEG 400 as precipitant, yielding monoclinic crystals that belonged to space group P21 (Table 1 ▸). Native AaTI crystals diffracted to 1.4 Å resolution (Table 2 ▸) at a synchrotron source, but initial attempts at structure determination using molecular-replacement techniques with the coordinates of a leech-derived tryptase inhibitor (PDB entry 1ldt; Stubbs et al., 1997 ▸), assuming that a single full-length AaTI molecule was present in the asymmetric unit (57.3% solvent content), as suggested by the probability distribution of the Matthews coefficient, failed. Therefore, the native crystals were soaked briefly in crystallization buffer containing sodium iodide and a complete, redundant data set to 1.8 Å resolution was collected using 8.5 keV synchrotron radiation (Table 2 ▸). Although maximization of the anomalous signal could have been achieved using a longer wavelength, which was within the reach of the experimental setup, at 8.5 keV a good compromise was found between anomalous signal and photon flux. The structure was solved by single-wavelength anomalous dispersion using the anomalous signal of iodide, and the experimental maps revealed the presence of two inhibitor molecules in the asymmetric unit. The molecular mass of the full-length inhibitor (7.4 kDa) and the small unit-cell volume (∼42 000 Å3) would result in an extremely low (<15%) solvent content for these samples. However, we observed unwanted processing of AaTI during heterologous expression, and the recombinant material contained a mixture of full-length AaTI and several truncated forms (data not shown). Mass-spectrometric analysis of the AaTI crystals identified an AaTI fragment spanning residues Glu1–Asp59, which is in good agreement with the segments that could be modelled in the electron-density maps: residues Gly3–Asp59 for AaTI molecule A and Arg2–Thr54 for molecule B. Considering the presence of this truncated form of AaTI in the asymmetric unit, the crystals displayed an unusually low solvent content of ∼22–23% and consequently tight crystal packing (Fig. 2 ▸; Offermann et al., 2015 ▸; Trillo-Muyo et al., 2013 ▸). The unusually tight packing of the AaTI crystals led to an erroneous initial assessment of the asymmetric unit composition, explaining why the attempts at phasing using molecular replacement failed. One of the three iodide ions that could be located (Table 3 ▸) is in the vicinity of molecule A (close to Tyr22 and Asn23), another is closer to molecule B (Lys44) and the third is at the interface between both molecules, close to the side chains of Ile10 and Cys26 of molecule A and of Met12 of molecule B.
Figure 2.

Stereoscopic view of the AaTI crystal packing. The two AaTI monomers in the asymmetric unit (molecule A in blue, molecule B in green) are displayed in ribbon representation, together with surrounding symmetry mates, highlighting the tight packing of the crystals. This figure was prepared with PyMOL (http://www.pymol.org).
3.2. Overall structure
AaTI displays a typical Kazal-domain structure, with central α-helical segments spanning residues 25–32 and 35–38 that, together with a short C-terminal 310-helix, border the small central three-stranded antiparallel β-sheet (Figs. 1 ▸ and 3 ▸). With only one amino acid separating the two first cysteine residues, AaTI can be unambiguously placed in the group of nonclassical Kazal-domain inhibitors (Fink et al., 1986 ▸), displaying highest sequence identity to other mosquito Kazal-domain proteins (Fig. 1 ▸). From a structural point of view, AaTI is most similar to three other insect Kazal inhibitors: infestins 1 and 4 from Triatoma infestans and rhodniin from Rhodnius prolixus (Campos et al., 2012 ▸; van de Locht et al., 1995 ▸; Fig. 3 ▸). The conservation of an arginine residue (Arg8; Fig. 1 ▸) at position P1 of the putative reactive loop of AaTI is also noteworthy, reinforcing its observed preference for trypsin-like proteinases (Watanabe et al., 2010 ▸).
Figure 3.

Structural alignment of AaTI monomers with other Kazal-type inhibitors. The two AaTI molecules (molecule A in green, molecule B in blue) in the asymmetric unit were superposed using LSQMAN (Kleywegt, 1996 ▸). The N- and C-termini of molecule A are labelled. The AaTI monomers are superposed on the closest structural homologues, as identified by the DALI server (Holm & Rosenström, 2010 ▸): infestin 1 (pink; PDB entry 2f3c; Campos et al., 2012 ▸), infestin 4 (white; PDB entry 2erw; Campos et al., 2012 ▸) and the N-terminal domain of rhodniin (orange; PDB entry 1tbq; van de Locht et al., 1995 ▸). This figure was prepared with PyMOL (http://www.pymol.org).
Unsurprisingly, the two AaTI molecules in the asymmetric unit are very similar, with an r.m.s.d. of 0.61 Å for 48 aligned Cα atoms (Fig. 3 ▸). Indeed, all observable differences can be attributed to intrinsic flexibility (for example the N- and C-terminal regions) or crystal-packing effects. The electron-density maps are of very good quality, with all built residues defined, except for the distal portions (past the Cγ atom) of a few side chains (for example Leu36, Asn41 and Asn58 of molecule A). Since the flexible C-terminal region is more ordered in AaTI molecule A, it was used for all comparisons with other Kazal-inhibitors described below.
3.3. Inhibition specificity
Although AaTI does not inhibit the trypsin-like serine proteinases human plasma kallikrein or coagulation factors Xa and XIIa (Watanabe et al., 2010 ▸), it behaves as a competitive inhibitor of plasmin (K i = 3.8 nM; Watanabe et al., 2010 ▸) and bovine trypsin (K i = 0.15 nM; Watanabe et al., 2010 ▸), with a potency comparable to that of infestin 1–2 (K i = 3.1 nM; Campos et al., 2004 ▸). Superposition of the three-dimensional model of AaTI with that of infestin 1 in complex with bovine trypsin (PDB entry 2f3c; Campos et al., 2012 ▸) highlights the full conservation of the enzyme–inhibitor contacts, which is in perfect agreement with the observed inhibitory activity of AaTI towards trypsin. In contrast, although AaTI displays anticoagulant activity in the form of prolongation of the prothrombin time, activated partial thromboplastin time and thrombin time (Watanabe et al., 2010 ▸), its affinity for thrombin (K i = 320 nM; Watanabe et al., 2011 ▸) is four orders of magnitude lower than that of infestin 1–2 (K i = 0.025 nM; Campos et al., 2004 ▸). Indeed, superposition of the AaTI model on the N-terminal domain of rhodniin in complex with thrombin (PDB entry 1tbq; van de Locht et al., 1995 ▸) reveals significant structural differences between the two Kazal domains that would result in important steric clashes between AaTI and the 60-loop of thrombin: Ala11 and Pro26 in rhodniin are replaced by the bulkier Ile10 and Asp25, respectively (Fig. 1 ▸). The side chains of both residues would clash significantly with that of Trp60D from the 60-loop of thrombin, impacting binding (Figs. 4 ▸ a and 4 ▸ b), which is in line with the observed lower affinity of AaTI towards thrombin. In contrast, in the thrombin-inhibiting infestin 1 a single noteworthy replacement is observed, with a slightly larger valine residue occupying the position equivalent to Ala11 in rhodniin. Further, there are two other nonconservative replacements in AaTI that could impact its affinity towards thrombin: Asn23 and Leu27, which replace Ser24 and Thr28, respectively, of both rhodniin and infestin 1 (Fig. 1 ▸). The former would require a readjustment of the positions of Trp148 and Asn143 from the protease, while the latter would not only clash with Glu192 from thrombin, but the observed polar interaction with this residue would also not be established (Fig. 4 ▸ c). The steric clashes of AaTI with the active-site region of thrombin resemble those identified for LDTI, in which the replacement of bulky residues potentially colliding with the protuberant 60-loop of the proteinase, including Ile9 and Ser24 (Ile10 and Asp25 in AaTI), increased the affinity for thrombin by several orders of magnitude (Morenweiser et al., 1997 ▸).
Figure 4.

Details of the interaction between AaTI and thrombin. (a) AaTI (green) was docked into the active site of thrombin (grey) by superposing it on the N-terminal domain of thrombin-bound rhodniin (PDB entry 1tbq; van de Locht et al., 1995 ▸). The N- and C-termini of AaTI are labelled. (b) Close-up of the interactions between AaTI and the 60-loop of thrombin, with a superposed rhodniin N-terminal domain [AaTI and thrombin colours as in (a); rhodniin is shown as an orange ribbon]. Relevant side chains are represented as sticks and labelled. (c) Close-up of the interactions between AaTI and Glu192 and the autolysis loop of thrombin, with a superposed rhodniin N-terminal domain [colours as in (b)]. Relevant side chains are represented as sticks and labelled. This figure was prepared with PyMOL (http://www.pymol.org).
In the conformation that is observed in these crystals, the C-terminal tail of AaTI would point away from the proteinase–inhibitor interaction surface upon canonical binding to thrombin. However, given its flexible nature, it is conceivable that the C-terminal segment of AaTI could extend along the surface of the proteinase, reaching the proximal region of exosite I. This hypothesis lacks experimental support, since it has previously been shown that AaTI inhibits α-thrombin and exosite I-disrupted γ-thrombin with similar efficiency (Watanabe et al., 2011 ▸), in contrast to what is observed for other exosite I-targeting inhibitors (for example hirudin), which display a much decreased inhibition potency towards γ-thrombin (Ascenzi et al., 1992 ▸). In agreement with this, in the bidentate binding of rhodniin to thrombin a significant contribution towards the observed affinity is attributable to the interactions established by the C-terminal, exosite I-targeting domain of the inhibitor (van de Locht et al., 1995 ▸). The considerable level of sequence identity between infestin 1–2 and rhodniin suggests that the mechanism of binding to thrombin is also conserved in the two inhibitors (Campos et al., 2002 ▸). It is worth noting that grafting the acidic C-terminal region of hirudin onto LDTI converts it into an efficient thrombin inhibitor (Morenweiser et al., 1997 ▸), an effect that is cumulative with the mutation of bulky amino acids within the reactive--site loop. Together, these changes convert LDTI from a mediocre (K i > 300 nM) to a very good (K i = 16.0 pM) thrombin inhibitor.
On the other hand, this seemingly plausible canonical binding of AaTI to thrombin does not support an interaction between the C-terminal region of the inhibitor and exosite II of thrombin, as observed for haemadin (Richardson et al., 2000 ▸). Considering that the structural features of the reactive loop and the surrounding regions of AaTI can satisfactorily explain its relatively low affinity for thrombin, canonical binding to the proteinase is indeed the most plausible mechanism for thrombin inhibition by AaTI. This is further underscored by the observed mild effect of disruption of thrombin exosite I in inhibition by AaTI, as well as by the significant anticlotting activity of C-terminally truncated AaTI, as measured by a thrombin-time assay (Watanabe et al., 2011 ▸). The seemingly minor contribution of the C-terminal acidic tail of AaTI to the interaction with thrombin does not explain the presence of a similar segment in related inhibitors from other species (Fig. 1 ▸), and therefore a different physiological (and hitherto unidentified) role cannot be ruled out because this segment alone displayed an observable effect in the prolongation of the clotting time of plasma (Watanabe et al., 2011 ▸).
Supplementary Material
PDB reference: Kazal-type serine protease inhibitor from the dengue vector Aedes aegypti, 5dae
Acknowledgments
This work was supported by the Brazilian Synchrotron Light Laboratory (LNLS). We thank Dr Alexandre Tashima (Department of Biochemistry, Universidade Federal de São Paulo, Brazil) for mass-spectrometric analyses.
References
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221.
- Aliota, M. T., Peinado, S. A., Velez, I. D. & Osorio, J. E. (2016). Sci. Rep. 6, 28792. [DOI] [PMC free article] [PubMed]
- Araujo, R. N., Campos, I. T., Tanaka, A. S., Santos, A., Gontijo, N. F., Lehane, M. J. & Pereira, M. H. (2007). Int. J. Parasitol. 37, 1351–1358. [DOI] [PMC free article] [PubMed]
- Ascenzi, P., Amiconi, G., Coletta, M., Lupidi, G., Menegatti, E., Onesti, S. & Bolognesi, M. (1992). J. Mol. Biol. 225, 177–184. [DOI] [PubMed]
- Bode, W. & Huber, R. (1992). Eur. J. Biochem. 204, 433–451. [DOI] [PubMed]
- Bond, C. S. & Schüttelkopf, A. W. (2009). Acta Cryst. D65, 510–512. [DOI] [PubMed]
- Campos, I. T. N., Amino, R., Sampaio, C. A. M., Auerswald, E. A., Friedrich, T., Lemaire, H.-G., Schenkman, S. & Tanaka, A. S. (2002). Insect Biochem. Mol. Biol. 32, 991–997. [DOI] [PubMed]
- Campos, I. T. N., Souza, T. A. C. B., Torquato, R. J. S., De Marco, R., Tanaka-Azevedo, A. M., Tanaka, A. S. & Barbosa, J. A. R. G. (2012). Acta Cryst. D68, 695–702. [DOI] [PubMed]
- Campos, I. T. N., Tanaka-Azevedo, A. M. & Tanaka, A. S. (2004). FEBS Lett. 577, 512–516. [DOI] [PubMed]
- Chouin-Carneiro, T., Vega-Rua, A., Vazeille, M., Yebakima, A., Girod, R., Goindin, D., Dupont-Rouzeyrol, M., Lourenço-de-Oliveira, R. & Failloux, A. B. (2016). PLoS Negl. Trop. Dis. 10, e0004543. [DOI] [PMC free article] [PubMed]
- Corral-Rodríguez, M. A., Macedo-Ribeiro, S., Pereira, P. J. B. & Fuentes-Prior, P. (2009). Insect Biochem. Mol. Biol. 39, 579–595. [DOI] [PubMed]
- Corral-Rodríguez, M. A., Macedo-Ribeiro, S., Pereira, P. J. B. & Fuentes-Prior, P. (2010). J. Med. Chem. 53, 3847–3861. [DOI] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Fink, E., Rehm, H., Gippner, C., Bode, W., Eulitz, M., Machleidt, W. & Fritz, H. (1986). Biol. Chem. Hoppe Seyler, 367, 1235–1242. [DOI] [PubMed]
- Friedrich, T., Kröger, B., Bialojan, S., Lemaire, H. G., Höffken, H. W., Reuschenbach, P., Otte, M. & Dodt, J. (1993). J. Biol. Chem. 268, 16216–16222. [PubMed]
- Grütter, M. G., Priestle, J. P., Rahuel, J., Grossenbacher, H., Bode, W., Hofsteenge, J. & Stone, S. R. (1990). EMBO J. 9, 2361–2365. [DOI] [PMC free article] [PubMed]
- Gubler, D. J. (2002). Trends Microbiol. 10, 100–103. [DOI] [PubMed]
- Guimarães, B. G., Sanfelici, L., Neuenschwander, R. T., Rodrigues, F., Grizolli, W. C., Raulik, M. A., Piton, J. R., Meyer, B. C., Nascimento, A. S. & Polikarpov, I. (2009). J. Synchrotron Rad. 16, 69–75. [DOI] [PubMed]
- Hemmi, H., Kumazaki, T., Yoshizawa-Kumagaye, K., Nishiuchi, Y., Yoshida, T., Ohkubo, T. & Kobayashi, Y. (2005). Biochemistry, 44, 9626–9636. [DOI] [PubMed]
- Holm, L. & Rosenström, P. (2010). Nucleic Acids Res. 38, W545–W549. [DOI] [PMC free article] [PubMed]
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Kleywegt, G. J. (1996). Acta Cryst. D52, 842–857. [DOI] [PubMed]
- Langer, G., Cohen, S. X., Lamzin, V. S. & Perrakis, A. (2008). Nature Protoc. 3, 1171–1179. [DOI] [PMC free article] [PubMed]
- Laskowski, M. Jr & Kato, I. (1980). Annu. Rev. Biochem. 49, 593–626. [DOI] [PubMed]
- Locht, A. van de, Lamba, D., Bauer, M., Huber, R., Friedrich, T., Kröger, B., Höffken, W. & Bode, W. (1995). EMBO J. 14, 5149–5157. [DOI] [PMC free article] [PubMed]
- Lovato, D. V., Nicolau de Campos, I. T., Amino, R. & Tanaka, A. S. (2006). Biochimie, 88, 673–681. [DOI] [PubMed]
- Morenweiser, R., Auerswald, E. A., van de Locht, A., Fritz, H., Stürzebecher, J. & Stubbs, M. T. (1997). J. Biol. Chem. 272, 19938–19942. [DOI] [PubMed]
- Mühlhahn, P., Czisch, M., Morenweiser, R., Habermann, B., Engh, R. A., Sommerhoff, C. P., Auerswald, E. A. & Holak, T. A. (1994). FEBS Lett. 355, 290–296. [DOI] [PubMed]
- Noriega, F. G. & Wells, M. A. (1999). J. Insect Physiol. 45, 613–620. [DOI] [PubMed]
- Offermann, L. R., Bublin, M., Perdue, M. L., Pfeifer, S., Dubiela, P., Borowski, T., Chruszcz, M. & Hoffmann-Sommergruber, K. (2015). J. Agric. Food Chem. 63, 9150–9158. [DOI] [PMC free article] [PubMed]
- Paim, R. M. M., Araújo, R. N., Soares, A. C., Lemos, L. C., Tanaka, A. S., Gontijo, N. F., Lehane, M. J. & Pereira, M. H. (2011). Int. J. Parasitol. 41, 765–773. [DOI] [PubMed]
- Pape, T. & Schneider, T. R. (2004). J. Appl. Cryst. 37, 843–844.
- Rawlings, N. D., Barrett, A. J. & Finn, R. (2016). Nucleic Acids Res. 44, D343–D350. [DOI] [PMC free article] [PubMed]
- Ribeiro, J. M. (1995). Infect. Agents Dis. 4, 143–152. [PubMed]
- Richardson, J. L., Kröger, B., Hoeffken, W., Sadler, J. E., Pereira, P., Huber, R., Bode, W. & Fuentes-Prior, P. (2000). EMBO J. 19, 5650–5660. [DOI] [PMC free article] [PubMed]
- Rydel, T. J., Ravichandran, K. G., Tulinsky, A., Bode, W., Huber, R., Roitsch, C. & Fenton, J. W. II (1990). Science, 249, 277–280. [DOI] [PubMed]
- Sheldrick, G. M. (2010). Acta Cryst. D66, 479–485. [DOI] [PMC free article] [PubMed]
- Soares, T. S., Watanabe, R. M., Tanaka-Azevedo, A. M., Torquato, R. J., Lu, S., Figueiredo, A. C., Pereira, P. J. & Tanaka, A. S. (2012). Vet. Parasitol. 187, 521–528. [DOI] [PubMed]
- Sommerhoff, C. P., Söllner, C., Mentele, R., Piechottka, G. P., Auerswald, E. A. & Fritz, H. (1994). Biol. Chem. Hoppe Seyler, 375, 685–694. [DOI] [PubMed]
- Stubbs, M. T., Morenweiser, R., Stürzebecher, J., Bauer, M., Bode, W., Huber, R., Piechottka, G. P., Matschiner, G., Sommerhoff, C. P., Fritz, H. & Auerswald, E. A. (1997). J. Biol. Chem. 272, 19931–19937. [DOI] [PubMed]
- Takác, P., Nunn, M. A., Mészáros, J., Pechánová, O., Vrbjar, N., Vlasáková, P., Kozánek, M., Kazimírová, M., Hart, G., Nuttall, P. A. & Labuda, M. (2006). J. Exp. Biol. 209, 343–352. [DOI] [PubMed]
- Trillo-Muyo, S., Jasilionis, A., Domagalski, M. J., Chruszcz, M., Minor, W., Kuisiene, N., Arolas, J. L., Solà, M. & Gomis-Rüth, F. X. (2013). Acta Cryst. D69, 464–470. [DOI] [PMC free article] [PubMed]
- Vega-Rúa, A., Zouache, K., Girod, R., Failloux, A. B. & Lourenço-de-Oliveira, R. (2014). J. Virol. 88, 6294–6306. [DOI] [PMC free article] [PubMed]
- Waisberg, M. et al. (2014). PLoS Pathog. 10, e1004338. [DOI] [PMC free article] [PubMed]
- Watanabe, R. M., Soares, T. S., Morais-Zani, K., Tanaka-Azevedo, A. M., Maciel, C., Capurro, M. L., Torquato, R. J. & Tanaka, A. S. (2010). Biochimie, 92, 933–939. [DOI] [PubMed]
- Watanabe, R. M., Tanaka-Azevedo, A. M., Araujo, M. S., Juliano, M. A. & Tanaka, A. S. (2011). Biochimie, 93, 618–623. [DOI] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: Kazal-type serine protease inhibitor from the dengue vector Aedes aegypti, 5dae