

Abstract
A goal of human genetics studies is to determine the mechanisms by which genetic variation produces phenotypic differences that affect human health. Efforts in this respect have previously focused on genetic variants that affect mRNA levels by altering epigenetic and transcriptional regulation. Recent studies show that genetic variants that affect RNA processing are at least equally as common as, and are largely independent from, those variants that affect transcription. We highlight the impact of genetic variation on pre-mRNA splicing and polyadenylation, and on the stability, translation and structure of mRNAs as mechanisms that produce phenotypic traits. These results emphasize the importance of including RNA processing signals in analyses to identify functional variants.
The effects of natural genetic variation on gene expression are a major determinant of phenotypic variability between individuals. A surprising early result from genome-wide association studies (GWAS) was that approximately 88% of the associations found between genetic variation and heritable features were within non-coding regions of the genome, indicating that the genetic variants have effects primarily on regulatory sequences rather than on protein-coding sequences1. Until recently, investigations have largely focused on genetic variants that are associated with differences in mRNA levels as a result of effects on epigenetic and transcriptional regulation. However, studies that have analysed the effects of genetic variants on the full cascade of gene regulation have found equal contributions from variants that affect transcriptional mechanisms and those that affect post-transcriptional mechanisms2–4.
A role for RNA processing in translating genotype to phenotype is not surprising given that genes contain huge amounts of information in cis that functions at the levels of pre-mRNA processing (splicing and polyadenylation) and regulation of mRNA dynamics (translation, stability and mRNA localization) (FIG. 1). All pre-mRNAs of protein-coding genes undergo a basal level of RNA processing that requires cis-acting sequences that are recognized by the appropriate processing machinery. The majority of human genes contain introns, which are removed during splicing of the transcribed pre-mRNA, and all genes produce defined mRNA 3′ ends that require cis-acting element-dependent processing. In addition to this basal RNA processing, the vast majority of human genes have the capacity to undergo alternative splicing and selection of alternative polyadenylation sites to express multiple mRNAs that encode different protein isoforms and contain different 5′ and 3′ untranslated regions (5′ and 3′ UTRs). More than 30% of human genes have multiple first exons owing to alternative transcription start sites, up to 70% of genes have multiple polyadenylation sites, and more than 90% of genes undergo alternative splicing5–7. A large proportion of this differential pre-mRNA processing is regulated in a temporal and cell-specific manner or in response to physiological cues by using additional cis-acting regulatory sequences within introns and exons5–7.
Figure 1. Genetic variation alters gene output by affecting RNA processing.

Genetic variants affect multiple steps of RNA processing and mRNA dynamics, including splicing, 3′ end processing, mRNA structure and stability, translation efficiency and regulation by RNA-binding proteins and by microRNAs (mi RNAs). Genetic variants that disrupt either cis-acting elements or key RNA secondary structures have downstream consequences in terms of protein composition and expression levels. UTR, untranslated region.
The spliced and polyadenylated mRNA is exported to the cytoplasm, where it undergoes dynamic regulation by factors that modulate translation, decay and intracellular localization to determine quantitative, temporal and spatial control of protein output. Each aspect of post-transcriptional regulation is mediated by cis-acting elements in mRNA 5′ UTRs, 3′ UTRs and coding regions8 (BOX 1). The sequence of the cis-acting elements within the pre-mRNA and mRNA determines the binding affinity of trans-acting factors such as RNA-binding proteins or complementary microRNAs (miRNAs). RNA structure also has a major role in determining RNA function by creating or blocking binding sites for trans-acting factors or by increasing the proximity of crucial distal nucleotides within the linear molecule. All of these cis-acting elements, whether binding sites or structural, and whether required for basal processing or for modulating alternative pathways, are potential targets of genetic variants that modify phenotypic traits.
Box 1. Identifying the sequence requirements for RNA processing.
To fully understand the impact of genetic variation on phenotype, a completely annotated human genome needs to include the cis-acting elements that mediate basal and regulated mRNA formation, function and fate. Computational and experimental efforts to annotate the cis-acting elements that are required for RNA processing are ongoing, such as in the ENCODE (Encyclopedia of DNA Elements) project, and include enhanced approaches to map genome-wide interactions of RNA-binding proteins120. However, a comprehensive accounting of RNA processing elements with predictive capabilities remains a challenge. Individual RNA processing elements have little information content, as RNA-binding proteins typically recognize variable binding motifs of only 5–6 nucleotides. Similarly, trans-acting RNAs bind to short complementary regions (6–7 nucleotides for small nuclear RNAs and 7–8 nucleotides for microRNA (miRNA) seed sequences). This low-information content of individual RNA processing elements is compensated for by the clustering of elements and combinatorial recognition by different trans-acting factors through multiple weak interactions, leading to the assembly of an active RNA processing complex. Predictive capabilities are further challenged by the fact that different transcripts can vary widely in the dominant features that determine the efficiency of the same processing events. For example, efficient splicing of some exons relies largely on splice site strength, whereas other exons require auxiliary splicing elements within the exon or flanking introns.
Despite these challenges, a huge variety of genome-wide, computational and experimental high-throughput approaches continue to identify RNA cis-acting elements, RNA secondary structures and trans-acting factors that mediate pre-mRNA processing and mRNA function. In particular, deep-sequencing approaches have provided a genome-wide accounting of the cis-acting elements that are required for basal and regulated splicing, polyadenylation, and mRNA stability, translation efficiency and secondary structure80,121–123. In addition, deep-sequencing approaches using the same cells or tissue to correlate genomic variation and processing differences within the transcriptome can pinpoint genetic variants that affect RNA processing. These studies provide mutually beneficial information that identifies both novel RNA processing elements and potential functional variants.
Defective RNA processing is a well-recognized cause of disease owing to mutations that either disrupt cis-acting elements within individual genes or affect components of the RNA processing machinery in trans to alter the expression of multiple genes9,10. The RNA processing mutations that cause highly penetrant Mendelian diseases are at the severe end of a spectrum of functional variants in RNA processing that produce a gradation of phenotypic effects (FIG. 2). Recent studies indicate that RNA processing variants have a substantial impact on more common and less severe differences between individuals in terms of disease risk, disease severity, prognosis and therapeutic response, while not directly causing disease. This Review highlights the prevalence of genetic variation that acts through effects on RNA processing as a major driver of phenotypic variability. The finding that genetic variants that affect RNA processing are as abundant as those that affect transcription suggests that both types of variant make an equal contribution to phenotypic diversity. We focus on protein-coding genes, with an emphasis on germline variants that are associated with phenotypic variability rather than overt disease causation. Another area of interest that is not discussed in this Review, owing to space limitations, is somatic variants associated with transformation and metastasis that affect RNA processing.
Figure 2. Genetic variants that affect RNA processing can have a range of effects on human health.

These effects range from benign or unclassified variants to pathological variants that cause monogenic diseases. Selected examples of functional genetic variants that affect RNA processing are depicted, which have a range of consequences, such as causing disease50,132,133, altering prognosis or therapeutic response92,134,135, modifying disease severity136–138 or modifying disease risk67,139,140. BMP1, bone morphogenetic protein 1; CDKAL1, CDK5 regulatory subunit-associated protein 1-like 1; CFTR, cystic fibrosis transmembrane conductance regulator; ERCC5, ERCC excision repair 5, endonuclease; GWAS, genome-wide association studies; HMBS, hydroxymethylbilane synthase; HNRNPH1, heterogeneous nuclear ribonucleoprotein H1; ICAM1, intercellular adhesion molecule 1; IRGM1, immunity-realted GTPase M member 1; miRNA, microRNA; MPZ, myelin protein zero; OLR1, oxidized low-density lipoprotein receptor 1; OPRM1, opioid receptor mu 1; pri, primary; SNV, single-nucleotide variant.
Impact of genetic variation on pre-mRNA splicing
Ninety-four per cent of human protein-coding genes contain introns that must be spliced during pre-mRNA processing to generate functional mRNAs. In addition, the majority of human genes produce multiple mRNA isoforms via alternative splicing. Therefore, most genes are susceptible to individual variation of pre-mRNA splicing or its regulation. In this section, we review the cis-acting elements that are required for splicing, and highlight the basis for individual differences in splicing that are due to common allelic variants and the consequences for phenotypic diversity.
Cis-acting elements for basal and regulated splicing
Splicing efficiency is determined by multiple features of pre-mRNAs that are potential targets for functional genetic variants, including the sequence and secondary structure of cis-acting elements that are bound by trans-acting factors. Approximately 50% of the information required for exon recognition during splicing is determined by the consensus 5′ and 3′ splice sites11 (FIG. 3). The consensus splice sites include the first and last two nucleotides of individual introns (GT–AG, functioning as GU–AG in the pre-mRNA), which are highly conserved and essential for splicing. Analyses to identify the effects of sequence variants on splicing are typically limited to changes in one of the four GT–AG nucleotides. However, variation at other positions of the consensus splice site, including the branch site, polypyrimidine tract and the additional nucleotides of the 5′ splice site (FIG. 3), can also affect splicing efficiency. The effects of such variants are generally less severe than the effects of GT–AG variants, and they typically have residual splicing activity, which makes them prime candidates to produce phenotypic consequences that are below the threshold for penetrant disease12.
Figure 3. Cis-acting splicing elements are not restricted to exon–intron boundaries.

Consensus and auxiliary splicing elements are required for efficient and regulated pre-mRNA splicing. The consensus splice site sequences are shown below the pre-mRNA. The GT (GU in the pre-mRNA) and AG dinucleotides at either end of introns are crucial for splicing, but substitutions of other nucleotides within the consensus splicing elements, such as in the branch site or polypyrimidine tract, can also have an effect on splicing. Examples of auxiliary splicing element motifs within exons and introns are shown above the pre-mRNA, together with the RNA-binding proteins that bind to these motifs. ESE, exonic splicing enhancer; HNRNPA1, heterogeneous nuclear ribonucleoprotein A1; ISE, intronic splicing enhancer; ISS, intronic splicing silencer; RBFOX1, RNA-binding protein, fox-1 homologue 1; SRSF, serine/arginine-rich splicing factor. Sequence logos for auxiliary splicing elements for the RNA-binding protein SRSF1, SRSF2, HNRNPA1 and RBFOX1 are from REF. 141, and for the consensus splice sites are from REF. 142 and http://weblogo.berkeley.edu/examples.html.
In addition to the consensus splice sites, exons and introns contain auxiliary splicing elements of 6–8 nucleotides that either enhance (exonic splicing enhancer (ESE) and intronic splicing enhancer (ISE)) or repress (exonic splicing silencer (ESS) and intronic splicing silencer (ISS)) use of the associated splice site (or sites) and prevent inappropriate splicing to cryptic splice sites (FIG. 3). Computational studies predict that most exons contain auxiliary splicing elements that are enriched near the exon–intron boundaries13. Analysis of the cis-acting elements that are involved in tissue-specific splicing by computational approaches has delineated a splicing code capable of predicting cell-specific patterns14,15. However, our understanding of cis-acting splicing elements remains incomplete, particularly elements that contribute to the ubiquitous recognition of constitutive exons.
As exonic splicing elements overlap with protein-coding information, both synonymous and non-synonymous variants can have phenotypic effects owing to altered splicing16–18. A phenotypic effect of a non-synonymous variant is most likely to be due to the consequences of the amino acid substitution rather than an effect on splicing. However, a recent analysis concluded that 22% of human disease alleles that were designated as missense mutations could affect splicing, just as other studies have shown that synonymous variants can cause disease by altering splicing19,20. Given that exome sequencing studies typically identify more than 30,000 variants per individual, half of which are equally split between non-synonymous and synonymous changes21–23, there are likely to be extensive individual differences in splicing efficiency owing to exonic variation that subsequently lead to phenotypic differences.
Extensive splicing variation is associated with genetic variants
It is well established that rare genetic variants cause inherited disease by affecting cis-acting elements that are required for splicing12,20. For example, an estimated 35% of disease-causing point mutations disrupt splicing; 15% of these mutations are in the consensus splice sites and 20% are in exonic or intronic auxiliary splicing elements19,24. In addition to the marked and penetrant phenotypes conferred by pathological splicing mutations, recent studies indicate that genetic variants affecting splicing with more moderate consequences contribute to a broad range of phenotypic variation. Investigations using matched transcriptome data (RNA sequencing (RNA-seq) or splicing-sensitive microarrays) and genome or exome sequencing to investigate splicing as a mechanism for generating functional variants have led to two unexpected conclusions. First, the extent to which genetic variants that alter splicing ratios (splicing quantitative trait loci (sQTLs)) affect phenotypic traits is equal to or even greater than the effects of expression variants (expression QTLs (eQTLs)). Second, although eQTLs can have secondary effects on splicing owing to the co-transcriptional nature of splicing, the majority of genetic variants that affect splicing are distinct from those that affect gene expression2–4,25. Importantly, a large fraction of sQTLs are within the open reading frame — 89% in one study3 — and thereby potentially affect protein function. The sQTLs identified thus far primarily affect cis-acting elements within individual genes rather than the trans-acting splicing machinery. Disease-causing variants within the splicing machinery have been identified10,12, and a variant affecting transcription of an alternative splicing factor has been shown to affect the splicing of multiple genes26, but genome-wide identification of trans-acting sQTLs must await future systematic analyses.
Several earlier studies using splicing-sensitive micro arrays in HapMap-derived lymphoblastoid cell lines (HapMap-derived LCLs) showed widespread associations between genetic variants and splicing differences2,27–29. In particular, an analysis of family trios found that tens of thousands of exons showed heritable differences in splicing and that more than 1,000 of these exons were associated with cis-acting single-nucleotide variants (SNVs) that strongly correlated with splicing effects27.
These findings have been expanded in several recent analyses, using matched genome and transcriptome deep sequencing data to directly correlate specific genomic variants with splicing differences3,4,25. An analysis that compared genome sequence and RNA-seq data from LCLs and tissues from the GEUVADIS and GTEx projects found that a large number of splicing changes were due to variation in multiple regions of the splice site sequences as well as within exons30. In another study, analysis of ENCODE Encyclopedia of DNA Elements) RNA-seq data from human cell lines identified more than 600 exons for which splicing differences were associated with intronic and exonic SNVs31. Hypothesizing that a subset of SNVs affected binding of auxiliary splicing factors, the investigators found that the binding motif of serine/arginine-rich splicing factor 1 (SRSF1) had the highest frequency of disruptive SNVs that were associated with altered splicing. Gel shift assays of the intronic allelic variants showed that there were SNV-dependent differences in SRSF1 binding. In addition, analysis of global cross-linking immunoprecipitation (CLIP) data demonstrated a significant allelic bias of SRSF1 binding, which strongly suggests that individual variation in the intronic SRSF1-binding site affects binding affinity and thus results in altered splicing31. These results provide an example of genetic variants within auxiliary cis-acting elements, separate from the consensus splice sites, that promote individual differences in splicing regulation and the ratios at which alternative mRNAs are expressed.
Contributions of splicing variation to complex disease
Having established that there is extensive variability in pre-mRNA splicing that is linked to genetic variation, several studies have gone further to demonstrate associations between disease risk and genetic variants that affect splicing14,25,32–36. There is a growing number of individual genes for which variant-specific splicing patterns have been directly linked with disease susceptibility, including splice variants of oxidized low-density lipoprotein receptor 1 (OLR1) and low-density lipoprotein receptor (LDLR) that are linked with coronary artery disease and splice variants of interleukin-7 receptor (IL7R) that are linked with multiple sclerosis37–39. However, broad-based evidence for the role of pre-mRNA splicing in disease risk comes from GWAS that have identified common SNVs that affect splicing and disease trait loci. For example, the study noted above that identified SNV-dependent SRSF1 binding found that among the 600 exons for which genetic variants correlated with altered splicing, 100 were in linkage disequilibrium with disease-associated GWAS loci31. In the study cited above that identified inherited splicing differences in 1,000 exons, cross-referencing these exons to GWAS loci identified 73 SNVs that were consistently associated with disease traits27. Given that the data in this study were derived from LCLs, it was of particular interest that these splicing-associated SNVs were enriched in genes associated with immune cell function, such as protein tyrosine phosphatase non-receptor type 2 (PTPN2), C-type lectin domain family 2 member D (CLEC2D) and interferon regulatory factor 5 (IRF5), and were associated with autoimmune diseases, including Crohn disease, type 1 diabetes and rheumatoid arthritis, respectively. For several genes, the functions of the predicted protein isoforms were consistent with activities relevant to altered immune cell functions.
An expansive study using a machine-learning approach to predict the impact of SNVs on the splicing code identified widespread disease-associated effects14. The analysis identified more than 20,000 SNVs that are predicted to alter splicing. SNVs located close to the splice sites were most common but, importantly, not all of the identified SNVs were in the consensus splicing sequence; 465 intronic SNVs were more than 30 nucleotides from a consensus splice site, which is indicative of uncharacterized splicing elements. Intronic disease-causing SNVs were predicted to be nine times more likely to disrupt splicing compared with local SNVs not associated with disease. Similarly, exonic synonymous disease-causing SNVs were nine times more likely to disrupt splicing than common SNVs. The model was used to evaluate the potential roles of SNVs that affect splicing in autism spectrum disorder and predicted the mis-splicing of six new candidate genes. Although this study focused on the identification of SNVs that cause disease, it can be extrapolated from the results that a large number of splicing-associated variants have determinative effects on less severe complex traits.
A systematic analysis of the effects of genetic variation on the full gene expression cascade from chromatin to protein expression was the most recent study to conclude that common genetic variants that affect splicing are major contributors to complex traits3. The study used LCLs from more than 60 individuals for whom genomic sequences were available and quantified the individual steps of gene expression, including chromatin and DNA modification, transcription rate, pre-mRNA splicing, mRNA decay, translation rate and protein levels. By overlapping GWAS results with variants that affect each step of gene expression, they found that the association between disease risk and variants affecting splicing was equal to or even greater than the association observed between disease risk and variants affecting gene expression3.
Now that large-scale molecular and genetic association studies have established the impact of splicing on disease risk, the stage is set to identify the mechanisms by which common individual splicing differences translate to phenotypic variability that is relevant to human health.
Impact of genetic variation on mRNA 3′ ends
Pre-mRNA processing includes formation of the 3′ end of the mature mRNA, which is crucial for gene expression. For the majority of protein-coding genes, the 3′ end is formed by consecutive cleavage and polyadenylation that occurs co-transcriptionally. The two major consensus sequences that are required for interactions with the polyadenylation machinery are an essentially invariant hexanucleotide signal (AATAAA, functioning as AAUAAA in the pre-mRNA), which is located 20–40 nucleotides upstream of the co-transcriptional cleavage site, and a variable GT (GU)-rich element located within 50 nucleotides downstream of the cleavage site40. In addition to basal 3′ end processing, up to 70% of human genes express mRNAs with different 3′ ends owing to alternative polyadenylation (APA)5–7. APA occurs either by selection of a distal versus a proximal polyadenylation site within the same terminal exon (tandem 3′ APA), which affects the 3′ UTR length, or by alternative splicing to one of multiple terminal exons (intronic APA), which provides different 3′ UTRs and often different carboxy termini. In this section, we highlight the evidence that genetic variants affecting both mechanisms of APA are prevalent, have substantial consequences for gene output and are associated with disease risk.
mRNA 3′ end processing is sensitive to genetic variation
Although reported frequencies differ between studies, genome-wide analyses consistently find substantial effects of genetic variants on mRNA 3′ end formation. One study using combined microarrays and HapMap LCLs found that, of the top 20 SNVs that have heritable effects on the ratio of isoforms generated by APA, 16 (80%) affected 3′ end formation27. A separate analysis of LCLs from more than 450 individuals identified 639 genes in which SNVs were associated with altered transcript isoform ratios, and 43% of these SNVs affected the mRNA 3′ end4. Whole-genome sequencing analysis of 179 unrelated individuals indicates that the 3′ UTR is under higher selective pressure than other non-coding gene domains, including the 5′ UTR and introns41. Genetic variants in the 3′ UTR are significantly more likely to be associated with changes in mRNA levels than are variants within introns, which is consistent with the presence of functional cis-acting elements in the 3′ UTR41,42. Together, these studies indicate that mRNA 3′ end formation is a common target by which genetic variants alter terminal exon splicing or polyadenylation site usage, with subsequent effects on the inclusion or exclusion of regulatory motifs within 3′ UTRs.
Variants in polyadenylation and disease risk
Given the prevalence of regulatory cis-acting elements in the 3′ UTRs of mRNAs, it is not surprising that genetic variants affecting APA are often associated with phenotypic changes. A substantial proportion of genetic variants with demonstrated functional effects on 3′ UTR length function by disrupting the consensus hexanucleotide AATAAA. Except for the functional variant ATTAAA, any single-nucleotide substitution of the consensus hexa-nucleotide reduces the efficiency of mRNA 3′ end formation by at least 70%43. Changes in the hexanucleotide motif are well recognized as a cause of disease, such as the historic example of an inactivating mutation in the polyadenylation hexanucleotide of haemoglobin subunit alpha 2 (HBA2) that causes α-thalassemia44; furthermore, altered polyadenylation is increasingly recognized as a risk factor and modifier of complex diseases. A notable example involves susceptibility to facioscapulohumeral muscular dystrophy (FSHD), in which individual differences in the polyadenylation hexanucleotide sequence of the last exon of double homeobox 4 (DUX4) are determinative for disease (BOX 2). Analysis of allele-specific expression in LCLs from six individuals revealed that variants within the hexanucleotide are significantly more likely to have an effect on gene expression than variants in the rest of the 3′ UTR, and that single-nucleotide substitutions are sufficient to change the use of distal versus proximal poly adenylation sites45. A detailed analysis of blood samples from 94 individuals to find SNVs in linkage disequilibrium with a change in polyadenylation site usage identified 5 top candidates, all of which were in the AATAAA motif. This analysis also identified seven APA-associated SNVs with disease associations in the GWAS database46.
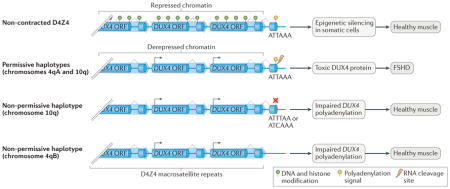
Box 2. The role of nucleotide polymorphisms in susceptibility to FSHD.
Facioscapulohumeral muscular dystrophy (FSHD) is a progressive, autosomal-dominant disorder that affects the facial and upper extremity muscles. Pathogenesis is due to chromatin relaxation at D4Z4 macrosatellite arrays located on chromosomes 4q and 10q. Each 3.3-kb D4Z4 repeat contains the open reading frame (ORF) of the double homeobox 4 (DUX4) retrogene. Only the last repeat in the array contains the terminal exon that provides the signals required for mRNA 3′ end formation and mRNA expression. DUX4 encodes a homeobox transcription factor that is expressed in germ cells and is epigenetically silenced in somatic cells. Derepression and expression of DUX4 is toxic to muscle and is the primary cause of muscle degeneration in FSHD124. In patients with FSHD1, the D4Z4 macrosatellite on chromosome 4q is derepressed owing to contraction of the repeat number to fewer than 11 (REF. 124). FSHD2 affects 5% of patients with FSHD and is caused by mutations in the structural maintenance of chromosomes flexible hinge domain-containing 1 (SMCHD1) gene, which is an epigenetic modifier of D4Z4 repeats, allowing DUX4 expression from non-contracted arrays on chromosomes 4q and 10q125. Importantly, derepressed chromatin alone is not sufficient for DUX4 expression, as there are permissive and non-permissive FSHD alleles on chromosomes 4q and 10q. Through rigorous genetic analyses, the primary determinant of permissive and non-permissive alleles was found to be the presence or absence, respectively, of a functional polyadenylation site in the last exon of the DUX4 gene126 (see the figure). The permissive 4q (designated 4qA) and 10q alleles contain an ATTAAA hexanucleotide polyadenylation signal, whereas non-permissive 10q alleles contain one nucleotide substitution within the hexanucleotide signal (ATTTAA or ATCAAA) that prevents DUX4 mRNA 3′ end chromosome 4q (designated 4qB) lacks a 68-bp region containing the polyadenylation signal. Therefore, a single-nucleotide variant within a polyadenylation site provides protection from DUX4 expression in individuals harbouring derepressed DUX4 chromatin.
Genetic variation acting on polyadenylation has emerged as a key risk factor and disease modifier in systemic lupus erythematosus (SLE). The SLE-associated risk allele rs10954213 disrupts the proximal polyadenylation hexanucleotide motif of the inflammation-associated gene IRF5. This SNV shifts polyadenylation from the proximal to the distal site and thereby produces a comparatively unstable IRF5 transcript with a longer 3′ UTR that contains a destabilizing AU-rich element (ARE). This results in reduced levels of IRF5 protein, the consequences of which are proposed to be relevant to increased SLE risk45,47,48. In another lupus-associated example, the rs6598 SNV in the proximal polyadenylation hexanucleotide site of GTPase IMAP family member 5 (GIMAP5) marks the risk haplotype for SLE and shifts polyadenylation to alternative sites49. Patients with SLE who carry this allele are at a higher risk of developing thrombocytopaenia, a complication of SLE that increases morbidity49. Similar SNVs that disrupt the conserved polyadenylation signals in bone morphogenetic protein 1 (BMP1) and insulin (INS) cause bone fragility50 and neonatal diabetes51, respectively. A germline SNV that disrupts the sole AATAAA polyadenylation signal in TP53 is associated with increased susceptibility to various cancers52.
Genetic variation in the GT (GU)-rich downstream element required for polyadenylation is also associated with disease risk. Fibrinogen gamma chain (FGG) mRNA undergoes APA to generate FGG-γA and FGG-γB isoforms, which differ in the carboxyl termini. An SNV within the downstream element strengthens the distal polyadenylation site that encodes FGG-γA, resulting in an altered FGG-γA:FGG-γB ratio that is associated with an increased risk of deep vein thrombosis53.
Whereas the consensus elements for basal polyadenylation are well established, the cis-acting elements that are required for regulated APA are largely unknown. Full annotation of the cis-acting elements that modulate APA is likely to uncover additional motifs associated with disease traits.
Variants affect miRNA-mediated regulation
An estimated 60% of protein-coding genes are targets of regulation by mi RNAs54. Furthermore, a single miRNA is estimated to alter the expression of more than 100 target mRNAs, and a single mRNA can be regulated by several different mi RNAs55–57. Here, we discuss evidence that genetic variation affects miRNA function and hence gene expression by two mechanisms — by altering miRNA expression levels or by altering miRNA–mRNA interactions — with subsequent effects on physiological traits.
Genetic variation modulates miRNA expression
Genome-wide analyses have revealed patterns of heritable variability in miRNA expression between individuals58. Cis-acting miRNA QTLs (cis-mirQTLs) are generally located in the transcription regulatory regions of the miRNA locus58,59. For example, an SNV that is associated with a risk of schizophrenia in the enhancer of mir-137 weakens binding of the transcription factor YY1, leading to decreased miR-137 expression60. The relatively high prevalence of cis-mirQTLs is illustrated by one study in which RNA-seq of small RNAs of more than 450 genetically diverse human LCLs from the 1000 Genomes Project revealed that 60 of the 644 expressed mi RNAs that were detected (approximately 9%) contained cis-mirQTLs4. A larger study of more than 5,000 individuals indicated that as many as 27% of mi RNAs expressed in whole blood are affected by cis-mirQTLs58. An estimated 20% of mirQTLs are also mRNA eQTLs, with a subset of cis-mirQTLs shown to be associated with altered mRNA expression of their target genes, which indicates that a proportion of individual variation in mRNA levels might be secondary to differences in miRNA expression4,58,61,62. In addition to cis-mirQTLs, association studies have provided evidence that SNVs affecting the expression of genes involved in primary miRNA (pri-miRNA) and precursor miRNA (pre-miRNA) processing — such as DICER1, SMAD3 and DGCR8 — can have trans-acting effects on miRNA expression61.
Individual differences in miRNA expression level that are due to mirQTLs are associated with complex disease traits. A large study of miRNA expression in the livers of more than 400 patients with obesity revealed a significant association between blood lipid levels and SNVs that alter the expression levels of mir-128-1 and mir-148a, which is similar to previous studies linking mirQTLs in mir-125b-5p and mir-339-3p to total cholesterol levels58,59. Additional mirQTLs are associated with body mass index, Parkinson disease and amyotrophic lateral sclerosis61. These GWAS-identified associations between disease traits and miRNA expression levels warrant additional study to identify the specific contributions of mirQTLs to pathogenesis.
Altered miRNA–mRNA interactions are associated with disease
Genetic variants can disrupt miRNA–mRNA interactions either by altering the presence of the miRNA-binding site in the mature mRNA as a result of APA, which affects 3′ UTR length or terminal exon identity, or by directly altering the sequence of the miRNA-binding site in the mRNA. Loss or gain of miRNA-mediated regulation of mRNAs has been proposed to link changes in APA with altered gene output63. Consistent with this proposal, regulatory cis-acting elements, including miRNA-binding sites, are enriched between proximal and distal polyadenylation sites45. In a GWAS using HapMap data across multiple populations, SNVs that shortened the mRNA 3′ UTR and caused a loss of miRNA-binding target sites were associated with increased gene expression63. Similarly, an analysis of seven mRNAs containing SNVs affecting 3′ UTR length found that more than 40% of miRNA-binding sites were lost in switching from a long to a short 3′ UTR64. The alternative scenario is also supported: an SNV associated with susceptibility to colorectal cancer that is found in the proximal polyadenylation site of the DNA methylation-associated gene DIP2B shifts poly adenylation to favour the distal polyadenylation site, creating a longer 3′ UTR45,65. The distal region contains a miR-101-binding site, which has been shown by mutation analysis to mediate degradation of the DIP2B long isoform45. As is the case for most examples of SNV–phenotype associations, the mechanistic relationship between cancer susceptibility and polyadenylation site selection of DIP2B remains to be established.
In addition to variants that alter 3′ UTR length, SNVs that alter miRNA-binding sites affect both mRNA and protein levels. The Crohn disease risk allele c.313C>T is a synonymous SNV in the immunity- related GTPase M (IRGM) gene, which encodes a protein involved in autophagy, and this SNV disrupts a miR-196-binding site66,67. Expression of mir-196 is upregulated in actively inflamed mucosa from individuals with Crohn disease, and this is associated with downregulation of IRGM expression in individuals homozygous for the protective allele (c.313C) but not in individuals possessing the risk allele (c.313T). This inflammation-dependent, allele-specific loss of miRNA-mediated regulation of IRGM expression leads to increased levels of intracellular bacteria that are associated with Crohn disease, an effect that is secondary to IRGM-mediated autophagy defects67. Similarly, a single-nucleotide insertion or deletion (indel) polymorphism disrupts binding of miR-148a in the 3′ UTR of the mRNA encoding human leukocyte antigen C (HLA-C), a member of the highly polymorphic HLA family that has a key role in the immune response to HIV. HLA-C alleles containing this indel escape miRNA-mediated repression, leading to higher cell surface levels of HLA-C that are associated with significantly lower viral load68. Other miRNA–mRNA interactions that are affected by genetic variants that cause reduced miRNA binding and increased target protein expression include: the miR-485-5p–APOA5 interaction as a risk factor for hyper triglyceridaemia, the hsa-miR-671-5p–BCL2L12 interaction as a risk factor for melanoma, the miR-433–FGF20 interaction as a risk factor for Parkinson disease in some populations and various miRNA–mRNA interactions that are associated with cardiometabolic phenotypes62,69–72.
Variants shift the balance of RNA dynamics
In addition to miRNA-mediated repression, mRNAs undergo regulation that is mediated by RNA-binding proteins to modulate gene output. In this section, we discuss evidence that genetic variants can alter RNA stability, translation efficiency and localization, and we highlight the importance of RNA structure in each of these processes (FIG. 4).
Figure 4. Genetic variants that create or abolish key RNA secondary structures affect multiple aspects of post-transcriptional regulation.

RNA-binding proteins rely on sequence and structural information for binding to their target cis-acting elements. a | Secondary RNA structures created by a genetic variant can prevent access of a microRNA (miRNA) within the RNA-induced silencing complex (RISC), thereby affecting the level of miRNA-mediated repression. b | Similarly, the creation of a novel RNA structure by a single-nucleotide variant can prevent binding of an RNA-binding protein to its cognate site. Alternatively, the affinities of some RNA-binding proteins require a specific RNA structure, and genetic variants that disrupt this structure can decrease binding of the trans-acting factor. As outlined in the text, sequence-specific interactions of RNA with RNA-binding proteins are crucial for appropriate basal and regulated RNA processing steps; their disruption can result in altered RNA splicing, stability or localization. c | Genetic variants that alter RNA structure within the coding region can affect the translation rate by impairing ribosome progression.
Variants in stability motifs affect mRNA half-life
mRNA levels are determined by the balance between transcription and decay. SNVs that alter transcription output have been investigated more than genetic variants that affect mRNA stability. However, a broad analysis in LCLs identified 195 genetic variants that are associated with differential rates of RNA decay, which indicates that RNA stability makes a substantial contribution to variation in gene output73. For example, an SNV in the 3′ UTR of the mRNA encoding the adhesion protein corneodesmosin (CDSN) confers a twofold increase in mRNA stability and is significantly associated with increased susceptibility to psoriasis74. This variant is immediately downstream of an RNA stability motif and results in reduced affinity of an unidentified 39 kDa cytoplasmic protein compared with the non-risk allele, which suggests that the SNV confers differential binding of a putative RNA destabilization factor74. SNVs within stability motifs are likely candidates to alter mRNA half-life, and variants that induce mechanisms of transcript surveillance, such as nonsense-mediated decay, nonstop decay or no-go decay, are also potential sources of functional variation75.
RNA stability is closely associated with RNA structure
RNA forms complex secondary structures through intramolecular base pairing that affect both RNA processing and RNA functionality76. Using various transcriptome-wide structural interrogation techniques — including psoralen analysis of RNA interactions and structures (PARIS), ligation of interacting RNA followed by high-throughput sequencing (LIGR–seq) and in vivo click selective 2′-hydroxyl acylation and profiling experiment (icSHAPE) — studies can effectively address the role of RNA structure in post-transcriptional gene regulation77–79. A seminal study identified distinct structural signatures of mRNAs that define splice junctions, CLIP-validated miRNA-binding sites and translation initiation sites, which suggests that RNA structure influences multiple steps in post-transcriptional gene regulation80. In this study, bioinformatic analysis of the RNA secondary structure landscape across a family trio indicated that 15% of transcribed SNVs alter local RNA structure and that the majority of these variant-induced structural changes were heritable80. SNVs that alter RNA structure are termed riboSNitches, signifying that the variant causes an allele-specific change in RNA structure that affects the function of the RNA and ultimately can contribute to disease81. Pathological SNVs that are associated with retinoblastoma, hyperferritinaemia cataract syndrome and cartilage-hair hypoplasia are thought to cause disease through their demonstrated effects on RNA structure82–84. An even larger number of riboSNitches are likely to be modifiers of disease severity or disease risk; SNVs with demonstrated effects on RNA structure are associated with an increased risk of various diseases, including Parkinson disease, asthma and metabolic syndrome80. Genome-wide analyses show that riboSNitches are depleted near the predicted binding sites for mi RNAs and RNA-binding proteins, whereas they are enriched near exon–exon junctions that exhibit altered splicing ratios. These results suggest that a subset of riboSNitches have functional consequences with a strong potential impact on phenotype80.
The primary effect of riboSNitches is often at the level of RNA stability. The synonymous SNV C957T in the coding sequence of dopamine receptor D2 (DRD2) causes a marked shift in the predicted folding of the DRD2 mRNA, which results in a decreased mRNA half-life and reduced DRD2 protein levels85. This effect was annulled in mRNA carrying another synonymous variant, SNV G1101A, that restores the RNA structure, which shows the compensatory effect of 1101A on RNA structure and mRNA stability. In another example, an SNV in the 3′ UTR of polo-like kinase 1 (PLK1), which encodes a cell cycle regulator, alters the predicted mRNA secondary structure and increases PLK1 mRNA stability86. Given that PLK1 is overexpressed in many types of cancer, this variant may be functionally relevant in terms of cancer risk. RiboSNitches can also affect the accessibility of miRNA-binding sites64,87,88. In a GWAS of 3′ UTR SNVs associated with mRNA expression levels screened against CLIP-validated miRNA-binding sites, about 5% of the variants (14 SNVs) were predicted to markedly alter the secondary structure of miRNA-binding sites, with corresponding effects on binding affinity of the RNA-induced silencing complex (RISC)64.
A link between SNVs, RNA structure and translation
Separate studies using GWAS and quantitative mass spectrometry of LCLs indicate that 10–50% of QTLs that alter protein levels (protein QTLs) do not affect mRNA expression levels, which suggests that genetic variation can independently affect translation efficiency or protein stability89,90. There are several examples of SNVs that create an upstream open reading frame within the 5′ UTR of an mRNA and affect translation efficiency of the encoded protein product91,92. An analysis in LCLs, which identified thousands of novel open reading frames, also identified more than 300 SNVs that influenced the ratio of translation at the alternative versus main open reading frames93. Global analysis of ribosome occupancy in LCLs has identified further SNVs that contribute to individual differences in translation by altering the presence of the upstream open reading frame or by disrupting the Kozak sequence, a key region near the start codon, often independently of effects on mRNA expression94. Genetic variants can also affect translation elongation by influencing both protein folding and the addition of post-translational modifications95,96.
Many SNVs that alter the rate of translation are thought to do so secondarily to changes in mRNA structure, as the introduction of RNA hairpins is known to decrease translation rate97. Although there are many examples of natural RNA structures involved in modulating translation in response to physiological cues96, only recently have studies emphasized that SNV-induced changes in RNA secondary structure can affect translation regulation97. RiboSNitches within synonymous sites are more likely to disrupt mRNA secondary structure than those within non-synonymous sites. This is because the redundant third codon position contributes more strongly to the generation of RNA secondary structures than the first two codon positions, probably owing to the third codon having more evolutionary flexibility98. In addition, synonymous variants that introduce non-preferred codons have been shown to affect protein folding owing to a decreased translation rate99. This is attributed to the longer time taken to incorporate amino acids at rare codons but may also reflect structural changes caused by synonymous site variants.
The relationship between synonymous substitutions, mRNA structure and human health is illustrated by the example of cystic fibrosis. The most common cause of cystic fibrosis is a three-nucleotide deletion in the coding region of the cystic fibrosis transmembrane conductance regulator (CFTR) gene, which has been designated ΔF508 (REF. 100). This mutation deletes the last nucleotide of the Ile507 (Ile507ATC) codon and the first two nucleotides of the Phe508 (Phe508TTT) codon. This results in the deletion of Phe and creates a synonymous substitution in the Ile codon (Ile507ATC>ATT). The resulting aberrant CFTR protein is misfolded and targeted for degradation, leading to cystic fibrosis101. Previously, CFTR misfolding was attributed solely to the loss of Phe508; however, the synonymous nucleotide change affects the CFTR mRNA secondary structure, which has no effect on mRNA half-life but significantly decreases the translation rate of ΔF508 CFTR transcripts, thereby potentially affecting protein folding102. Reverting the Ile507ATT synonymous mutation to the original Ile507ATC restored RNA structure and rescued normal levels of CFTRΔF508 protein by increasing resistance to the endoplasmic reticulum-associated degradation pathway (ERAD pathway) for misfolded proteins. These results suggest that the mRNA structural change induced by the synonymous Ile sub stitution causes a slower rate of translation, allowing time for the ERAD machinery to mark CFTR for degradation.
In a similar example, three haplotypes of catechol-O-methyltransferase (COMT) produce different amounts of COMT protein and are associated with high, average or low pain sensitivity, respectively103. These haplotypes differ in two synonymous nucleotide changes and one non-synonymous nucleotide change within the coding region. It was initially thought that the non-synonymous mutation was the sole cause of altered COMT protein levels, but it was found that the synonymous nucleotide changes are responsible for reduced translation of COMT mRNA by stabilizing hairpin structures within the coding region103. Finally, the missense SNV rs9840993 in myosin light-chain kinase (MYLK) is associated with asthma susceptibility and results in increased translation of MYLK104. This SNV alters RNA structure, making the translation initiation site more accessible, which is thought to increase translation efficiency. These examples show that common genetic variants affecting RNA secondary structure can have a marked effect on protein output and ultimately affect phenotypic variation.
SNV-mediated effects on mRNA localization
Subcellular localization of mRNAs is a key mechanism for spatial regulation of protein expression. Both the primary sequence and secondary structure of RNAs are important for proper RNA subcellular localization, and genetic variants that disrupt either RNA zipcode motifs or crucial RNA structural features can abolish proper transcript localization105–107. The G196A (Val66Met) SNV in brain-derived neurotrophic factor (BDNF) is associated with increased susceptibility to various psychiatric disorders, and mice with a BDNF G196A knock-in mutation exhibit anxiety-related behaviour108–110. This G–A substitution decreases trafficking of BDNF transcripts to the distal dendrites of neurons by disrupting the interaction between BDNF mRNA and the mRNA trafficking protein translin. In combination with altered protein function due to the Val66Met amino acid change, this SNV-induced loss of BDNF mRNA at the dendrites is thought to contribute to allele-specific defects in activity-dependent secretion of BDNF protein at dendrites105,108,111.
Conclusions and perspectives
A future goal of precision medicine is the ability to use genomic sequence to predict, with high confidence, health-related features including disease risk, success of prevention strategies, prognosis and positive or negative responses to specific therapeutics (BOX 3). To meet this goal, a multidisciplinary approach is needed to create a comprehensive knowledge base that fully integrates both functional variants mediated by RNA processing and those mediated by epigenetic and transcriptional mechanisms.
Box 3. RNA processing and pharmacogenetics.
More than 40% of patients with complex diseases such as depression, diabetes, asthma or Alzheimer disease will not respond to the drug used in their initial treatment, despite the drug having demonstrated benefit for other individuals127. In addition, drugs that are beneficial for some individuals can have adverse effects in others. Pharmacogenetics aims to understand how genetic variation leads to individual differences in drug response, particularly differences in drug metabolism or the risk of adverse events. A small but growing number of studies show that variation in RNA processing in a basis for differences in therapeutic response. For example, the rs751402 polymorphism in the 5′ underslated region (UTR) of the DNA damage response gene excision repair cross-complementation group 5 (ERCC5) is predictive of tumour resistance to platinum-based chemotherapy and of poor prognosis in paediatric ependymoma92. This single-nucleotide variant (SNV) creates an upstream open reading frame that promotes selective translation of pro-survival ERCC5 following platinum-based chemotherapy. Splicing is also a key mechanism for individual differences in drug response. A splice-site polymorphism in the neuronal sodium voltage-gated channel type 1 subunit alpha (SCN1A) gene is associated with an altered response to anti-epileptic drugs128. Similarly, the SNV rs3846662 in 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR), which encodes the rate-limiting enzyme in cholesterol synthesis, is associated with a reduced efficacy of statin treatment; this SNV results in skipping of exon 13 of HMGCR, producing a catalytically inactive protein in cell culture experiments129,130. The SNV is in the binding site of two antagonistic RNA splicing factors, serine/arginine-rich splicing factor 1 (SRSF1) and heterogeneous nuclear ribonucleoprotein A1 (HNRNPA1), and was shown to cause differential binding of HNRNPA1, with resulting effects on HMGCR splicing and the statin response131. Identifying associations between genotype, RNA processing and drug response is only the first step towards providing actionable data for clinical care, and there remains a large proportion of complex diseases for which pharmacogenetic data are unavailable or inconclusive. The above examples in which RNA processing affects pharmacogenetics illustrate the need to further study RNA metabolism as a mechanism of individual differences in drug response.
A first step is the annotation of genomic elements that function at the level of RNA processing. It is now clear that individuals differ in the ratios of mRNA isoforms, as well as total mRNA abundance, for a large number of genes owing to variations in RNA processing. The computational and experimental breakthroughs that allow genome to transcriptome sequence comparisons from an unprecedented number of individuals will provide an extensive accounting of the RNA processing effects of DNA variants, and thereby identify cis-acting elements involved in RNA processing. Particularly informative are analyses of allele-specific expression in which the mRNAs from the two alleles function as mutual internal controls for a shared cellular environment, such that allele-specific differences in terms of the ratio of mRNA splice variants or mRNA abundance most probably reflect cis-acting effects112. Analyses that directly link genetic variants with RNA processing differences will benefit the study of both RNA biology and human genetics through the discovery of novel RNA processing elements and the identification of causal variants in RNA processing that modify phenotypic traits.
This comprehensive annotation of cis-acting elements involved in RNA processing is the basis of efforts to identify the molecular mechanisms through which SNVs affect gene expression. Many SNVs probably function through combinatorial mechanisms, as the multiple steps of gene expression are highly dynamic and interdependent113,114. Pre-mRNA splicing and mRNA 3′ end formation are largely co-transcriptional; SNVs that affect chromatin structure can have a secondary effect on alternative splicing3, presumably owing to effects on the speed of transcription115. Changes in mRNA 3′ UTR length that are due to alternative splicing or alternative selection of polyadenylation sites can affect mRNA stability, translation efficiency or localization, owing to the gain or loss of binding sites for regulatory proteins or mi RNAs, and RNA structure has a crucial role in all aspects of RNA processing96,116,117. Up to 30% of cases of alternative splicing and alternative selection of polyadenylation sites can affect the translation efficiency of the processed mRNA118. RNA processing that disrupts the open reading frame often results in decreased protein expression through the actions of cytoplasmic mRNA surveillance systems such as nonsense-mediated decay119. This interdependence of the regulatory mechanisms of gene expression adds to the complexity of interpreting the primary effects of genetic variants on RNA processing.
The next steps will be to determine the functional relevance of variant-induced RNA processing differences and ultimately to understand the mechanisms by which prefer ential activity of one processing pathway over others has an impact on human health. Current technologies have provided the tools both to systematically identify functional variants within large data sets and to probe the molecular mechanisms responsible for phenotype alterations. In the near future, comparisons of whole-genome sequences and the transcriptome of cells from the same individual will be routine. Computational approaches to identify allele-specific expression are being refined to provide information correlating specific genetic variants with expression and processing patterns. Such analyses will be greatly enhanced by high-throughput, long-read sequencing to identify linkage of distal regions of a haplotype. Ideally, multidisciplinary investigations correlating DNA, RNA and protein variations will both computationally predict the effects of genetic variation and establish the molecular mechanisms through which specific genetic variants affect gene output. Systematic implementation of these methods to identify functional genetic variants will provide the basis for a broad mechanistic view of the odyssey from genotype to phenotype.
Acknowledgments
The authors thank members of the Cooper laboratory as well as the reviewers for valuable input. The work of the authors is supported by the US National Institutes of Health (NIH) (R01AR045653, R01AR060733 and R01HL045565 to T.A.C.), the Muscular Dystrophy Association (MDA276796 to T.A.C.), NIH training grant T32 GM008231 (to K.S.M.) and Baylor Research Advocates for Student Scientists (to K.S.M.).
Glossary
- Genome-wide association studies
(GWAS). Large studies across many individuals to determine whether the presence of a specific genotype is correlated with the manifestation of natural and pathological phenotypic traits or diseases.
- mRNA 3′ ends
The 3′ end of an mRNA is formed by endonucleolytic cleavage. The vast majority of protein-coding mRNAs have approximately 250 non-templated adenosines added to the last templated nucleotide at the site of endonucleolytic cleavage.
- Polyadenylation sites
Technically refers to the site of addition of the poly(A) tail at the last templated nucleotide of an mRNA. However, the term is sometimes used to refer to the AAUAAA hexanucleotide motif that is required for polyadenylation and that is typically located within 25 nucleotides of the last templated nucleotide.
- 5′ and 3′ untranslated regions
(5′ and 3′ UTRs). The open reading frames of protein-coding mRNAs can constitute less than half of their nucleotide sequence. The 5′ UTR is the mRNA region upstream of the translation start codon and the 3′ UTR is the region downstream of the translation stop codon.
- MicroRNAs
(miRNAs). Small RNAs (about 22 nucleotides long) that base pair to their mRNA targets and reduce protein expression by mRNA destabilization or translational repression.
- Branch site
A consensus mRNA splicing element typically located within 30 nucleotides of the 3′ splice site. It is recognized by sequential binding of branch point-binding protein followed by the U2 small nuclear ribonucleoprotein by base pairing.
- Exonic splicing enhancer
(ESE). One of several auxiliary mRNA splicing elements that are bound by splicing factors to promote (ESE or intronic splicing enhancer (ISE)) or inhibit (exonic splicing silencer (ESS) or intronic splicing silencer (ISS)) splicing. Disruption of these motifs and the resulting effects on splicing indicate that genetic variants outside exon–intron junctions can affect gene output.
- Synonymous and non-synonymous variants
Refers to variants in the coding region of a gene that either alter the encoded amino acid (non-synonymous) or do not affect the encoded amino acid (synonymous).
- Splicing quantitative trait loci
(sQTLs). Genomic regions containing variants that affect the splicing patterns of the associated pre-mRNA.
- Expression QTLs
(eQTLs). Genomic regions containing variants that affect the level of mRNA expression from one or more genes.
- HapMap-derived lymphoblastoid cell lines
(HapMap-derived LCLs). Refers to the International HapMap Project, which derived LCLs from 270 individuals of European, African and Asian ancestry for analysis of genotype, gene expression and pharmacological response.
- Single-nucleotide variants
(SNVs). We use the term SNV to refer to any genetic variant that changes one nucleotide, which may or may not have functional or pathological consequences. We largely focus on examples of common variants (also known as single-nucleotide polymorphisms (SNPs)) that are associated with either disease risk or disease severity.
- GEUVADIS and GTEx
Genetic European Variation in Health and Disease (GEUVADIS) and Genotype Tissue Expression (GTEx) are consortia for the high-throughput sequencing of human genomes and transcriptomes.
- Crosslinking immunoprecipitation
(CLIP). Used to identify the direct targets of an RNA-binding protein by covalently linking RNA and the protein in vivo by UV crosslinking, followed by immunoprecipitation and then high-throughput sequencing.
- Linkage disequilibrium
The nonrandom association of two or more genetic variants, usually owing to close proximity on the same chromosome and reflecting shared ancestry of alleles at flanking loci.
- Cis-acting miRNA QTLs
(Cis-mirQTLs). Genomic regions containing variants that affect the level of expression of individual microRNAs (miRNAs) located in cis with the variant.
- 1000 Genomes Project
This database was designed to identify genetic variants present in at least 1% of individuals across 26 populations, and contains DNA sequencing data for more than 2,500 individuals. This data set has been expanded by the addition of RNA sequencing data from the GEUVADIS consortium.
- Nonsense-mediated decay
An RNA surveillance mechanism that recognizes and degrades RNA transcripts containing premature termination codons.
- Nonstop decay
An RNA surveillance mechanism that recognizes and degrades RNA transcripts lacking a stop codon.
- No-go decay
An RNA and translation quality control mechanism that recognizes and degrades RNA -transcripts containing stalled ribosomes.
- RNA-induced silencing complex
(RISC). A ribonucleoprotein complex that mediates binding between a microRNA and its target mRNA.
- Protein QTLs
Genomic regions containing variants that affect the expression level of protein from one or more genes.
- Kozak sequence
A consensus sequence surrounding the start codon in eukaryotic mRNAs that promotes translation initiation.
- Endoplasmic reticulum-associated degradation pathway
(ERAD pathway). A quality control system for misfolded proteins in the endoplasmic reticulum that marks them for degradation by the ubiquitin–proteasome system.
- RNA zipcode motifs
Regulatory cis-acting RNA elements containing a specific sequence and secondary structure that are sufficient to confer mRNA subcellular localization.
Footnotes
Competing interests statement
The authors declare no competing interests.
FURTHER INFORMATION
ENCODE: https://www.encodeproject.org
GEUVADIS: http://www.geuvadis.org
GTEx: http://www.gtexportal.org/home
ALL LINKS ARE ACTIVE IN THE ONLINE PDF
References
- 1.Edwards SL, Beesley J, French JD, Dunning AM. Beyond GWASs: illuminating the dark road from association to function. Am J Hum Genet. 2013;93:779–797. doi: 10.1016/j.ajhg.2013.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kwan T, et al. Genome-wide analysis of transcript isoform variation in humans. Nat Genet. 2008;40:225–231. doi: 10.1038/ng.2007.57. One of the early genome-wide comparisons showed that genomic variation between individuals correlated with differences in mRNA levels and structure. [DOI] [PubMed] [Google Scholar]
- 3.Li YI, et al. RNA splicing is a primary link between genetic variation and disease. Science. 2016;352:600–604. doi: 10.1126/science.aad9417. A systematic analysis of LCLs from 68 individuals identified genomic variants that correlate with differences in up to eight molecular features of the gene regulatory cascade. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lappalainen T, et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature. 2013;501:506–511. doi: 10.1038/nature12531. This study identified the genomic causes of variation in the transcriptome using RNA-seq and genomic sequence data from LCLs of 462 individuals in the 1000 Genomes Project. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang ET, et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tian B, Hu J, Zhang H, Lutz CS. A large-scale analysis of mRNA polyadenylation of human and mouse genes. Nucleic Acids Res. 2005;33:201–212. doi: 10.1093/nar/gki158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Derti A, et al. A quantitative atlas of polyadenylation in five mammals. Genome Res. 2012;22:1173–1183. doi: 10.1101/gr.132563.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Halbeisen RE, Galgano A, Scherrer T, Gerber AP. Post-transcriptional gene regulation: from genome-wide studies to principles. Cell Mol Life Sci. 2008;65:798–813. doi: 10.1007/s00018-007-7447-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cooper TA, Wan L, Dreyfuss G. RNA and disease. Cell. 2009;136:777–793. doi: 10.1016/j.cell.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scotti MM, Swanson MS. RNA mis-splicing in disease. Nat Rev Genet. 2016;17:19–32. doi: 10.1038/nrg.2015.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lim LP, Burge CB. A computational analysis of sequence features involved in recognition of short introns. Proc Natl Acad Sci USA. 2001;98:11193–11198. doi: 10.1073/pnas.201407298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang GS, Cooper TA. Splicing in disease: disruption of the splicing code and the decoding machinery. Nat Rev Genet. 2007;8:749–761. doi: 10.1038/nrg2164. [DOI] [PubMed] [Google Scholar]
- 13.Wang Z, Burge CB. Splicing regulation: from a parts list of regulatory elements to an integrated splicing code. RNA. 2008;14:802–813. doi: 10.1261/rna.876308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xiong HY, et al. RNA splicing. The human splicing code reveals new insights into the genetic determinants of disease. Science. 2015;347:1254806. doi: 10.1126/science.1254806. A computational model was developed to predict splicing patterns based on genomic elements, which identified a large number of genetic variants among more than 650,000 SNVs that affect splicing and are associated with human disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Busch A, Hertel KJ. Splicing predictions reliably classify different types of alternative splicing. RNA. 2015;21:813–823. doi: 10.1261/rna.048769.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Supek F, Miñana B, Valcárcel J, Gabaldón T, Lehner B. Synonymous mutations frequently act as driver mutations in human cancers. Cell. 2014;156:1324–1335. doi: 10.1016/j.cell.2014.01.051. [DOI] [PubMed] [Google Scholar]
- 17.Takata A, Ionita-Laza I, Gogos JA, Xu B, Karayiorgou M. De novo synonymous mutations in regulatory elements contribute to the genetic etiology of autism and schizophrenia. Neuron. 2016;89:940–947. doi: 10.1016/j.neuron.2016.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bali V, Bebok Z. Decoding mechanisms by which silent codon changes influence protein biogenesis and function. Int J Biochem Cell Biol. 2015;64:58–74. doi: 10.1016/j.biocel.2015.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lim KH, Ferraris L, Filloux ME, Raphael BJ, Fairbrother WG. Using positional distribution to identify splicing elements and predict pre-mRNA processing defects in human genes. Proc Natl Acad Sci USA. 2011;108:11093–11098. doi: 10.1073/pnas.1101135108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cartegni L, Chew SL, Krainer AR. Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat Rev Genet. 2002;3:285–298. doi: 10.1038/nrg775. [DOI] [PubMed] [Google Scholar]
- 21.Zemojtel T, et al. Effective diagnosis of genetic disease by computational phenotype analysis of the disease-associated genome. Sci Transl Med. 2014;6:252ra123. doi: 10.1126/scitranslmed.3009262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li MX, et al. Predicting Mendelian disease-causing non-synonymous single nucleotide variants in exome sequencing studies. PLoS Genet. 2013;9:e1003143. doi: 10.1371/journal.pgen.1003143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cargill M, et al. Characterization of single-nucleotide polymorphisms in coding regions of human genes. Nat Genet. 1999;22:231–238. doi: 10.1038/10290. [DOI] [PubMed] [Google Scholar]
- 24.Wu X, Hurst LD. Determinants of the usage of splice-associated cis-motifs predict the distribution of human pathogenic SNPs. Mol Biol Evol. 2016;33:518–529. doi: 10.1093/molbev/msv251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang X, et al. Identification of common genetic variants controlling transcript isoform variation in human whole blood. Nat Genet. 2015;47:345–352. doi: 10.1038/ng.3220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ulirsch JC, et al. Systematic functional dissection of common genetic variation affecting red blood cell traits. Cell. 2016;165:1530–1545. doi: 10.1016/j.cell.2016.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fraser HB, Xie X. Common polymorphic transcript variation in human disease. Genome Res. 2009;19:567–575. doi: 10.1101/gr.083477.108. [DOI] [PubMed] [Google Scholar]
- 28.Zhang W, et al. Identification of common genetic variants that account for transcript isoform variation between human populations. Hum Genet. 2009;125:81–93. doi: 10.1007/s00439-008-0601-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.ElSharawy A, et al. Systematic evaluation of the effect of common SNPs on pre-mRNA splicing. Hum Mutat. 2009;30:625–632. doi: 10.1002/humu.20906. [DOI] [PubMed] [Google Scholar]
- 30.Rivas MA, et al. Human genomics. Effect of predicted protein-truncating genetic variants on the human transcriptome. Science. 2015;348:666–669. doi: 10.1126/science.1261877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hsiao Y-HE, et al. Alternative splicing modulated by genetic variants demonstrates accelerated evolution regulated by highly conserved proteins. Genome Res. 2016;26:440–450. doi: 10.1101/gr.193359.115. The authors identified more than 600 exons for which the splicing is affected by SNVs using a computational approach to measure allele-specific gene expression, including splicing differences, in ENCODE RNA-seq data sets. The results demonstrate the prominence of variant-driven splicing differences and present a powerful approach to analyse allele-specific expression. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matesanz F, et al. A functional variant that affects exon-skipping and protein expression of SP140 as genetic mechanism predisposing to multiple sclerosis. Hum Mol Genet. 2015;24:5619–5627. doi: 10.1093/hmg/ddv256. [DOI] [PubMed] [Google Scholar]
- 33.Paraboschi EM, et al. Functional variations modulating PRKCA expression and alternative splicing predispose to multiple sclerosis. Hum Mol Genet. 2014;23:6746–6761. doi: 10.1093/hmg/ddu392. [DOI] [PubMed] [Google Scholar]
- 34.Bojesen SE, et al. Multiple independent variants at the TERT locus are associated with telomere length and risks of breast and ovarian cancer. Nat Genet. 2013;45:371–384. doi: 10.1038/ng.2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Di Giacomo D, et al. Functional analysis of a large set of BRCA2 exon 7 variants highlights the predictive value of hexamer scores in detecting alterations of exonic splicing regulatory elements. Hum Mutat. 2013;34:1547–1557. doi: 10.1002/humu.22428. [DOI] [PubMed] [Google Scholar]
- 36.Monlong J, Calvo M, Ferreira PG, Guigó R. Identification of genetic variants associated with alternative splicing using sQTLseekeR. Nat Commun. 2014;5:4698. doi: 10.1038/ncomms5698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tejedor JR, Tilgner H, Iannone C, Guigó R, Valcárcel J. Role of six single nucleotide polymorphisms, risk factors in coronary disease, in OLR1 alternative splicing. RNA. 2015;21:1187–1202. doi: 10.1261/rna.049890.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gregory SG, et al. Interleukin 7 receptor α chain (IL7R) shows allelic and functional association with multiple sclerosis. Nat Genet. 2007;39:1083–1091. doi: 10.1038/ng2103. [DOI] [PubMed] [Google Scholar]
- 39.Gretarsdottir S, et al. A splice region variant in LDLR lowers non-high density lipoprotein cholesterol and protects against coronary artery disease. PLoS Genet. 2015;11:e1005379. doi: 10.1371/journal.pgen.1005379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shi Y, Manley JL. The end of the message: multiple protein-RNA interactions define the mRNA polyadenylation site. Genes Dev. 2015;29:889–897. doi: 10.1101/gad.261974.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mu XJ, Lu ZJ, Kong Y, Lam HYK, Gerstein MB. Analysis of genomic variation in non-coding elements using population-scale sequencing data from the 1000 Genomes Project. Nucleic Acids Res. 2011;39:7058–7076. doi: 10.1093/nar/gkr342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lu J, Clark AG. Impact of microRNA regulation on variation in human gene expression. Genome Res. 2012;22:1243–1254. doi: 10.1101/gr.132514.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sheets MD, Ogg SC, Wickens MP. Point mutations in AAUAAA and the poly (A) addition site: effects on the accuracy and efficiency of cleavage and polyadenylation in vitro. Nucleic Acids Res. 1990;18:5799–5805. doi: 10.1093/nar/18.19.5799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Higgs DR, et al. α-Thalassaemia caused by a polyadenylation signal mutation. Nature. 1983;306:398–400. doi: 10.1038/306398a0. [DOI] [PubMed] [Google Scholar]
- 45.Yoon OK, Hsu TY, Im JH, Brem RB. Genetics and regulatory impact of alternative polyadenylation in human B-lymphoblastoid cells. PLoS Genet. 2012;8:e1002882. doi: 10.1371/journal.pgen.1002882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhernakova DV, et al. DeepSAGE reveals genetic variants associated with alternative polyadenylation and expression of coding and non-coding transcripts. PLoS Genet. 2013;9:e1003594. doi: 10.1371/journal.pgen.1003594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Graham RR, et al. Three functional variants of IFN regulatory factor 5 (IRF5) define risk and protective haplotypes for human lupus. Proc Natl Acad Sci USA. 2007;104:6758–6763. doi: 10.1073/pnas.0701266104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cunninghame Graham DS, et al. Association of IRF5 in UK SLE families identifies a variant involved in polyadenylation. Hum Mol Genet. 2007;16:579–591. doi: 10.1093/hmg/ddl469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hellquist A, et al. The human GIMAP5 gene has a common polyadenylation polymorphism increasing risk to systemic lupus erythematosus. J Med Genet. 2007;44:314–321. doi: 10.1136/jmg.2006.046185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fahiminiya S, et al. A polyadenylation site variant causes transcript-specific BMP1 deficiency and frequent fractures in children. Hum Mol Genet. 2015;24:516–524. doi: 10.1093/hmg/ddu471. [DOI] [PubMed] [Google Scholar]
- 51.Garin I, et al. Recessive mutations in the INS gene result in neonatal diabetes through reduced insulin biosynthesis. Proc Natl Acad Sci USA. 2010;107:3105–3110. doi: 10.1073/pnas.0910533107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stacey SN, et al. A germline variant in the TP53 polyadenylation signal confers cancer susceptibility. Nat Genet. 2011;43:1098–1103. doi: 10.1038/ng.926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Uitte de Willige S, Rietveld IM, De Visser MCH, Vos HL, Bertina RM. Polymorphism 10034C>T is located in a region regulating polyadenylation of FGG transcripts and influences the fibrinogen γ′/γA mRNA ratio. J Thromb Haemost. 2007;5:1243–1249. doi: 10.1111/j.1538-7836.2007.02566.x. [DOI] [PubMed] [Google Scholar]
- 54.Friedman RC, Farh KKH, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19:92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.John B, et al. Human microRNA targets. PLoS Biol. 2004;2:e363. doi: 10.1371/journal.pbio.0020363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microRNA targets. Cell. 2003;115:787–798. doi: 10.1016/s0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
- 57.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 58.Huan T, et al. Genome-wide identification of microRNA expression quantitative trait loci. Nat Commun. 2015;6:6601. doi: 10.1038/ncomms7601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wagschal A, et al. Genome-wide identification of microRNAs regulating cholesterol and triglyceride homeostasis. Nat Med. 2015;21:1290–1297. doi: 10.1038/nm.3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Duan J, et al. A rare functional noncoding variant at the GWAS-implicated MIR137/MIR2682 locus might confer risk to schizophrenia and bipolar disorder. Am J Hum Genet. 2014;95:744–753. doi: 10.1016/j.ajhg.2014.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gamazon ER, et al. Genetic architecture of microRNA expression: implications for the transcriptome and complex traits. Am J Hum Genet. 2012;90:1046–1063. doi: 10.1016/j.ajhg.2012.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ghanbari M, et al. Genetic variations in microRNA-binding sites affect microRNA-mediated regulation of several genes associated with cardio-metabolic phenotypes. Circ Cardiovasc Genet. 2015;8:473–486. doi: 10.1161/CIRCGENETICS.114.000968. [DOI] [PubMed] [Google Scholar]
- 63.Thomas LF, Sætrom P. Single nucleotide polymorphisms can create alternative polyadenylation signals and affect gene expression through loss of microRNA-regulation. PLoS Comput Biol. 2012;8:e1002621. doi: 10.1371/journal.pcbi.1002621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Arnold M, Ellwanger DC, Hartsperger ML, Pfeufer A, Stümpflen V. Cis-acting polymorphisms affect complex traits through modifications of microRNA regulation pathways. PLoS ONE. 2012;7:e36694. doi: 10.1371/journal.pone.0036694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Houlston RS, et al. Meta-analysis of three genome-wide association studies identifies susceptibility loci for colorectal cancer at 1q41, 3q26.2, 12q13.13 and 20q13.33. Nat Genet. 2010;42:973–977. doi: 10.1038/ng.670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Parkes M, et al. Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn’s disease susceptibility. Nat Genet. 2007;39:830–832. doi: 10.1038/ng2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Brest P, et al. A synonymous variant in IRGM alters a binding site for miR-196 and causes deregulation of IRGM-dependent xenophagy in Crohn’s disease. Nat Genet. 2011;43:242–245. doi: 10.1038/ng.762. [DOI] [PubMed] [Google Scholar]
- 68.Kulkarni S, et al. Differential microRNA regulation of HLA-C expression and its association with HIV control. Nature. 2011;472:495–498. doi: 10.1038/nature09914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Caussy C, et al. An APOA5 3′ UTR variant associated with plasma triglycerides triggers APOA5 downregulation by creating a functional miR-485-5p binding site. Am J Hum Genet. 2014;94:129–134. doi: 10.1016/j.ajhg.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gartner JJ, et al. Whole-genome sequencing identifies a recurrent functional synonymous mutation in melanoma. Proc Natl Acad Sci USA. 2013;110:13481–13486. doi: 10.1073/pnas.1304227110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang G, et al. Variation in the miRNA-433 binding site of FGF20 confers risk for Parkinson disease by overexpression of α-synuclein. Am J Hum Genet. 2008;82:283–289. doi: 10.1016/j.ajhg.2007.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Haghnejad L, et al. Variation in the miRNA-433 binding site of FGF20 is a risk factor for Parkinson’s disease in Iranian population. J Neurol Sci. 2015;355:72–74. doi: 10.1016/j.jns.2015.05.020. [DOI] [PubMed] [Google Scholar]
- 73.Pai AA, et al. The contribution of RNA decay quantitative trait loci to inter-individual variation in steady-state gene expression levels. PLoS Genet. 2012;8:e1003000. doi: 10.1371/journal.pgen.1003000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Capon F, et al. A synonymous SNP of the corneodesmosin gene leads to increased mRNA stability and demonstrates association with psoriasis across diverse ethnic groups. Hum Mol Genet. 2004;13:2361–2368. doi: 10.1093/hmg/ddh273. [DOI] [PubMed] [Google Scholar]
- 75.van Hoof A, Wagner EJ. A brief survey of mRNA surveillance. Trends Biochem Sci. 2011;36:585–592. doi: 10.1016/j.tibs.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jacobs E, Mills JD, Janitz M. The role of RNA structure in posttranscriptional regulation of gene expression. J Genet Genomics. 2012;39:535–543. doi: 10.1016/j.jgg.2012.08.002. [DOI] [PubMed] [Google Scholar]
- 77.Lu Z, et al. RNA duplex map in living cells reveals higher-order transcriptome structure. Cell. 2016;165:1267–1279. doi: 10.1016/j.cell.2016.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sharma E, Sterne-Weiler T, O’Hanlon D, Blencowe BJ. Global mapping of human RNA–RNA interactions. Mol Cell. 2016;62:618–626. doi: 10.1016/j.molcel.2016.04.030. [DOI] [PubMed] [Google Scholar]
- 79.Spitale RC, et al. Structural imprints in vivo decode RNA regulatory mechanisms. Nature. 2015;519:486–490. doi: 10.1038/nature14263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wan Y, et al. Landscape and variation of RNA secondary structure across the human transcriptome. Nature. 2014;505:706–709. doi: 10.1038/nature12946. This study identified more than 1,900 SNVs that affected local RNA structure using high-throughput analysis of RNA structure in LCLs from a family trio. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Halvorsen M, Martin JS, Broadaway S, Laederach A. Disease-associated mutations that alter the RNA structural ensemble. PLoS Genet. 2010;6:e1001074. doi: 10.1371/journal.pgen.1001074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rogler LE, et al. Small RNAs derived from lncRNA RNase MRP have gene-silencing activity relevant to human cartilage–hair hypoplasia. Hum Mol Genet. 2014;23:368–382. doi: 10.1093/hmg/ddt427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Martin JS, et al. Structural effects of linkage disequilibrium on the transcriptome. RNA. 2012;18:77–87. doi: 10.1261/rna.029900.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kutchko KM, et al. Multiple conformations are a conserved and regulatory feature of the RB1 5′ UTR. RNA. 2015;21:1274–1285. doi: 10.1261/rna.049221.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Duan J, et al. Synonymous mutations in the human dopamine receptor D2 (DRD2) affect mRNA stability and synthesis of the receptor. Hum Mol Genet. 2003;12:205–216. doi: 10.1093/hmg/ddg055. [DOI] [PubMed] [Google Scholar]
- 86.Akdeli N, et al. A 3′UTR polymorphism modulates mRNA stability of the oncogene and drug target polo-like kinase 1. Mol Cancer. 2014;13:87. doi: 10.1186/1476-4598-13-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Niu T, et al. Identification of a novel FGFRL1 microRNA target site polymorphism for bone mineral density in meta-analyses of genome-wide association studies. Hum Mol Genet. 2015;24:4710–4727. doi: 10.1093/hmg/ddv144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sabarinathan R, et al. Transcriptome-wide analysis of UTRs in non-small cell lung cancer reveals cancerrelated genes with SNV-induced changes on RNA secondary structure and miRNA target sites. PLoS ONE. 2014;9:e82699. doi: 10.1371/journal.pone.0082699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wu L, et al. Variation and genetic control of protein abundance in humans. Nature. 2013;499:79–82. doi: 10.1038/nature12223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Battle A, et al. Genomic variation. Impact of regulatory variation from RNA to protein. Science. 2015;347:664–667. doi: 10.1126/science.1260793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Suhl JA, et al. A 3′ untranslated region variant in FMR1 eliminates neuronal activity-dependent translation of FMRP by disrupting binding of the RNA-binding protein HuR. Proc Natl Acad Sci USA. 2015;112:E6553–E6561. doi: 10.1073/pnas.1514260112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Somers J, et al. A common polymorphism in the 5′ UTR of ERCC5 creates an upstream ORF that confers resistance to platinum-based chemotherapy. Genes Dev. 2015;29:1891–1896. doi: 10.1101/gad.261867.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Raj A, et al. Thousands of novel translated open reading frames in humans inferred by ribosome footprint profiling. eLife. 2016;5:e13328. doi: 10.7554/eLife.13328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cenik C, et al. Integrative analysis of RNA, translation, and protein levels reveals distinct regulatory variation across humans. Genome Res. 2015;25:1610–1621. doi: 10.1101/gr.193342.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhang F, Saha S, Shabalina SA, Kashina A. Differential arginylation of actin isoforms is regulated by coding sequence-dependent degradation. Science. 2010;329:1534–1537. doi: 10.1126/science.1191701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mortimer SA, Kidwell MA, Doudna JA. Insights into RNA structure and function from genome-wide studies. Nat Rev Genet. 2014;15:469–479. doi: 10.1038/nrg3681. [DOI] [PubMed] [Google Scholar]
- 97.Wen JD, et al. Following translation by single ribosomes one codon at a time. Nature. 2008;452:598–603. doi: 10.1038/nature06716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Shabalina SA, Ogurtsov AY, Spiridonov NA. A periodic pattern of mRNA secondary structure created by the genetic code. Nucleic Acids Res. 2006;34:2428–2437. doi: 10.1093/nar/gkl287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yu CH, et al. Codon usage influences the local rate of translation elongation to regulate co-translational protein folding. Mol Cell. 2015;59:744–754. doi: 10.1016/j.molcel.2015.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Riordan JR, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- 101.Ward CL, Kopito RR. Intracellular turnover of cystic fibrosis transmembrane conductance regulator. Inefficient processing and rapid degradation of wild-type and mutant proteins. J Biol Chem. 1994;269:25710–25718. [PubMed] [Google Scholar]
- 102.Bartoszewski RA, et al. A synonymous single nucleotide polymorphism in ΔF508 CFTR alters the secondary structure of the mRNA and the expression of the mutant protein. J Biol Chem. 2010;285:28741–28748. doi: 10.1074/jbc.M110.154575. This study proposes that the effects on CFTR mRNA secondary structure induced by the ΔF508 mutation, rather than the deletion of Phe, destabilize the protein, leading to loss of its function. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Nackley AG, et al. Human catechol-O-methyltransferase haplotypes modulate protein expression by altering mRNA secondary structure. Science. 2006;314:1930–1933. doi: 10.1126/science.1131262. [DOI] [PubMed] [Google Scholar]
- 104.Wang T, Zhou T, Saadat L, Garcia JGN. A MYLK variant regulates asthmatic inflammation via alterations in mRNA secondary structure. Eur J Hum Genet. 2015;23:874–876. doi: 10.1038/ejhg.2014.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chiaruttini C, et al. Dendritic trafficking of BDNF mRNA is mediated by translin and blocked by the G196A (Val66Met) mutation. Proc Natl Acad Sci USA. 2009;106:16481–16486. doi: 10.1073/pnas.0902833106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bergalet J, Lécuyer E. The functions and regulatory principles of mRNA intracellular trafficking. Adv Exp Med Biol. 2014;825:57–96. doi: 10.1007/978-1-4939-1221-6_2. [DOI] [PubMed] [Google Scholar]
- 107.Chabanon H, Mickleburgh I, Burtle B, Pedder C, Hesketh J. An AU-rich stem-loop structure is a critical feature of the perinuclear localization signal of c-myc mRNA. Biochem J. 2005;392:475–483. doi: 10.1042/BJ20050812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chen ZY, et al. Genetic variant BDNF (Val66Met) polymorphism alters anxiety-related behavior. Science. 2006;314:140–143. doi: 10.1126/science.1129663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Egan MF, et al. The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell. 2003;112:257–269. doi: 10.1016/s0092-8674(03)00035-7. [DOI] [PubMed] [Google Scholar]
- 110.Notaras M, Hill R, van den Buuse M. The BDNF gene Val66Met polymorphism as a modifier of psychiatric disorder susceptibility: progress and controversy. Mol Psychiatry. 2015;20:916–930. doi: 10.1038/mp.2015.27. [DOI] [PubMed] [Google Scholar]
- 111.Mallei A, et al. Expression and dendritic trafficking of BDNF-6 splice variant are impaired in knock-in mice carrying human BDNF Val66Met polymorphism. Int J Neuropsychopharmacol. 2015;18:pyv069. doi: 10.1093/ijnp/pyv069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Li G, et al. Identification of allele-specific alternative mRNA processing via transcriptome sequencing. Nucleic Acids Res. 2012;40:e104. doi: 10.1093/nar/gks280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Maniatis T, Reed R. An extensive network of coupling among gene expression machines. Nature. 2002;416:499–506. doi: 10.1038/416499a. [DOI] [PubMed] [Google Scholar]
- 114.Braunschweig U, Gueroussov S, Plocik AM, Graveley BR, Blencowe BJ. Dynamic integration of splicing within gene regulatory pathways. Cell. 2013;152:1252–1269. doi: 10.1016/j.cell.2013.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Naftelberg S, Schor IE, Ast G, Kornblihtt AR. Regulation of alternative splicing through coupling with transcription and chromatin structure. Annu Rev Biochem. 2015;84:165–198. doi: 10.1146/annurev-biochem-060614-034242. [DOI] [PubMed] [Google Scholar]
- 116.Mayr C, Bartel DP. Widespread shortening of 3′ UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell. 2009;138:673–684. doi: 10.1016/j.cell.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Taliaferro JM, et al. Distal alternative last exons localize mRNAs to neural projections. Mol Cell. 2016;61:821–833. doi: 10.1016/j.molcel.2016.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sterne-Weiler T, et al. Frac-seq reveals isoform-specific recruitment to polyribosomes. Genome Res. 2013;23:1615–1623. doi: 10.1101/gr.148585.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Simms CL, Thomas EN, Zaher HS. Ribosome-based quality control of mRNA and nascent peptides. Wiley Interdiscip Rev RNA. 2016 doi: 10.1002/wrna.1366. http://dx.doi.org/10.1002/wrna.1366. [DOI] [PMC free article] [PubMed]
- 120.Van Nostrand EL, et al. Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP) Nat Methods. 2016;13:508–514. doi: 10.1038/nmeth.3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.de Klerk E, ‘t Hoen PAC. Alternative mRNA transcription, processing, and translation: insights from RNA sequencing. Trends Genet. 2015;31:128–139. doi: 10.1016/j.tig.2015.01.001. [DOI] [PubMed] [Google Scholar]
- 122.Weng L, Li Y, Xie X, Shi Y. Poly(A) code analyses reveal key determinants for tissue-specific mRNA alternative polyadenylation. RNA. 2016;22:813–821. doi: 10.1261/rna.055681.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Brar GA, Weissman JS. Ribosome profiling reveals the what, when, where and how of protein synthesis. Nat Rev Mol Cell Biol. 2015;16:651–664. doi: 10.1038/nrm4069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.van der Maarel SM, Tawil R, Tapscott SJ. Facioscapulohumeral muscular dystrophy and DUX4: breaking the silence. Trends Mol Med. 2011;17:252–258. doi: 10.1016/j.molmed.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lemmers RJLF, et al. Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nat Genet. 2012;44:1370–1374. doi: 10.1038/ng.2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lemmers RJLF, et al. A unifying genetic model for facioscapulohumeral muscular dystrophy. Science. 2010;329:1650–1653. doi: 10.1126/science.1189044. The authors demonstrate the role of allelic differences in a polyadenylation signal that allows expression of a toxic protein in muscle tissue of patients with FSHD. This followed a detailed genetic analysis to identify permissive and non-permissive alleles within complex genomic regions. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Spear BB, Heath-Chiozzi M, Huff J. Clinical application of pharmacogenetics. Trends Mol Med. 2001;7:201–204. doi: 10.1016/s1471-4914(01)01986-4. [DOI] [PubMed] [Google Scholar]
- 128.Tate SK, et al. Genetic predictors of the maximum doses patients receive during clinical use of the anti-epileptic drugs carbamazepine and phenytoin. Proc Natl Acad Sci USA. 2005;102:5507–5512. doi: 10.1073/pnas.0407346102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Burkhardt R, et al. Common SNPs in HMGCR in Micronesians and whites associated with LDL-cholesterol levels affect alternative splicing of exon13. Arterioscler Thromb Vasc Biol. 2008;28:2078–2084. doi: 10.1161/ATVBAHA.108.172288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Medina MW, Gao F, Ruan W, Rotter JI, Krauss RM. Alternative splicing of 3-hydroxy-3-methylglutaryl coenzyme A reductase is associated with plasma low-density lipoprotein cholesterol response to simvastatin. Circulation. 2008;118:355–362. doi: 10.1161/CIRCULATIONAHA.108.773267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Yu CY, et al. HNRNPA1 regulates HMGCR alternative splicing and modulates cellular cholesterol metabolism. Hum Mol Genet. 2014;23:319–332. doi: 10.1093/hmg/ddt422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Corrado L, et al. A novel synonymous mutation in the MPZ gene causing an aberrant splicing pattern and Charcot–Marie–Tooth disease type 1b. Neuromuscul Disord. 2016;26:516–520. doi: 10.1016/j.nmd.2016.05.011. [DOI] [PubMed] [Google Scholar]
- 133.Llewellyn DH, et al. Acute intermittent porphyria caused by defective splicing of porphobilinogen deaminase RNA: a synonymous codon mutation at -22 bp from the 5′ splice site causes skipping of exon 3. J Med Genet. 1996;33:437–438. doi: 10.1136/jmg.33.5.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Thanopoulou E, et al. The single nucleotide polymorphism g.1548A>G (K469E) of the ICAM-1 gene is associated with worse prognosis in non-small cell lung cancer. Tumour Biol. 2012;33:1429–1436. doi: 10.1007/s13277-012-0393-4. [DOI] [PubMed] [Google Scholar]
- 135.Boni V, et al. Role of primary miRNA polymorphic variants in metastatic colon cancer patients treated with 5-fluorouracil and irinotecan. Pharmacogenomics J. 2010;11:429–436. doi: 10.1038/tpj.2010.58. [DOI] [PubMed] [Google Scholar]
- 136.Xu J, et al. A heroin addiction severity-associated intronic single nucleotide polymorphism modulates alternative pre-mRNA splicing of the μ opioid receptor gene OPRM1 via hnRNPH Interactions. J Neurosci. 2014;34:11048–11066. doi: 10.1523/JNEUROSCI.3986-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Chiba-Falek O, et al. The molecular basis of disease variability among cystic fibrosis patients carrying the 3849 + 10 kb C>T mutation. Genomics. 1998;53:276–283. doi: 10.1006/geno.1998.5517. [DOI] [PubMed] [Google Scholar]
- 138.Hinzpeter A, et al. Alternative splicing at a NAGNAG acceptor site as a novel phenotype modifier. PLoS Genet. 2010;6:e1001153. doi: 10.1371/journal.pgen.1001153. This study demonstrates that selection of the upstream AG in the NAGNAG sequence at the 3′ splice site introduces a termination codon in the CFTR mRNA, thereby modifying disease severity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Morini E, et al. The human rs1050286 polymorphism alters LOX-1 expression through modifying miR-24 binding. J Cell Mol Med. 2016;20:181–187. doi: 10.1111/jcmm.12716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Zhou B, et al. Identification of a splicing variant that regulates type 2 diabetes risk factor CDKAL1 level by a coding-independent mechanism in human. Hum Mol Genet. 2014;23:4639–4650. doi: 10.1093/hmg/ddu184. [DOI] [PubMed] [Google Scholar]
- 141.Ray D, et al. A compendium of RNA-binding motifs for decoding gene regulation. Nature. 2013;499:172–177. doi: 10.1038/nature12311. This study identified the preferred binding motifs of hundreds of RNA-binding proteins with broad implications regarding roles in post-transcriptional regulation. A high-throughput approach was used to interrogate more than 200 RNA-binding protein motifs from vertebrate and invertebrate organisms using a randomized pool of 30–41-nucleotide RNAs containing all possible 9-nucleotide combinations. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Stephens RM, Schneider TD. Features of spliceosome evolution and function inferred from an analysis of the information at human splice sites. J Mol Biol. 1992;228:1124–1136. doi: 10.1016/0022-2836(92)90320-j. [DOI] [PubMed] [Google Scholar]