

Abstract
Coordinated cardiomyocyte growth, differentiation, and morphogenesis are essential for heart formation. We demonstrate that the bHLH transcription factors Hand1 and Hand2 play critical regulatory roles for left ventricle (LV) cardiomyocyte proliferation and morphogenesis. Using an LV-specific Cre allele (Hand1LV-Cre), we ablate Hand1-lineage cardiomyocytes, revealing that DTA-mediated cardiomyocyte death results in a hypoplastic LV by E10.5. Once Hand1-linage cells are removed from the LV, and Hand1 expression is switched off, embryonic hearts recover by E16.5. In contrast, conditional LV loss-of-function of both Hand1 and Hand2 results in aberrant trabeculation and thickened compact zone myocardium resulting from enhanced proliferation and a breakdown of compact zone/trabecular/ventricular septal identity. Surviving Hand1;Hand2 mutants display diminished cardiac function that is rescued by concurrent ablation of Hand-null cardiomyocytes. Collectively, we conclude that, within a mixed cardiomyocyte population, removal of defective myocardium and replacement with healthy endogenous cardiomyocytes may provide an effective strategy for cardiac repair.
Author summary
The left ventricle of the heart drives blood flow throughout the body. Impaired left ventricle function, associated either with heart failure or with certain, severe cardiac birth defects, constitutes a significant cause of mortality. Understanding how heart muscle grows is vital to developing improved treatments for these diseases. Unfortunately, genetic tools necessary to study the left ventricle have been lacking. Here we engineer the first mouse line to enable specific genetic study of the left ventricle. We show that, unlike in the adult heart, the embryonic left ventricle is remarkably tolerant of cell death, as remaining cells have the capacity to proliferate and to restore heart function. Conversely, disruption of two related genes, Hand1 and Hand2, within the left ventricle causes cells to assume the wrong identity, and to consequently overgrow and impair cardiac function. Ablation of these mutant cells rescues heart function. We conclude that selective removal of defective heart muscle and replacement with healthy cells may provide an effective therapy to treat heart failure.
Introduction
The left ventricle (LV) of the heart drives systemic circulation. Because the LV must be large enough to support adequate cardiac output but not hypertrophic or hypoplastic, such that it obstructs blood flow, congenital heart defects (CHDs) and acquired diseases that impact LV morphology or cell number present a significant cause of morbidity and mortality in the human population [1]. These CHDs include left ventricular noncompaction or hypertrabeculation (LVNC; OMIM: 604169), which is a cardiomyopathy characterized by prominent trabeculations occluding the ventricular lumen and associated with a high risk of heart failure and sudden death [2, 3] and hypoplastic left heart syndrome (HLHS; OMIM: 614435), which presents an underdeveloped LV unable to sustain sufficient blood flow [4, 5]. From the perspective of adult cardiac disease, especially cardiomyopathies, it is of particular importance to establish the capacity of proliferating cardiomyocytes to replace dead or dysfunctional cardiomyocytes [1]. Critical breakthroughs in patient outcomes demand a better understanding of the etiology of cardiac growth, differentiation and morphogenic patterning.
The embryonic heart forms from two distinct cardiomyocyte progenitor populations, termed the primary (PHF) and secondary (SHF) heart fields, which give rise to the LV and right ventricle (RV), respectively [6]. The bHLH transcription factor Hand1 is predominantly expressed within the LV myocardium, with minimal expression in SHF derivatives [7]. Interestingly, HAND1 mutations have been identified in HLHS patients [8]; however, cardiac-specific Hand1 ablation in mice does not recapitulate HLHS [9]. Previous studies suggest that the related bHLH transcription factor Hand2 can functionally cooperate with Hand1 during murine LV development [9]. Although Cre-loxP technology has been used to great effect in mice to genetically model SHF myocardium defects, the genetic tools to specifically interrogate genetic loss-of-function in LV cardiomyocytes have, thus far, not been available.
We have isolated a conserved 744 bp enhancer 5’ to the Hand1 transcription start site that is sufficient to drive reporter and Cre recombinase gene expression specifically within the LV. Hand1LV-Cre recapitulates endogenous Hand1 expression within an estimated 80–90% of LV cardiomyocytes between embryonic stages (E) E8.5-E13.5. Ablation of the Hand1-lineage LV myocardium results in a markedly smaller LV by E10.5; however, LV chamber size and adult cardiac function is ultimately rescued via proliferation of non-Hand1-lineage LV cardiomyocytes.
In contrast to this LV cell ablation model, Hand1;Hand2 loss-of-function within the LV results in increased cardiomyocyte proliferation resulting in morphogenic defects that lead to the occlusion of the LV lumen. LV cardiomyocytes invade the LV chamber and show mis-expression of compact zone, trabeculae, and intraventricular septum (IVS) restricted genes. We reveal a cooperative Hand factor function that is required to morphogenetically specify subpopulations of ventricular myocardium, and to thereby regulate cardiomyocyte growth. Functional analysis of surviving Hand1;Hand2 double conditional knockouts (CKOs) reveals impaired LV function that can be rescued by ablating the mutant Hand1 LV-lineage cells. Taken together, these data suggest that, due to its inherent proliferative capacity, the developing LV tolerates myocardial cell death, whereas developmentally abnormal cardiomyocytes adversely impact cardiac function.
Results
A distal Hand1 enhancer recapitulates gene expression within the LV
We have identified a cis-regulatory element(s) that drives gene expression specifically within the LV, and not within other cardiac tissues (Vincentz and Firulli, manuscript in preparation). We used this Hand1 enhancer to make an LV-specific Cre driver. Lineage analysis using the Hand1eGFPCre knock-in allele revealed that Hand1-lineage cells are restricted to the LV myocardium and to a ring of SHF-derived myocardium occupying the OFT, termed the myocardial cuff [7, 10]. Using the 2.7kb Hand1 basal promoter to provide the eGFPCre with a transcriptional start site, we cloned the Hand1 LV-enhancer 5’ (Fig 1A) and generated several F0 transgenic lines. In two of these lines, Cre activity, as assessed via the R26RlacZ reporter allele, was detectable within the forming LV at E9.0 (Fig 1E; white arrow) specifically marking only the Hand1 LV cardiomyocyte-lineage (Fig 1E, 1G, 1I, 1K and 1M). Hand1-lineage epicardium (black arrowhead; Fig 1H and 1I) and OFT tissues (Fig 1J and 1K; black arrows), detectable from the Hand1eGFPCre allele, were not observed in the Hand1LV-Cre transgenic. Immunohistochemistry using β-galactosidase to mark Cre-lineage cells and Mlc2v to mark ventricular cardiomyocytes showed total co-localization of these two markers (S1A, S1C, S1E and S1G Fig), whereas expression of β-galactosidase and PECAM, an endothelial/endocardial marker, appears mutually exclusive (S1A, S1C, S1E and S1G Fig). Together, these data validate that this Hand1LV-Cre driver recombines specifically within LV cardiomyocytes.
Fig 1. The Hand1LV-Cre transgenic allele recombines within the embryonic LV.
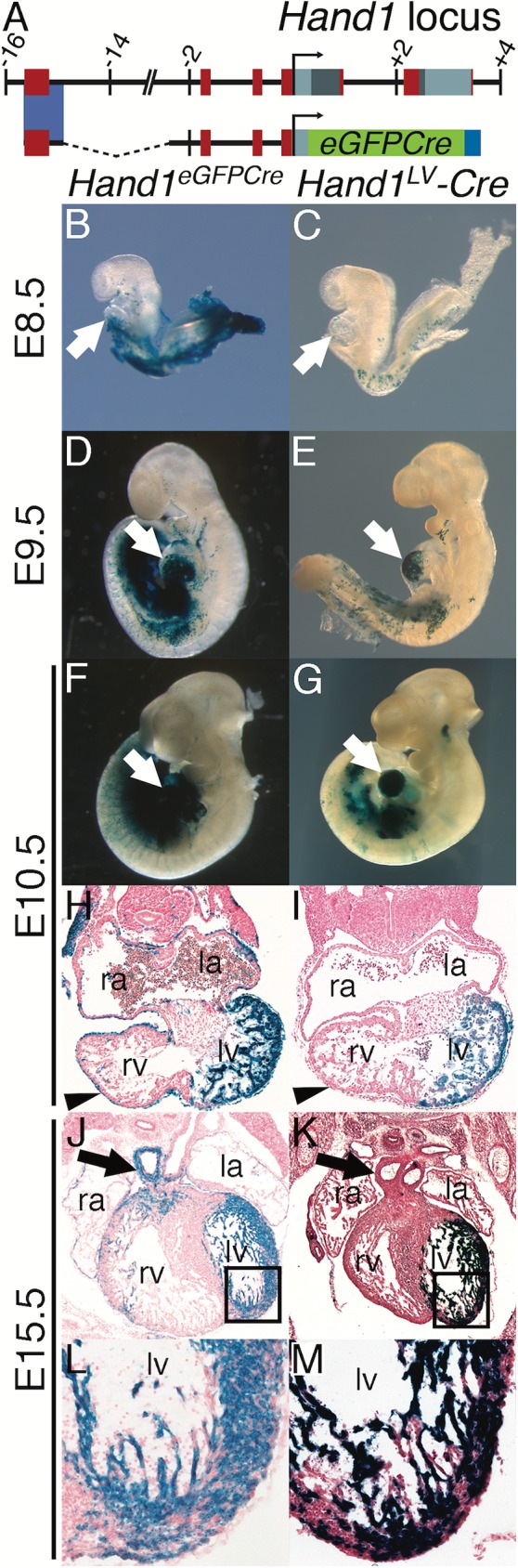
A) Schematic of the mouse Hand1LV-Cre transgene, in which a 744bp Hand1 LV-specific enhancer and ~2.7Kb Hand1 endogenous promoter regulate an eGFPCre expression cassette. B-M) X-gal staining to detect the R26RlacZ reporter allele demonstrates that, like the Hand1eGFPCre knock-in allele (B, D, F, H, J, L), Hand1LV-Cre activity initiates in the LV (white arrows) between E8.5 and E9.5 (C, E). Cre-lineage cells are detectable within the LV at E10.5 (G, I) and E15.5 (K, M), but not within the epicardium (H, I; black arrowheads) or the outflow tract (H, I; black arrows). la–left atrium, ra–right atrium, lv–left ventricle, rv–right ventricle.
Cellular ablation of the Hand1LV-Cre embryonic LV myocardium results in hypoplastic LV at E10.5 that fully recovers in size and function
To address the importance of the Hand1LV lineage during cardiac development, we utilized the conditionally active Rosa26 (R26R) Diphtheria Toxin A chain (R26RDTA) allele to conditionally ablate Hand1-expressing LV cardiomyocytes. Analysis of cell death via TUNEL on E9.5 sections revealed that the LVs of Hand1LV-Cre; R26RlacZ/+ control embryos displayed few apoptotic cells (Fig 2A), whereas Hand1LV-Cre; R26RlacZ/DTA LVs contained scattered TUNEL-positive cells (Fig 2B, quantified in Fig 2C). However, given the robust LV expression of the Hand1LV-Cre, fewer TUNEL-positive cells than would be predicted were detected in E9.5 R26RDTA-ablated LVs. Additional TUNEL staining, (Fig 2D) and whole mount lysotracker staining at E10.5 revealed markedly increased cell death within the LV of Hand1LV-Cre; R26RlacZ/DTA embryos when compared to controls (white arrows Fig 2E and 2F). At both time points, no difference in cell death was observed in the RV myocardium (Fig 2A–2D, 2G and 2H), and cell death in the pharyngeal arches served as a positive control (white arrowheads Fig 2A, 2B and 2E–2H).
Fig 2. Hand1LV-Cre-mediated DTA activation results in LV cardiomyocyte cell death and hypoplastic LV, which, through cardiomyocyte proliferation, recovers fully by E16.5.

A, B) TUNEL on E9.5 sections of Hand1LV-Cre(+);R26RlacZ/+ control (A) and Hand1LV-Cre(+);R26RlacZ/DTA embryos. Sections are counterstained with propidium iodide (PI). C, D) Quantification of the number of TUNEL-positive cells per heart in control and Hand1LV-Cre(+);R26RlacZ/DTA embryos at E9.5 (C) and E10.5 (D). E-H) Whole mount lysotracker staining detecting cell death confirms that conditional activation of R26ReGFPDTA/+ via intercross to Hand1LV-Cre causes pronounced LV cardiomyocyte death as assayed at E10.5 (white arrow; n = 3). White arrows denote TUNEL or lysotracker-positive cells in the LV. White arrowheads denote TUNEL or lysotracker-positive cells in the pharyngeal arches. I-P) X-Gal staining to detect the R26RlacZ reporter allele demonstrates that activation of the R26ReGFPDTA allele nearly completely ablates the Hand1LV-Cre lineage (black arrow) by E9.5 (I, J; n = 5); however, LV hypoplasia is not apparent until E10.5 (K, L; n = 3) and persists at E12.5 (M, N; n = 4). Despite DTA-mediated ablation of the Hand1LV-Cre lineage, by E16.5, the initially hypoplastic LV shows a pronounced recovery, and LV size is comparable to controls (O, P; n = 3). Q-T) Immunohistochemistry for pHH3 at E12.5 (Q, R) and E14.5 (S, T) in Hand1LV-Cre(+);R26R+/+ control (Q, S) and Hand1LV-Cre(+);R26R+/DTA embryos (R, T). U) Quantification of pHH3+ cells relative to the number of DAPI+ pixels show that proliferation is not altered at E12.5, but is elevated specifically within the LV at E14.5. Data are represented as mean ± standard error of mean. Asterisks denote significance (p ≤ 0.05) as determined by student’s t-test.
We then bred the R26RlacZ reporter onto the R26RDTA allele to monitor the loss of Hand1-expressing LV cardiomyocytes in our ablation model. At E9.5, the Hand1-LV lineage is largely absent from the heart, but the LV is not grossly hypoplastic (Fig 2J). By E10.5, the lack of Hand1-LV lineage cells results in a markedly hypoplastic LV (Fig 2L). At E12.5, the LVs of Hand1LV-Cre; R26RlacZ/DTA embryos were visibly smaller than Hand1LV-Cre; R26RlacZ/+ controls (Fig 2M and 2N) and showed few X-gal-stained cells when compared to littermates that do not carry the DTA allele. Interestingly, these mid-gestation LVs, from which the Hand1-lineage has been ablated, do not display perturbed expression of the LV markers Nppa and Gja5 (S2 Fig). Thus, ablation of Hand1-lineage LV myocardium between E8.5, when the Hand1LV enhancer is first upregulated, and E12.5, when the Hand1LV enhancer begins downregulation, results in a hypoplastic LV.
To follow the biological impact of Hand1-LV lineage ablation, we assayed the phenotypes of Hand1LV-Cre; R26RlacZ/DTA and control embryos at E16.5. To our surprise, Hand1LV-Cre; R26RlacZ/DTA hearts were indistinguishable in size from controls that lacked the DTA allele (Fig 2O and 2P; arrows). X-gal staining confirmed that embryos that contain the DTA allele exhibit minimal Hand1-lineage cells within their LVs. Although adult cardiomyocytes are refractory to reentering the cell cycle [1, 11–13], embryonic and neonatal cardiomyocytes retain proliferative potential [14–16]. To confirm that the restoration of LV size was simply due to an enhanced cardiomyocyte proliferation from non-Hand1-lineage LV myocardium, phospho-Histone-H3 immunostaining was carried out at E12.5 and E14.5 (Fig 2Q–2U). Increased proliferation was observed within the LVs of Hand1LV-Cre; R26RlacZ/DTA hearts, compared to controls, at E14.5, but not at E12.5 (Fig 2U). Hand1LV-Cre; R26RlacZ/DTA mice were born at Mendelian ratios (S3A Fig) and were viable and fertile. These hearts lacked gross structural cardiac abnormalities, exhibiting only a subtle rounding of the free LV wall (S3B and S3C Fig). These hearts exhibited cardiac function, as assayed by ejection fraction (EF), fractional shortening (FS), and other echocardiographic parameters, that was indistinguishable from Cre-negative controls (S3D–S3O Fig), and within the normal range for adult mice under isoflurane anesthesia [17]. Taken together, these data demonstrate that embryonic ablation of the Hand1-lineage is not sufficient to permanently disrupt LV development.
Myocardial deletion of Hand1 and Hand2 within the Hand1LV-Cre lineage results in proliferative, morphological, and molecular abnormalities that result in a hypoplastic LV lumen
As we have established that the Hand1-LV myocardial lineage is not required for cardiogenesis, we next investigated whether the expression of Hand1 and Hand2 within the embryonic LV is required for normal heart development. Although early embryonic expression analysis shows that the majority of Hand2 expression within the heart is restricted to the endocardium, epicardium, and SHF derived myocardium [10, 18, 19], at later embryonic stages, Hand2 mRNA becomes detectable within E11.5 LV myocardium in a pattern overlapping with Hand1 (S4C and S4D Fig). We subsequently generated compound heterozygous Hand1LV-Cre;Hand1fx/+;Hand2fx/+ male mice and crossed them to Hand1fx/fx;Hand2fx/fx females to generate Hand1LV-Cre;Hand1fx/fx;Hand2fx/fx offspring (Fig 3). Hand1LV-Cre;Hand1fx/+;Hand2fx/+ (Fig 3A–3D) embryos undergo normal cardiac development and are indistinguishable from wild type controls. Similarly, embryos that delete Hand2 from the LV but retain a single copy of Hand1 (Hand1LV-Cre;Hand1fx/+;Hand2fx/fx) also exhibit largely normal cardiac development (Fig 3E–3H). Interestingly, consistent with previous studies [9], deletion of Hand1 from the LV (Hand1LV-Cre;Hand1fx/fx) results in a morphologically disorganized LV, wherein Hand1-lineage cells appear to localize more to trabeculations compared to the compact zone (Fig 3I–3L). At E17.5, abnormal cardiomyocytes localize within the LV lumen (Fig 3L) and ventricular septal defects are also observed (S5H Fig). Deletion of Hand1 from the LV leaving a single copy of Hand2 (Hand1LV-Cre;Hand1fx/fx;Hand2fx/+) results in a similar phenotype (Fig 3M–3P, S5J–S5L Fig). Complete deletion of Hand factors from the LV (Hand1LV-Cre;Hand1fx/fx;Hand2fx/fx) caused a pronounced internalization of the Hand1-lineage cells where X-gal staining in whole mount is noticeably opaque and in section reveals Hand1-lineage cells occluding the LV lumen (Fig 3Q–3S). Histological analysis at E17.5 revealed severely occluded LV lumen and poorly formed IVS (Fig 3T, S5M–S5O Fig), as well as a double outlet right ventricle (black arrowheads, S5N Fig), and hyperplastic mitral valves (S5O Fig), although the aorta and OFT valves are phenotypically indistinguishable from controls (S5M Fig). These phenotypes are summarized in Table 1. These data suggest that a Hand factor loss-of-function within the LV myocardium alters chamber morphology via a Hand gene dosage dependent mechanism, in which Hand1-lineage cells are found less frequently in the compact zone at the expense of increased cells within the LV lumen representing trabecular and papillary muscle cardiomyocytes.
Fig 3. Hand1LV-Cre-mediated ablation of Hand1 and Hand2 results in a dysmorphic LV, and a single ventricle phenotype perinatally.

A-T) R26RlacZ reporter staining reveals that ablation of both Hand1 and Hand2 results in an abnormally thickened ventricular wall (R-T, white asterisks) that reduces the LV chamber lumen at E12.5, E14.5, and E17.5 (n = 4, 4, and 8, respectively).
Table 1. Cardiac defects in Hand1;Hand2 conditional knockouts at E17.5.
| Genotype | n | VSD | hyperplastic mitral valves | abnormal LV trabeculae | overgrown LV myocardium | Phenotypically Normal |
|---|---|---|---|---|---|---|
| H1fx/fx;H2fx/fx;(-) | 2 | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 2 (100%) |
| H1+/fx;H2+/fx;Cre(+) | 6 | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 6 (100%) |
| H1+/fx;H2fx/fx;Cre(+) | 4 | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 4 (100%) |
| H1fx/fx;H2+/+;Cre(+) | 4 | 2 (50%) | 0 (0%) | 1 (25%) | 0 (0%) | 2 (50%) |
| H1fx/fx;H2+/fx;Cre(+) | 5 | 1 (20%) | 3 (60%) | 3 (60%) | 0 (0%) | 1 (20%) |
| H1fx/fx;H2fx/fx;Cre(+) | 8 | 6 (75%) | 7 (87.5%) | 3 (37.5%) | 5 (62.5%) | 0 (0%) |
VSD–ventricular septal defect.
Surviving myocardial Hand1LV-Cre;Hand1fx/fx;Hand2fx/fx exhibit compromised cardiac function
Despite the morphological defects observed in Hand1LV-Cre;Hand1fx/fx;Hand2fx/fx embryos, Hand loss-of-function mice survive perinatally at approximately 50% of the expected Mendelian ratio (Table 2). Indeed, although Hand1LV-Cre;Hand1fx/fx;Hand2fx/fx pups are underrepresented, this underrepresentation is not statistically significant (Table 2). Echocardiographic analysis of P56 Hand1LV-Cre;Hand1fx/fx;Hand2fx/fx survivors (S6 Fig) revealed that systolic function (fractional shortening and ejection fraction) is significantly compromised in Hand1LV-Cre;Hand1fx/fx;Hand2fx/fx mice (Fig 4), whereas other measures of LV morphology and function, such as chamber dimensions and wall thickness, were not significantly altered (S7 Fig).
Table 2. Expected and actual survival data of intercrosses of Hand1LV-Cre; Hand1fx/+;Hand2fx/+ and Hand1fx/fx;Hand2fx/fx mice.
| Genotype | Expected (n = 123) | Recovered |
|---|---|---|
| H1+/fx;H2+/fx;(-) | 15.4 | 13 (1) |
| H1+/fx;H2fx/fx;(-) | 15.4 | 20 |
| H1fx/fx;H2+/fx;(-) | 15.4 | 12 |
| H1fx/fx;H2fx/fx;(-) | 15.4 | 14 |
| H1+/fx;H2+/fx;Cre(+) | 15.4 | 25 |
| H1+/fx;H2fx/fx;Cre(+) | 15.4 | 13 |
| H1fx/fx;H2+/fx;Cre(+) | 15.4 | 17 |
| H1fx/fx;H2fx/fx;Cre(+) | 15.4 | 9 (1) |
Numbers in parentheses denote pups that died prior to weaning.
Chi squared equals 11.829 with 7 degrees of freedom.
The two-tailed P value equals 0.1063
Fig 4. Hand1LV-Cre-mediated Hand1;Hand2 CKOs have impaired cardiac function as adults.

Mice at P56 reveal significant decreases in fractional shortening (A) and ejection fraction (B) in Hand1LV-Cre;Hand1fx/fx;Hand2fx/fx mutants (n = 8) relative to Hand1LV-Cre;Hand1fx/+;Hand2fx/+ littermates (n = 8). Data are represented as mean ± standard error of mean.
LV cardiomyocytes are mis-specified in Hand1LV-Cre;Hand1fx/fx;Hand2fx/fx mice
We next sought to characterize the etiology of the luminal cardiomyocyte overgrowth in Hand1LV-Cre;Hand1fx/fx;Hand2fx/fx embryos. To this end, we performed marker analyses to characterize distinct subpopulations of cardiomyocytes in the heart. The following expression studies revealed no appreciable difference between (-);Hand1fx/fx;Hand2fx/fx, Hand1LV-Cre(+); Hand1fx/+;Hand2fx/+, and Hand1LV-Cre(+);Hand1fx/+;Hand2fx/ fx embryos. For ease of presentation, Hand1LV-Cre(+); Hand1fx/+;Hand2fx/+ embryos are presented as controls. Tbx20 and Hey2 expression marks compact and IVS myocardium [20, 21]. Section in situ hybridization of E11.5 hearts showed that Tbx20 and Hey2 are both expressed throughout the presumptive compact myocardium and excluded from the trabecular myocardium in control hearts (Fig 5A and 5B). This sharp delineation between compact and trabecular myocardium is lost in the LVs of Hand1LV-Cre(+); Hand1fx/fx;Hand2fx/+ hearts (Fig 5E and 5F; asterisks). In Hand1LV-Cre;Hand1fx/fx;Hand2fx/fx hearts, most of the trabeculae within the LV ectopically express compact myocardium markers, whereas the RV trabeculae do not (Fig 5I and 5J). Conversely, the trabecular markers Bmp10 and Nppb show robust expression throughout the trabeculae, and expression is largely excluded from the compact myocardium in control hearts (Fig 5C and 5D). In Hand1LV-Cre;Hand1fx/fx;Hand2fx/fx hearts, Bmp10 and Nppb expression is downregulated within the LV trabeculae when compared to RV trabeculae (Fig 5K and 5L; asterisks). We conclude that Hand1LV-Cre;Hand1fx/fx;Hand2fx/fx hearts display ectopic compact myocardium marker expression and reduced trabecular marker expression within the LV.
Fig 5. Hand1LV-Cre-mediated Hand1;Hand2 CKOs display abnormal myocardial gene expression and trabecular proliferation in the LV.

A, B, E, F, I, J) Section in situ hybridization of E11.5 hearts showing that the transcription factors Tbx20 and Hey2 are both expressed throughout both the presumptive compact myocardium and IVS, but excluded from the trabecular myocardium in control hearts (A, B). This sharp delineation between compact and trabecular myocardium is lost in the LVs of Hand1LV-Cre;Hand1fx/fx;Hand2fx/+ hearts (E, F). In Hand1LV-Cre;Hand1fx/fx;Hand2fx/fx hearts, most of the trabeculae in the LV ectopically express compact myocardium markers, whereas the RV trabeculae do not (I, J; n = 4). C, D, G, H, K, L) Section in situ hybridization of E11.5 hearts showing that Bmp10 and Nppb are both expressed throughout the trabeculae, and are largely excluded from the compact myocardium in control hearts (C, D). In Hand1LV-Cre;Hand1fx/fx;Hand2fx/fx hearts, Bmp10 and Nppb expression is downregulated within the trabeculae (K, L; n = 4). M-T) Immunohistochemistry for Mki67 on E11.5 Control (M-P) and Hand1LV-Cre;Hand1fx/fx;Hand2fx/fx hearts (Q-T). Dashed lines delimit the boundary of trabeculae and compact zone. Boxes in M and Q denote high magnification images in N-P and R-T, respectively. White arrowheads in R, S and T highlight Mki67-positive nuclei. la–left atrium, ra–right atrium, rv–right ventricle, PI–propidium iodide. Asterisks denote the left ventricle.
Hand1LV-Cre;Hand1fx/fx;Hand2fx/fx hearts display abnormal trabecular proliferation in the LV
By Carnegie Stage 16 in the human embryo (equivalent to E11.5 in mice) proliferation in the ventricular trabeculae has declined [22]. We next sought to correlate differences in the proliferative capacity of compact and trabecular myocardium with the overgrowth seen in Hand1LV-Cre;Hand1fx/fx;Hand2fx/fx LVs. Mki67 marks cells actively undergoing cell cycle, but is excluded from cells in G0 [23]. Mki67 immunohistochemistry revealed that, in control embryos, nuclear Mki67 is robustly detected within compact myocardium and the IVS, but is largely excluded from trabecular myocardium (Fig 5M–5P). In contrast, Hand1LV-Cre;Hand1fx/fx;Hand2fx/fx trabecular cardiomyocytes show robust Mki67 staining (Fig 5Q–5T; arrowheads). These findings indicate that cardiomyocyte proliferation is dysregulated within the LV of Hand1LV-Cre;Hand1fx/fx;Hand2fx/fx embryos.
Hand1LV-Cre;Hand1fx/fx;Hand2fx/fx hearts display abnormal expansion of ventricular septum markers into the LV
Previous studies have reported that ectopic HAND1 expression throughout the heart disrupts IVS formation [24]. We reasoned that the abnormal proliferation and marker expression seen in Hand1LV-Cre;Hand1fx/fx;Hand2fx/fx hearts may reflect aberrant ventricular septogenesis. Section in situ hybridization of E11.5 hearts showed that expression of the chemokine Cxcl12 is strong in the free walls of the ventricles, but is largely excluded from the IVS (Fig 6A). In both Hand1LV-Cre;Hand1fx/fx;Hand2fx/+. and Hand1LV-Cre;Hand1fx/fx;Hand2fx/fx hearts, LV Cxcl12 expression is excluded specifically from the trabecular myocardium, in addition to the left side of the ventricular septum (Fig 6D and 6G). The secreted Wnt inhibitor Dkk3 and the transcription factor Irx2 are both markers of the IVS [25]. Expression of both Dkk3 (Fig 6H; black arrowhead) and Irx2 (Fig 6I; black arrow) expands toward the atrioventricular canal in Hand1LV-Cre;Hand1fx/fx;Hand2fx/fx hearts. Together, these data indicate that a loss of both Hand1 and Hand2 function within the LV causes an expansion of the IVS gene expression program.
Fig 6. Hand1LV-Cre-mediated Hand1;Hand2 CKOs display abnormal expansion of ventricular septum markers into the LV.

A-I) Section in situ hybridization of E11.5 hearts showing expression of the chemokine Cxcl12 (A, D, G), the secreted Wnt inhibitor Dkk3 (B, E, H), and the transcription factor Irx2 (C, F, I) in control (A-C), Hand1 heterozygous; Hand2 CKO (D-F), and Hand1LV-Cre;Hand1fx/fx;Hand2fx/fx hearts (G-I; n = 4). Black arrowhead (H) indicates the expansion of the Dkk3 expression domain. Black arrow (I) indicates the expansion of the Irx2 expression domain. la–left atrium, ra–right atrium, rv–right ventricle. Asterisks denote the left ventricle.
DTA ablation rescues the lethality and cardiac dysfunction seen in Hand1;Hand2 conditional knockouts
Given that the embryonic heart can recover from DTA-mediated ablation of Hand1-lineage cardiomyocytes, we tested whether ablation of Hand1;Hand2-null cardiomyocytes can rescue the phenotypes associated with their loss-of-function. The R26RDTA allele was bred onto a Hand1LV-Cre;Hand1fx/fx;Hand2fx/fx background. E14.5 section in situ hybridization of control hearts (Fig 7A–7C) showed expected expression patterns for Bmp10, Irx2, and Dkk3 within the trabeculae and IVS respectively. As expected Hand1LV-Cre;Hand1fx/fx;Hand2fx/fx hearts showed reduced Bmp10 LV expression along with expanded Dkk3- and Irx2 LV cardiomyocyte expression (Fig 7E–7G). These changes in gene expression are reversed in Hand1fx/fx;Hand2fx/fx;Hand1LV-Cre(+);R26RDTA embryos (Fig 7I–7K). Morphological examination of X-gal-stained bisected P56 hearts revealed heart morphology indistinguishable from controls (Fig 7D, S8A Fig), in contrast to the overgrowth of Hand1-lineage cardiomyocytes that occlude the lumen of Hand1LV-Cre;Hand1fx/fx;Hand2fx/fx hearts (Fig 7H, X-gal stained myocardium; S8B Fig). Occluded LV lumens are absent in Hand1LV-Cre;Hand1fx/fx;Hand2fx/fx;R26RDTA embryos (Fig 7L, S8C and S8D Fig). Lineage tracing reveals that the majority of lacZ-positive cells are ablated in these hearts; however, consistent with DTA-ablation embryos (Fig 2P), small populations of lacZ-positive cells were sometimes detectable within these hearts (S8D Fig), which we interpret as cells that have recombined only the lacZ reporter, but not the DTA allele. Hand1LV-Cre;Hand1fx/fx;Hand2fx/fx;R26RDTA pups survive at Mendelian ratios (Table 3) and display restored ejection fraction (Fig 7M) and fractional shortening (Fig 7N). Again, other measures of cardiac function were not significantly altered (S8E–S8J Fig).Together, these data demonstrate that ablation of mutant cardiomyocytes from the developing LV restores cardiac function.
Fig 7. DTA ablation rescues the lethality and cardiac dysfunction seen in Hand1LV-Cre Hand1;Hand2 CKOs.

A-K) Section in situ hybridization showing expression of Bmp10 (A, E, I), Irx2 (B, F, J), Dkk3 (C, G, K) in E14.5 embryonic hearts, and X-gal-stained bisected P56 Control (D), Hand1LV-Cre;Hand1fx/fx;Hand2fx/fx (H), and Hand1LV-Cre;Hand1fx/fx;Hand2fx/fx;R26RDTA-rescued hearts (L). Note that the RV of the heart in panel L has been mechanically damaged. (M, N) Echocardiographic analyses of P56 mice revealed restoration of ejection fraction (M) and fractional shortening (N) in Hand1LV-Cre(+);Hand1fx/fx;Hand2fx/fx;R26RDTA mice (n = 15) compared to Hand1LV-Cre(+);Hand1fx/fx;Hand2fx/fx CKOs. Hand1LV-Cre(-);R26RDTA mice, denoted as DTA(+), are included as controls. Data are represented as mean ± standard error of mean.
Table 3. Expected and actual survival data of intercrosses of Hand1LV-Cre; Hand1fx/fx;Hand2fx/fx and Hand1fx/fx;Hand2fx/fx;R26R+/eGFPDTA mice.
| Genotype | Expected (n = 77) | Recovered |
|---|---|---|
| H1fx/fx;H2fx/fx;(-);+/+ | 19.25 | 19 |
| H1fx/fx;H2fx/fx;(-);DTA/+ | 19.25 | 31 |
| H1fx/fx;H2fx/fx;Cre(+);+/+ | 19.25 | 8 |
| H1fx/fx;H2fx/fx;Cre(+);DTA/+ | 19.25 | 19 |
Chi squared equals 13.753 with 3 degrees of freedom.
The two-tailed P value equals 0.0033.
Discussion
CHDs that alter the LV exhibit poor clinical outcomes [5, 26]. The inability to interrogate LV gene expression in isolation has limited the ability to understand the molecular mechanisms that drive LV morphogenesis. This study reports the generation of a novel LV-restricted Cre driver line that allows for such focused interrogation. First, we ablated Hand1-lineage cardiomyocytes within an E9.0-E13.5 developmental window. As expected, LV size was greatly reduced by E10.5, and this reduced size remains clear at E14.5 (Fig 2). Hand1 cardiac expression is downregulated by E13.5 [27]. We observed a significant increase in LV cardiomyocyte cell proliferation at E14.5 (Fig 2U). By E16.5, LV size is indistinguishable from control hearts (Fig 2O and 2P). These findings support published data showing that embryonic and up to 7-day postnatal cardiomyocytes retain regenerative potential [14–16], and further demonstrate that the Hand1-lineage can be ablated from the developing LV, and the heart can nonetheless undergo full regenerative repair. It is also clear that these replacement cardiomyocytes do not express Hand1—if they did, Cre would also be expressed, thereby activating DTA expression and killing the replacement cell. That said, conclusive identification of the origin(s) of the cells that replace the ablated Hand1-lieage cells will require further study employing, for example, dual lineage tracing systems. A candidate for this progenitor population is the SHF. One of the most well studied markers of the SHF is the Isl1-Cre [28]. Although it is excluded from the majority of LV cardiomyocytes, Isl1-Cre-lineage cells do appear in the LV [28–31], and it is possible that this relatively minor population expands in the absence of Hand1-lineage cells. Indeed, this would explain why early stage DTA-ablation embryos, despite almost completely lacking Cre-lineage cells, are grossly similar to non-ablated littermates (Fig 2I and 2J). If this were the case, it would indicate that SHF-derived cardiomyocytes are sufficient to replace PHF-derived cardiomyocytes. Regardless of potential contributions from other cardiomyocyte lineages, including the SHF, these observations indicate that Hand1-lineage cells are not required for cardiac development, and that, through elevated proliferation, a Hand1-negative fated cardiomyocyte population within the developing heart is sufficient to generate a functional LV.
In contrast, Hand1 and Hand2 are required for normal LV morphogenesis. By E11.5, both Hand genes are expressed within the LV myocardium (S4 Fig). Loss of Hand1, and, more severely, Hand1 and Hand2, leads to abnormal LV morphology characterized by expanded compact and IVS marker expression (Tbx20, Hey2, Dkk3, Irx2; Figs. 5 and 6) and correspondingly reduced trabecular marker expression. An increase in Mki67-positive nuclei (Fig 5M–5T) within the LV trabecular zone indicates that aberrant cell proliferation causes the observed LV hyperplasia. Taking these observations together, we conclude that the expanding cardiomyocyte population observed in Hand1LV-Cre;Hand1fx/fx;Hand2fx/fx mutants is derived from myocardium that has ectopically activated the IVS gene expression program. The Hand1-lineage marks very little of the IVS [7]. Proliferation in Hand1LV-Cre;Hand1fx/fx;Hand2fx/fx mutant hearts is not restricted to this small domain of Hand1-expressing IVS cells on the inner wall near the cardiac apex. This suggests that some Hand1-lineage compact zone myocardium adopts an IVS cell-like fate. The observation that the Hand1-lineage within Hand1LV-Cre;Hand1fx/fx;Hand2fx/fx mutant hearts is largely excluded from the LV compact zone, and is instead localized to the inner curvature supports this finding. These results are especially interesting in light of recent studies, which propose that LVNC likely results from abnormal growth of compact myocardium [32]. It would be informative to test whether IVS markers also expand into luminal domains in such models.
In addition to LVNC, these Hand1LV-Cre;Hand1fx/fx;Hand2fx/fx mutants share certain phenotypic similarities with HLHS patients; however, they lack the aortic valve phenotypes characteristic of HLHS (S5M Fig). A recently published mouse model of HLHS [33] posits a digenic etiology of HLHS, in which dysfunction of one gene disrupts cardiomyocyte proliferation and differentiation to cause LV hypoplasia, while a second mutation causes aortic valve abnormalities. Importantly, these findings provide evidence that the aortic phenotypes are not secondary to the LV phenotypes, and therefore, the lack of Hand1LV-Cre;Hand1fx/fx;Hand2fx/fx aortic defects is not surprising. As mentioned, a subset of HLHS patients displays HAND1 mutations [8]. Hand1 is expressed in the OFT [27] as well as the LV, and as such could have pleiotropic functions in both aortic valve and LV development; however, ablation of Hand1 in the neural crest progenitors of the aortic valves is not pathogenic [34]. Given that this study reports the unexpected finding that Hand1 and Hand2 have overlapping roles in LV development, it would be of interest to reevaluate Hand1 function in the OFT considering potential functional redundancy with Hand2.
Increased cardiomyocyte proliferation can negatively impact specification [14, 15]. The Hand1LV-Cre;Hand1fx/fx;Hand2fx/fx LV hyperplasia occludes so much of the LV lumen that it results, in severe cases, in a single ventricle phenotype (Fig 3T, S5N Fig). In spite of the significant LV hyperplasia, 50% of Hand1LV-Cre;Hand1fx/fx;Hand2fx/fx mutants survive to birth and live to adulthood. These surviving individuals exhibit compromised systolic function (Fig 4). It is clear that both Hand1 and Hand2 contribute to this phenotype, and that Hand1 plays a more significant role. Hand1LV-Cre;Hand1fx/+;Hand2fx/fx mutants display no observable cardiac phenotypes, whereas Hand1LV-Cre;Hand1fx/fx;Hand2+/+ and Hand1LV-Cre;Hand1fx/x;Hand2fx/+ mutants show both morphological and molecular changes in gene expression. Given that Hand2 LV expression is not detectable until E11.5, and its LV expression is not dependent upon Hand1, Hand1 mutants are less severe likely due to the later expression of Hand2.
Finally, we observe a functional rescue of Hand1LV-Cre;Hand1fx/fx;Hand2fx/fx mutants when mutant cells are ablated via co-activation of DTA expression (Fig 7). Given that heart development does not require the Hand1-lineage (Fig 2) this may not be surprising. Nevertheless, this observation suggests that removal of molecularly abnormal cardiomyocytes from a developing heart could be beneficial if molecularly normal cells are present and are still proliferative.
Materials and methods
All relevant data are within the manuscript and its Supporting Information files.
Transgenic mice
The Indiana University Transgenic and Knock-Out Mouse Core generated the Hand1LV-Cre transgenic mouse line on a C3HeB/FeJ background. Genotyping of the Hand1tm2Eno, Hand2tm1Cse, Gt(ROSA)26Sortm1(DTA)Jpmb, and Gt(ROSA)26Sortm1Sor alleles has previously been described [9, 10, 35, 36]. These mice were maintained on a mixed C57Bl/6;129S background. Embryos were not selected for sex, and were evaluated blindly for all analyses. Mice and other reagents are available from the authors upon request.
Cloning
To generate the Hand1LV-Cre transgene, the genomic sequence corresponding to chr11:57660605–57661348 (in the mm9 assembly) was cloned 5’ to a modified eGFP-Cre vector driven by the basal Hand1 2.7kb promoter. Mice were genotyped for the Cre allele either via Southern blot or PCR using the primers Cre(F) 5’-CGTACTGACGGTGGGAGAAT-3’ and Cre(R) 5’-TGCATGATCTCCGGTATTGA-3’, with the internal controls Smad4(F) 5’-TAAGAGCCACAGGGTCAAGC-3’ and Smad4(R) 5’-TTCCAGGAAAAACAGGGCTA-3’.
X-gal staining and histology
X-gal staining was performed as previously described [7, 19, 37, 38].
Lysotracker and TUNEL
Cell death analysis on control and mutant embryos was performed as described [39]. Lysotracker (Life Technologies) was incubated with embryos as per the manufacturer's instructions. Embryos were imaged in a well slide on a Leica DM5000 B compound florescent microscope. TUNEL analyses were performed upon sectioned embryos using the ApopTag Plus Fluorescein in situ Apoptosis detection kit (S7111 Chemicon International) as per manufacturer’s instructions. TUNEL-positive cells occupying the free wall of the LV and RV (including the myocardial outflow tract) were counted every other section. Significance was determined by student’s t-test.
Immunohistochemistry and quantification of cell proliferation
Immunohistochemistry was performed as previously described [19, 40–42]. β-galactosidase (Aves BGL-1010; 1:200 dilution), Mlc2v (Synaptic Systems 310 111; 1:500), PECAM (BD Pharmingen 550274; 1:200), Phospho-Histone H3 (Abcam 4797; 1:500), and Mki67 (Dako; 1:500) antibodies were used with biotinylated secondary antibodies and streptavidin-conjugated DyLight 488 or 594 fluorophores (ThermoFisher). Images were collected on a Leica DM5000 B microscope and Leica Application Suite software.
Cell proliferation was assayed via counting of phospho-histone H3 positive nuclei. Left and right ventricles from E12.5 and E14.5 immunostained images were manually isolated in Adobe Photoshop, and nuclei were manually counted using Image J software. For E12.5, n≥9 sections, and for E14.5, n≥10 sections per heart were counted. Total samples analyzed were as follows: E12.5, control embryos (Hand1LV-Cre(-); R26RlacZ/DTA)–n = 2, RV, 35057 cells, total, LV, 35250 cells, total; LV-ablated embryos (Hand1LV-Cre(+); R26RlacZ/DTA)–n = 5, RV, 81471 cells, total, LV, 67618 cells, total; E14.5, control embryos (Hand1LV-Cre(-); R26RlacZ/DTA or Hand1LV-Cre(+); R26RlacZ/+)–n = 3, RV, 76254 cells, total, LV, 85121 cells, total; LV-ablated embryos–n = 4, RV, 167897 cells, total, LV, 80894 cells, total.
Echocardiography
Mice were lightly anesthetized with mixture of 1% to 1.5% isoflurane and 100% oxygen while supine on a heated platform. The heart rate were stabilized at 400 to 500 beats per minute before image recording. Images were obtained with a high resolution Micro-Ultrasound system (Vevo 2100, VisualSonics Inc, Toronto, Canada) equipped with a 40-MHz mechanical scan probe. Two-dimensional images were recorded in the parasternal long and short-axis to guide M-mode recordings in the mid-ventricular level. LV systolic function was computed from M-mode measurement according to the recommendations of American Society of Echocardiography committee [43].
Ethics statement
Animal work was approved by the Indiana University School of Medicine Animal Care and use committee (IACUC) via protocol 10809 Issued to ABF.
Supporting information
A-H) Immunohistochemistry on E15.5 Hand1LV-Cre(+);R26R+/lacZ hearts for β-galactosidase (C, D), Mlc2v (E), and PECAM (F) show that the Hand1LV-Cre lineage is predominantly myocardial (yellow co-localization in G), and not endocardial (H). The apical free wall of the LV is shown. Sections are counterstained with DAPI (A, B).
(TIFF)
A-D) Section in situ hybridization of E11.5 hearts showing expression of LV markers Nppa (A, B) and Gja5 (C, D). la–left atrium, lv–left ventricle, ra–right atrium, rv–right ventricle.
(TIFF)
A) Hand1LV-Cre; R26RlacZ/DTA pups are recovered with expected Mendelian distribution at P28. B, C) Bisected P56 Hand1LV-Cre(+); R26RlacZ/DTA hearts (B) display no gross structural abnormalities compared to control, (-); R26RlacZ/DTA hearts (C). D-O) Echocardiography of these mice at P56 revealed no significant difference in echocardiographic parameters between Hand1LV-Cre; R26RlacZ/DTA (n = 12) and control littermates (n = 8). d–diastole, s–systole, C–corrected, ID–internal diameter, PW–posterior wall.
(TIFF)
In situ hybridization of E11.5 hearts to detect Hand1 (A, B) and Hand2 (C, D). Hand1 cardiac expression is restricted to the LV myocardium, whereas Hand2 at this stage of development is robustly expressed in the endocardium, epicardium and RV and LV myocardium.
(TIFF)
A-O) H&E staining reveals that, by E17.5, Hand1LV-Cre;Hand1fx/fx;Hand2+/+ (G-I), Hand1LV-Cre;Hand1fx/fx;Hand2fx/+ (J-L), and Hand1LV-Cre;Hand1fx/fx;Hand2fx/fx (M-O) hearts display ventricular septal defects (black arrowheads), an RV that communicates with both the pulmonary trunk and the aorta, and mitral valve hyperplasia. Asterisks denote the LV.
(TIFF)
A-F) B-mode (A-C) and M-mode (D-F) echocardiographic analyses of control (A, D), Hand1;Hand2 CKO (B, E) and of Hand1;Hand2 CKO DTA-rescued (C, F) mice at P56 reveals that the obstructive cardiomyocytes (E, yellow arrowhead) characteristic of Hand1LV-Cre;Hand1fx/fx;Hand2fx/fx hearts are absent from Hand1LV-Cre;Hand1fx/fx;Hand2fx/fx;R26R+/eGFPDTA hearts.
(TIFF)
A-J) Other than EF and FS, shown in Fig 4, echocardiography of Hand1LV-Cre;Hand1fx/+;Hand2fx/+, Hand1LV-Cre;Hand1fx/+;Hand2fx/fx, Hand1LV-Cre;Hand1fx/fx;Hand2fx/+, and Hand1LV-Cre;Hand1fx/fx;Hand2fx/fx mice at P56 revealed no significant difference in additional echocardiographic parameters. Data are represented as mean ± standard error of mean. d–diastole, s–systole, C–corrected, ID–internal diameter, PW–posterior wall.
(TIFF)
A-C) Color photos of the X-gal-stained bisected P56 of Control (A), Hand1LV-Cre;Hand1fx/fx;Hand2fx/fx (B), and Hand1LV-Cre;Hand1fx/fx;Hand2fx/fx;R26RDTA-rescued hearts (C) shown in Fig 7D) Section of an X-gal stained Hand1LV-Cre;Hand1fx/fx;Hand2fx/fx;R26RDTA-rescued heart showing persistent, lacZ-positive cells. E-J) Echocardiography of these mice at P56 revealed no significant difference in additional echocardiographic parameters between Hand1LV-Cre(-);R26RDTA controls, denoted as DTA(+), Hand1LV-Cre(+);Hand1fx/fx;Hand2fx/fx CKOs, and Hand1LV-Cre(+);Hand1fx/fx;Hand2fx/fx;R26RDTA rescue mice. Data are represented as mean ± standard error of mean. d–diastole, s–systole, ID–internal diameter.
(TIFF)
The supplemental raw data speed sheet (excel file) presents the data and calculations for these figures in separate tabs. Fig 2C and 2D tabs show sections that are counterstained with propidium iodide (Quantification of the number of TUNEL-positive cells per heart in control and Hand1LV-Cre(+);R26RlacZ/DTA embryos at E9.5 (C) and E10.5 (D) were performed and Data are represented as mean ± standard error of mean. Asterisks denote significance (p ≤ 0.05) as determined by student’s t-test. Fig 2U tab shows) Quantification of pHH3+ cells relative to the number of DAPI+ pixels show that proliferation is not altered at E12.5, but is elevated specifically within the LV at E14.5. Data are represented as mean ± standard error of mean. Asterisks denote significance (p ≤ 0.05) as determined by student’s t-test. Fig 4 tab shows evaluation of mice at P56 reveal significant decreases in fractional shortening (A) and ejection fraction (B) in Hand1LV-Cre;Hand1fx/fx;Hand2fx/fx mutants relative to Hand1LV-Cre;Hand1fx/+;Hand2fx/+ littermates. Data are represented as mean ± standard error of mean. Asterisks denote significance (p ≤ 0.05) as determined by student’s t-test. Fig 7M and 7N tab shows calculations for the functional analysis (EF and FS) of DTA ablation rescue in Fig 7. Data are represented as mean ± standard error of mean.
(XLSX)
Acknowledgments
We thank Danny Carney, Hannah Millar, and Laura Bess for technical assistance. We thank the Herman B Wells Center Cardiac Developmental Biology Group for helpful discussions. Infrastructural support at the Herman B Wells Center is partially supported by the Riley Children’s Foundation and the Carleton Buehl McCulloch Chair of Pediatrics.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work is also supported by NIH Grants 1R01 HL122123-03, 1R01 HL120920-03, 1R0 AR061392-05 (to ABF); the Carleton Buehl McCulloch chair (to ABF); by an award from the American Heart Association and The Children’s Heart Foundation (16SDG27260072 to JWV), and by the Indiana University Health–Indiana University School of Medicine Strategic Research Initiative. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Xin M, Olson EN, Bassel-Duby R. Mending broken hearts: cardiac development as a basis for adult heart regeneration and repair. Nat Rev Mol Cell Biol. 2013;14(8):529–41. doi: 10.1038/nrm3619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Towbin JA, Lorts A, Jefferies JL. Left ventricular non-compaction cardiomyopathy. Lancet. 2015;386(9995):813–25. doi: 10.1016/S0140-6736(14)61282-4 [DOI] [PubMed] [Google Scholar]
- 3.Hussein A, Karimianpour A, Collier P, Krasuski RA. Isolated Noncompaction of the Left Ventricle in Adults. Journal of the American College of Cardiology. 2015;66(5):578–85. doi: 10.1016/j.jacc.2015.06.017 [DOI] [PubMed] [Google Scholar]
- 4.Tchervenkov CI, Walters HL, 3rd, Chu VF. Congenital Heart Surgery Nomenclature and Database Project: double outlet left ventricle. Ann Thorac Surg. 2000;69(4 Suppl):S264–9. [DOI] [PubMed] [Google Scholar]
- 5.Gordon BM, Rodriguez S, Lee M, Chang RK. Decreasing number of deaths of infants with hypoplastic left heart syndrome. J Pediatr. 2008;153(3):354–8. doi: 10.1016/j.jpeds.2008.03.009 [DOI] [PubMed] [Google Scholar]
- 6.Rochais F, Mesbah K, Kelly RG. Signaling pathways controlling second heart field development. Circ Res. 2009;104(8):933–42. doi: 10.1161/CIRCRESAHA.109.194464 [DOI] [PubMed] [Google Scholar]
- 7.Barnes RM, Firulli BA, Conway SJ, Vincentz JW, Firulli AB. Analysis of the Hand1 cell lineage reveals novel contributions to cardiovascular, neural crest, extra-embryonic, and lateral mesoderm derivatives. Developmental dynamics: an official publication of the American Association of Anatomists. 2010;239(11):3086–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reamon-Buettner SM, Ciribilli Y, Inga A, Borlak J. A loss-of-function mutation in the binding domain of HAND1 predicts hypoplasia of the human hearts. Human molecular genetics. 2008;17(10):1397–405. doi: 10.1093/hmg/ddn027 [DOI] [PubMed] [Google Scholar]
- 9.McFadden DG, Barbosa AC, Richardson JA, Schneider MD, Srivastava D, Olson EN. The Hand1 and Hand2 transcription factors regulate expansion of the embryonic cardiac ventricles in a gene dosage-dependent manner. Development. 2005;132(1):189–201. doi: 10.1242/dev.01562 [DOI] [PubMed] [Google Scholar]
- 10.Barnes RM, Firulli BA, VanDusen NJ, Morikawa Y, Conway SJ, Cserjesi P, et al. Hand2 loss-of-function in Hand1-expressing cells reveals distinct roles in epicardial and coronary vessel development. Circulation research. 2011;108(8):940–9. doi: 10.1161/CIRCRESAHA.110.233171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zaruba MM, Soonpaa M, Reuter S, Field LJ. Cardiomyogenic potential of C-kit(+)-expressing cells derived from neonatal and adult mouse hearts. Circulation. 2010;121(18):1992–2000. doi: 10.1161/CIRCULATIONAHA.109.909093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maillet M, van Berlo JH, Molkentin JD. Molecular basis of physiological heart growth: fundamental concepts and new players. Nat Rev Mol Cell Biol. 2013;14(1):38–48. doi: 10.1038/nrm3495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reuter S, Soonpaa MH, Firulli AB, Chang AN, Field LJ. Recombinant neuregulin 1 does not activate cardiomyocyte DNA synthesis in normal or infarcted adult mice. PLoS One. 2014;9(12):e115871 doi: 10.1371/journal.pone.0115871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Porrello ER, Mahmoud AI, Simpson E, Hill JA, Richardson JA, Olson EN, et al. Transient regenerative potential of the neonatal mouse heart. Science. 2011;331(6020):1078–80. doi: 10.1126/science.1200708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Drenckhahn JD, Schwarz QP, Gray S, Laskowski A, Kiriazis H, Ming Z, et al. Compensatory growth of healthy cardiac cells in the presence of diseased cells restores tissue homeostasis during heart development. Developmental cell. 2008;15(4):521–33. doi: 10.1016/j.devcel.2008.09.005 [DOI] [PubMed] [Google Scholar]
- 16.Sturzu AC, Rajarajan K, Passer D, Plonowska K, Riley A, Tan TC, et al. Fetal Mammalian Heart Generates a Robust Compensatory Response to Cell Loss. Circulation. 2015;132(2):109–21. doi: 10.1161/CIRCULATIONAHA.114.011490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vinhas M, Araujo AC, Ribeiro S, Rosario LB, Belo JA. Transthoracic echocardiography reference values in juvenile and adult 129/Sv mice. Cardiovasc Ultrasound. 2013;11:12 doi: 10.1186/1476-7120-11-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tsuchihashi T, Maeda J, Shin CH, Ivey KN, Black BL, Olson EN, et al. Hand2 function in second heart field progenitors is essential for cardiogenesis. Developmental biology. 2011;351(1):62–9. doi: 10.1016/j.ydbio.2010.12.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.VanDusen NJ, Casanovas J, Vincentz JW, Firulli BA, Osterwalder M, Lopez-Rios J, et al. Hand2 is an essential regulator for two Notch-dependent functions within the embryonic endocardium. Cell reports. 2014;9(6):2071–83. doi: 10.1016/j.celrep.2014.11.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koibuchi N, Chin MT. CHF1/Hey2 plays a pivotal role in left ventricular maturation through suppression of ectopic atrial gene expression. Circ Res. 2007;100(6):850–5. doi: 10.1161/01.RES.0000261693.13269.bf [DOI] [PubMed] [Google Scholar]
- 21.Singh MK, Christoffels VM, Dias JM, Trowe MO, Petry M, Schuster-Gossler K, et al. Tbx20 is essential for cardiac chamber differentiation and repression of Tbx2. Development. 2005;132(12):2697–707. doi: 10.1242/dev.01854 [DOI] [PubMed] [Google Scholar]
- 22.Sizarov A, Ya J, de Boer BA, Lamers WH, Christoffels VM, Moorman AF. Formation of the building plan of the human heart: morphogenesis, growth, and differentiation. Circulation. 2011;123(10):1125–35. doi: 10.1161/CIRCULATIONAHA.110.980607 [DOI] [PubMed] [Google Scholar]
- 23.Scholzen T, Gerdes J. The Ki-67 protein: from the known and the unknown. J Cell Physiol. 2000;182(3):311–22. doi: 10.1002/(SICI)1097-4652(200003)182:3<311::AID-JCP1>3.0.CO;2-9 [DOI] [PubMed] [Google Scholar]
- 24.Togi K, Kawamoto T, Yamauchi R, Yoshida Y, Kita T, Tanaka M. Role of Hand1/eHAND in the dorso-ventral patterning and interventricular septum formation in the embryonic heart. Mol Cell Biol. 2004;24(11):4627–35. doi: 10.1128/MCB.24.11.4627-4635.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koshiba-Takeuchi K, Mori AD, Kaynak BL, Cebra-Thomas J, Sukonnik T, Georges RO, et al. Reptilian heart development and the molecular basis of cardiac chamber evolution. Nature. 2009;461(7260):95–8. doi: 10.1038/nature08324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oechslin EN, Attenhofer Jost CH, Rojas JR, Kaufmann PA, Jenni R. Long-term follow-up of 34 adults with isolated left ventricular noncompaction: a distinct cardiomyopathy with poor prognosis. Journal of the American College of Cardiology. 2000;36(2):493–500. [DOI] [PubMed] [Google Scholar]
- 27.Cserjesi P, Brown D, Lyons GE, Olson EN. Expression of the novel basic helix-loop-helix gene eHAND in neural crest derivatives and extraembryonic membranes during mouse development. Developmental biology. 1995;170(2):664–78. doi: 10.1006/dbio.1995.1245 [DOI] [PubMed] [Google Scholar]
- 28.Cai CL, Liang X, Shi Y, Chu PH, Pfaff SL, Chen J, et al. Isl1 identifies a cardiac progenitor population that proliferates prior to differentiation and contributes a majority of cells to the heart. Developmental cell. 2003;5(6):877–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sun Y, Liang X, Najafi N, Cass M, Lin L, Cai CL, et al. Islet 1 is expressed in distinct cardiovascular lineages, including pacemaker and coronary vascular cells. Developmental biology. 2007;304(1):286–96. doi: 10.1016/j.ydbio.2006.12.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moretti A, Caron L, Nakano A, Lam JT, Bernshausen A, Chen Y, et al. Multipotent embryonic isl1+ progenitor cells lead to cardiac, smooth muscle, and endothelial cell diversification. Cell. 2006;127(6):1151–65. doi: 10.1016/j.cell.2006.10.029 [DOI] [PubMed] [Google Scholar]
- 31.Milgrom-Hoffman M, Harrelson Z, Ferrara N, Zelzer E, Evans SM, Tzahor E. The heart endocardium is derived from vascular endothelial progenitors. Development. 2011;138(21):4777–87. doi: 10.1242/dev.061192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jensen B, van der Wal AC, Moorman AF, Christoffels VM. Excessive trabeculations in noncompaction do not have the embryonic identity. Int J Cardiol. 2017;227:325–30. doi: 10.1016/j.ijcard.2016.11.089 [DOI] [PubMed] [Google Scholar]
- 33.Liu X, Yagi H, Saeed S, Bais AS, Gabriel GC, Chen Z, et al. The complex genetics of hypoplastic left heart syndrome. Nature genetics. 2017;49(7):1152–9. doi: 10.1038/ng.3870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barbosa AC, Funato N, Chapman S, McKee MD, Richardson JA, Olson EN, et al. Hand transcription factors cooperatively regulate development of the distal midline mesenchyme. Developmental biology. 2007;310(1):154–68. doi: 10.1016/j.ydbio.2007.07.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ivanova A, Signore M, Caro N, Greene ND, Copp AJ, Martinez-Barbera JP. In vivo genetic ablation by Cre-mediated expression of diphtheria toxin fragment A. Genesis. 2005;43(3):129–35. doi: 10.1002/gene.20162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nature genetics. 1999;21(1):70–1. doi: 10.1038/5007 [DOI] [PubMed] [Google Scholar]
- 37.Firulli AB, McFadden DG, Lin Q, Srivastava D, Olson EN. Heart and extra-embryonic mesodermal defects in mouse embryos lacking the bHLH transcription factor Hand1. Nature genetics. 1998;18(3):266–70. doi: 10.1038/ng0398-266 [DOI] [PubMed] [Google Scholar]
- 38.Vincentz JW, VanDusen NJ, Fleming AB, Rubart M, Firulli BA, Howard MJ, et al. A Phox2- and Hand2-dependent Hand1 cis-regulatory element reveals a unique gene dosage requirement for Hand2 during sympathetic neurogenesis. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2012;32(6):2110–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Firulli BA, Fuchs RK, Vincentz JW, Clouthier DE, Firulli AB. Hand1 phosphoregulation within the distal arch neural crest is essential for craniofacial morphogenesis. Development. 2014;141(15):3050–61. doi: 10.1242/dev.107680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vincentz JW, Firulli BA, Lin A, Spicer DB, Howard MJ, Firulli AB. Twist1 controls a cell-specification switch governing cell fate decisions within the cardiac neural crest. PLoS genetics. 2013;9(3):e1003405 doi: 10.1371/journal.pgen.1003405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vincentz JW, Barnes RM, Firulli BA, Conway SJ, Firulli AB. Cooperative interaction of Nkx2.5 and Mef2c transcription factors during heart development. Developmental dynamics: an official publication of the American Association of Anatomists. 2008;237(12):3809–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vincentz JW, Casasnovas JJ, Barnes RM, Que J, Clouthier DE, Wang J, et al. Exclusion of Dlx5/6 expression from the distal-most mandibular arches enables BMP-mediated specification of the distal cap. Proceedings of the National Academy of Sciences of the United States of America. 2016;113(27):7563–8. doi: 10.1073/pnas.1603930113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schiller NB, Shah PM, Crawford M, DeMaria A, Devereux R, Feigenbaum H, et al. Recommendations for quantitation of the left ventricle by two-dimensional echocardiography. American Society of Echocardiography Committee on Standards, Subcommittee on Quantitation of Two-Dimensional Echocardiograms. J Am Soc Echocardiogr. 1989;2(5):358–67. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A-H) Immunohistochemistry on E15.5 Hand1LV-Cre(+);R26R+/lacZ hearts for β-galactosidase (C, D), Mlc2v (E), and PECAM (F) show that the Hand1LV-Cre lineage is predominantly myocardial (yellow co-localization in G), and not endocardial (H). The apical free wall of the LV is shown. Sections are counterstained with DAPI (A, B).
(TIFF)
A-D) Section in situ hybridization of E11.5 hearts showing expression of LV markers Nppa (A, B) and Gja5 (C, D). la–left atrium, lv–left ventricle, ra–right atrium, rv–right ventricle.
(TIFF)
A) Hand1LV-Cre; R26RlacZ/DTA pups are recovered with expected Mendelian distribution at P28. B, C) Bisected P56 Hand1LV-Cre(+); R26RlacZ/DTA hearts (B) display no gross structural abnormalities compared to control, (-); R26RlacZ/DTA hearts (C). D-O) Echocardiography of these mice at P56 revealed no significant difference in echocardiographic parameters between Hand1LV-Cre; R26RlacZ/DTA (n = 12) and control littermates (n = 8). d–diastole, s–systole, C–corrected, ID–internal diameter, PW–posterior wall.
(TIFF)
In situ hybridization of E11.5 hearts to detect Hand1 (A, B) and Hand2 (C, D). Hand1 cardiac expression is restricted to the LV myocardium, whereas Hand2 at this stage of development is robustly expressed in the endocardium, epicardium and RV and LV myocardium.
(TIFF)
A-O) H&E staining reveals that, by E17.5, Hand1LV-Cre;Hand1fx/fx;Hand2+/+ (G-I), Hand1LV-Cre;Hand1fx/fx;Hand2fx/+ (J-L), and Hand1LV-Cre;Hand1fx/fx;Hand2fx/fx (M-O) hearts display ventricular septal defects (black arrowheads), an RV that communicates with both the pulmonary trunk and the aorta, and mitral valve hyperplasia. Asterisks denote the LV.
(TIFF)
A-F) B-mode (A-C) and M-mode (D-F) echocardiographic analyses of control (A, D), Hand1;Hand2 CKO (B, E) and of Hand1;Hand2 CKO DTA-rescued (C, F) mice at P56 reveals that the obstructive cardiomyocytes (E, yellow arrowhead) characteristic of Hand1LV-Cre;Hand1fx/fx;Hand2fx/fx hearts are absent from Hand1LV-Cre;Hand1fx/fx;Hand2fx/fx;R26R+/eGFPDTA hearts.
(TIFF)
A-J) Other than EF and FS, shown in Fig 4, echocardiography of Hand1LV-Cre;Hand1fx/+;Hand2fx/+, Hand1LV-Cre;Hand1fx/+;Hand2fx/fx, Hand1LV-Cre;Hand1fx/fx;Hand2fx/+, and Hand1LV-Cre;Hand1fx/fx;Hand2fx/fx mice at P56 revealed no significant difference in additional echocardiographic parameters. Data are represented as mean ± standard error of mean. d–diastole, s–systole, C–corrected, ID–internal diameter, PW–posterior wall.
(TIFF)
A-C) Color photos of the X-gal-stained bisected P56 of Control (A), Hand1LV-Cre;Hand1fx/fx;Hand2fx/fx (B), and Hand1LV-Cre;Hand1fx/fx;Hand2fx/fx;R26RDTA-rescued hearts (C) shown in Fig 7D) Section of an X-gal stained Hand1LV-Cre;Hand1fx/fx;Hand2fx/fx;R26RDTA-rescued heart showing persistent, lacZ-positive cells. E-J) Echocardiography of these mice at P56 revealed no significant difference in additional echocardiographic parameters between Hand1LV-Cre(-);R26RDTA controls, denoted as DTA(+), Hand1LV-Cre(+);Hand1fx/fx;Hand2fx/fx CKOs, and Hand1LV-Cre(+);Hand1fx/fx;Hand2fx/fx;R26RDTA rescue mice. Data are represented as mean ± standard error of mean. d–diastole, s–systole, ID–internal diameter.
(TIFF)
The supplemental raw data speed sheet (excel file) presents the data and calculations for these figures in separate tabs. Fig 2C and 2D tabs show sections that are counterstained with propidium iodide (Quantification of the number of TUNEL-positive cells per heart in control and Hand1LV-Cre(+);R26RlacZ/DTA embryos at E9.5 (C) and E10.5 (D) were performed and Data are represented as mean ± standard error of mean. Asterisks denote significance (p ≤ 0.05) as determined by student’s t-test. Fig 2U tab shows) Quantification of pHH3+ cells relative to the number of DAPI+ pixels show that proliferation is not altered at E12.5, but is elevated specifically within the LV at E14.5. Data are represented as mean ± standard error of mean. Asterisks denote significance (p ≤ 0.05) as determined by student’s t-test. Fig 4 tab shows evaluation of mice at P56 reveal significant decreases in fractional shortening (A) and ejection fraction (B) in Hand1LV-Cre;Hand1fx/fx;Hand2fx/fx mutants relative to Hand1LV-Cre;Hand1fx/+;Hand2fx/+ littermates. Data are represented as mean ± standard error of mean. Asterisks denote significance (p ≤ 0.05) as determined by student’s t-test. Fig 7M and 7N tab shows calculations for the functional analysis (EF and FS) of DTA ablation rescue in Fig 7. Data are represented as mean ± standard error of mean.
(XLSX)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.