

Abstract
Objective
To investigate the neuroprotective effects of Ginkgolide B (GB) against ischemic stroke-induced injury in vivo and in vitro, and further explore the possible mechanisms concerned.
Methods
Transient middle cerebral artery occlusion (tMCAO) mice and oxygen-glucose deprivation/reoxygenation (OGD/R)-treated N2a cells were used to explore the neuroprotective effects of GB. The expression of brain-derived neurotrophic factor (BDNF) was detected via Western blot and qRT-PCR.
Results
GB treatment (4 mg/kg, i. p., bid) significantly reduced neurological deficits, water content, and cerebral infarct volume in tMCAO mice. GB also significantly increased Bcl-2/Bax ratio, reduced the expression of caspase-3, and protected against OGD/R-induced neuronal apoptosis. Meanwhile, GB caused the up-regulation of BDNF protein in vivo and in vitro.
Conclusion
Our data suggest that GB might protect the brain against ischemic insult partly via modulating BDNF expression.
Keywords: Stroke, Ginkgolide B, Brain-derived neurotrophic factor, Apoptosis
INTRODUCTION
Ischemic stroke is the most common acute neurologic disease and a leading cause of death, disability, and human suffering6). Over decades, many neuroprotective agents have been proven effective in a variety of animal ischemic stroke models. However, none has been shown to work in clinical trials. The use of recombinant tissue plasminogen activator has been the only exception for treatment of acute ischemic stroke1). Considering that ischemic stroke is a complicated vascular and metabolic process that evolves over minutes to hours, a more detailed understanding of the mechanisms involved will have a substantial effect in the optimization and development of treatment strategies.
As a member of the neurotrophin family, brain-derived neurotrophic factor (BDNF) is present in mature brain where it is stored and released from neurons in a use-dependent fashion and has been implicated in long term potentiation, learning and memory formation3,13). It also plays a fundamental role in protecting against ischemia stroke4). By activating intracellular tyrosine receptor kinase B, mitogen-activated protein kinases, and the extracellular signal-regulated kinases16), BDNF can decrease infarct volume and improve neurological outcome via either exogenously supplied17,18) or overexpression in vivo11,12) in experimental stroke. Thus, pharmacological strategies aimed at increasing post-ischemic cerebral BDNF production appear to be a promising option in the treatment of stroke.
Ginkgo biloba L (Ginkgoaceae) has been used as a traditional Chinese medicine for about 5000 years, and now Ginkgo biloba extract (GBE) is used widely in the treatment of acute ischemic stroke in China and Europe23). Ginkgolide B (GB) is one of the primary constituents of the terpenoid fraction in GBE. In the past decades, the pharmacological studies have showed that GB is a potent antagonist against platelet activating factor7,22). It can exert inhibition of platelet aggregation, prevention of thrombus19), protection of ischemia20), anti-inflammation21), and anti-allergy5) and so on. However, it remains unclear whether GB modulates the level of BDNF after ischemic stroke.
In the present study, we evaluated the therapeutic effect of GB in transient middle cerebral artery occlusion (tMCAO) mice and oxygen-glucose deprivation/reoxygenation (OGD/R)-treated N2a cells. Then, we found that GB caused the up-regulation of BDNF protein in vivo and in vitro. Therefore, we suggest that GB modulates BDNF expression in ischemic stroke.
MATERIALS AND METHODS
Animals and surgery
All experimental procedures were approved by Institutional Animal Care and Use Committee of Anhui Medical University. Mice (male, 10–12 weeks) were kept on a 12/12 hours day and night cycle and with food and water available Ad libitum. Mice were anesthetized with isoflurane using anaesthesia machine (Matrx, Orchard Park, NY, USA). Transient focal cerebral ischemia was induced by intraluminal occlusion of the right middle cerebral artery via silicone-coated nylon monofilament (6023PK; Doccol Corp, Redlands, CA, USA). Briefly, for the transient model, reperfusion was produced by withdrawal of the monofilament 1 hour after occlusion in mice. The regional cerebral blood flow was continuously assessed by recording laser Doppler flow (Moor VSM-LDF; Moor Instruments, Wilmington, DE, USA) to ensure appropriate ischemia and reperfusion. Body temperature was regularly checked and maintained between 36.5°C and 37.5°C with a heating pad to avoid hypothermia. The sham-operated animals underwent the same anesthesia and surgical procedures except tMCAO.
In vivo drug administration and experimental groups
GB (from National Institutes for Food and Drug Control, China) was freshly diluted to the concentrations as needed with 0.9% saline and administered intraperitoneally 60 minutes after tMCAO. All mice were randomly assigned to the following experimental treatment groups: a sham-operated group (saline, 10 mL/kg), a GB-only group (GB at doses of 4 mg/kg, i. p., bid.), a tMCAO group (mice suffered from tM-CAO and saline, 10 mL/kg), and tMCAO+GB groups (mice suffered from tMCAO, followed by GB at doses of 1, 2, and 4 mg/kg, i. p., bid.). The doses of GB were comparable with therapeutic concentration in humans.
Assessment of neurological deficits
Neurological deficits were assessed 72 hours after tMCAO described using a 5-point-scale neurological score2): 0, normal motor function; 1, flexion of the contralateral torso and forearm on lifting the animal by the tail; 2, spontaneous circling to the contralateral side but normal posture at rest; 3, leaning/falling to the contralateral side; 4, no spontaneous movement; 5, death.
Measurement of cerebral infarct volume and cerebral edema
At 72 hours after tMCAO, the mice were deeply anaesthetized with pentobarbital sodium (80 mg/kg) and euthanized by decapitation. The brains were quickly removed and sectioned coronally into slices of 1 mm thickness. Slices were incubated in a 1% solution of 2, 3, 5-triphenyltetrazolium chloride (TTC; Sigma, St Louis, MO, USA) in phosphate buffered saline at 37°C (water bath) for 15 minutes and then fixed by 4% buffered formaldehyde solution. TTC stained viable brain tissue dark red based on intact mitochondrial function, whereas infarcted tissue areas remain unstained (white). Images were acquired by a digital camera (Nikon Coolpix; Nikon Corporation, Tokyo, Japan), and areas of both hemispheres and the infarcted regions were quantified for each slice using Image J software (National Institutes of Health, Bethesda, MD, USA). The actual infarct volume with correction of edema was calculated as the volume of the contralateral hemisphere minus the noninfarcted volume of the ipsilateral hemisphere. Edema was measured using the wet/dry weight method with water content expressed as the percent of tissue wet weight as previously described8).
Cell culture, generation of OGD/R model and drug treatment
N2a cell, an immortalized cell line of murine nerve cells, was purchased from American Type Culture Collection. Cells were maintained in Dulbecco’s modified Eagle’s medium (Gibco, Waltham, MA, USA) containing 10% fetal bovine serum (Millipore, Billerica, MA, USA), and 1% penicillin and streptomycin in a 37°C humidified incubator with 21% O2 and 5% CO2. To generate OGD/R model, cells were incubated with glucose-free medium (Gibco) in a 37°C incubator chamber (Thermo Scientific Forma® Series II Water Jacketed CO2 Incubators model 3131; Thermo Fisher, Waltham, MA, USA) aerated with an anaerobic gas mixture (1% O2, 5% CO2, 94% N2). After 3 hours, cells were moved out from the anaerobic chamber and then the glucose-free medium was replaced with normal medium. The cells were cultured for 24 hours in normal incubator for re-oxygenation. GB (0.1–40 μM) was pre-incubated 2 hours before OGD/R injury.
Cell assays
Cell viability was determined by 3-[4, 5-dimethylthiazol-2-yl]-2, 5-diphenyltetrazolium bromide incorporation. The optical density value was measured at 570 nm and 630 nm using micro-plate reader (Bio-Rad, Hercules, CA, USA). Cell injury was assessed by measuring the activity of lactate dehydrogenase (LDH) in the culture medium. Data were presented as percentage of the maximal LDH release.
Western blotting
For analysis of specific proteins abundance 72 hours after stroke or 24 hours after OGD/R, ischemic penumbra of individual mice or cell homogenate was complemented with lysis buffer according to instructions of the protein extraction kit (Nanjing KeyGen Biotech. Co. Ltd., Nanjing, China). The supernatants were used for sodium dodecyl sulfate polyacrylamide gel electrophoresis, and transferred to polyvinylidene fluoride membranes with the electrophoretic transfer system (Transblot Semi-dry Transfer Cell; Bio-Rad). The membranes were blocked with blocking solution (10% milk in 0.1% Tween 20 Tris-buffered saline; 1 hour at room temperature) and incubated with primary antibodies including BDNF (AB6201; Abcam, Cambridge, MA, USA), Bcl-2, Bax, Caspase-3 (1: 1000; Cell Signaling Technology, Danvers, MA, USA), and β-actin (1: 2000; Sigma) overnight at 4°C. Thereafter, membranes were incubated with corresponding secondary antibodies (1: 1000; Invitrogen, Waltham, MA, USA) at room temperature. Finally, immunoblots were scanned and determined by Omega16IC (Ultra-Lum, Inc., Claremont, CA, USA).
Statistical analysis
Results were presented as mean±standard error of mean and analyzed with one-way analysis of variance (ANOVA). Neurological scores (median value) were analyzed by Wilcox-on-Mann-Whitney test. For all analyses, p<0.05 was considered statistically significant.
RESULTS
GB alleviated tMCAO-induced neurological deficits and brain water content
To examine the neuroprotective effect of GB in vivo, GB (1, 2 or 4 mg/kg, i. p., bid) was administered 1 hour after onset of reperfusion in tMCAO mice. Neurological deficits and brain water contents were measured 72 hours after tMCAO. As shown in Fig. 1, GB could significantly reduce ischemia-induced neurological deficits (Fig. 1A) and brain water content (Fig. 1B) at dose of 4 mg/kg.
Fig. 1.
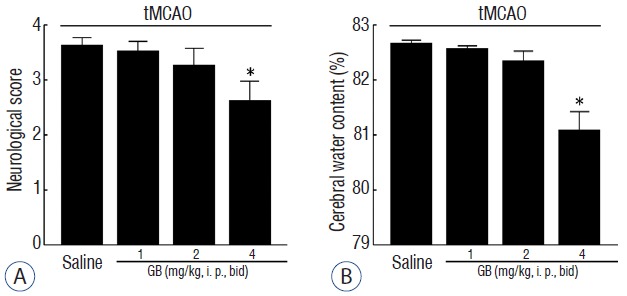
GB attenuated neurological deficits (A) and brain edema (B) 72 hours of reperfusion after 1 hour of tMCAO. All of the mice were suffered from tMCAO. *p<0.05 vs. saline group, mean±standard error of mean, n=10. GB: Ginkgolide B, tMCAO: transient middle cerebral artery occlusion.
GB reduced tMCAO-induced infarct volume
As shown in Fig. 2, GB significantly decreased total infarct volume at dose of 1, 2, and 4 mg/kg (by 21.2%, 34.5%, and 52.6% respectively, p<0.05) in comparison with mice received saline treatment 72 hours after reperfusion. These results demonstrated that GB significantly alleviated cerebral ischemia/reperfusion-induced brain damage in vivo.
Fig. 2.
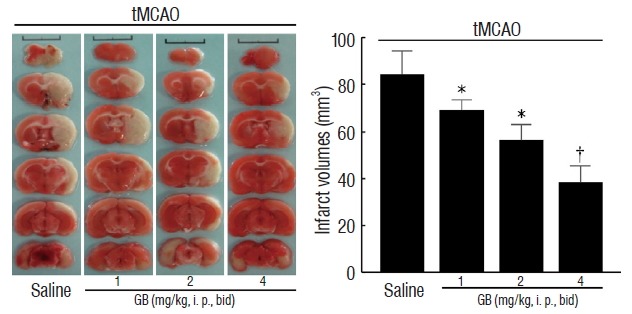
GB alleviated infarction 72 hours of reperfusion after 1 hour of tMCAO. All of the mice were suffered from tMCAO. Six consecutive TTC-stained coronal brain slices arranged in cranial to caudal order are shown. The white brain area represents infarcted tissue. *p<0.05, †p<0.01 vs. saline group, mean±standard error of mean, n=6. GB: Ginkgolide B, tMCAO: transient middle cerebral artery occlusion.
GB caused the up-regulation of BDNF protein in vivo
To assess whether GB can regulate BDNF expression, we evaluated the protein expression of BDNF in the ischemic penumbra of individual mice. As shown in Fig. 3, we observed that GB (4 mg/kg) significantly increased the expression of BDNF protein (p<0.05).
Fig. 3.
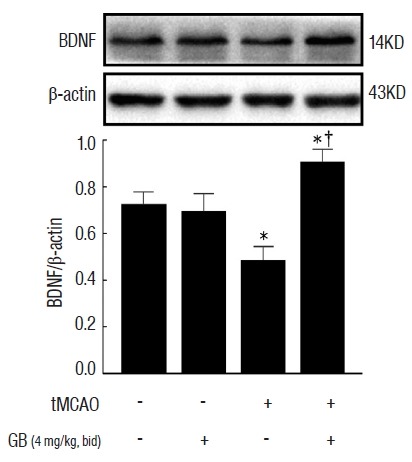
GB upregulated the expression of BDNF protein in the per-infarct area forming ipsilateral cerebral cortex. *p<0.05 vs. sham group, †p<0.05 vs. tMCAO group, mean±standard error of mean, n=4. GB: Ginkgolide B, BDNF: brain-derived neurotrophic factor, tMCAO: transient middle cerebral artery occlusion.
GB inhibited OGD/R-induced cell death in N2a cells
To further investigate whether GB produce a cytoprotective effect against ischemic insult, we generated an OGD/R model using N2a cells. Pre-treatment with GB significantly promoted the viability of cells (Fig. 4A), and decreased the release of LDH (Fig. 4B) in a concentration-dependent manner after OGD/R.
Fig. 4.
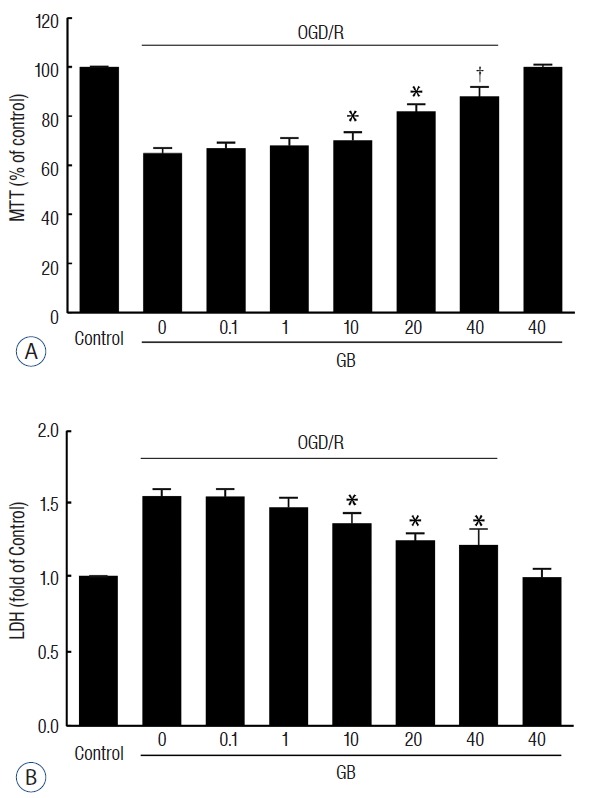
GB decreased cell death after OGD/R in N2a cells. Pre-treatment with GB significantly promoted the viability of cells (A), and decreased the release of LDH (B) in a concentration-dependent manner after OGD/R stimulation. *p<0.05, †p<0.01 vs. OGD/R group, mean±standard error of mean of 4 independent experiments. GB: Ginkgolide B, OGD/R: oxygen-glucose deprivation/reoxygenation, LDH: lactate dehydrogenase.
GB increased Bcl-2/Bax ratio and reduced caspase-3 expression via upregulating the expression of BDNF protein
To determine the effects of GB on OGD/R-induced cell apoptosis, the proteins Bcl-2, Bax, and Caspase-3 were calculated in the presence or absence of OGD/R injury. As shown in Fig. 5, GB significantly increased Bcl-2/Bax ratio, and reduced the expression of caspase-3. Furthermore, in accordance with the results in vivo, OGD/R stimulation decreased the expression of BDNF, while the administration of GB induced a conspicuous increase. These observations indicated that GB may effectively alleviate OGD/R-induced apoptosis via upregulating BDNF expression.
Fig. 5.
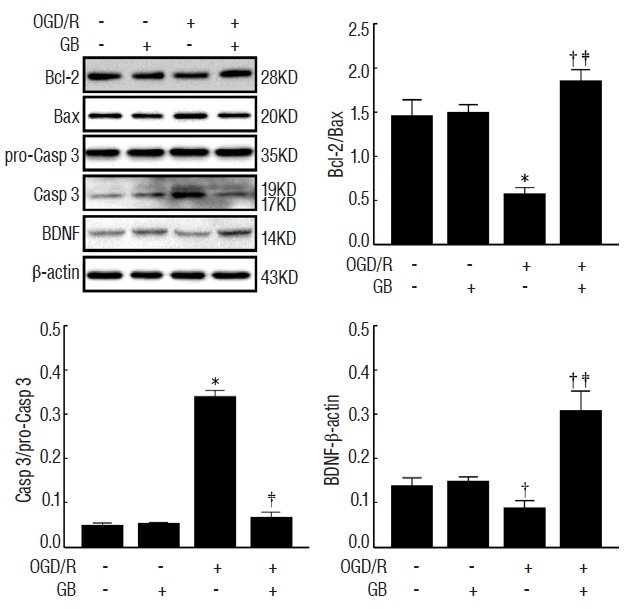
Effects of GB on the protein levels of BDNF, Bcl-2, Bax, pro-Casp 3, and Casp-3 after OGD/R in N2a cells. *p<0.01, †p<0.05 vs. control group, ‡p<0.05 vs. OGD/R group, mean±standard error of mean of 4 independent experiments. GB: Ginkgolide B, BDNF: brain-derived neurotrophic factor, OGD/R: oxygen-glucose deprivation/reoxygenation.
DISCUSSION
GB is one primary constituents of the terpenoid fraction from GBE, which show neuroprotective effects during cerebral ischemia. Here we demonstrate that it can act as a modulator of BDNF in mice and N2a cells. Our findings indicate a novel function for GB in BDNF biogenesis, which may be linked, in part, to its mechanism for neuroprotection.
Extensive researches have demonstrated that GBE can reduce ischemia damage20,23). In our study, GB decreased infarct volume, ameliorated brain edema, and improved neurological function. To further verify these effects on the survival of neuron cells after ischemia/reperfusion-induced injury, GB was used in N2a cells with and without OGD/R injury. We found that it significantly increased Bcl-2/Bax ratio, reduced the expression of caspase-3 after OGD/R. These results confirmed that GB could protect against OGD/R-induced neuronal cell death.
BDNF-Bcl-2 anti-apoptotic signaling pathway could be a fundamental mechanism at least partially responsible for the pathological changes in ischemic stroke4,12,17,18). In addition, the related mechanisms of neuroprotective effect induced by GBE can also be achieved through the up-regulation of BDNF expression in depression15) and Alzheimer’s disease10). As previously reported9,14,24), we found that the levels of BDNF protein were significantly decreased not only after tMCAO in mice but also after OGD/R in N2a cells. As expected, we observed that GB protected against the OGD/R-induced apoptosis of N2a cell in association with increasing levels of BDNF. However, it is clear that further experiments are needed to examine how GB modulate the expression of BDNF.
CONCLUSION
In the present study, we found that GB attenuated ischemic insult induced by tMCAO and OGD/R. It can increase Bcl-2/Bax ratio and reduce the expression of caspase-3 partly via modulating BDNF expression. Thus, these data demonstrates a potential mechanism of GB in the neuroprotection of ischemic stroke.
References
- 1.Albers GW, Goldstein LB, Hess DC, Wechsler LR, Furie KL, Gorelick PB, et al. Stroke Treatment Academic Industry Roundtable (STAIR) recommendations for maximizing the use of intravenous thrombolytics and expanding treatment options with intra-arterial and neuroprotective therapies. Stroke. 2011;42:2645–2650. doi: 10.1161/STROKEAHA.111.618850. [DOI] [PubMed] [Google Scholar]
- 2.Bederson JB, Pitts LH, Tsuji M, Nishimura MC, Davis RL, Bartkowski H. Rat middle cerebral artery occlusion: evaluation of the model and development of a neurologic examination. Stroke. 1986;17:472–476. doi: 10.1161/01.str.17.3.472. [DOI] [PubMed] [Google Scholar]
- 3.Bekinschtein P, Cammarota M, Medina JH. BDNF and memory processing. Neuropharmacology. 2014;76 Pt C:677–683. doi: 10.1016/j.neuropharm.2013.04.024. [DOI] [PubMed] [Google Scholar]
- 4.Berretta A, Tzeng YC, Clarkson AN. Post-stroke recovery: the role of activity-dependent release of brain-derived neurotrophic factor. Expert Rev Neurother. 2014;14:1335–1344. doi: 10.1586/14737175.2014.969242. [DOI] [PubMed] [Google Scholar]
- 5.Braquet P. Proofs of involvement of PAF-acether in various immune disorders using BN 52021 (ginkgolide B): a powerful PAF-acether antagonist isolated from Ginkgo biloba L. Adv Prostaglandin Thromboxane Leukot Res. 1986;16:179–198. [PubMed] [Google Scholar]
- 6.Chen PH, Gao S, Wang YJ, Xu AD, Li YS, Wang D. Classifying ischemic stroke, from TOAST to CISS. CNS Neurosci Ther. 2012;18:452–456. doi: 10.1111/j.1755-5949.2011.00292.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Desquand S, Touvay C, Randon J, Lagente V, Vilain B, Maridonneau-Parini I, et al. Interference of BN 52021 (ginkgolide B) with the broncho-pulmonary effects of PAF-acether in the guinea-pig. Eur J Pharmacol. 1986;127:83–95. doi: 10.1016/0014-2999(86)90208-6. [DOI] [PubMed] [Google Scholar]
- 8.Hatashita S, Hoff JT, Salamat SM. Ischemic brain edema and the osmotic gradient between blood and brain. J Cereb Blood Flow Metab. 1988;8:552–559. doi: 10.1038/jcbfm.1988.96. [DOI] [PubMed] [Google Scholar]
- 9.Hirouchi M, Itoh Y, Aoki Y, Oka M, Ukai Y. Reversal by NS-7, a neuroprotective compound, of the decrease in transcription factor CREB mRNA expression in rat brain after permanent middle cerebral artery occlusion. Neurosci Lett. 2001;305:193–196. doi: 10.1016/s0304-3940(01)01852-3. [DOI] [PubMed] [Google Scholar]
- 10.Hou Y, Aboukhatwa MA, Lei DL, Manaye K, Khan I, Luo Y. Anti-depressant natural flavonols modulate BDNF and beta amyloid in neurons and hippocampus of double TgAD mice. Neuropharmacology. 2010;58:911–920. doi: 10.1016/j.neuropharm.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ke Z, Yip SP, Li L, Zheng XX, Tong KY. The effects of voluntary, involuntary, and forced exercises on brain-derived neurotrophic factor and motor function recovery: a rat brain ischemia model. PLoS One. 2011;6:e16643. doi: 10.1371/journal.pone.0016643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Larsson E, Nanobashvili A, Kokaia Z, Lindvall O. Evidence for neuroprotective effects of endogenous brain-derived neurotrophic factor after global forebrain ischemia in rats. J Cereb Blood Flow Metab. 1999;19:1220–1228. doi: 10.1097/00004647-199911000-00006. [DOI] [PubMed] [Google Scholar]
- 13.Panja D, Bramham CR. BDNF mechanisms in late LTP formation: a synthesis and breakdown. Neuropharmacology. 2014;76 Pt C:664–676. doi: 10.1016/j.neuropharm.2013.06.024. [DOI] [PubMed] [Google Scholar]
- 14.Qi D, Ouyang C, Wang Y, Zhang S, Ma X, Song Y, et al. HO-1 attenuates hippocampal neurons injury via the activation of BDNF-TrkB-PI3K/Akt signaling pathway in stroke. Brain Res. 2014;1577:69–76. doi: 10.1016/j.brainres.2014.06.031. [DOI] [PubMed] [Google Scholar]
- 15.Qin XS, Jin KH, Ding BK, Xie SF, Ma H. Effects of extract of Ginkgo biloba with venlafaxine on brain injury in a rat model of depression. Chin Med J (Engl) 2005;118:391–397. [PubMed] [Google Scholar]
- 16.Reichardt LF. Neurotrophin-regulated signalling pathways. Philos Trans R Soc Lond B Biol Sci. 2006;361:1545–1564. doi: 10.1098/rstb.2006.1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schäbitz WR, Sommer C, Zoder W, Kiessling M, Schwaninger M, Schwab S. Intravenous brain-derived neurotrophic factor reduces infarct size and counterregulates Bax and Bcl-2 expression after temporary focal cerebral ischemia. Stroke. 2000;31:2212–2217. doi: 10.1161/01.str.31.9.2212. [DOI] [PubMed] [Google Scholar]
- 18.Schäbitz WR, Steigleder T, Cooper-Kuhn CM, Schwab S, Sommer C, Schneider A, et al. Intravenous brain-derived neurotrophic factor enhances poststroke sensorimotor recovery and stimulates neurogenesis. Stroke. 2007;38:2165–2172. doi: 10.1161/STROKEAHA.106.477331. [DOI] [PubMed] [Google Scholar]
- 19.Seth P, Kumari R, Dikshit M, Srimal RC. Effect of platelet activating factor antagonists in different models of thrombosis. Thromb Res. 1994;76:503–512. doi: 10.1016/0049-3848(94)90279-8. [DOI] [PubMed] [Google Scholar]
- 20.Smith PF, Maclennan K, Darlington CL. The neuroprotective properties of the Ginkgo biloba leaf: a review of the possible relationship to platelet-activating factor (PAF) J Ethnopharmacol. 1996;50:131–139. doi: 10.1016/0378-8741(96)01379-7. [DOI] [PubMed] [Google Scholar]
- 21.Teather LA, Afonso VM, Wurtman RJ. Inhibition of platelet-activating factor receptors in hippocampal plasma membranes attenuates the inflammatory nociceptive response in rats. Brain Res. 2006;1097:230–233. doi: 10.1016/j.brainres.2006.03.036. [DOI] [PubMed] [Google Scholar]
- 22.Vargaftig BB, Lefort J, Chignard M, Benveniste J. Platelet-activating factor induces a platelet-dependent bronchoconstriction unrelated to the formation of prostaglandin derivatives. Eur J Pharmacol. 1980;65:185–192. doi: 10.1016/0014-2999(80)90391-x. [DOI] [PubMed] [Google Scholar]
- 23.Yin B, Xu Y, Wei R, Luo B. Ginkgo biloba on focal cerebral ischemia: a systematic review and meta-analysis. Am J Chin Med. 2014;42:769–783. doi: 10.1142/S0192415X14500499. [DOI] [PubMed] [Google Scholar]
- 24.Zhou J, Ping FF, Lv WT, Feng JY, Shang J. Interleukin-18 directly protects cortical neurons by activating PI3K/AKT/NF-κB/CREB pathways. Cytokine. 2014;69:29–38. doi: 10.1016/j.cyto.2014.05.003. [DOI] [PubMed] [Google Scholar]