

Abstract
Endometrial carcinoma (EC) exhibits the strongest association with obesity of all cancers. Growth of these tumors is driven by PI3K/AKT activation, and opposed by tumor suppressors, including the tuberous sclerosis complex 2 (TSC-2) and p27, with inactivation of TSC2 and loss or cytoplasmic mislocalization of p27 both being linked to PI3K/AKT activation. However, little is known about the involvement of p27 in the development of EC arising in the setting of obesity, especially its role early in disease progression. Using a panel of EC cell lines, in vitro studies using PI3K inhibitors provided evidence that p27 rescue contributes to the efficacy of interventions that inhibit endometrial cell growth. In “at risk” obese patients, and in an animal model of obesity-associated EC (Tsc2-deficient Eker rats), p27 was moderately-to-severely reduced in both “normal” endometrial glands as well as in endometrial complex atypical hyperplasia (obese women), and endometrial hyperplasia (obese rats). In obese Eker rats, an energy balance intervention; caloric restriction from 2–4 months of age, reduced weight, increased adiponectin and lowered leptin to produce a favorable leptin:adiponectin ratio, and reduced circulating insulin levels. Caloric restriction also increased p27 levels, relocalized this tumor suppressor to the nucleus, and significantly decreased hyperplasia incidence. Thus, dietary and pharmacologic interventions that inhibit growth and decrease risk for development of endometrial lesions are associated with increased expression and nuclear (re)localization of p27. These data suggest that p27 levels and localization may be useful as a biomarker, and possible determinant, of risk for EC arising in the setting of obesity.
Keywords: benign endometrium, PI3Kinase, Eker rat, endometrial hyperplasia, endometrial carcinoma, mTORC1, obesity, p27
Introduction
Of all cancers, evidence is strongest for the association between obesity and increased risk for Type I endometrial endometrioid adenocarcinoma (EC) incidence and associated mortality (1). While an average woman has a 3% lifetime risk of EC, the risk for obese women is increased to 9–10%. Obese women account for up to 46% of all postmenopausal endometrial cancers, and a higher proportion of pre-menopausal EC than normal weight women (2–4). In addition, obese women have a 6-fold increased risk of dying from EC as compared to women of normal weight (5). Despite this clear-cut association, and obvious importance for women’s health, the molecular mechanism(s) promoting the development of EC in the setting of obesity remain unclear.
Dysregulation in the production of adipokines that either promote (i.e. leptin) or inhibit (i.e. adiponectin) IGF-IR/AKT/mTOR driven proliferation may play a role in the development of EC in obese women (6). Interestingly, increased serum levels of leptin have been reported in women with EC in addition to decreased serum levels of adiponectin (7–10). Low levels of adiponectin, which correlate with high insulin resistance, are a strong predictor of EC risk; women with EC are more likely to have low adiponectin levels than controls (11). In 2007, a WHO nested case-control study of adiponectin levels from prospectively collected, pre-diagnosis samples also found that low adiponectin levels were associated with increased endometrial cancer risk (12).
The cell cycle regulator and tumor suppressor p27 plays a key role in the development of EC. p27 is a member of the Cip/Kip family of cyclin-dependent kinase inhibitors (CKIs), which function to negatively regulate cell cycle progression. The cell-cycle and phosphorylation-dependent events that control p27 levels and subcellular localization are catalyzed by different kinases that regulate degradation and nuclear-cytoplasmic shuttling. Nuclear p27 is targeted for degradation by Skp2 E3 ligase (6). Phosphorylation of p27 by several kinases including AKT (7) causes nuclear p27 to be exported to the cytoplasm (8, 9). In the cytoplasm, p27 is targeted for degradation by a different E3-ligase, KPC1/KPC2 (10). In addition to targeting p27 for degradation, phosphorylation also can stabilize this CKI and sequester it in the cytoplasm. In the cytoplasm, p27 has been shown to be anti-apoptotic (11) and mediate cellular migration and metastasis by blocking Rho stress fiber formation (12–14). Dysregulation of p27 has been reported in EC, most commonly as a result of decreased p27 expression (15–21). During progression of EC, normal regulation of p27 is lost, with p27 being absent or undetectable in a significant proportion of endometrial hyperplasia (EndoHP) (22). Alternatively, in some EC, p27 expression is retained, but is mislocalized to the cytoplasm (17).
In addition to aberrant p27, alterations in the PTEN tumor suppressor and the PI3K/AKT signaling pathway comprise an important group of “driver” mutations for EC (23). Loss of function of TSC2, a tumor suppressor gene that regulates mTORC1, also occurs in a subset of EC (24–26). As a result, the PI3K/AKT/mTORC1 signaling pathway is thought to be the central driver for a subset of EC, particularly endometrioid EC, and an important target for new therapeutic approaches for this disease (27).
We have recently reported that aged Eker rats carrying a defect in the Tsc2 tumor suppressor gene develop endometrial hyperplasia (HP) with a high frequency with the potential to progress to EC (26). Similar to the human disease, IGFI-R and PI3K/AKT signaling pathways participate in development of endometrial lesions in this model. IGFI-R signaling to PI3K/AKT and downstream activation of mTORC1, drives development of these lesions, with loss of inhibitory feedback to the IGFI-R adapter protein IRS linked to progression to EH following loss of the normal Tsc2 allele and activation of IGFI-R signaling (26). Thus, development of EH in Eker rats mimics the human disease at both the genetic (Tsc2) and biochemical (activation of IGF and mTORC1) level.
Although obesity is an established risk factor for EC, to date little or no data are available on how interventions, such as caloric restriction, will affect the development of endometrial lesions arising in the setting of obesity. We asked whether p27 was modulated in association with increased risk for EC in the setting of obesity, and in response to interventions that decreased risk and inhibited growth of endometrial cells. We found that p27 expression was severely reduced and/or mislocalized to the cytoplasm in histologically “normal” endometrial glands and CAH of obese women compared to normal weight women. We also demonstrated in the Eker rat model of obesity-associated EC, that similar to the human disease, loss of p27 occurs early in association with development of EH. Importantly, in this model, caloric restriction reduced weight, induced a favorable adiponectin:leptin ratio, increased nuclear p27 levels and significantly reduced the incidence of EH. Similarly, efficacy of pharmacologic PI3K inhibitors that inhibited EC growth was associated with rescue of p27 function. These data point to p27 as a modifiable biomarker and possible determinant of risk for EC in the setting of obesity.
Methods
Endometrial tissue samples
For Western blot analysis, normal luteal (secretory) phase endometrium (L; n = 6) and normal follicular (proliferative) phase endometrium (F; n= 3) from endometrial biopsies were flash frozen and stored at −80°C. For immunohistochemistry, formalin-fixed paraffin-embedded sections of normal luteal phase endometrium (n = 6; mean BMI = 26.9 kg/m2), normal follicular phase endometrium from normal weight women (n=4; mean BMI = 26.0 kg/m2), normal follicular phase endometrium from obese women (n=6; 36.8 kg/m2), endometrial complex hyperplasia with atypia from obese women (CAH; n = 11; mean BMI = 34.7 kg/m2), and endometrial endometrioid adenocarcinoma grade 1 (EEC grade 1; n = 9; mean BMI = 39.6 kg/m2) were derived from hysterectomy surgical specimens submitted to the Department of Pathology, U.T. MD Anderson Cancer Center. In addition, 51 endometrial biopsies were voluntarily collected from postmenopausal women on days 5–10 of the menstrual cycle from asymptomatic lean (BMI < 25 kg/m2; n=10) and obese (BMI > 30 kg/m2; n=41) women. For normal weight women the mean BMI was 23.4 kg/m2. For obese women the mean BMI was 36.3 kg/m2. H&E-stained slides were microscopically evaluated by two gynecologic pathologists (R.R.B. and B.D.) to verify the presence of histologically normal follicular (proliferative) phase endometrium, normal luteal (secretory) phase endometrium or to confirm the diagnosis of complex atypical endometrial hyperplasia or endometrioid endometrial adenocarcinoma. Of the 51 timed endometrial biopsies collected voluntarily from lean and obese postmenopausal women there was no histological difference between lean and obese biopsies. The additional 51 endometrial biopsies were formalin-fixed paraffin embedded and evaluated for immunohistochemical protein expression.
Animals
The care and handling of rats were in accord with NIH guidelines in Association for the Assessment and Accreditation of Laboratory Animal Care–accredited facilities. All protocols involving the use of these animals were approved by the U.T. MD Anderson Animal Care and Use Committee. Fifteen-month-old Eker (Tsc2Ek/+) rats were treated with vehicle (n = 15) or a rapamycin analogue (n=14) as previously described (26). Body composition was measured in Eker rats (n = 5) and Long-Evans rats (n = 5) fed a standard AIN diet (Cat #D1001; Research Diets, New Brunswick, NJ) at 16 months of age by quantitative magnetic resonance (qMR; Echo Medical Systems, Houston, TX). Endpoints for qMR analysis included fat mass, total mass, and percent body fat.
For caloric restriction studies, all rats were housed in a semibarrier facility at the University of Texas at Austin Animal Resource Center. All experimentation was approved by the Institutional Animal Care and Use Committee at the University of Texas (Austin, TX). Female Eker rats were treated on postnatal days 10, 11, and 12 with GEN 50mg/kg (Sigma) or sesame oil vehicle, using a total of 50 l of this vehicle for each subcutaneous injection. VEH controls and GEN treated animals were randomized littermates. Upon weaning, all animals were genotyped for the presence of the Eker mutation (Tsc-2+/+ vs. Tsc-2Ek/+). Eker carriers were then singly housed in an enriched environment (shepherd shacks) in preparation for dietary interventions.
Beginning at 2 months of age, rats were subjected to dietary intervention with either control AIN-76A diet (#D12450B, Research Diets) fed ad libitum (which, from our previous experience, results in overweight to moderately obese female Eker rats, ~30% body fat by 12 months of age); or 30% calorie restricted (CR) diet (#D03020702, Research Diets) administered as a daily aliquot equivalent to 70% of the daily amount of total energy consumed by the control group. The CR diet is a modified version of the AIN-76A diet and is adjusted to provide 100% of all vitamins, minerals, fatty acids, and amino acids relative to the control group. The reduction in calorie intake was entirely due to reduced carbohydrates, with all other nutrients equivalent to those in the control group.
Rats were euthanized at 4 months of age with serum obtained at sacrifice by cardiac puncture. Serum was stored at −80°C for subsequent analysis of energy balance-related hormones. Uteri were fixed before paraffin embedding and staining with hematoxylin-eosin or incubated with antibody directed against p27. H&E-stained slides of left and right uterine horns collected from all animals were microscopically evaluated for the presence of endometrial hyperplastic lesions. Microscopic criteria for identifying rat endometrial hyperplasia (EH) have been previously published by our group (28). Incidence was defined as percent of rats with EH, with a minimum of 6 cross sections scored/animal. Multiplicity was defined as the number of uterine cross sections with EH/total number of uterine cross sections scored. Serum levels of insulin, leptin, and adiponectin were measured using Lincoplex bead-based multiplexed assays (Millipore Corporation, Billerica, MA) on a BioRad Bioplex 200 analyzer according to manufacturer’s directions.
Protein isolation, subcellular fractionation and Western blot analysis
To prepare lysates from tumors or benign endometrium, a small portion (0.5 cm3) of tissue was isolated and crushed with a mortar and pestle over dry ice and the resulting pulverized tissues were collected with liquid nitrogen and transferred into a microcentrifuge tube. Protein lysates were collected from EC cell lines (AN3CA, KLE, RL95-2 and ECC1), which were confirmed by DNA fingerprinting analysis. Radioimmunoprecipitation assay (RIPA) buffer (0.5–1.0 mL; 1% NP40, 1% sodium deoxycholate, 0.1% SDS, 0.15 mol/L NaCl, 0.01 mol/L NaPO4, 2 mmol/L EDTA, 200 mmol/L phenylmethylsulfonyl fluoride, 100 mmol/L activated sodium orthovanadate, and 1X Roche complete protease inhibitor) was added and samples were rotated at 4°C for 2 h. After rotations, the samples were centrifuged at 14,000 rpm at 4°C for 10 min. The resulting supernatant was collected, aliquoted, and stored at −80°C for future analysis. Lysate protein concentrations were determined with BCA Protein Assay Kit (Pierce Biotechnology).
Subcellular fractionation was performed as described previously (35). In brief, cells were washed with ice-cold PBS and scraped into chilled hypotonic buffer and broken by a Dounce homogenizer. After centrifugation at 3,000 rpm, the supernatant was collected (cytoplasmic fraction) and the crude nuclei pellet was further homogenized in hypotonic buffer and washed in nuclear washing buffer and centrifuged at 3,000 rpm. Purified nuclei were lysed in high-salt lysis buffer. All buffers contained 1X complete protease inhibitor cocktail and phosphatase inhibitor cocktails 1 and 2.
Samples of protein from each sample (30 μg) were size separated by SDS-PAGE and transferred overnight at 4°C onto polyvinylidene diflouride (PVDF) membranes (Pierce Biotechnology). Membranes were blocked in a TBST (TBS plus 0.05% Tween 20) + 5% nonfat milk solution for 1 h. Membranes were incubated in a primary antibody solution for 2 h at room temperature with varying antibody (1:500–1:2,000) and milk (3–5%) concentrations. Membranes were then washed three times with TBST solution for 10 min each and incubated for 1 h with the appropriate horseradish peroxidase–conjugated secondary antibody (Santa Cruz Biotechnology) at room temperature. Membranes were washed and visualized with LumiGLO chemiluminescent reagents (Kirkegaard and Perry Laboratories) or the enhanced chemiluminescence plus kit (Amersham Pharmacia Biotech) for more sensitive detection. Membrane immunoreactivity was detected by X-ray film (BioMax, Eastman Kodak). Primary antibodies for S6 ribosomal protein, phosphorylated (S235/236) S6 ribosomal protein, Akt, phosphorylated (S473) Akt, AMPK, phosphorylated (T172) AMPK, ACC and phosphorylated (S79) ACC were purchased from Cell Signaling. Primary antibodies for phosphorylated (T157) p27 and total p27 were purchased from R&D Systems. Primary antibody for glyceraldehyde-3-phosphate dehydrogenase, β-Actin and lamin A/C were purchased from Santa Cruz Biotechnology. The anti-LDH antibody was from Chemicon and Ki67 antibody was purchased from DAKO. LY294002 (20 μmol/L), and rapamycin (0.2–2 μmol/L) were purchased from Sigma and resuspended in DMSO as the vehicle.
Immunohistochemistry
Formalin-fixed paraffin-embedded tissue sections were deparaffinized and endogenous peroxidases were quenched by incubation in 1% H2O2. Antigen retrieval was done by microwaving the slides in 10 mmol/L citrate buffer (pH 6.0). Slides were incubated with primary antibody (1:50) in PBS containing 10% normal goat or horse serum overnight at 4°C. Primary antibodies were purchased from Cell Signaling for S6 ribosomal protein and phosphorylated (S235/236) S6 ribosomal protein. Primary antibody for Ki67 was purchased from DAKO North America, Inc. Primary antibodies for p27 were purchased from R&D Systems. A biotin-labeled secondary antibody was conjugated for 30 min at 37°C. The sections were stained using avidin-biotinylated horseradish peroxidase complex from the DAKO Cytomation LSAB2 System according to the manufacturer’s instructions. Diaminobenzidine reagent plus chromagen (DAKO) was incubated with the sections for up to 30 min. The sections were counterstained with hematoxylin, dehydrated, and mounted. Controls that lacked primary antibody were incubated in 1X PBS with 10% goat serum in each experiment. Immunostained sections were examined by light microscopy by two investigators (A.S.M. and B.D.), one of whom is a gynecologic pathologist (B.D.). S6 and phosphorylated S6 were scored semiquantitatively according to the intensity of staining on a scale of 0 (no staining) to 3+ (strong staining). Tissues with 2+ or 3+ staining in >10% of cells were considered positive for protein expression. Ki67 was scored by assessing the percentage of positive cells per 100 epithelial cells evaluated for normal weight and obese proliferative endometrium. p27 expression in the endometrium was scored semiquantitatively according to four categories (Figure 1B):
“Intact” p27 levels (Figure 1a): diffuse nuclear expression present in >75% of all endometrial glands, with the remaining glands showing a reduced number of cells with p27 expression per gland, but no completely negative glands.
“Slightly reduced”p27 levels (Figure 1b): diffuse nuclear expression present in <75% of all glands, with the remaining glands showing a reduced number of cells with p27 expression per gland, but no completely negative glands.
“moderately reduced” p27 (Figure 1c): diffuse nuclear expression present in <75% of all glands, with remaining glands showing either a reduced number of cells with p27 expression per gland or a complete absence of p27 expression.
“severely reduced” p27 levels (Figure 1d): were defined as diffuse nuclear expression present in <75% of all glands, with remaining glands showing either a reduced number of cells with p27 expression per gland or a complete absence of p27 expression.
Figure 1. Interventions that inhibit growth of EC cell lines rescue mislocalization of p27 to the cytoplasm.
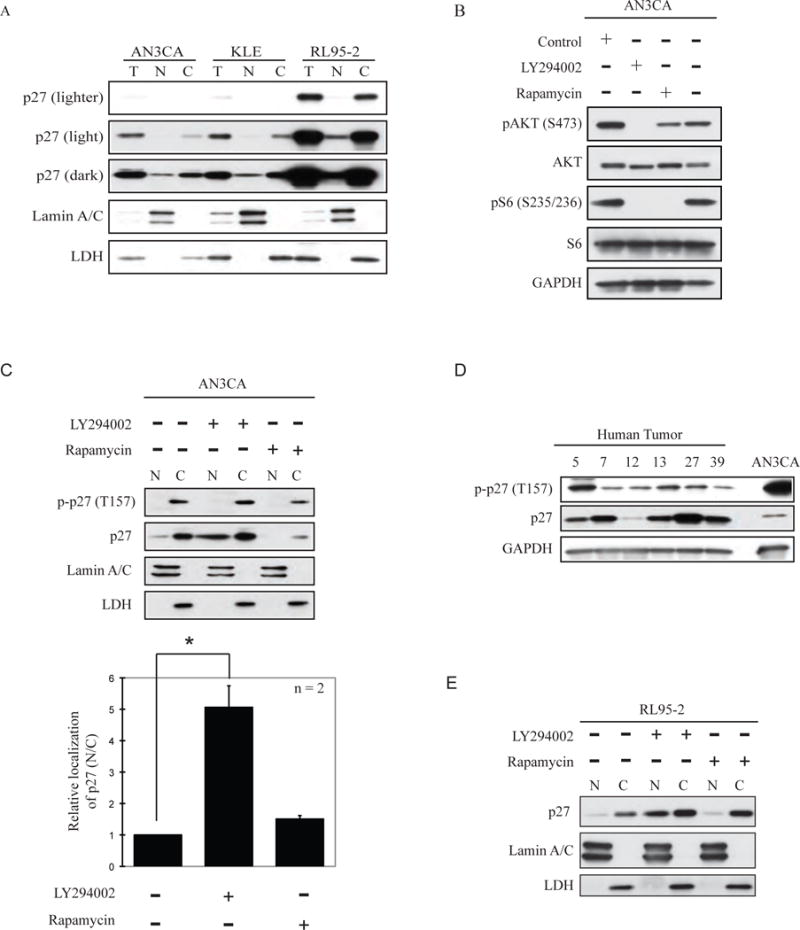
(A) Cell fractionation of a panel of EC cell lines showed increased cytoplasmic (C) localization as compared to nuclear (N) localization of p27. Total p27 is indicated (T). Lamin A/C was used as a control to identify the nuclear fraction. LDH was used as a control to identify the cytosolic fraction. (B) PI3K signaling (phosphorylation of (S473) Akt) and mTOR signaling (phosphorylation of (S235/236) S6) was activated in AN3CA cells. PI3K inhibition by LY294002 reduced Akt and mTOR activation. mTOR inhibition by rapamycin reduced phosphorylation of S6. In AN3CA (C) p27 localization was predominantly cytosolic and was phosphorylated at T157 by Akt. Treatment with LY294002 caused re-localization of p27 to the cytosol, although T157 was still phosphorylated in the remaining cytosolic pool of p27. Treatment with rapamycin did not relocalize p27 to the nucleus. The relative localization of p27 in nuclear and cytosolic fractions was quantitated by densitometry (bottom panel). (D) The expression of p27 and cytosolic localization (as indicated by phosphorylation of T157 by Akt) was investigated in 6 primary tumors and AN3CA cells for comparison. Some tumors showed severely reduced expression of p27 while the majority showed phosphorylation of T157 indicative of mislocalization of p27 to the cytoplasm. In RL95-2 (E) inhibition of PI3K by LY294002, but not mTORC1 inhibition with rapamycin, re-localized p27 to the nucleus.
Immunocytochemistry
ECC1 cells were plated onto glass coverslips 18 to 24 h before treatment and treated as indicated. Cells were then fixed in 4% paraformaldehyde in PME for 30 min at 4°C and permeabilized in 0.5% Triton X-100/PME for 30 min at room temperature. Nonspecific antigens were blocked in TBST containing 1% bovine serum albumin. Endogenous p27 was detected by anti-p27 antibody and subsequent Alexa Fluor 488 goat anti mouse antibody (Invitrogen, A-11001). The cell nuclei were detected by DAPI staining. P27 antibody was purchased from BD Biosciences (554069). Lamin A/C was purchased from Cell Signaling (2032). α–tubulin antibody was purchased from Santa Cruz Biotechnology (sc-32293). Wortmannin was purchased from Cell Signaling (9951).
RNA isolation and real-time quantitative reverse transcriptase-polymerase chain reaction (qRT-PCR)
Total RNA was extracted from frozen endometrial biopsies using Tri-reagent (Molecular Research Center, Inc.) as previously described (36). For the p27 transcript, specific polymerase chain reaction (PCR) primer pairs and a dual fluorochrome-labeled hybridization probe (TaqMan probe) were designed using Primer Express (Applied Biosystems, Foster City, CA) or Beacon Designer (Premier Biosoft International, Palo Alto, CA). Reverse transcription and qRT-PCR were performed as previously described (36). The sequence of forward and reverse primers and TaqMan Probes for the p27 assay are: 1338(+) GTAGCACATAAACTTTGGGGAAGG; 1407(−) CTTCATACCCCGCTCCAC and 1388(−) FAM-TCAGTTCCTCAGCCCCACCCTGCC-BHQ1 (based on hp27 NM_004064). Data were normalized to the geometric mean of 18S, βActin and 36B4 transcript levels as previously described (36).
Statistical analysis
For continuous variables, the differences between groups were examined by unpaired Student’s t tests, ANOVA, or nonparametric Mann-Whitney test. Fisher’s exact test and χ2 analysis were used to compare the immunostaining levels of p27 between groups. Statistical significance was defined as P < 0.05.
Results
Interventions that rescue p27 inhibit EC growth
Both loss and mislocalization of p27 can abrogate p27 function as a CKI, and in EC, both decreased p27 expression and cytoplasmic sequestration have been observed (15–21). PI3K/AKT and mTORC1 activation are high frequency events in EC, and PI3K/AKT pathway activation is known to both sequester p27 in the cytoplasm and induce degradation. We asked if therapeutic interventions targeting PI3K/AKT and/or mTORC1 could rescue p27, specifically, relocalize this CKI to the nucleus in EC cell lines in which p27 was predominantly cytoplasmic. Rapalogues are currently in use in the clinic for treatment of EC, but have shown only modest efficacy (29–33), and PI3K inhibitors are being evaluated as investigational drugs for EC (34, 35). We evaluated pharmacologic interventions targeting the PI3K pathway upstream of p27 (LY294002 and wortmannin), or downstream with an mTORC1 inhibitor (rapamycin), to obtain proof-of-concept that interventions that were effective at inhibit ing EC cell growth rescued p27.
We examined p27 localization in AN3CA, KLE and RL95-2 EC cell lines using subcellular fractionation, with lamin A/C and LDH as controls for nuclear and cytoplasmic fractions, respectively, and found that p27 was low (AN3CA and KLE) and/or predominantly cytoplasmic in all three cell lines (Figure 1A). p27 phosphorylation at T157 in the p27 nuclear localization signal (NLS) by AKT is associated with sequestration in the cytoplasm due to 14-3-3 binding (12). As a control, we confirmed that when AKT was activated in EC cells (as assessed by phosphorylation at S473), PI3K inhibition by LY294002 reduced AKT activation and signaling by its down-stream effector, mTORC1 (Figure 1B). As expected, the specific mTORC1 inhibitor rapamycin also blocked mTORC1 signaling (reduced phospho-S6) in EC cells, and reduced AKT phosphorylation at S473, the mTORC1 phosphorylation site (Figure 1B).
Using an antibody specific for p27 phosphorylated at T157, we observed phosphorylation of cytoplasmic (but not nuclear) p27 at T157 in EC cells, concordant with activation of AKT in these cells (Figure 1C). Consistent with high frequency of activation of PI3K/AKT signaling in EC, phosphorylation of p27 at T157 was also a feature of primary tumors (Figure 1D). Importantly, treatment with the PI3K inhibitor LY294002 significantly increased the total amount of p27 in the nuclear fraction (Figure 1C and 1E) in EC cells. In contrast, inhibition of mTORC1 signaling by rapamycin did not promote re-localization of p27 to the nucleus (Figures 1C and 1E). Densitometric analysis showed a ~ 4-fold increase of nuclear p27 compared to that observed in the cytoplasm with PI3K/AKT inhibition, but not with mTOR inhibition (Figure 1C, bottom). Overall, these data indicate that PI3K/AKT signaling contributes to cytoplasmic mislocalization of p27 in EC, and that targeting PI3K/AKT signaling can relocalize p27 to the nucleus in EC cells.
These biochemical data were replicated in AN3CA (data not shown) and an additional EC cell line, ECC1 using another PI3K inhibitor, wortmannin. As shown in Figure 2A, similar to what was observed with LY294002, wortmannin increased the ratio of nuclear to cytoplasmic p27 compared to vehicle control by cell fractionation. Immunocytochemistry confirmed that in wortmannin-treated ECC1 cells, levels of nuclear p27 increased, with a commensurate decrease in cytoplasmic p27 (Figure 2B).
Figure 2. Pharmacologic interventions that relocalize p27 to the nucleus inhibit EC proliferation.
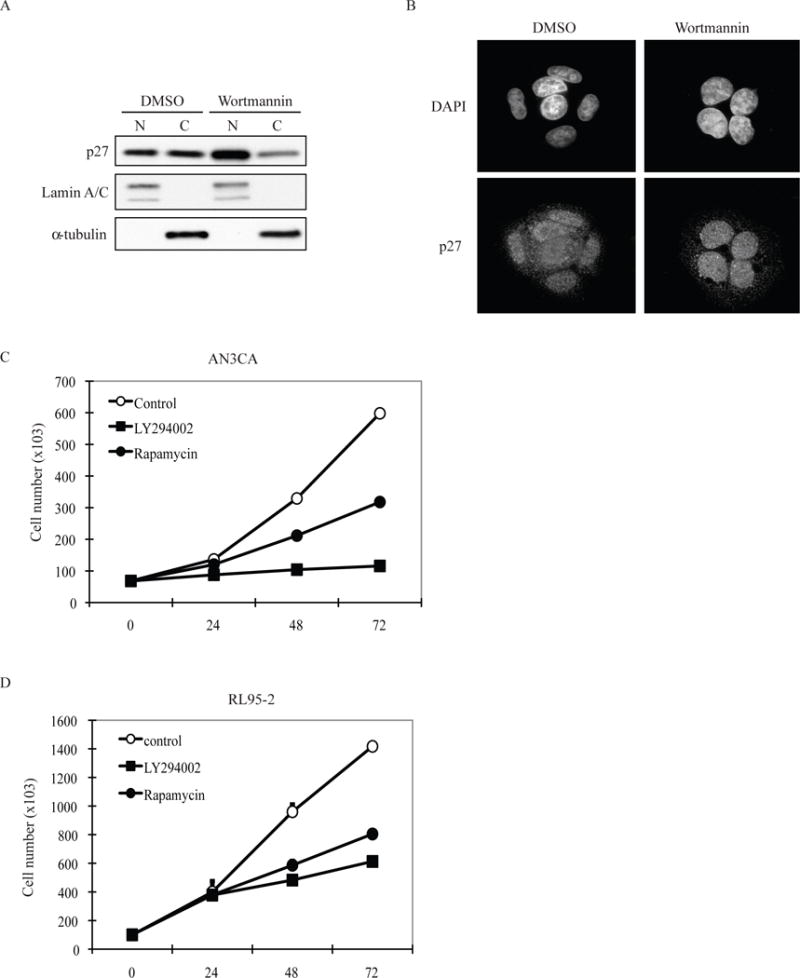
(A) Inhibition of Akt by wortmannin relocalizes p27 to cell nuclei by subcellular fractionation. ECC1 cells were treated with DMSO or wortmannin (200nM, 24 hours). The cells were then fractionated for cytosolic (C) and nuclear (N) fractions. Equal amount samples were applied to SDS-PAGE for western blot analysis with specific antibodies. Lamin A/C was used as nuclear protein marker; α-tubulin was used as cytosolic protein marker. The western blot is representative for 3 independent experiments. (B) Inhibition of Akt by wortmannin re-localizes p27 to cell nuclei by immunocytochemistry. ECC1 cells were treated with DMSO or wortmannin (200nM, 24hours). The cells were then stained with p27 antibody and DAPI. The images are representative for 3 independent experiments. Both PI3K inhibition by LY294002 and mTORC1 inhibition by rapamycin treatment significantly decreased cell proliferation in AN3CA (C) and RL95-2 (D) cells, although PI3K inhibition decreased proliferation to a greater degree than mTOR inhibition. The growth curves were performed in triplicate. Data represented is the mean ± standard error for each data point.
To confirm that p27 rescue had functional consequences, we examined the growth kinetics of EC cell lines treated with LY294002, which relocalized this CKI to the nucleus, versus rapamycin, which did not. While both PI3K (LY294002) and mTORC1 (rapamycin) inhibitors blocked mTORC1 signaling (Figure 1B), in AN3CA and RL95-2 cells, LY294002, which relocalized p27 to the nucleus, was more efficacious at inhibiting cell growth than rapamycin (Figure 2C and 2D).
Loss of p27 is an early event in progression of EC in the setting of obesity
In normal weight women, p27 expression levels are lowest in follicular endometrium and highest in luteal phase endometrium as assessed by endometrial biopsies and western analysis, with p27 levels inversely correlated with activation of mitogenic PI3K/mTORC1 signaling (phosphorylation of S6 ribosomal protein) ((37, 38) and Supplemental Figure S1).
Remarkably, in obese women, we found that endometrial p27 expression was significantly lower than even follicular phase endometrium of normal weight women (Table 1 and Figure 3A and 3B). Immunohistochemistry was performed on endometrial biopsies collected voluntarily from normal weight (BMI < 25 kg/m2) and obese (BMI > 30 kg/m2) women. Control luteal phase (high p27) biopsies from asymptomatic normal weight women (N=6) and follicular phase (low p27) biopsies from asymptomatic normal weight (N=14) and obese (N=47) women were collected on days 5–10 of the menstrual cycle, with the diagnosis of histologically normal endometrium verified by two pathologists (RRB and BD). As shown in Figure 3B and 3C and Table 1, although expressed at lower levels than luteal phase endometrium, p27 expression was intact or slightly-moderately reduced, but could be detected immunohistochemically in all follicular phase endometrial glands of normal weight women. Strong, predominantly nuclear expression of p27 was observed in >75% of all endometrial glands of normal weight women.
Table 1.
p27 expression is low in human normal obese endometrium, endometrial CAH and carcinoma.
| p27 expression | Normal Weight-Luteal Endometrium, n (%) | Normal Weight-Follicular Endometrium, n (%) | Obese-Follicular Endometrium, n (%) | Obese Endometrial CAH, n (%) | Obese-Endometrial Carcinoma Grade 1, n (%) |
|---|---|---|---|---|---|
| Intact | 4 (67%)* | 1 (7%) | 10 (21%) | 1 (9%) | 1 (11%) |
| Slightly Reduced | 2 (33%) | 9 (64%) | 16 (34%) | 2 (18%) | 0 (0%) |
| Moderately Reduced | 0 (0%) | 4 (29%) | 12 (26%) | 0 (0%) | 0 (0%) |
| Severely Reduced | 0 (0%) | 0 (0%) | 9 (19%) | 8 (73%)** | 8 (89%)*** |
| No. samples | 6 | 14 | 47 | 11 | 9 |
Fishers exact test is significant for intact p27 expression in normal weight-luteal endometrium as compared to normal weight-follicular endometrium, obese-follicular endometrium, obese-endometrial CAH and obese-EC grade 1at the p<0.05 level.
Fishers exact test is significant for severely reduced p27 expression in normal weight luteal endometrium as compared to obese endometrial CAH and obese EC at the p<0.025 level.
Fishers exact test is significant for severely reduced p27 expression in normal weight follicular endometrium as compared to obese endometrial CAH and obese EC at the p<0.001 level.
Figure 3. Reduced or mislocalized p27 is a common feature in normal follicular phase endometrium, endometrial CAH and primary endometrial carcinoma (EC) from obese women.
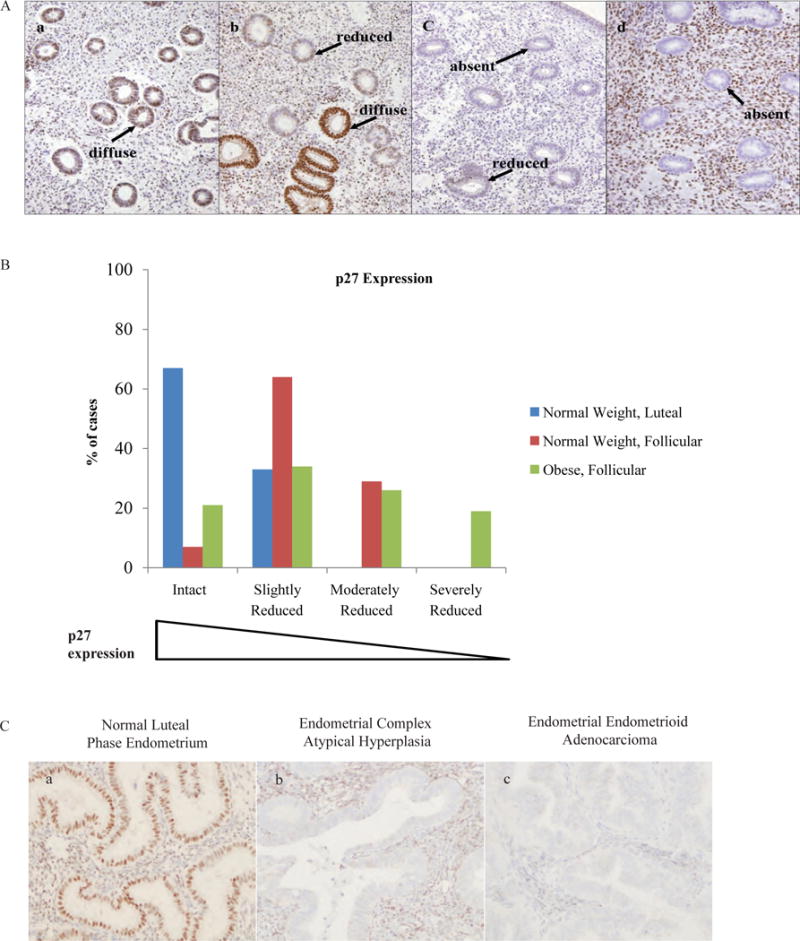
(A) The scoring system used to define immunohistochemical expression of p27 is defined as (a) intact, (b) slightly reduced, (c) moderately reduced or (d) severely reduced. More specifically, intact p27 expression is defined as diffuse nuclear expression present in >75% of all endometrial glands; expression may be reduced in <25% of glands but is never completely absent. Slightly reduced p27 expression is defined as diffuse nuclear expression present in <75% of all endometrial glands; expression may be slightly reduced in >25% of glands but is never absent. Moderately reduced p27 expression is defined as diffuse nuclear expression present in <50% of all endometrial glands; expression is reduced in >25% of glands but is never absent. Severely reduced p27 expression is defined as diffuse nuclear expression present in <75% of endometrial glands and expression is completely absent in >25% of all glands. Benign follicular phase endometrium from lean women (n=14) showed intact (a), slightly reduced (b) or moderately reduced (c) p27 levels with nuclear p27 expression never absent in endometrial glands. 19% of benign follicular phase endometrium from obese women had severely reduced (d) p27 levels including >25% of glands with entirely absent nuclear p27 expression. Arrows indicate endometrial glandular epithelium with either “diffuse”, “reduced” or “absent” nuclear p27 expression. Original magnification, ×200. (B) p27 expression was intact or only slightly reduced in a majority of cases of normal luteal phase endometrium (6 out of 6 biopsies; 100%), moderately reduced in normal follicular endometrium from normal weight women (4 out of 14 biopsies; 29%) and moderately or severely reduced in normal follicular endometrium from obese women (21 out of 47 biopsies; 45%). (C) Nuclear p27 was severely reduced with >25% of endometrial glands completely absent of p27 expression in 73% of endometrial CAH biopsies and in 89% of primary endometrial carcinoma biopsies. Original magnification, ×200.
In contrast, p27 expression was moderately to severely reduced in > 50% of endometrial glands from stage-matched histologically normal endometrium from obese women, with 19% of biopsies from obese women exhibiting severely reduced or absent p27 (Figure 3A and 3B; Table 1). Notably, and in contrast to differing p27 protein levels, there was no significant difference in p27 transcript levels between normal weight and obese endometrium (mean transcripts ± standard error for normal weight endometrium was (3.33 ± 0.37) × 10−2 p27/gNorm and obese endometrium (3.34 ± 0.28) × 10−2 p27/gNorm), consistent with the wealth of data that p27 is predominantly regulated post-translationally.
To determine if decreased p27 was a reflection of differences in the amount of cell proliferation in follicular phase endometrium from normal versus obese women, Ki67 immunostaining was performed. Remarkably, the percentage of Ki67 positive (proliferating) cells was significantly higher in the glandular epithelium of endometrium from normal weight women as compared to endometrium from obese women (48.8% ± 6.5 in normal weight versus 24.5% ± 3.6 in obese; p<0.025), indicating that lower p27 expression in obese endometrium was not a secondary effect of increased endometrial cell proliferation in obese women. Overall, these data indicate that p27 is suppressed in normal endometrial glands of obese women.
We next compared expression and localization of p27 in CAH and primary EC during luteal phase where p27 levels in normal endometrial glands are at their highest. As reported previously, both endometrial CAH and EC exhibited high levels of phospho-S6, consistent with activation of PI3K signaling in these lesions (39). As shown in Figure 3C, in luteal phase endometrium, p27 expression is intact (high) and primarily nuclear. In contrast, in the majority of endometrial CAH, p27 expression was severely reduced or absent in >70% of these early lesions, and was severely reduced or absent in 89% of primary EC (Figure 3C, Table 1). Thus, the phenotype of p27 loss observed in “normal” endometrium of obese women is retained as a feature of early (CAH) and neoplastic (EC) endometrial lesions arising in the setting of obesity,.
Caloric restriction blocks obesity-associated loss of p27 and development of preneoplastic lesions
Eker rats carrying a defect in the Tsc2 tumor suppressor gene are a genetically-defined model for endometrial hyperplasia (EH) that progresses to EC by 16 months of age (26, 28). The germ line Tsc-2 mutation is maintained on the Long-Evans background, a strain commonly used as a model for dietary-induced obesity, and we found that on a standard ad-libitum AIN diet, Eker rats became obese, with ~30% body fat as determined by quantitative magnetic resonance using an EchoMRI whole body magnetic resonance analysis system (18.3 ± 1.7% mean body fat in young Eker rats as compared to 29.0 ± 2.9% mean body fat in aged Eker rats; p<0.01); non-obese rats are typically < 20% body fat at comparable age. Aged Eker rats also had a 2.6 fold increase in mean fat mass as compared to young Eker rats (57.1 ± 11.3 mean fat mass in young Eker rats as compared to 148.0 ± 31.0 mean fat mass in aged Eker rats; p<0.01).
An early feature of this model, associated with loss of the wild-type Tsc2 allele, is the appearance of normal appearing “pre-hyperplastic” glands that show activated mTORC1 signaling (phosphorylation of the downstream effector S6) in 6% of glands that retain negative feedback to PI3K via serine phorphorylation of IRS-1, which are the earliest histologically discernible lesions in this model (26, 36). Figure 4A shows a normal endometrial gland with very few phospho-S6 positive cells, and an adjacent “pre-hyperplastic” gland that shows activation of mTORC1 signaling (phospho-S6) in all glandular epithelial cells. Figure 4B shows that the expression of p27 is severely reduced in “pre-hyperplastic” glandular cells that had lost Tsc2 (mTORC1 active and phospho-S6 positive) indicating that loss of p27 expression is an early event in the development of EH. Consistent with the Tsc2-deficiency that drives progression in this model, EH were also strongly positive for phospho-S6, indicative of mTORC1 activation (Figure 4C).
Figure 4. Expression of p27 is decreased in obese Tsc2-deficient rats and reversed by caloric restriction.
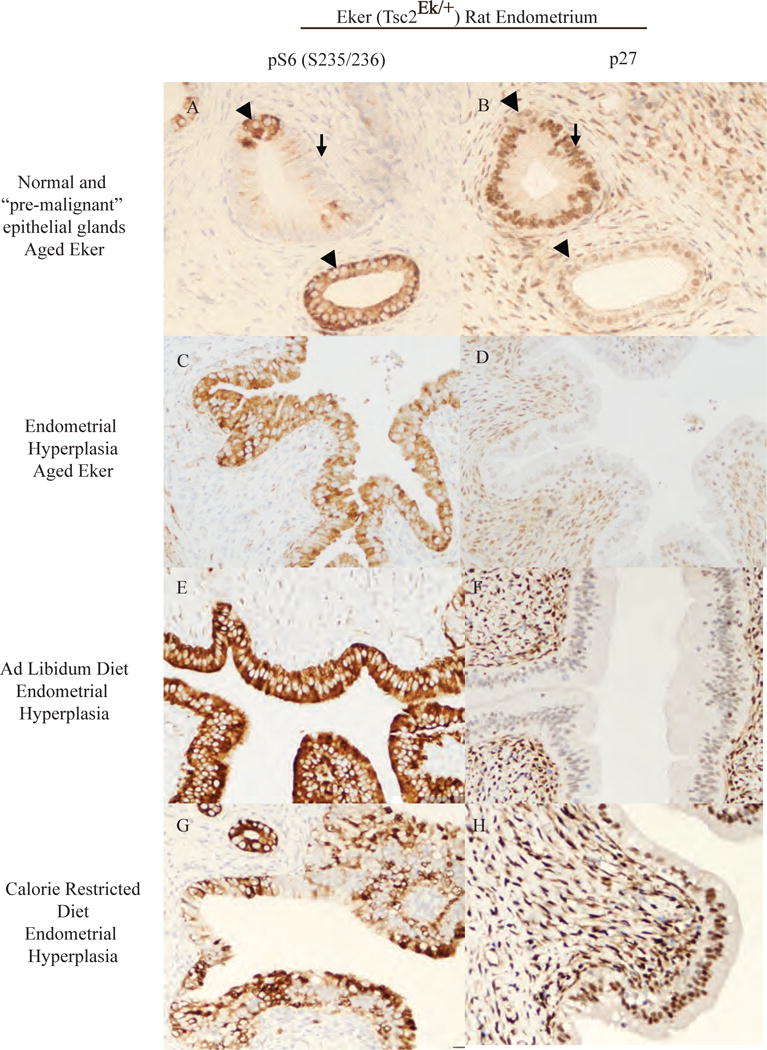
mTORC1 activation (phosphorylation at (S235/236) of S6 ribosomal protein) was present in 6% of normal appearing glands (A) and in 100% of endometrial hyperplastic (EH) lesions (C). p27 nuclear localization was intact in normal endometrial epithelium that do not have activated mTORC1 signaling (B) and was decreased or absent in endometrial hyperplasia (EH)(D). Eker rats treated with genestein neonatally developed EH exhibiting mTORC1 activation in ad libitum fed rats (E) and in calorie restriction (CR) rats (G). ad libitum fed rats had decreased or absent nuclear p27 in EH lesions (F). Caloric restriction restores and increases the nuclear expression of p27 in EH lesions (H). Arrows indicate glandular epithelial cells with mTORC1 activation that was inversely correlated with nuclear p27 expression. Original magnification, ×200.
Similar to what was observed in normal endometrium from obese women (Table 1) in obese aged Eker rats, 24% of normal endometrial glands had severely reduced or absent expression of p27 (Table 2A). Reduced p27 expression was also observed in the “pre-hyperplastic” glandular cells that had lost Tsc2 and EH that develop in obese Eker rats, with 82% of EH exhibiting moderately to severely reduced p27 (Figure 4B and 4D and Table 2A). Thus in the setting of obesity in women and the Eker rat model, loss of p27 occurs very early, being observed with a high frequency in histologically “normal” obese endometrium and “pre-hyperplastic” lesions with aberrant PI3K signaling, a phenotype that persists in preneoplastic EH/CAH and EC, identifying dysregulation of p27 as an early event in the progression of EC.
Table 2A.
p27 immunohistochemical expression in Aged Eker rats.
| p27 expression | Aged Eker rats | |
|---|---|---|
| ad libidum diet | ||
| Normal Endometrial Glands, n (%) | Endometrial Hyperplastic Glands, n (%) | |
| Intact | 214 (57%)* | 0 (0%) |
| Slightly Reduced | 74 (20%)* | 16 (19%) |
| Moderately Reduced | 0 (0%)* | 5 (6%) |
| Severely Reduced | 89 (24%)* | 65 (76%) |
| Total | 377 | 86 |
Fishers exact test is significant at the p<0.001 level for comparison of p27 expression in normal glands as compared to endometrial hyperplastic glands.
We next utilized this model to ask whether a dietary intervention to reduce weight gain could affect p27 expression, localization and/or development of endometrial lesions. We previously demonstrated in this model that a brief, neonatal xenoestrogen treatment exposure accelerated the development of EH in adult female rats (28), allowing us to conduct caloric restriction studies in normally cycling animals. In the present study we utilized the xenoestrogen genestein, a soy phytoestrogen found in soy-based infant formulas. For our dietary intervention studies, Eker rats were exposed neonatally to the soy phytoestrogen genistein, and then randomized at weaning into two groups: ad libitum fed or or 30% calorie restricted (CR) diet, and maintained on these diets from 2–4 months (see Methods for details). Both ad libitum fed and CR rats exhibited normal estrus cycles and had indistinguishable levels of ovarian hormones (Supplemental Table 1).
Rats on the CR diet gained significantly less weight compared to ad-libitum fed rats: CR rats had a median body weight of 176.3 ± 5.2g while ad-libitum fed rats had a median body weight of 234.5 ± 14.9 g (median ± S.E.M) (p<0.025). We also measured serum levels of insulin, leptin and adiponectin, and observed decreased serum insulin levels in CR rats (0.78 ± 0.07) × 103 ng/ml relative to ad libitum fed rats (1.48 ± 0.15 ×103 ng/ml) (median ± S.E.M.). Serum leptin levels decreased in CR rats (0.93 ± 0.06) × 103 ng/ml) (median ± S.E.M.) relative to ad libitum fed rats (2.98 ± 0.17) × 103 ng/ml), and adiponectin levels increased in CR rats (3.82 ± 1.57) × 107 μg/ml) (median ± S.E.M.) relative to ad libitum fed rats ((2.79 ± 0.13) × 107 μg/ml). Thus, even a short (2 mo) calorie-restricted diet significantly reduced weight and produced a significant and favorable change in the ratio of leptin:adiponectin from 0.11 ng/μg in ad-libitum fed rats to 0.02 ng/μg in CR fed rats (p<0.025).
The ad libitum fed Eker rats had an EH incidence of 100% by 4 months of age (12/12). In contrast, in CR rats, the incidence of EH was reduced to 70% (7/10). The multiplicity of EH lesions was also significantly reduced by CR: in ad-libitum fed rats EH were present in 123/179 (69%) of uterine cross sections as compared to CR rats with EH present in 48/146 (33%) of the uterine cross sections (p<0.001).
In both CR and ad-libitum rats, EH exhibited mTORC1 activation and were positive for phospho-S6, consistent with loss of Tsc2 function driving lesion development (Figure 4E and 4G). However differences were observed in both levels and localization of p27 between obese (ad-libitum) and lean (CR) rats. p27 was moderately or severely reduced in 100% of EH of obese rats, but was moderately or severely reduced in on 15% of EH of lean rats (Table 2B). Figure 4F illustrates loss of nuclear p27 in EH of obese rats compared EH of lean rats, which had strong nuclear expression of p27 (Figure 4H). Thus, caloric restriction reduced body weight, induced a favorable adiponectin:leptin ratio, and inhibited development of preneoplastic endometrial lesions in conjunction with increased p27 expression and localization to the nucleus.
Table 2B.
p27 immunohistochemical expression in Intervention Study Rats.
| p27 expression | Intervention Study Rats | |||
|---|---|---|---|---|
| ad libidum diet | calorie restricted diet | |||
| Normal Endometrial Glands, n (%) | Endometrial Hyperplastic Glands, n (%) | Normal Endometrial Glands, n (%) | Endometrial Hyperplastic Glands, n (%) | |
| Intact | 56 (100%)* | 0 (0%)** | 91 (93%)* | 11 (23%) |
| Slightly Reduced | 0 (0%) | 0 (0%) | 0 (0%)* | 30 (63%) |
| Moderately Reduced | 0 (0%)* | 63 (51%)** | 0 (0%) | 0 (0%) |
| Severely Reduced | 0 (0%)* | 60 (49%)** | 7 (7%) | 7 (15%) |
| Total | 56 | 123 | 98 | 48 |
Fishers exact test is significant at the p<0.001 level for comparison of p27 expression in normal glands as compared to endometrial hyperplastic glands.
Fishers exact test is significant at the p<0.001 level for comparison of p27 expression in ad libidum diet endometrial hyperplastic glands as compared to calorie restricted diet endometrial hyperplastic glands.
Together, our in vitro data demonstrating that interventions that rescue p27 inhibit growth of EC cells, clinical data demonstrating that in the setting of obesity loss of p27 occurs early in association with increased risk of CAH and EC, and experimental data in a rodent model of obesity demonstrating that an intervention (CR) that rescues p27 is effective at reducing risk of EH, point to p27 as a potential biomarker, and possible determinant, of efficacious interventions for EC that arise in the setting of obesity.
Discussion
Loss of p27 cell cycle regulation via loss of expression or cytoplasmic mislocalization of this CKI is a consistent feature of EC (17, 37). Here we report that loss of p27 function is an early event in the development of EC, occurring in histologically “normal” endometrium of obese women at increased risk of EC, in EH in obese rats, with progression to preneoplastic lesions (CAH), and primary EC and EC-derived cell lines with activated PI3K/AKT/mTORC1 signaling.
Our observation that a significant proportion of obese women had severely reduced p27 expression in histologically “normal” endometrium when compared to stage-matched endometrium from normal weight women is important, because it suggests that loss of p27-function contributes to early stages of disease progression. Dellas et al. observed that aberrant PI3K signaling (loss of PTEN) (and p27) was associated with obesity, and prognosis of EC (38). Tumors of obese women expressed significantly less PTEN (which would presumably activate PI3K signaling) and less p27 than tumors from non-obese patients. It is well known that loss of PTEN expression is an early event in the progression of EC, often observed in histologically normal endometrial glands (39). In this study, we identified loss of p27 as an early event in endometrial tumorigenesis. Confirming this hypothesis was the observed loss of p27 that occurred in normal appearing endometrial glands of obese Eker rats that have lost Tsc2, another tumor suppressor like PTEN whose function is to repress mitogenic PI3K/AKT/mTORC1 signaling. Thus, p27 status may be a better indicator of endometrial cancer risk than body mass index alone, and thus, has the potential to be developed into a biomarker for triaging asymptomatic obese women for increased endometrial cancer surveillance.
Obesity is a result of an energy imbalance that occurs when caloric intake exceeds caloric expenditure. At the organismal and cellular level, energy balance is regulated by AMPK signaling to TSC2 in the PI3K/AKT signaling pathway to suppress mTORC1 when energy levels are low (40). Several tumor suppressors are known to be lost or mutated in EC that regulate this pathway, including LKB1 (a kinase that activates AMPK), PTEN, and TSC2 along with activating mutations of PI3K (41). Identification of defects in LKB1, PTEN, PI3K, and TSC2 in preneoplastic lesions and/or EC opens up opportunities for preventative and/or therapeutic interventions targeting these defects.
In primary tumors, and EC cell lines, we found that phosphorylation of p27 at T157 was associated with cytoplasmic mislocalization. Inhibition of PI3K reduced T157 phosphorylation and relocalized p27 to the nucleus, whereas inhibition of mTORC1 by rapamycin (which is downstream of p27), neither reduced phosphorylation of T157 nor reversed cytoplasmic mislocalization of this CKI in human EC cells. To date, both PI3K inhibitors and mTORC1 inhibitors have been evaluated for therapeutic efficacy in preclinical studies and clinical trials. However, single agent rapalogs that inhibit mTORC1 signaling have only achieved modest anticancer efficacy in clinical trials (42), and the field is now moving toward combination therapies for this disease. Yang et al. evaluated a combinatorial approach using both mTORC1 and PI3K inhibitors in endometrial cancer cell lines and reported improved efficacy over use of single-agents alone (43). We found that although inhibition of PI3K and mTORC1 signaling both can decrease EC cell proliferation, inhibition of PI3K is more potent, and in contrast to mTORC1 inhibition, relocalizes p27 to the nucleus. Taken together, our data provide a mechanistic underpinning for the use of PI3K inhibitors to relocalize p27 to the nucleus to restore function of this cell-cycle inhibitor as therapeutic intervention for EC, and possibly, prevention of progression of CAH and EC in at-risk obese women.
Importantly, these studies demonstrate that p27 expression and localization is modifiable by dietary intervention. Caloric restriction in the Eker rat resulted in decreased body weight, decreased endometrial hyperplasia multiplicity, favorable modification of serum insulin and cytokines and increased expression and nuclear localization of p27 in endometrial hyperplastic lesions. We and others have previously shown that serum levels of leptin and adiponectin are significantly deregulated in obese women at risk of developing EC and can be reversed by weight loss (44–46), however, tissue specific biomarkers of risk that are modifiable by changes in body weight have not been identified. The identification of tissue specific biomarkers of EC risk that are modifiable with weight loss is of great utility in developing therapeutic approaches to EC prevention. In addition, these studies indicate that weight loss alone or in combination with PI3K inhibitors may be a good therapeutic option for the prevention or treatment of EC.
Supplementary Material
Supplemental Figure 1
p27 expression was significantly decreased in follicular (proliferative phase) (F; n=3) endometrium as compared to luteal (secretory phase) (L; n=6) endometrium and inversely correlated with activation of PI3K/mTOR (phosphorylation of (S235/236) S6 ribosomal protein) as indicated by Western analysis.
Acknowledgments
Grant support: American Cancer Society Mentored Research Scholar Grant MRSG-11-221-01-CNE to A.S.M.; NIH/National Cancer Institute grants Gynecologic SPORE in Uterine Cancer 1P50CA098258-01 and 2P50 CA098258-06A2 awarded to C.L.W. and K.H.L.; Cancer Prevention and Research Institute of Texas RP120855-P1; Welch Foundation BE-0023 to C.L.W.
Footnotes
The authors disclose no potential conflicts of interest.
References
- 1.Schmandt RE, Iglesias DA, Co NN, Lu KH. Understanding obesity and endometrial cancer risk: opportunities for prevention. Am J Obstet Gynecol. Dec;205(6):518–25. doi: 10.1016/j.ajog.2011.05.042. Epub 2011/08/02. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Soliman PT, Oh JC, Schmeler KM, Sun CC, Slomovitz BM, Gershenson DM, et al. Risk factors for young premenopausal women with endometrial cancer. Obstet Gynecol. 2005 Mar;105(3):575–80. doi: 10.1097/01.AOG.0000154151.14516.f7. [DOI] [PubMed] [Google Scholar]
- 3.Lu KH, Schorge JO, Rodabaugh KJ, Daniels MS, Sun CC, Soliman PT, et al. Prospective determination of prevalence of lynch syndrome in young women with endometrial cancer. J Clin Oncol. 2007 Nov 20;25(33):5158–64. doi: 10.1200/JCO.2007.10.8597. Epub 2007/10/11. eng. [DOI] [PubMed] [Google Scholar]
- 4.Schottenfeld D. Epidemiology of endometrial neoplasia. J Cell Biochem Suppl. 1995;23:151–9. doi: 10.1002/jcb.240590920. Epub 1995/01/01. eng. [DOI] [PubMed] [Google Scholar]
- 5.Abu-Abid S, Szold A, Klausner J. Obesity and cancer. J Med. 2002;33(1–4):73–86. Epub 2003/08/27. eng. [PubMed] [Google Scholar]
- 6.Bloom J, Pagano M. Deregulated degradation of the cdk inhibitor p27 and malignant transformation. Seminars in cancer biology. 2003 Feb;13(1):41–7. doi: 10.1016/s1044-579x(02)00098-6. [DOI] [PubMed] [Google Scholar]
- 7.Fujita N, Sato S, Katayama K, Tsuruo T. Akt-dependent phosphorylation of p27Kip1 promotes binding to 14-3-3 and cytoplasmic localization. J Biol Chem. 2002 Aug 9;277(32):28706–13. doi: 10.1074/jbc.M203668200. [DOI] [PubMed] [Google Scholar]
- 8.Ishida N, Kitagawa M, Hatakeyama S, Nakayama K. Phosphorylation at serine 10, a major phosphorylation site of p27(Kip1), increases its protein stability. J Biol Chem. 2000 Aug 18;275(33):25146–54. doi: 10.1074/jbc.M001144200. [DOI] [PubMed] [Google Scholar]
- 9.Rodier G, Montagnoli A, Di Marcotullio L, Coulombe P, Draetta GF, Pagano M, et al. p27 cytoplasmic localization is regulated by phosphorylation on Ser10 and is not a prerequisite for its proteolysis. EMBO J. 2001 Dec 3;20(23):6672–82. doi: 10.1093/emboj/20.23.6672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kamura T, Hara T, Matsumoto M, Ishida N, Okumura F, Hatakeyama S, et al. Cytoplasmic ubiquitin ligase KPC regulates proteolysis of p27(Kip1) at G1 phase. Nat Cell Biol. 2004 Dec;6(12):1229–35. doi: 10.1038/ncb1194. Epub 2004/11/09. eng. [DOI] [PubMed] [Google Scholar]
- 11.Liang J, Shao SH, Xu ZX, Hennessy B, Ding Z, Larrea M, et al. The energy sensing LKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat Cell Biol. 2007 Feb;9(2):218–24. doi: 10.1038/ncb1537. Epub 2007/01/24. eng. [DOI] [PubMed] [Google Scholar]
- 12.Shin I, Yakes FM, Rojo F, Shin NY, Bakin AV, Baselga J, et al. PKB/Akt mediates cell-cycle progression by phosphorylation of p27(Kip1) at threonine 157 and modulation of its cellular localization. Nat Med. 2002 Oct;8(10):1145–52. doi: 10.1038/nm759. Epub 2002/09/24. eng. [DOI] [PubMed] [Google Scholar]
- 13.Viglietto G, Motti ML, Bruni P, Melillo RM, D’Alessio A, Califano D, et al. Cytoplasmic relocalization and inhibition of the cyclin-dependent kinase inhibitor p27(Kip1) by PKB/Akt-mediated phosphorylation in breast cancer. Nat Med. 2002 Oct;8(10):1136–44. doi: 10.1038/nm762. Epub 2002/09/24. eng. [DOI] [PubMed] [Google Scholar]
- 14.Denicourt C, Dowdy SF. Cip/Kip proteins: more than just CDKs inhibitors. Genes Dev. 2004 Apr 15;18(8):851–5. doi: 10.1101/gad.1205304. [DOI] [PubMed] [Google Scholar]
- 15.Erkanli S, Kayaselcuk F, Kuscu E, Bagis T, Bolat F, Haberal A, et al. Expression of survivin, PTEN and p27 in normal, hyperplastic, and carcinomatous endometrium. Int J Gynecol Cancer. 2006 May-Jun;16(3):1412–8. doi: 10.1111/j.1525-1438.2006.00541.x. Epub 2006/06/29. eng. [DOI] [PubMed] [Google Scholar]
- 16.Abargel A, Avinoach I, Kravtsov V, Boaz M, Glezerman M, Menczer J. Expression of 27 and 53: comparative analysis of uterine carcinosarcoma and endometrial carcinoma. Int J Gynecol Cancer. 2004 Mar-Apr;14(2):354–9. doi: 10.1111/j.1048-891x.2004.014221.x. Epub 2004/04/17. eng. [DOI] [PubMed] [Google Scholar]
- 17.Masciullo V, Susini T, Zamparelli A, Bovicelli A, Minimo C, Massi D, et al. Frequent loss of expression of the cyclin-dependent kinase inhibitor p27(Kip1) in estrogen-related Endometrial adenocarcinomas. Clin Cancer Res. 2003 Nov 1;9(14):5332–8. [PubMed] [Google Scholar]
- 18.Pavlides SC, Huang KT, Reid DA, Wu L, Blank SV, Mittal K, et al. Inhibitors of SCF-Skp2/Cks1 E3 ligase block estrogen-induced growth stimulation and degradation of nuclear p27kip1: therapeutic potential for endometrial cancer. Endocrinology. 2013 Nov;154(11):4030–45. doi: 10.1210/en.2013-1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bamberger AM, Riethdorf L, Milde-Langosch K, Bamberger CM, Thuneke I, Erdmann I, et al. Strongly reduced expression of the cell cycle inhibitor p27 in endometrial neoplasia. Virchows Arch. 1999 May;434(5):423–8. doi: 10.1007/s004280050361. Epub 1999/07/02. eng. [DOI] [PubMed] [Google Scholar]
- 20.Oshita T, Shigemasa K, Nagai N, Ohama K. p27, cyclin E, and CDK2 expression in normal and cancerous endometrium. Int J Oncol. 2002 Oct;21(4):737–43. doi: 10.3892/ijo.21.4.737. Epub 2002/09/20. eng. [DOI] [PubMed] [Google Scholar]
- 21.Nycum LR, Smith LM, Farley JH, Kost ER, Method MW, Birrer MJ. The role of p27 in endometrial carcinoma. Gynecol Oncol. 2001 May;81(2):242–6. doi: 10.1006/gyno.2001.6144. Epub 2001/05/02. eng. [DOI] [PubMed] [Google Scholar]
- 22.Horree N, van Diest PJ, van der Groep P, Sie-Go DM, Heintz AP. Progressive derailment of cell cycle regulators in endometrial carcinogenesis. J Clin Pathol. 2008 Jan;61(1):36–42. doi: 10.1136/jcp.2006.043794. [DOI] [PubMed] [Google Scholar]
- 23.Cancer Genome Atlas Research N. Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, et al. Integrated genomic characterization of endometrial carcinoma. Nature. 2013 May 2;497(7447):67–73. doi: 10.1038/nature12113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu KH, Broaddus RR. Gynecological tumors in hereditary nonpolyposis colorectal cancer: We know they are common–now what? Gynecol Oncol. 2001 Aug;82(2):221–2. doi: 10.1006/gyno.2001.6332. Epub 2001/09/05. eng. [DOI] [PubMed] [Google Scholar]
- 25.Tanwar PS, Kaneko-Tarui T, Zhang L, Tanaka Y, Crum CP, Teixeira JM. Stromal liver kinase B1 [STK11] signaling loss induces oviductal adenomas and endometrial cancer by activating mammalian Target of Rapamycin Complex 1. PLoS genetics. 2012;8(8):e1002906. doi: 10.1371/journal.pgen.1002906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McCampbell AS, Harris HA, Crabtree JS, Winneker RC, Walker CL, Broaddus RR. Loss of inhibitory insulin receptor substrate-1 phosphorylation is an early event in mammalian target of rapamycin-dependent endometrial hyperplasia and carcinoma. Cancer Prev Res (Phila) 2010 Mar;3(3):290–300. doi: 10.1158/1940-6207.CAPR-09-0199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Slomovitz BM, Coleman RL. The PI3K/AKT/mTOR pathway as a therapeutic target in endometrial cancer. Clin Cancer Res. 2012 Nov 1;18(21):5856–64. doi: 10.1158/1078-0432.CCR-12-0662. [DOI] [PubMed] [Google Scholar]
- 28.McCampbell AS, Walker CL, Broaddus RR, Cook JD, Davies PJ. Developmental reprogramming of IGF signaling and susceptibility to endometrial hyperplasia in the rat. Lab Invest. 2008 Jun;88(6):615–26. doi: 10.1038/labinvest.2008.29. Epub 2008/04/23. eng. [DOI] [PubMed] [Google Scholar]
- 29.Meyer LA, Slomovitz BM, Djordjevic B, Westin SN, Iglesias DA, Munsell MF, et al. The search continues: looking for predictive biomarkers for response to Mammalian target of rapamycin inhibition in endometrial cancer. Int J Gynecol Cancer. 2014 May;24(4):713–7. doi: 10.1097/IGC.0000000000000118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Slomovitz BM, Lu KH, Johnston T, Coleman RL, Munsell M, Broaddus RR, et al. A phase 2 study of the oral mammalian target of rapamycin inhibitor, everolimus, in patients with recurrent endometrial carcinoma. Cancer. Aug 2; doi: 10.1002/cncr.25515. Epub 2010/08/04. Eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Milam MR, Celestino J, Wu W, Broaddus RR, Schmeler KM, Slomovitz BM, et al. Reduced progression of endometrial hyperplasia with oral mTOR inhibition in the Pten heterozygote murine model. Am J Obstet Gynecol. 2007 Mar;196(3):247e1–5. doi: 10.1016/j.ajog.2006.10.872. Epub 2007/03/10. eng. [DOI] [PubMed] [Google Scholar]
- 32.Husseinzadeh N, Husseinzadeh HD. mTOR inhibitors and their clinical application in cervical, endometrial and ovarian cancers: A critical review. Gynecol Oncol. 2014 May;133(2):375–81. doi: 10.1016/j.ygyno.2014.02.017. [DOI] [PubMed] [Google Scholar]
- 33.Oza AM, Elit L, Tsao MS, Kamel-Reid S, Biagi J, Provencher DM, et al. Phase II study of temsirolimus in women with recurrent or metastatic endometrial cancer: a trial of the NCIC Clinical Trials Group. J Clin Oncol. 2011 Aug 20;29(24):3278–85. doi: 10.1200/JCO.2010.34.1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Janku F, Wheler JJ, Westin SN, Moulder SL, Naing A, Tsimberidou AM, et al. PI3K/AKT/mTOR inhibitors in patients with breast and gynecologic malignancies harboring PIK3CA mutations. J Clin Oncol. 2012 Mar 10;30(8):777–82. doi: 10.1200/JCO.2011.36.1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen J, Zhao KN, Li R, Shao R, Chen C. Activation of PI3K/Akt/MTOR Pathway and Dual Inhibitors of PI3K and MTOR in Endometrial Cancer. Current medicinal chemistry. 2014 Apr 13; doi: 10.2174/0929867321666140414095605. [DOI] [PubMed] [Google Scholar]
- 36.McCampbell AS, Broaddus RR, Walker CL. Loss of inhibitory insulin receptor substrate-1 phosphorylation: An early event in endometrial hyperplasia and progression to carcinoma. Cell Cycle. 2010 Jul 15;9(14):2698–9. doi: 10.4161/cc.9.14.12618. [DOI] [PubMed] [Google Scholar]
- 37.Watanabe J, Sato H, Kanai T, Kamata Y, Jobo T, Hata H, et al. Paradoxical expression of cell cycle inhibitor p27 in endometrioid adenocarcinoma of the uterine corpus -correlation with proliferation and clinicopathological parameters. Br J Cancer. 2002 Jul 1;87(1):81–5. doi: 10.1038/sj.bjc.6600434. Epub 2002/06/27. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dellas A, Jundt G, Sartorius G, Schneider M, Moch H. Combined PTEN and p27kip1 protein expression patterns are associated with obesity and prognosis in endometrial carcinomas. Clin Cancer Res. 2009 Apr 1;15(7):2456–62. doi: 10.1158/1078-0432.CCR-08-1732. Epub 2009/03/19. eng. [DOI] [PubMed] [Google Scholar]
- 39.Mutter GL, Lin MC, Fitzgerald JT, Kum JB, Baak JP, Lees JA, et al. Altered PTEN expression as a diagnostic marker for the earliest endometrial precancers. J Natl Cancer Inst. 2000 Jun 7;92(11):924–30. doi: 10.1093/jnci/92.11.924. [DOI] [PubMed] [Google Scholar]
- 40.Dzamko NL, Steinberg GR. AMPK-dependent hormonal regulation of whole-body energy metabolism. Acta Physiol (Oxf) 2009 May;196(1):115–27. doi: 10.1111/j.1748-1716.2009.01969.x. Epub 2009/02/28. eng. [DOI] [PubMed] [Google Scholar]
- 41.Lu KH, Wu W, Dave B, Slomovitz BM, Burke TW, Munsell MF, et al. Loss of tuberous sclerosis complex-2 function and activation of mammalian target of rapamycin signaling in endometrial carcinoma. Clin Cancer Res. 2008 May 1;14(9):2543–50. doi: 10.1158/1078-0432.CCR-07-0321. Epub 2008/05/03. eng. [DOI] [PubMed] [Google Scholar]
- 42.Breuleux M, Klopfenstein M, Stephan C, Doughty CA, Barys L, Maira SM, et al. Increased AKT S473 phosphorylation after mTORC1 inhibition is rictor dependent and does not predict tumor cell response to PI3K/mTOR inhibition. Mol Cancer Ther. 2009 Apr;8(4):742–53. doi: 10.1158/1535-7163.MCT-08-0668. Epub 2009/04/18. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang S, Xiao X, Meng X, Leslie KK. A mechanism for synergy with combined mTOR and PI3 kinase inhibitors. PLoS One. 6(10):e26343. doi: 10.1371/journal.pone.0026343. Epub 2011/11/01. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Petridou E, Mantzoros C, Dessypris N, Koukoulomatis P, Addy C, Voulgaris Z, et al. Plasma adiponectin concentrations in relation to endometrial cancer: a case-control study in Greece. J Clin Endocrinol Metab. 2003 Mar;88(3):993–7. doi: 10.1210/jc.2002-021209. [DOI] [PubMed] [Google Scholar]
- 45.Soliman PT, Wu D, Tortolero-Luna G, Schmeler KM, Slomovitz BM, Bray MS, et al. Association between adiponectin, insulin resistance, and endometrial cancer. Cancer. 2006 Jun 1;106(11):2376–81. doi: 10.1002/cncr.21866. [DOI] [PubMed] [Google Scholar]
- 46.Cust AE, Kaaks R, Friedenreich C, Bonnet F, Laville M, Lukanova A, et al. Plasma adiponectin levels and endometrial cancer risk in pre- and postmenopausal women. J Clin Endocrinol Metab. 2007 Jan;92(1):255–63. doi: 10.1210/jc.2006-1371. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1
p27 expression was significantly decreased in follicular (proliferative phase) (F; n=3) endometrium as compared to luteal (secretory phase) (L; n=6) endometrium and inversely correlated with activation of PI3K/mTOR (phosphorylation of (S235/236) S6 ribosomal protein) as indicated by Western analysis.