

Cryo-EM structures of ClpB complexes reveal a pathway for substrate transfer and translocation during protein disaggregation.
Abstract
Refolding aggregated proteins is essential in combating cellular proteotoxic stress. Together with Hsp70, Hsp100 chaperones, including Escherichia coli ClpB, form a powerful disaggregation machine that threads aggregated polypeptides through the central pore of tandem adenosine triphosphatase (ATPase) rings. To visualize protein disaggregation, we determined cryo–electron microscopy structures of inactive and substrate-bound ClpB in the presence of adenosine 5′-O-(3-thiotriphosphate), revealing closed AAA+ rings with a pronounced seam. In the substrate-free state, a marked gradient of resolution, likely corresponding to mobility, spans across the AAA+ rings with a dynamic hotspot at the seam. On the seam side, the coiled-coil regulatory domains are locked in a horizontal, inactive orientation. On the opposite side, the regulatory domains are accessible for Hsp70 binding, substrate targeting, and activation. In the presence of the model substrate casein, the polypeptide threads through the entire pore channel and increased nucleotide occupancy correlates with higher ATPase activity. Substrate-induced domain displacements indicate a pathway of regulated substrate transfer from Hsp70 to the ClpB pore, inside which a spiral of loops contacts the substrate. The seam pore loops undergo marked displacements, along with ordering of the regulatory domains. These asymmetric movements suggest a mechanism for ATPase activation and substrate threading during disaggregation.
INTRODUCTION
Aggregation of misfolded proteins is a hallmark of stressed, aged, and disease states of cells, and its reversal is essential for cell health. In bacteria, fungi, and plants, disaggregation involves cooperation between Hsp100 chaperones (ClpB in Escherichia coli and Hsp104 in Saccharomyces cerevisiae) and the Hsp70 chaperone system (DnaK system in E. coli) (1). Hsp100 belongs to the AAA+ superfamily of adenosine triphosphatases (ATPases), hexameric ring proteins often involved in disassembling and translocating substrates (2). Little is known about how Hsp100 disaggregases transfer adenosine 5′-triphosphate (ATP) hydrolysis power into mechanical movements to engage and translocate polypeptide substrates. For protein disaggregation, Hsp70 recruits aggregated polypeptides and delivers them to Hsp100, concomitantly activating the ATPase and enabling Hsp100 to thread extracted polypeptides through a central pore via conserved tyrosine loops (3–5).
The Hsp100 hexamer is arranged in three tiers: a highly mobile N-terminal domain (NTD) involved in substrate recruitment, followed by two distinct, conserved AAA+ ATPase domains (AAA1 and AAA2) that power substrate threading. A regulatory, coiled-coil middle domain (MD) forms a belt around the AAA1 tier (6). Different orientations of MDs are associated with repressed or activated states of the chaperone: horizontal head-to-tail orientations of MDs confer the repressed ATPase state, whereas tilted conformations allow interaction with DnaK and derepression (6–8). Full activation of ClpB requires both binding of DnaK to tilted MDs and substrate interaction (7, 9–11), but structural changes in the AAA+ domains associated with these events have not yet been described, and little is known about substrate transfer from DnaK to ClpB and subsequent threading of it through the central Hsp100 pore.
RESULTS AND DISCUSSION
To visualize the disaggregation process, we determined cryo–electron microscopy (cryo-EM) structures of a homohexameric ClpB variant in the presence of adenosine 5′-O-(3-thiotriphosphate) (ATP-γ-S), in empty and substrate-bound forms at a resolution of about 4.5 to 5 Å (fig. S1). For stable substrate trapping, we used a ClpB variant, BAP (5), with mutated Walker B motifs of both AAA+ domains (E279A/E678A) to prevent ATP hydrolysis (12), in the presence of ATP-γ-S. The BAP variant is fully functional in disaggregation (5) and shows an HX protection pattern identical to that of ClpB (6). As a model substrate, we used casein, a natively unfolded protein that requires neither DnaK nor active unfolding to engage with ClpB. The use of a double Walker B variant and ATP-γ-S causes the substrate to be stalled. However, the register and polarity of the substrate are not fixed. To interpret our reconstructions, we performed flexible refinement of the E. coli ClpB protomer atomic coordinates (6, 13) in the EM densities. Difference maps were generated between the density maps and the fits to isolate nucleotide and substrate densities.
The hexameric structure of ClpB–ATP-γ-S in the absence of the substrate, hereafter termed ground state, shows a remarkably asymmetric arrangement with different degrees of spiral distortion in the two AAA+ rings, capped by a largely undetectable NTD part with only one single visible and ordered subunit (Fig. 1A). The two AAA+ rings adopt different spiral arrangements, resulting in a tilt of the AAA1 relative to the AAA2 ring and extended AAA1-to-AAA2 linkers for protomers on one side of the hexamer and more compact stacking on the other (Fig. 1A). A 60° rotation between the AAA+ rings places the AAA1 domain of one protomer on top of AAA2 of the next one (Fig. 1B), providing a pathway for intersubunit communication.
Fig. 1. Domain and protomer organization of ClpB.
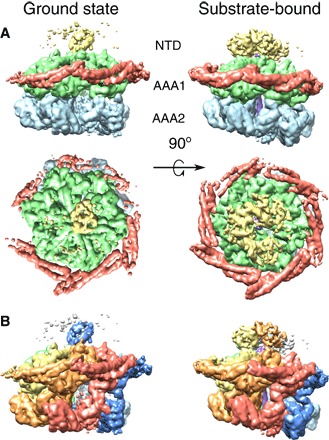
(A) Side and top views of the ClpB maps in the ground (left) and substrate-bound (right) states. Maps are segmented and colored according to the domain organization of ClpB protomers: NTD (yellow), MD (red), AAA1 (green), and AAA2 (blue). (B) Side views of the ClpB maps in the ground (left) and substrate-bound (right) states, segmented and colored by protomer. The substrate is shown in purple.
The hand of the helical distortions in our structures is opposite to the recent cryo-EM structure of the Hsp104-AMPPNP hexamer (14) and to the filaments formed in crystal forms of ClpB and Hsp104 (8, 13). Our structure is also different from the Hsp104-AMPPNP cryo-EM map, showing a continuous spiral arrangement of both AAA+ rings involving noncanonical interactions between neighboring AAA1 and AAA2 domains (14). Although our substrate-free, ground-state ClpB–ATP-γ-S structure is very asymmetric, it does not form a continuous spiral, and the two AAA+ rings are separated. ClpB does not bind the substrate in the presence of AMPPNP, in contrast to ATP-γ-S, as shown previously (15) and confirmed here for two different substrates and Hsp104 (fig. S2). Therefore, the ClpB–ATP-γ-S structure presented here shows the ATP-induced substrate acceptor state.
The AAA1 subunits in the ground state are organized in a canonical AAA+ arrangement, with nucleotide density only in four adjacent protomers and two subunits (A and F) less tightly packed and nucleotide-free (Fig. 2C). The most asymmetric feature of the AAA1 layer is the density of regulatory MDs, most of which are well resolved. Three of six MDs show both blades of the coiled coil, known as motifs 1 and 2, forming inhibitory head-to-tail contacts (6–8), with motif 2 contacting motif 1 of the adjacent MD as well as the AAA1 of its own subunit (Fig. 2, A and C, and fig. S3A). Residues Y503 and E432, whose mutation give hyperactive and repressed phenotypes, respectively (7), are involved in the head-to-tail contacts (fig. S3A). MD contacts with the AAA1 domain of the same protomer include a large salt bridge network that controls ATPase and chaperone activity (fig. S3B) (8, 16), suggesting a pathway for allosteric communication. Of the remaining three MDs, motif 2 is not visible, whereas motif 1 is only visible in two subunits at a lower density threshold indicating increased dynamics, and interacts with AAA1 of the neighboring protomer as in Hsp104 (fig. S3C) (14). Tilting of motif 1 to interact with the adjacent AAA1 frees motif 2 for DnaK binding. This primes three of six MDs for DnaK interaction and activation of the ClpB ground state.
Fig. 2. Fitted atomic models of ClpB–ATP-γ-S, empty (left) and substrate-bound (right), colored by subunit.
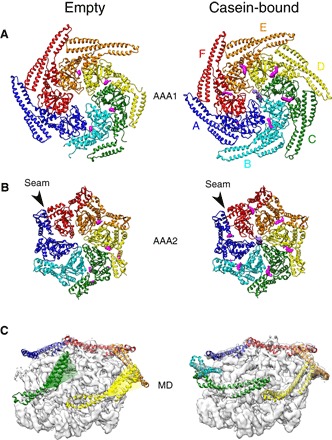
Difference density between map and model shows extra density for occupied nucleotide-binding sites in magenta and casein substrate in purple. (A) Top views of AAA1 and MD. (B) Top views of AAA2. (C) MD conformational changes in ClpB–ATP-γ-S (NTDs were masked for clarity). Left: Tilted MDs of protomers C (green) and D (yellow) in the ground state. The motif 2 segment of the coiled coil, absent in the density map, is shown as a transparent ribbon and colored to indicate delocalization. Seam subunits A (blue) and F (red) have horizontal and docked MDs. Right: Horizontal and docked MDs in the substrate-bound state.
In the AAA2 ring, four subunits are tightly packed together, but the other two (A and F) are more mobile (fig. S1F) and displaced, creating a pronounced seam in the ring (Fig. 2B). The gaps created by their displacement leave three empty nucleotide pockets. The side of the ring corresponding to the seam is the one with the ordered and locked MDs, suggesting an allosteric interaction between MD conformation and nucleotide occupancy in AAA2, in which only protomers with a tilted, “active-like” MD conformation are nucleotide-bound and competent for ATP binding and hydrolysis.
Only one NTD of the ground state is visible and sufficiently defined for rigid body fitting (fig. S1F). The resolution is lower in this region (8 Å compared to 4 to 5 Å in the AAA+ domains), indicating that the NTD is more mobile than other regions. This single NTD plugs the channel entrance, preventing substrate engagement (Fig. 3A), thereby explaining a previous biochemical observation that the NTD controls substrate access to the ClpB pore (17). The NTD plug has its substrate-binding cleft (17) oriented toward the more tilted MDs, suggesting a regulated pathway for substrate handover from DnaK to the NTD and from there into the translocation channel (Fig. 3A). This conformation is radically different from that of hexameric Hsp104-AMPPNP, where NTDs form a large C-shaped surface available for substrate interaction at the entrance of a wide-open channel (14). Density for the other NTDs is ill-defined and only visible at a lower threshold, indicating that they are highly mobile and do not adopt a fixed conformation.
Fig. 3. NTD positions and substrate interactions.
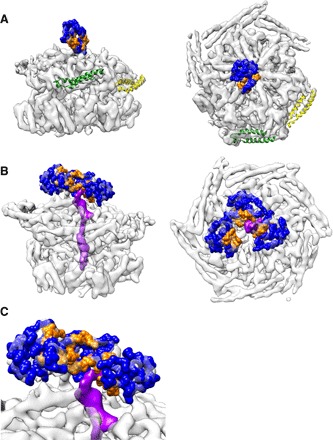
(A) The NTD of protomer A blocks the pore of empty ClpB. The hydrophobic cleft colored in orange is oriented toward protomers C (green) and D (yellow), with tilted MDs (Hsp70 acceptor state). (B and C) Substrate density (purple) adjacent to the hydrophobic sites in the best refining of 10 classes, focused on the NTDs.
Our ClpB–ATP-γ-S structure represents a low-activity ground state with reduced nucleotide occupancy, explaining why this chaperone is an inefficient ATPase, with lower hydrolysis power compared to related Hsp100 chaperones (7). In addition, one NTD blocks the central pore of the ground state, rendering it inaccessible to substrates. Nevertheless, this state is concurrently primed for DnaK interaction and substrate delivery and thereby is an acceptor state competent for regulated activation.
Upon substrate interaction, ClpB–ATP-γ-S undergoes substantial conformational changes in the NTD region, AAA+ rings, and MDs, as well as global rigidification (fig. S1 and movie S1). Movement of seam protomers A and F narrows both AAA+ rings around the engaged substrate. Additionally, ClpB switches from a partially loaded (7 ATPs) to an almost fully loaded (11 ATPs) nucleotide state, consistent with ATPase activation by the substrate (Fig. 2, A and B, and fig. S4B) (18). This change is most pronounced in the AAA2 ring, which becomes fully occupied, in agreement with previous biochemical data indicating that AAA2 is the main ClpB motor and predominantly stimulated by the substrate (fig. S4B) (19). This is accompanied by rearrangements of the nucleotide pockets, effected by a corkscrew-like motion tightening the AAA2 ring in the region of subunits E, F, A, and B (movie S2).
Substrate binding also results in global ordering of MDs, which adopt a more repressed-like, horizontal, and docked conformation (Fig. 2, A and C) that favors DnaK dissociation upon substrate handover to ClpB. This could serve to avoid DnaK rebinding and further substrate recruitment during ongoing translocation. Furthermore, maximal ClpB activation, requiring DnaK and substrate binding, will be restricted to the initial phase of substrate handover, and ClpB will work at lower force during subsequent translocation. This is consistent with the weaker threading power of ClpB compared to the related unfoldases ClpA and ClpX, which has been linked to increased refolding yields of aggregated substrates (7, 20).
The NTDs of substrate-bound ClpB–ATP-γ-S are mobile (fig. S1F), but focused three-dimensional (3D) classification reveals diverse arrangements, with two to five NTDs visible and substrate interaction with one or two domains (fig. S5). In the best refining of 10 classes, three NTDs adopt a helical arrangement, with their hydrophobic substrate-binding cleft oriented toward the channel and the substrate interacting with two of them (Fig. 3, B and C). Thus, the NTDs move independently of the rest of the protein and interact with the substrate in a dynamic manner to funnel it toward the pore.
The difference map between the reconstruction of substrate-bound ClpB and the fit reveals a strong and well-defined density for casein in the central channel, making specific interactions from top to bottom of both AAA+ rings, over a distance of 70 Å (~18 substrate residues). The conserved tyrosine pore loops of AAA1 (L1; residues 247 to 253) adopt a staggered arrangement over 18 Å (Fig. 4). L1 of protomers B to E spiral around casein with ~6 Å axial steps between successive contacts. L1 loops of protomers A and F do not follow the regular spiral step (Fig. 4) but undergo large movements upon casein binding (10 and 5 Å for L1 of protomers A and F, respectively), closing the channel around the substrate. In the AAA2 ring, the conserved tyrosine loops (L2; residues 648 to 660) form a spiral arrangement of five staggered loops over 24 Å, with 6 Å downward steps between successive L2 loops. Key roles of L1 and L2 in substrate interaction are confirmed by reduced casein binding of L1 and L2 mutants, which also exhibit reduced stimulation of ATPase activity by the substrate (fig. S4, A to C).
Fig. 4. Track of pore loop interactions with the substrate and movements upon substrate binding.
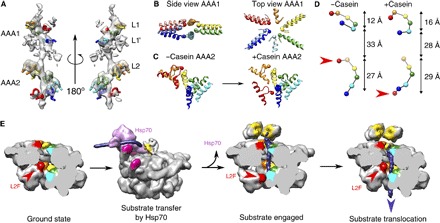
Cα positions of Tyr251 and Tyr653 are indicated by spheres. (A) EM density of the casein substrate and interacting pore loops. Conserved tyrosine pore loops (L1 and L2) and conserved AAA1 loop (L1′) form a well-defined spiral track wrapping around the substrate. (B) Conformational changes in tyrosine pore loops of AAA1. Models for ground-state and substrate-bound ClpB are shown in pale and bright colors, respectively. (C) Conformational changes in the tyrosine pore loops of AAA2 and 90° rotation of L2F upon substrate binding. (D) Spiral tracks of tyrosines in ground and substrate-bound states. Red arrowheads indicate the positions of the seam subunit AAA2 tyrosine in the two states. (E) Model of substrate engagement and threading by ClpB. The model was generated using the ClpB models from the present study and a Protein Data Bank (PDB) model of DnaK (4B9Q), docked to motif 2 of a tilted MD of ClpB using available structural information (10). In the case of empty ClpB–ATP-γ-S, the position of motif 2 of tilted MDs was extrapolated using the MD conformation of a horizontal MD docked into tilted motif 1.
L1 and L2 are separated by 28 Å from one another and do not form a continuous staircase as described for Hsp104-AMPPNP (14). A second spiral of charged AAA1 loops underneath L1 loops (L1′; residues 284 to 294) interacts more weakly with casein (Fig. 4A). These additional loops provide continuity of interaction with the substrate in the gap between the L1 and L2 spirals. This loop is conserved in ClpA and ClpB and was shown to be crucial for substrate binding and processing in ClpA by mutation of key residues (A293 and G294) (21).
The most marked conformational change induced by the substrate is observed for the L2 loop of protomer F (L2F) at the seam. In the ground state, L2F is greatly displaced relative to the other L2 loops (fig. S6), whereas, upon substrate binding, it undergoes a 90° rotation and 28 Å downward movement (Fig. 4C and movie S3) and joins the spiral path of the other L2s to bind the substrate (Fig. 4). Changes in subunit packing, nucleotide occupancy, and L2F movement correlate with relief of repression of the ATPase by L2 loops in the adjacent subunit (fig. S4). The longer track and large movement of L2 loops can explain their dominant function in substrate threading and protein disaggregation (fig. S4, D and E) (5).
The huge movements of the seam L2F and the AAA2 domain described in this work suggest a mechanism for substrate threading, in which five subunits pull the substrate down the channel while one repositions to grasp the substrate before the next pulling stroke. This movement suggests a spring loading mechanism in which ClpB, similar to its Hsp100 homologs (22–25), threads substrate in 6 Å steps (~2–amino acid residues). We envisage a spiral distortion propagating around the ring with each ATPase cycle, in which the L2 loop of the seam subunit goes to the top of the spiral of L2 loops while the other five loops move the substrate down (Fig. 4E).
Asymmetric structures of the VAT (24) and Vps4 (25) unfoldases and of the Hsp104 disaggregase (26), all in complex with a substrate, were recently obtained by cryo-EM. As in the case of ClpB, these structures show right-handed helical distortions and partial nucleotide occupancy, suggesting that sequential nucleotide hydrolysis around the ring is a common feature of AAA+ unfoldases and disaggregases. Marked pore loop movements were also observed at the seam of the Vps4 structure (27), as seen here for the ClpB AAA2 ring upon casein binding.
Several double AAA+ ring complexes, such as VAT, Hsp104, and NSF, show split spiral to ring transitions, which are postulated to effect translocation via global changes in helical distortion (24, 26, 28). However, our ClpB structures both have closed rings with seams, with the two rings in different conformations. They directly show the conformational effects of substrate binding. In particular, the large pore loop movement and distortion of the seam subunit in AAA2 are consistent with biochemical data showing that AAA2 is the main motor of ClpB (fig. S4).
Together, these AAA+ protein unfoldases and disaggregases show common features of sequentially propagated translocation mechanisms involving seams or dislocations of the homohexameric rings, with specificities linked to a variety of different substrates requiring different pulling forces. Our ClpB structural snapshots illuminate key steps of protein disaggregation and substrate threading. They provide a pathway for regulated and directional transfer of aggregated substrates from DnaK to ClpB NTDs and subsequently to the ClpB pore, suggesting how the energy of ATP is used for regulated translocation of the extracted polypeptides during the reversal of aggregation.
MATERIALS AND METHODS
Strains, plasmids, and proteins
E. coli strains used were derivatives of MC4100. ClpB was amplified by polymerase chain reaction (PCR), inserted into pDS56, and verified by sequencing. Mutant derivatives of clpB were generated by PCR mutagenesis and standard cloning techniques in pDS56 and were verified by sequencing. ClpB was purified after overproduction from E. coli ΔclpB::kan cells. ClpB wild-type and mutant variants were purified using Ni-IDA (Macherey-Nagel), following standard protocols, and size exclusion chromatography (Superdex S200) in MDH buffer [50 mM tris (pH 7.5), 150 mM KCl, 20 mM MgCl2, 2 mM dithiothreitol (DTT)] supplemented with 5% (v/v) glycerol.
Purifications of DnaK, DnaJ, GrpE, and ClpP were performed as described previously (20). Pyruvate kinase of rabbit muscle, malate dehydrogenase of pig heart muscle, casein, and fluorescein isothiocyanate (FITC)–casein were purchased from Sigma. Protein concentrations were determined with the Bio-Rad Bradford assay.
Biochemical methods
Disaggregation of 1 μM MDH aggregates, generated by incubation at 47°C for 30 min, was monitored by turbidity measurements using a PerkinElmer LS55 spectrofluorimeter. The disaggregation activity was analyzed by determining initial slopes of turbidity decrease in the presence of the DnaK chaperone system (1 μM DnaK, 0.2 μM DnaJ, and 0.1 μM GrpE) and 0.5 μM ClpB (wild type or mutants) and an ATP-regenerating system [2 mM ATP, 3 mM phosphoenolpyruvate (PEP), and pyruvate kinase (20 ng/μl)] in MDH buffer.
ATPase activities of ClpB (wild type and mutants; 0.5 μM; 2 mM ATP) were determined in MDH buffer in the absence or presence of the substrate (10 μM casein) using a NADH (reduced form of nicotinamide adenine dinucleotide)–coupled colorimetric assay (Sigma) by measuring the decrease of NADH absorption at 340 nm in a BMG Labtech FLUOstar Omega plate reader. Casein degradation assays were performed in the presence of BAP (1 μM), ClpP (1.5 μM), casein (20 μM), and an ATP-regenerating system [2 mM ATP, 3 mM PEP, and pyruvate kinase (20 ng/μl)] in buffer A [25 mM tris (pH 7.5), 75 mM KCl, 10 mM MgCl2, 2 mM DTT] and analyzed by SDS–polyacrylamide gel electrophoresis and Coomassie staining.
Interaction of ClpB and Hsp104 (1.2 μM each) with IAANS (2-[4′-(iodoacetamido)anilino]naphthalene-6-sulfonic acid)–conjugated peptide B1 (AHAWQHQGKTLFISRKTYRIC; 100 nM) was monitored after incubation in buffer A for 15 min at 30°C in the absence or presence of nucleotides (2 mM). The fluorescence emission spectra of IAANS-labeled peptide B1 were recorded between 350 and 550 nm on a PerkinElmer LS55 spectrofluorimeter at a fixed excitation wavelength of 335 nm.
Interaction of ClpB and Hsp104 with FITC-casein (100 nM) was monitored by fluorescence anisotropy measurements using a BMG Biotech CLARIOstar plate reader. Samples were incubated in buffer A for 30 min at room temperature in the absence or presence of nucleotides (2 mM), and polarization of FITC-casein was determined in black 384-well plates (excitation, 482 nm; emission, 530 nm; target millipolarization units, 35). A sample containing FITC-casein only served as reference.
Cryo–electron microscopy
Purified BAP-DWB (0.7 mg/ml) was incubated in 25 mM tris-HCl (pH 7.5), 25 mM KCl, 10 mM MgCl2, 1 mM DTT, and 2 mM ATP-γ-S for 15 min at room temperature. For the casein-bound state, an excess of α-casein (0.6 mg/ml; Sigma) was added to the solution to maximize occupancy. Lacey grids coated with a thin carbon layer were glow-discharged in the presence of amylamine to improve the distribution of orientations adopted by the protein on the grids before application of the sample (3.5 μl). The vitrification was performed with Vitrobot (FEI), and the sample was blotted for 3 to 3.5 s and plunge-frozen in liquid ethane.
Images were collected using EPU software on a Titan Krios transmission electron microscope (FEI) operating at 300 kV, equipped with a Gatan K2 Summit direct electron detector and bioquantum energy filter with 20-eV slit. The defocus range was set between −1 and −3 μm, and the total dose was 50 electrons/Å2. Pixel size was 1.39 Å/pixel for unbound BAP-DWB and 1.37 Å/pixel for casein-bound BAP-DWB.
Image processing
After initial sorting of the collected images, movie frames of each image were aligned using MotionCor2 (29) with 25 patches per image, and dose compensation was applied. The contrast transfer function was estimated with CTFFIND4 (30). Particles were picked with Gautomatch, and a data set of ~250,000 particles from 1800 micrographs was generated for unbound BAP-DWB (~500,000 particles from 3600 micrographs for casein-bound BAP-DWB). The initial data sets were subjected to reference-free 2D classification in Relion2.0 (31), and poorly resolved class averages were removed. For 3D processing of casein-bound BAP-DWB, the initial model was a 40 Å low pass–filtered map obtained from the PDB model of a previously determined EM reconstruction of a ClpB variant (4D2Q), where the flexible NTDs were removed. Several rounds of 3D classification were performed in Relion, yielding a more homogeneous set of ~230,000 particles. The resulting map was refined to an estimated resolution of 4.5 Å with the 0.143 Fourier shell correlation (FSC) criterion. Particles of the final map were transferred into cryoSPARC software (32) for ab initio 3D reconstruction and 3D refinement, yielding a comparable map at an estimated resolution of 4.5 Å with the 0.143 FSC criterion. For 3D processing of empty BAP-DWB, the initial model was the map obtained for casein-bound BAP-DWB low pass–filtered at 40 Å. Several rounds of 3D classification were performed in Relion, yielding a more homogeneous set of ~120,000 particles. The resulting map was refined to an estimated resolution of 6 Å with the 0.143 FSC criterion. Further classification and refinement were performed, which yielded the selection of ~60,000 particles that resulted in a 5 Å refined map. Both data sets of 120,000 and 60,000 particles were transferred into the cryoSPARC software for ab initio 3D reconstruction and 3D refinement, yielding a map at an estimated 4.6 Å resolution with the 0.143 FSC criterion, containing ~60,000 particles. A flowchart summarizes image processing for empty and substrate-bound BAP-DWB in fig. S7. The local resolution was evaluated using the local resolution tool of Relion. There were marked variations in local resolution, especially in the empty complex, with lower resolution in the NTDs and MDs, as well as in the AAA+ domains on one side of the ring. Extensive 3D classification and particle sorting failed to improve the resolution, suggesting that it was limited by the flexibility of the complex rather than by the presence of several distinct conformations. Map segmentation for figures was done in UCSF Chimera using the Segger tool.
Model building and refinement
Initial models of the BAP-DWB monomer were generated using MODELLER 9.17 (33), with previously determined crystal structures of ClpB or ClpB domains as templates (PDB IDs: 4CIU, 1QVR, 4HSE, and 4LJ9). The crystal structure of E. coli ClpB (4CIU) was used as the main template, the crystal structure of Thermus thermophilus (1QVR) was used to model positions of the NTD, and the crystal structures of AAA1 and AAA2 of E. coli ClpB (4HSE and 4LJ9, respectively) were used to model the pore loops disordered in other crystal structures. Initial rigid body fitting of the monomers in the map was manually done in Chimera using the Fit in Map tool. iMODFIT (34) was used for fitting involving large domain motions. Flex-EM (35) was then used for refinement of secondary structures and loops in the map. Energy minimization to fix geometry was performed in Phenix (36). Quality and improvement of the fit were assessed with TEMPy (37) using the segment-based Manders’ overlap coefficient scores and segment-based cross-correlation scores (38). Quality of the fit in different regions is highlighted in fig. S8. For the ground state of ClpB, NTDs of subunits B to F, the linker between the NTD and AAA1 of subunit A, MD of subunit B, and motif 2 of the MDs of subunits C and D were not included in the final model because these regions are not defined in the map. For the substrate-bound state of ClpB, the NTDs were not included in the final model.
Acknowledgments
Funding: We are grateful for support from the Hartmut Hoffmann-Berling International Graduate School of Molecular and Cellular Biology (to K.B.F.); Deutsche Forschungsgemeinschaft grants BB617/17-2 and MO 970/4-2 (to B.B. and A.M.); Wellcome Trust grants 106252, 101488, and 079605 (to H.R.S.); Biotechnology and Biological Sciences Research Council grant BB/L014211/1 (to H.R.S.); Medical Research Council project grant MR/M019292/1 (M.T.); the Knut and Alice Wallenberg Foundation; and the Erling-Persson Family Foundation (Science for Life Laboratory, University of Stockholm). We acknowledge Diamond for access and support of the cryo-EM facilities at the UK national electron bio-imaging centre (eBIC), (proposal EM14704), funded by the Wellcome Trust, MRC and BBSRC, and thank D. Clare and A. Siebert for support; N. Lukoyanova for microscope support; D. Houldershaw and S. Fleischmann for information technology support; and E. Morris for advice on grid treatment. Author contributions: C.D. and M.C. prepared the cryo-EM samples, collected the data, and did the image processing and model building. K.B.F. and A.M. prepared samples and performed biochemical characterization. H.R.S. supervised the cryo-EM and image processing. A.M. and B.B. supervised the biochemical work. M.T. supervised the model building and fitting. H.R.S., A.M., and B.B. supervised the project. C.D., M.C., H.R.S., A.M., and B.B. wrote the paper. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. EM maps were deposited in the Electron Microscopy Data Bank: EMD-3776 and EMD-3777. Molecular models were deposited in the PDB: 5OFO and 5OG1.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/8/e1701726/DC1
fig. S1. EM of empty and substrate-bound ClpB in the presence of ATP-γ-S.
fig. S2. AMPPNP does not induce the substrate-binding ATP state of ClpB and Hsp104.
fig. S3. MD interactions.
fig. S4. ClpB pore loop mutants are affected in substrate binding and threading.
fig. S5. Focused 3D classification of the NTDs.
fig. S6. Refinement of the L2 tyrosine pore loop region of AAA2 of protomer F in the ground state.
fig. S7. Tabulation of the steps in data processing for ClpB and ClpB-casein data sets.
fig. S8. Details of the model fitted into the cryo-EM maps.
movie S1. Conformational changes of ClpB upon substrate binding.
movie S2. Corkscrew motion of the ClpB AAA2 hexamer to engage with the substrate.
movie S3. Pore loop motion upon substrate binding.
REFERENCES AND NOTES
- 1.Doyle S. M., Wickner S., Hsp104 and ClpB: Protein disaggregating machines. Trends Biochem. Sci. 34, 40–48 (2009). [DOI] [PubMed] [Google Scholar]
- 2.Olivares A. O., Baker T. A., Sauer R. T., Mechanistic insights into bacterial AAA+ proteases and protein-remodelling machines. Nat. Rev. Microbiol. 14, 33–44 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim Y.-I., Burton R. E., Burton B. M., Sauer R. T., Baker T. A., Dynamics of substrate denaturation and translocation by the ClpXP degradation machine. Mol. Cell 5, 639–648 (2000). [DOI] [PubMed] [Google Scholar]
- 4.Lum R., Tkach J. M., Vierling E., Glover J. R., Evidence for an unfolding/threading mechanism for protein disaggregation by Saccharomyces cerevisiae Hsp104. J. Biol. Chem. 279, 29139–29146 (2004). [DOI] [PubMed] [Google Scholar]
- 5.Weibezahn J., Tessarz P., Schlieker C., Zahn R., Maglica Z., Lee S., Zentgraf H., Weber-Ban E. U., Dougan D. A., Tsai F. T. F., Mogk A., Thermotolerance requires refolding of aggregated proteins by substrate translocation through the central pore of ClpB. Cell 119, 653–665 (2004). [DOI] [PubMed] [Google Scholar]
- 6.Carroni M., Kummer E., Oguchi Y., Wendler P., Clare D. K., Sinning I., Kopp J., Mogk A., Bukau B., Saibil H. R., Head-to-tail interactions of the coiled-coil domains regulate ClpB activity and cooperation with Hsp70 in protein disaggregation. eLife 3, e02481 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oguchi Y., Kummer E., Seyffer F., Berynskyy M., Anstett B., Zahn R., Wade R. C., Mogk A., Bukau B., A tightly regulated molecular toggle controls AAA+ disaggregase. Nat. Struct. Mol. Biol. 19, 1338–1346 (2012). [DOI] [PubMed] [Google Scholar]
- 8.Heuck A., Schitter-Sollner S., Suskiewicz M. J., Kurzbauer R., Kley J., Schleiffer A., Rombaut P., Herzog F., Clausen T., Structural basis for the disaggregase activity and regulation of Hsp104. eLife, e21516 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seyffer F., Kummer E., Oguchi Y., Winkler J., Kumar M., Zahn R., Sourjik V., Bukau B., Mogk A., Hsp70 proteins bind Hsp100 regulatory M domains to activate AAA+ disaggregase at aggregate surfaces. Nat. Struct. Mol. Biol. 19, 1347–1355 (2012). [DOI] [PubMed] [Google Scholar]
- 10.Rosenzweig R., Moradi S., Zarrine-Afsar A., Glover J. R., Kay L. E., Unraveling the mechanism of protein disaggregation through a ClpB-DnaK interaction. Science 339, 1080–1083 (2013). [DOI] [PubMed] [Google Scholar]
- 11.Lee J., Kim J.-H., Biter A. B., Sielaff B., Lee S., Tsai F. T. F., Heat shock protein (Hsp) 70 is an activator of the Hsp104 motor. Proc. Natl. Acad. Sci. U.S.A. 110, 8513–8518 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weibezahn J., Schlieker C., Bukau B., Mogk A., Characterization of a trap mutant of the AAA+ chaperone ClpB. J. Biol. Chem. 278, 32608–32617 (2003). [DOI] [PubMed] [Google Scholar]
- 13.Lee S., Sowa M. E., Watanabe Y.-h., Sigler P. B., Chiu W., Yoshida M., Tsai F. T. F., The structure of ClpB: A molecular chaperone that rescues proteins from an aggregated state. Cell 115, 229–240 (2003). [DOI] [PubMed] [Google Scholar]
- 14.Yokom A. L., Gates S. N., Jackrel M. E., Mack K. L., Su M., Shorter J., Southworth D. R., Spiral architecture of the Hsp104 disaggregase reveals the basis for polypeptide translocation. Nat. Struct. Mol. Biol. 23, 830–837 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee S., Choi J.-M., Tsai F. T. F., Visualizing the ATPase cycle in a protein disaggregating machine: Structural basis for substrate binding by ClpB. Mol. Cell 25, 261–271 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lipińska N., Ziętkiewicz S., Sobczak A., Jurczyk A., Potocki W., Morawiec E., Wawrzycka A., Gumowski K., Ślusarz M., Rodziewicz-Motowidło S., Chruściel E., Liberek K., Disruption of ionic interactions between the nucleotide binding domain 1 (NBD1) and middle (M) domain in Hsp100 disaggregase unleashes toxic hyperactivity and partial independence from Hsp70. J. Biol. Chem. 288, 2857–2869 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rosenzweig R., Farber P., Velyvis A., Rennella E., Latham M. P., Kay L. E., ClpB N-terminal domain plays a regulatory role in protein disaggregation. Proc. Natl. Acad. Sci. U.S.A. 112, E6872–E6881 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Woo K. M., Kim K. I., Goldberg A. L., Ha D. B., Chung C. H., The heat-shock protein ClpB in Escherichia coli is a protein-activated ATPase. J. Biol. Chem. 267, 20429–20434 (1992). [PubMed] [Google Scholar]
- 19.Franke K. B., Bukau B., Mogk A., Mutant analysis reveals allosteric regulation of ClpB disaggregase. Front. Mol. Biosci. 4, 6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haslberger T., Zdanowicz A., Brand I., Kirstein J., Turgay K., Mogk A., Bukau B., Protein disaggregation by the AAA+ chaperone ClpB involves partial threading of looped polypeptide segments. Nat. Struct. Mol. Biol. 15, 641–650 (2008). [DOI] [PubMed] [Google Scholar]
- 21.Hinnerwisch J., Fenton W. A., Furtak K. J., Farr G. W., Horwich A. L., Loops in the central channel of ClpA chaperone mediate protein binding, unfolding, and translocation. Cell 121, 1029–1041 (2005). [DOI] [PubMed] [Google Scholar]
- 22.Maillard R. A., Chistol G., Sen M., Righini M., Tan J., Kaiser C. M., Hodges C., Martin A., Bustamante C., ClpX(P) generates mechanical force to unfold and translocate its protein substrates. Cell 145, 459–469 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rodriguez-Aliaga P., Ramirez L., Kim F., Bustamante C., Martin A., Substrate-translocating loops regulate mechanochemical coupling and power production in AAA+ protease ClpXP. Nat. Struct. Mol. Biol. 23, 974–981 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ripstein Z. A., Huang R., Augustyniak R., Kay L. E., Rubinstein J. L., Structure of a AAA+ unfoldase in the process of unfolding substrate. eLife 6, e25754 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Monroe N., Han H., Shen P. S., Sundquist W. I., Hill C. P., Structural basis of protein translocation by the Vps4-Vta1 AAA ATPase. eLife 6, e24487 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gates S. N., Yokom A. L., Lin J. B., Jackrel M. E., Rizo A. N., Kendsersky N. M., Buell C. E., Sweeny E. A., Mack K. L., Chuang E., Torrente M. P., Su M., Shorter J., Southworth D. R., Ratchet-like polypeptide translocation mechanism of the AAA+ disaggregase Hsp104. Science, eaan1052 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Su M., Guo E. Z., Ding X., Li Y., Tarrasch J. T., Brooks C. L. III, Xu Z., Skiniotis G., Mechanism of Vps4 hexamer function revealed by cryo-EM. Sci. Adv. 3, e1700325 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao M., Wu S., Zhou Q., Vivona S., Cipriano D. J., Cheng Y., Brunger A. T., Mechanistic insights into the recycling machine of the SNARE complex. Nature 518, 61–67 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zheng S. Q., Palovcak E., Armache J.-P., Verba K. A., Cheng Y., Agard D. A., MotionCor2: Anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rohou A., Grigorieff N., CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scheres S. H. W., RELION: Implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 180, 519–530 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Punjani A., Rubinstein J. L., Fleet D. J., Brubaker M. A., cryoSPARC: Algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017). [DOI] [PubMed] [Google Scholar]
- 33.Webb B., Sali A., Comparative protein structure modeling using MODELLER. Curr. Protoc. Bioinform. 54, 5.6.1–5.6.37 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lopéz-Blanco J. R., Chacón P., iMODFIT: Efficient and robust flexible fitting based on vibrational analysis in internal coordinates. J. Struct. Biol. 184, 261–270 (2013). [DOI] [PubMed] [Google Scholar]
- 35.Topf M., Lasker K., Webb B., Wolfson H., Chiu W., Sali A., Protein structure fitting and refinement guided by cryo-EM density. Structure 16, 295–307 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H., PHENIX: A comprehensive Python-based system for macromolecular structure solution research papers. Acta Crystallogr. Sect. D Struct. Biol. 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Farabella I., Vasishtan D., Joseph A. P., Pandurangan A. P., Sahota H., Topf M., TEMPy: A Python library for assessment of three-dimensional electron microscopy density fits. J. Appl. Crystallogr. 48, 1314–1323 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Joseph A. P., Malhotra S., Burnley T., Wood C., Clare D. K., Winn M., Topf M., Refinement of atomic models in high resolution EM reconstructions using Flex-EM and local assessment. Methods 100, 42–49 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/8/e1701726/DC1
fig. S1. EM of empty and substrate-bound ClpB in the presence of ATP-γ-S.
fig. S2. AMPPNP does not induce the substrate-binding ATP state of ClpB and Hsp104.
fig. S3. MD interactions.
fig. S4. ClpB pore loop mutants are affected in substrate binding and threading.
fig. S5. Focused 3D classification of the NTDs.
fig. S6. Refinement of the L2 tyrosine pore loop region of AAA2 of protomer F in the ground state.
fig. S7. Tabulation of the steps in data processing for ClpB and ClpB-casein data sets.
fig. S8. Details of the model fitted into the cryo-EM maps.
movie S1. Conformational changes of ClpB upon substrate binding.
movie S2. Corkscrew motion of the ClpB AAA2 hexamer to engage with the substrate.
movie S3. Pore loop motion upon substrate binding.