

Abstract
IL-18 and IL-1β, which are cytokines of the IL-1 family, are synthesized as precursor proteins and activated by the inflammasome via proteolytic processing. IL-1β is induced only in response to inflammatory stimuli, but IL-18 is constitutively expressed. However, how IL-18 and IL-1β expression is regulated by different inflammatory signals remains poorly studied. In this study, we found that IL-18 and IL-1β are differentially regulated. Despite being constitutively expressed, IL-18 expression was increased and sustained after stimulation of Toll-like receptors. In contrast, IL-1β was induced but not sustained after chronic treatment. Furthermore, type I IFN signaling was essential for induction of IL-18 and macrophages lacking type I IFN signaling were impaired in their ability to promote IL-18 induction. Thus, our findings reveal a fundamental difference in IL-18 and IL-1β regulation and uncover novel mechanisms that are relevant to the inflammatory settings where these proinflammatory cytokines play a critical role.
Keywords: IL-18, IL-1β, type I IFN, IFNAR1, IFNAR2, IRF9, STAT1
Introduction
IL-18 and IL-1β, members of the IL-1 cytokine family, are important mediators of inflammatory diseases and play critical roles in infection and cancer (1–4). Unlike other cytokines, cellular IL-18 and IL-1β are synthesized as precursor proteins and need to be cleaved to generate their biologically active forms. Inflammasome, a multimeric protein complex, is a central regulator of this process by which bioactive IL-18 and IL-1β are generated. The inflammasome comprises an innate immune sensor that includes the nucleotide-binding domain, leucine-rich-repeat–containing protein (NLR), AIM2-like receptor, and pyrin; the adaptor protein ASC; and the cysteine protease caspase 1 (5). Inflammasome assembly induces the activation of caspase-1, which mediates the proteolytic processing of pro-IL-18 and pro-IL-1β to generate their bioactive forms and their release from the cell (5).
IL-18 is constitutively expressed in blood monocytes and intestinal epithelial cells of healthy humans (6, 7). It is also expressed in murine macrophages, dendritic cells, endothelial cells, intestinal epithelial cells, and keratinocytes under steady state (8–10). However, IL-1β is not constitutively expressed under homeostasis. IL-1β expression is induced in blood mononuclear cells, macrophages, and dendritic cells during stimulation with Toll-like receptor (TLR) ligands and other cytokines (e.g., tumor necrosis factor) (1, 2, 4). Unlike IL-1β, a cellular pool for IL-18 already exists before an inflammatory stimulus, and is ready to be activated and released by the inflammasome (11). It must however be noted that LPS-mediated priming of NLRP3 is required for inflammasome activation and subsequent release of both IL-18 and IL-1β (12). However, several studies suggest that despite being constitutively expressed, IL-18 can also be induced under certain circumstances. Treatment with LPS (TLR4 agonist) has been shown to upregulate the mRNA levels of Il18 (7, 13). Incubation of CpG oligonucleotides (TLR9 agonist) with dendritic cells or infection of monocytes with the Sendai virus can also increase the expression of Il18 (14, 15).
The mechanisms by which IL-18 and IL-1β levels are regulated by different inflammatory signals remain unclear. In this study, we showed that expression of IL-18 is induced and that the increased level is sustained during TLR4, TLR2, or TLR7 ligands stimulation. In contrast, IL-1β expression declines soon after reaching its peak level. The TLR3 and cyclic GMP–AMP synthase (cGAS)–stimulator of interferon genes (STING) pathways induced IL-18 but were modest at inducing IL-1β expression. Importantly, type I IFN signaling was required to upregulate IL-18 in response to all stimuli tested. Thus, in macrophages lacking components of type I IFN signaling, IL-18 was not induced by any inflammatory stimuli. Together, our findings demonstrate that IL-18 and IL-1β expression are differentially regulated. Expression of these inactive proinflammatory cytokines is an essential step for their maturation into bioactive forms by caspase-1 inflammasomes. Overall, our data demonstrating differential regulation of IL-18 and IL-1β will be fundamental to understanding several inflammatory disease settings.
Materials and Methods
Mice
Ifnar1−/− mice (16), Ifnar2−/− mice (17), Irf1−/− mice (18), Irf3−/− mice (19), Irf7−/− mice (18), and Irf9−/− mice (20) were generated as described previously. C57BL/6J mice [wide type (WT)], Irf4tm1Rdf/J mice (Stock Number 009380), Irf8 tm1.2Hm/J mice (also known as Irf8−/−; Stock Number 018298) and Irf5tm1Ppr/J mice (Stock Number 017311) were purchased from The Jackson Laboratory. Irf4tm1Rdf/J mice and Irf5tm1Ppr/J mice were crossed with mice expressing LsyM-Cre and Cre recombinase, respectively, to generate Irf4fl/fl-Lysm-Cre mice and Irf5−/− mice. Stat1−/− mice were provided by Dr. Abhay Satoskar (Ohio State University). All mice were bred at the Animal Resource Center at St. Jude. Animal studies were conducted according to protocols approved by the St. Jude Animal Care and Use Committee.
Cell culture and stimulation
Bone marrow–derived macrophages were prepared as described previously (21). Cells were stimulated with LPS (500 ng/mL; InvivoGen), Pam3CSK4 (1 μg/mL), gardiquimod (1 μg/mL; InvivoGen), Poly(I:C) (10 μg/mL; InvivoGen), or IFNβ (400 U/mL; PBL Assay Science). For transfection of DNA or 2´3´-cyclic guanosine monophosphate–adenosine monophosphate (cGAMP), 0.25 μg of poly(dA:dT) (InvivoGen) or 1 μg of 2´3´-cGAMP (InvivoGen) was mixed with 0.3 mL of Xfect polymer in Xfect reaction buffer (Clontech Laboratories) for 10 min and then added to BMDMs in Opti-MEM (ThermoFisher Scientific).
Quantitative real-time PCR
cDNA was prepared as described previously (22). Primers used were Hprt-forward 5´-CTC ATG GAC TGA TTA TGG ACA GGA C-3´ and Hprt-reverse 5´-GCA GGT CAG CAA AGA ACT TAT AGC C-3´; Il18-forward-F 5´-GCC TCA AAC CTT CCA AAT CA-3´ and Il18-reverse-R 5´-TGG ATC CAT TTC CTC AAA GG-3´; Il1b-forward-F 5´-GAC CTT CCA GGA TGA GGA CA-3´ and Il1b-reverse-R 5´-AGC TCA TAT GGG TCC GAC AG-3´. Gene expression levels were normalized to Hprt.
Western blotting
Protein samples for western blotting were prepared as previously described (21). Primary antibodies used were anti-IL-18 (#D046-3, MBL International), anti-IL-1β (#AF-401-NA, R&D Systems), and anti-β-actin (#8457, Cell Signaling Technology).
Results and Discussion
IL-18 and IL-1β are differentially induced in response to inflammatory stimuli
TLRs, one of the major types of pattern recognition receptors in the cell (23), induce expression of pro-inflammatory cytokines and type I IFNs in response to pathogen-associated molecular patterns and damage-associated molecular patterns (23). We first confirmed that IL-18 was constitutively expressed in untreated WT BMDMs, similar to previous reports (7, 8). Interestingly, we observed that its expression was upregulated by the TLR4 ligand LPS (Fig. 1A). As expected, Il1b was not expressed in untreated BMDMs but was induced shortly after LPS treatment (Fig. 1A). Interestingly, although gene and protein expression of IL-18 was sustained 24 h after stimulation, that of IL-1β was induced initially but reduced substantially at 24 h (Fig. 1A). These results suggest that the expressions of IL-18 and IL-1β are differentially regulated after LPS stimulation.
Figure 1. IL-18 and IL-1β are differentially expressed in response to inflammatory stimuli.
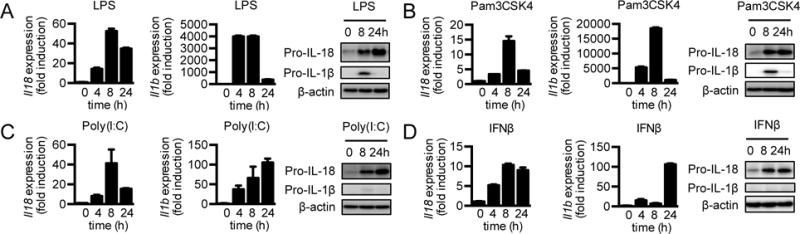
(A–D) Real-time qPCR ana1ysis of genes encoding IL-18 and IL-1β and immunoblot analysis of pro-IL-18, pro-IL-1β, and β-actin (loading control) in WT BMDMs at various time points after LPS (A), Pam3CSK4 (B), Poly(I:C) (C), or IFNβ (D) treatment. Data are representative of 3 independent experiments.
After binding to TLR4, LPS signals through both the MyD88 and TRIF pathways (23). To determine the specific roles of MyD88 and TRIF pathways in regulating IL-18 and IL-1β expression, we treated cells with ligands that signal through either the MyD88 or TRIF pathway. Interestingly, Pam3CSK4, which signals through the MyD88 pathway downstream of TLR2 induced sustained expression of IL-18 over the treatment period in WT BMDMs (Fig. 1B). On the other hand, IL-1β was upregulated 4 h after treatment but diminished dramatically at later time points (Fig. 1B). Gardiquimod induces the MyD88 pathway downstream of TLR7. Induction kinetics of mRNA expression and protein levels of IL-18 and IL-1β in WT BMDMs treated with gardiquimod were similar to those treated with Pam3CSK4 (Supplemental Fig. 1A). Poly(I:C) is sensed by TLR3 to activate the TRIF pathway. Poly(I:C) treatment induced Il18 expression in WT BMDMs. It also induced Il1b expression, although at a much lower fold induction than by LPS, Pam3CSK4, and gardiquimod (Fig. 1C). These findings suggest that the TRIF pathway moderately induces Il1b expression, and both the MyD88 and TRIF pathways induce Il18 expression. Taken together, our results show that IL-18 and IL-1β can be induced by various TLR ligands. Further, during chronic TLR stimulation, IL-18 expression is sustained but IL-1β expression is downregulated. These data suggest distinct regulatory pathways that control the expression of pro-IL-18 and pro-IL-1β levels during chronic TLR stimulation.
One of the prominent mediators induced by the TRIF pathway are type I IFNs, including IFNβ (23). Poly(I:C) induced the expression of Il18 comparable to LPS stimulation, which led us to hypothesize that the induction of Il18 requires type I IFNs. To determine the role of type I IFNs in modulating the expressions of Il18 and Il1b, we treated WT BMDMs with recombinant IFNβ and measured Il18 and Il1b expression. IL-18 mRNA and protein levels were induced after IFNβ treatment, but IL-1β mRNA and protein levels were only modestly induced at levels similar to those seen for Poly(I:C) stimulation (Fig. 1C and 1D). Overall, these data suggest that treatment of type I IFNs upregulates IL-18 expression.
Induction of IL-18 but not IL-1β is dependent on type I IFN signaling
To test whether type I IFN signaling was required for induction of Il18 expression by inflammatory stimuli, we treated WT BMDMs and cells lacking the subunit of type I IFN receptor IFNAR1 (Ifnar1−/−) with LPS and monitored the expression of Il18 and Il1b over the treatment period. LPS treatment led to upregulation of IL-18 in WT BMDMs as early as 4 h after treatment. IL-18 expression further increased and was sustained during chronic LPS treatment (Fig. 2A). Surprisingly, this induction of IL-18 by LPS was impaired in Ifnar1−/− cells (Fig. 2A), indicating that type I IFN signaling is crucial to induce IL-18 during LPS treatment. In WT BMDMs, IL-1β expression increased 4 h and 8 h after treatment but diminished at later time points (Fig. 2A). Unlike Il18, induction of IL-1β remained largely intact in Ifnar1−/− cells (Fig. 2A). Moreover, expression of Il1b mRNA and protein was higher in Ifnar1−/− BMDMs than in WT BMDMs 16 h and 24 h after stimulation (Fig. 2A), suggesting that type I IFN signaling may negatively regulate Il1b expression during chronic LPS stimulation.
Figure 2. Type I IFN signaling is essential for induction of IL-18 but not IL-1β.
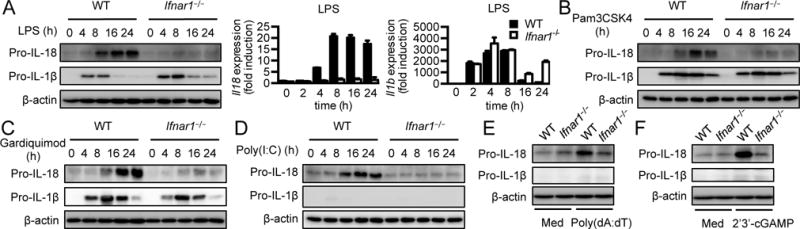
(A) Immunoblot analysis of pro-IL-18, pro-IL-1β, and β-actin (loading control) and real-time qPCR analysis of genes encoding IL-18 and IL-1β in WT or Ifnar1−/− BMDMs at various time points after LPS treatment. (B–D) Immunoblot analysis of pro-IL-18, pro-IL-1β, and β-actin (loading control) in WT or Ifnar1−/− BMDMs at various time points after Pam3CSK4 (B), gardiquimod (C), or Poly(I:C) (D) treatment. (E and F) Immunoblot analysis of pro-IL-18, pro-IL-1β, and β-actin (loading control) in untreated WT or Ifnar1−/− BMDMs (medium alone [Med]) or BMDMs at 8h after transfected with poly(dA:dT) (E) or 2´3´-cGAMP (F). Data are representative of 3 independent experiments.
To further test whether the induction of IL-18 by stimuli that trigger only the MyD88 or TRIF pathway also depends on type I IFN signaling, we treated WT and Ifnar1−/− BMDMs with Pam3CSK4, gardiquimod, or Poly(I:C). Induction of IL-18 was abolished in Ifnar1−/− BMDMs compared with WT BMDM when MyD88 signaling was triggered by Pam3CSK4 or gardiquimod treatment (Fig. 2B and 2C). However, IL-1β expression remained unchanged in Ifnar1−/− BMDMs after either stimulation (Fig. 2B and 2C). Further, during treatment with Poly(I:C), which signals via the TRIF pathway, IL-18 induction was impaired in Ifnar1−/− BMDMs compared with WT BMDMs (Fig. 2D). Recognition of cytosolic DNA by the cGAS-STING pathway results in the production of type I IFNs (24). We hypothesized that stimuli that induce the production of type I IFNs will promote IL-18 expression that is dependent on type I IFN signaling. Therefore, we activated the cGAS-STING pathway by transfecting double-stranded DNA, poly(dA:dT), or the STING ligand 2´3´-cGAMP in BMDMs. Expression of Il18 was increased by activation of the cGAS-STING pathway (Fig. 2E and 2F), but that of Il1b was not robustly induced (Fig. 2E and 2F). As expected, the induction of Il18 was lower in Ifnar1−/− BMDMs in response to poly(dA:dT) or 2´3´-cGAMP than in WT BMDMs (Fig. 2E and 2F). Collectively, these results suggest that type I IFN signaling is critical for induction of IL-18, but not IL-1β after inflammatory stimulations.
Components of type I IFN signaling are essential to induce IL-18
The type I IFN receptor is a heterodimeric complex formed by IFNAR1 and IFNAR2 (25). Binding of type I IFN to IFNAR1 and IFNAR2 induces the formation of interferon stimulating gene factor 3 (ISGF3) by signal transducer and activator of transcription (STAT) members STAT1, STAT2, and IRF9, which function as transcriptional factors to induce the expression various interferon-stimulated genes (ISGs) (25). To test whether other components of type I IFN signaling are also required to induce IL-18, we first measured IL-18 and IL-1β levels in WT BMDMs and cells lacking IFNAR2, IRF9, or STAT1 (Ifnar2−/−, Irf9−/− and Stat1−/−, respectively) after LPS treatment. Similar to Ifnar1−/− BMDMs, induction of IL-18 was also impaired in Ifnar2−/−, Irf9−/−, and Stat1−/− BMDMs (Fig. 3A). However, expression of Il1b mRNA and protein was not compromised but showed a slight increase at 16 h and 24 h after stimulation in Ifnar2−/−, Irf9−/−, and Stat1−/− BMDMs (Fig. 3A). Moreover, pro-IL-18 was not upregulated in Ifnar2−/−, Irf9−/−, and Stat1−/− BMDMs treated with Pam3CSK4 or Poly(I:C) compared with WT BMDMs (Fig. 3B and 3C). ISGF3 directly binds to promoters of ISGs to induce their expression, some of which include IFN regulatory factors (IRFs) that function as transcriptional factors to drive the expression of additional genes (26). To test whether IL-18 was induced via ISGF3 or other IRFs downstream of the type I IFN receptor, we stimulated Ifnar1−/−, Ifnar2−/−, Irf9−/−, and Stat1−/− BMDMs with recombinant IFNβ and observed that IFNβ–induced expression of Il18 was compromised in Ifnar1−/−, Ifnar2−/−, Irf9−/−, and Stat1−/− BMDMs compared with WT cells (Supplemental Fig. 2A). On the other hand, the induction of Il18 by IFNβ was not affected in BMDMs lacking other IRFs, including IRF1, IRF3, IRF4, IRF5, IRF7, or IRF8 (Irf1−/−, Irf3−/−, Irf4fl/fl-Lysm-Cre, Irf5−/−, Irf7−/− and Irf8−/−, respectively; Supplemental Fig. 2B and 2C), suggesting that induction of Il18 is dependent on ISGF3 but not other IRFs.
Figure 3. Components of type I IFN signaling are required for induction of IL-18.
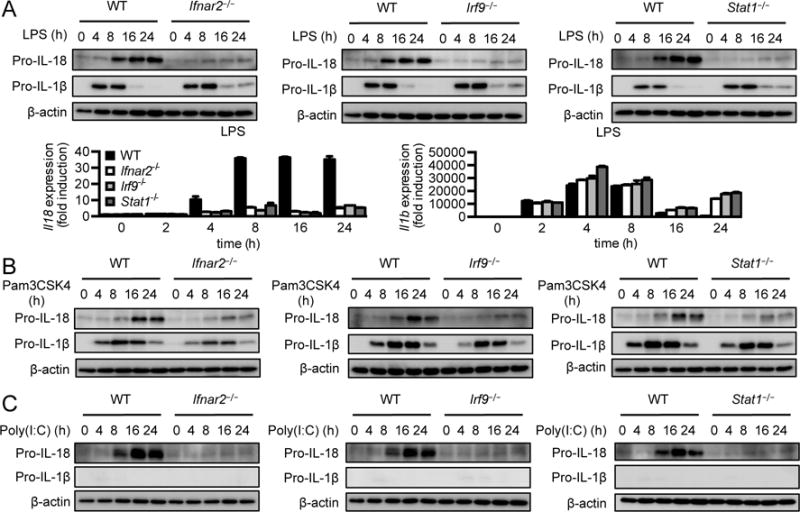
(A) Immunoblot analysis of pro-IL-18, pro-IL-1β, and β-actin (loading control) and real-time qPCR analysis of genes encoding IL-18 and IL-1β in WT, Ifnar2−/−, Irf9−/−, or Stat1−/− BMDMs at various time points after LPS treatment. (B and C) Immunoblot analysis of pro-IL-18, pro-IL-1β, and β-actin (loading control) in WT, Ifnar2−/−, Irf9−/−, or Stat1−/− BMDMs at various time points after Pam3CSK4 (B) or Poly(I:C) treatment (C). Data are representative of 3 independent experiments.
We have established that although both IL-18 and IL-1β belong to the IL-1 family and are activated by the inflammasome, IL-18 and IL-1β are differentially regulated (Supplemental Fig. 2D). Indeed, previous report has shown that secretion of IL-1β but not IL-18 from murine dendritic cells in response to Listeria monocytogenes p60 protein requires ROS production and caspase 11 (27). Distinctive expression patterns of IL-18 and IL-1β are observed in the intestine. IL-18 is expressed at high level in colon and confers protection against inflammation by promoting epithelial cell proliferation and tissue repair (28). IL-1β, on the other hand is expressed at lower levels basally, but enhanced during acute inflammation (28). Given that type I IFNs are protective against DSS-induced colitis (29), it is possible that the type I IFNs contribute to protection by upregulating the expression of Il18 in the colon. Activation of the cGAS-STING pathway leads to the production of type I IFN (24). Colon tissues from mice lacking STING also have decreased Il18 mRNA and protein expression after DSS treatment (22, 30). Therefore, type I IFN signaling can be explored as a therapeutic target in IL-18–associated diseases. We further demonstrated that Il1b gene and protein expression was reduced during chronic TLR stimulation in WT BMDMs, indicating that the transcription of Il1b or stability of Il1b mRNA may be compromised. Reduced translational activity or protein stability can also contribute to decreased levels of pro-IL-1β. Indeed, there is translational inhibition of Il1b expression in LPS-stimulated macrophages (31). Moreover, pro-IL-1β is ubiquinated by E2 conjugase UBE2L3 and subsequently degraded during chronic LPS stimulation (32). This observation is in line with several studies showing that when macrophages are treated with TLR ligands, pro-IL-1β is sequestered in autophagosomes for degradation (33, 34). Of note, we found that although increases in mRNA levels were similar between Il18 and Il1b in response to Poly(I:C) in WT BMDMs, upregulation of IL-1β protein levels was much less robust compared with that of IL-18 levels (Fig. 1C). This could be resulted from decreased translational activity of Il1b mRNA or lower stability of pro-IL-1β due to proteasome or autophagy-mediated degradation, revealing another layer of differential regulation between IL-18 and IL-1β. In addition, the modest expression of IL-1β in response to Poly(I:C) is supported by a previous study showing that pro-IL-1β is much less induced by Poly(I:C) than by LPS or gardiquimod (35). Our study also found that downregulation of both Il1b mRNA and protein expression was ameliorated in cells lacking type I IFN signaling at later time points of LPS stimulation. An intriguing theory is that type I IFN signaling may be involved in the negative control of Il1b expression. Indeed, macrophages lacking tyrosine kinase 2, which is downstream of the type I IFN receptor, have increased translation of Il1b and hence high levels of pro-IL-1β in response to LPS (36). Previous study has also demonstrated that type I IFN signaling suppresses IL-1β expression via IL-10-STAT3 signaling axis and inhibits proteolytic processing of IL-1β by NLRP3 inflammasome (37). Interestingly, downregulation of IL-1β expression was not affected in BMDMs lacking type I IFN signaling compared with WT cells at later time points of Pam3CSK4 or gardiquimod stimulation (Fig. 2B, 2C, 3B and 3C). The MyD88 pathway might induce lower production of type I IFNs and consequently the negative control on IL-1β is less robust as compared to the TRIF pathway that is activated by LPS.
IL-1 family cytokines have pleiotropic functions and are involved in several inflammatory and autoinflammatory disease settings. Interestingly, IL-1α, IL-1β and IL-18 have specific functions in regulating disease outcomes (5). Autoinflammatory models associated with overt pyrin inflammasome activation are differentially mediated through IL-18 (38) or IL-1β (39) production. While levels of circulating IL-1β and IL-18 in the serum are upregulated in patients bearing activating NLRC4 or NLRP3 mutation, IL-18 is induced to a greater extent in patients harboring mutation in NLRC4 (40). IL-18 is also chronically elevated in several of the autoinflammatory diseases and inflammasomepathies (41). However, the cause and effect of this chronically sustained elevated level of IL-18 is not studied well. Our current studies demonstrate that targeting type I IFN signaling pathway could be a way to regulate the pathologic level of IL-18 in the affected patients. In summary, our study demonstrated that during chronic stimulation, IL-18 expression is induced and sustained. In contrast, during chronic stimulations IL-1β is robustly induced but not sustained. Finally, we show that type I IFN signaling is necessary for the induction of IL-18, but not IL-1β, which points to a critical and differential role for type I IFN in regulating IL-18 signaling.
Supplementary Material
Acknowledgments
We thank the members of the Kanneganti lab for their comments and suggestions. We also thank Department of Scientific Editing at St. Jude for editing the manuscript.
Grant: This work was supported by grants from the National Institutes of Health (AI101935, AI124346, AR056296, and CA163507) and ALSAC (to T.-D.K).
Abbreviations
- BMDMs
bone marrow–derived macrophages
- cGAMP
cyclic guanosine monophosphate–adenosine monophosphate
- cGAS
cyclic GMP–AMP synthase
- DSS
dextran sulfate sodium
- IFNAR
interferon-α/β receptor
- ISGs
interferon-stimulated genes
- IRF
interferon regulatory factor
- ISGF3
interferon stimulated gene factor 3
- STING
stimulator of interferon genes
- STAT
signal transducer and activator of transcription
- TRIF
TIR-domain-containing adapter-inducing interferon-β
- WT
wild type
References
- 1.Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annual review of immunology. 2009;27:519–550. doi: 10.1146/annurev.immunol.021908.132612. [DOI] [PubMed] [Google Scholar]
- 2.Garlanda C, Dinarello CA, Mantovani A. The interleukin-1 family: back to the future. Immunity. 2013;39:1003–1018. doi: 10.1016/j.immuni.2013.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dinarello CA, Simon A, van der Meer JW. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nature reviews Drug discovery. 2012;11:633–652. doi: 10.1038/nrd3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sims JE, Smith DE. The IL-1 family: regulators of immunity. Nature reviews Immunology. 2010;10:89–102. doi: 10.1038/nri2691. [DOI] [PubMed] [Google Scholar]
- 5.Man SM, Kanneganti TD. Converging roles of caspases in inflammasome activation, cell death and innate immunity. Nature reviews Immunology. 2016;16:7–21. doi: 10.1038/nri.2015.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pizarro TT, Michie MH, Bentz M, Woraratanadharm J, Smith MF, Jr, Foley E, Moskaluk CA, Bickston SJ, Cominelli F. IL-18, a novel immunoregulatory cytokine, is up-regulated in Crohn’s disease: expression and localization in intestinal mucosal cells. Journal of immunology. 1999;162:6829–6835. [PubMed] [Google Scholar]
- 7.Puren AJ, Fantuzzi G, Dinarello CA. Gene expression, synthesis, and secretion of interleukin 18 and interleukin 1beta are differentially regulated in human blood mononuclear cells and mouse spleen cells. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:2256–2261. doi: 10.1073/pnas.96.5.2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stoll S, Jonuleit H, Schmitt E, Muller G, Yamauchi H, Kurimoto M, Knop J, Enk AH. Production of functional IL-18 by different subtypes of murine and human dendritic cells (DC): DC-derived IL-18 enhances IL-12-dependent Th1 development. European journal of immunology. 1998;28:3231–3239. doi: 10.1002/(SICI)1521-4141(199810)28:10<3231::AID-IMMU3231>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 9.Matsui K, Yoshimoto T, Tsutsui H, Hyodo Y, Hayashi N, Hiroishi K, Kawada N, Okamura H, Nakanishi K, Higashino K. Propionibacterium acnes treatment diminishes CD4+ NK1.1+ T cells but induces type I T cells in the liver by induction of IL-12 and IL-18 production from Kupffer cells. Journal of immunology. 1997;159:97–106. [PubMed] [Google Scholar]
- 10.Stoll S, Muller G, Kurimoto M, Saloga J, Tanimoto T, Yamauchi H, Okamura H, Knop J, Enk AH. Production of IL-18 (IFN-gamma-inducing factor) messenger RNA and functional protein by murine keratinocytes. Journal of immunology. 1997;159:298–302. [PubMed] [Google Scholar]
- 11.Kupz A, Guarda G, Gebhardt T, Sander LE, Short KR, Diavatopoulos DA, Wijburg OL, Cao H, Waithman JC, Chen W, Fernandez-Ruiz D, Whitney PG, Heath WR, Curtiss R, 3rd, Tschopp J, Strugnell RA, Bedoui S. NLRC4 inflammasomes in dendritic cells regulate noncognate effector function by memory CD8(+) T cells. Nature immunology. 2012;13:162–169. doi: 10.1038/ni.2195. [DOI] [PubMed] [Google Scholar]
- 12.Mehta VB, Hart J, Wewers MD. ATP-stimulated release of interleukin (IL)-1beta and IL-18 requires priming by lipopolysaccharide and is independent of caspase-1 cleavage. The Journal of biological chemistry. 2001;276:3820–3826. doi: 10.1074/jbc.M006814200. [DOI] [PubMed] [Google Scholar]
- 13.Marshall JD, Aste-Amezaga M, Chehimi SS, Murphy M, Olsen H, Trinchieri G. Regulation of human IL-18 mRNA expression. Clinical immunology. 1999;90:15–21. doi: 10.1006/clim.1998.4633. [DOI] [PubMed] [Google Scholar]
- 14.Bohle B, Jahn-Schmid B, Maurer D, Kraft D, Ebner C. Oligodeoxynucleotides containing CpG motifs induce IL-12, IL-18 and IFN-gamma production in cells from allergic individuals and inhibit IgE synthesis in vitro. European journal of immunology. 1999;29:2344–2353. doi: 10.1002/(SICI)1521-4141(199907)29:07<2344::AID-IMMU2344>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 15.Pirhonen J, Sareneva T, Kurimoto M, Julkunen I, Matikainen S. Virus infection activates IL-1 beta and IL-18 production in human macrophages by a caspase-1-dependent pathway. Journal of immunology. 1999;162:7322–7329. [PubMed] [Google Scholar]
- 16.Muller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, Aguet M. Functional role of type I and type II interferons in antiviral defense. Science. 1994;264:1918–1921. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- 17.Fenner JE, Starr R, Cornish AL, Zhang JG, Metcalf D, Schreiber RD, Sheehan K, Hilton DJ, Alexander WS, Hertzog PJ. Suppressor of cytokine signaling 1 regulates the immune response to infection by a unique inhibition of type I interferon activity. Nature immunology. 2006;7:33–39. doi: 10.1038/ni1287. [DOI] [PubMed] [Google Scholar]
- 18.Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, Shimada N, Ohba Y, Takaoka A, Yoshida N, Taniguchi T. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature. 2005;434:772–777. doi: 10.1038/nature03464. [DOI] [PubMed] [Google Scholar]
- 19.Sato M, Suemori H, Hata N, Asagiri M, Ogasawara K, Nakao K, Nakaya T, Katsuki M, Noguchi S, Tanaka N, Taniguchi T. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity. 2000;13:539–548. doi: 10.1016/s1074-7613(00)00053-4. [DOI] [PubMed] [Google Scholar]
- 20.Kimura T, Kadokawa Y, Harada H, Matsumoto M, Sato M, Kashiwazaki Y, Tarutani M, Tan RS, Takasugi T, Matsuyama T, Mak TW, Noguchi S, Taniguchi T. Essential and non-redundant roles of p48 (ISGF3 gamma) and IRF-1 in both type I and type II interferon responses, as revealed by gene targeting studies. Genes to cells : devoted to molecular & cellular mechanisms. 1996;1:115–124. doi: 10.1046/j.1365-2443.1996.08008.x. [DOI] [PubMed] [Google Scholar]
- 21.Man SM, Karki R, Sasai M, Place DE, Kesavardhana S, Temirov J, Frase S, Zhu Q, Malireddi RK, Kuriakose T, Peters JL, Neale G, Brown SA, Yamamoto M, Kanneganti TD. IRGB10 Liberates Bacterial Ligands for Sensing by the AIM2 and Caspase-11-NLRP3 Inflammasomes. Cell. 2016;167:382–396 e317. doi: 10.1016/j.cell.2016.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu Q, Man SM, Gurung P, Liu Z, Vogel P, Lamkanfi M, Kanneganti TD. Cutting edge: STING mediates protection against colorectal tumorigenesis by governing the magnitude of intestinal inflammation. Journal of immunology. 2014;193:4779–4782. doi: 10.4049/jimmunol.1402051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.O’Neill LA, Golenbock D, Bowie AG. The history of Toll-like receptors – redefining innate immunity. Nature reviews Immunology. 2013;13:453–460. doi: 10.1038/nri3446. [DOI] [PubMed] [Google Scholar]
- 24.Wu J, Chen ZJ. Innate immune sensing and signaling of cytosolic nucleic acids. Annual review of immunology. 2014;32:461–488. doi: 10.1146/annurev-immunol-032713-120156. [DOI] [PubMed] [Google Scholar]
- 25.Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nature reviews Immunology. 2014;14:36–49. doi: 10.1038/nri3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Honda K, Taniguchi T. IRFs: master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nature reviews Immunology. 2006;6:644–658. doi: 10.1038/nri1900. [DOI] [PubMed] [Google Scholar]
- 27.Schmidt RL, Lenz LL. Distinct licensing of IL-18 and IL-1beta secretion in response to NLRP3 inflammasome activation. PloS one. 2012;7:e45186. doi: 10.1371/journal.pone.0045186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zaki MH, Lamkanfi M, Kanneganti TD. The Nlrp3 inflammasome: contributions to intestinal homeostasis. Trends in immunology. 2011;32:171–179. doi: 10.1016/j.it.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Katakura K, Lee J, Rachmilewitz D, Li G, Eckmann L, Raz E. Toll-like receptor 9-induced type I IFN protects mice from experimental colitis. The Journal of clinical investigation. 2005;115:695–702. doi: 10.1172/JCI22996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ahn J, Konno H, Barber GN. Diverse roles of STING-dependent signaling on the development of cancer. Oncogene. 2015;34:5302–5308. doi: 10.1038/onc.2014.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schott J, Reitter S, Philipp J, Haneke K, Schafer H, Stoecklin G. Translational regulation of specific mRNAs controls feedback inhibition and survival during macrophage activation. PLoS genetics. 2014;10:e1004368. doi: 10.1371/journal.pgen.1004368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eldridge MJ, Sanchez-Garrido J, Hoben GF, Goddard PJ, Shenoy AR. The Atypical Ubiquitin E2 Conjugase UBE2L3 Is an Indirect Caspase-1 Target and Controls IL-1beta Secretion by Inflammasomes. Cell reports. 2017;18:1285–1297. doi: 10.1016/j.celrep.2017.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Giegerich AK, Kuchler L, Sha LK, Knape T, Heide H, Wittig I, Behrends C, Brune B, von Knethen A. Autophagy-dependent PELI3 degradation inhibits proinflammatory IL1B expression. Autophagy. 2014;10:1937–1952. doi: 10.4161/auto.32178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Harris J, Hartman M, Roche C, Zeng SG, O’Shea A, Sharp FA, Lambe EM, Creagh EM, Golenbock DT, Tschopp J, Kornfeld H, Fitzgerald KA, Lavelle EC. Autophagy controls IL-1beta secretion by targeting pro-IL-1beta for degradation. The Journal of biological chemistry. 2011;286:9587–9597. doi: 10.1074/jbc.M110.202911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Duong BH, Onizawa M, Oses-Prieto JA, Advincula R, Burlingame A, Malynn BA, Ma A. A20 restricts ubiquitination of pro-interleukin-1beta protein complexes and suppresses NLRP3 inflammasome activity. Immunity. 2015;42:55–67. doi: 10.1016/j.immuni.2014.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Radwan M, Stiefvater R, Grunert T, Sharif O, Miller I, Marchetti-Deschmann M, Allmaier G, Gemeiner M, Knapp S, Kovarik P, Muller M, Strobl B. Tyrosine kinase 2 controls IL-1ss production at the translational level. Journal of immunology. 2010;185:3544–3553. doi: 10.4049/jimmunol.0904000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guarda G, Braun M, Staehli F, Tardivel A, Mattmann C, Forster I, Farlik M, Decker T, Du Pasquier RA, Romero P, Tschopp J. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity. 2011;34:213–223. doi: 10.1016/j.immuni.2011.02.006. [DOI] [PubMed] [Google Scholar]
- 38.Kim ML, Chae JJ, Park YH, De Nardo D, Stirzaker RA, Ko HJ, Tye H, Cengia L, DiRago L, Metcalf D, Roberts AW, Kastner DL, Lew AM, Lyras D, Kile BT, Croker BA, Masters SL. Aberrant actin depolymerization triggers the pyrin inflammasome and autoinflammatory disease that is dependent on IL-18, not IL-1beta. The Journal of experimental medicine. 2015;212:927–938. doi: 10.1084/jem.20142384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sharma D, Sharma BR, Vogel P, Kanneganti TD. IL-1beta and Caspase-1 Drive Autoinflammatory Disease Independently of IL-1alpha or Caspase-8 in a Mouse Model of Familial Mediterranean Fever. The American journal of pathology. 2017;187:236–244. doi: 10.1016/j.ajpath.2016.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Canna SW, de Jesus AA, Gouni S, Brooks SR, Marrero B, Liu Y, DiMattia MA, Zaal KJ, Sanchez GA, Kim H, Chapelle D, Plass N, Huang Y, Villarino AV, Biancotto A, Fleisher TA, Duncan JA, O’Shea JJ, Benseler S, Grom A, Deng Z, Laxer RM, Goldbach-Mansky R. An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nature genetics. 2014;46:1140–1146. doi: 10.1038/ng.3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Canna SW, Girard C, Malle L, de Jesus A, Romberg N, Kelsen J, Surrey LF, Russo P, Sleight A, Schiffrin E, Gabay C, Goldbach-Mansky R, Behrens EM. Life-threatening NLRC4-associated hyperinflammation successfully treated with IL-18 inhibition. The Journal of allergy and clinical immunology. 2016 doi: 10.1016/j.jaci.2016.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.