

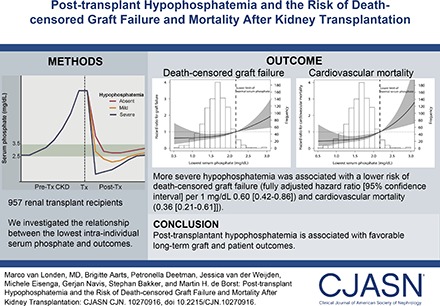
Abstract
Background and objectives
Hypophosphatemia is common in the first year after kidney transplantation, but its clinical implications are unclear. We investigated the relationship between the severity of post-transplant hypophosphatemia and mortality or death-censored graft failure in a large cohort of renal transplant recipients with long-term follow-up.
Design, setting, participants, & measurements
We performed a longitudinal cohort study in 957 renal transplant recipients who were transplanted between 1993 and 2008 at a single center. We used a large real-life dataset containing 28,178 phosphate measurements (median of 27; first to third quartiles, 23–34) serial measurements per patient) and selected the lowest intraindividual phosphate level during the first year after transplantation. The primary outcomes were all-cause mortality, cardiovascular mortality, and death-censored graft failure.
Results
The median (interquartile range) intraindividual lowest phosphate level was 1.58 (1.30–1.95) mg/dl, and it was reached at 33 (21–51) days post-transplant. eGFR was the main correlate of the lowest serum phosphate level (model R2 =0.32). During 9 (5–12) years of follow-up, 181 (19%) patients developed graft failure, and 295 (35%) patients died, of which 94 (32%) deaths were due to cardiovascular disease. In multivariable Cox regression analysis, more severe hypophosphatemia was associated with a lower risk of death-censored graft failure (fully adjusted hazard ratio, 0.61; 95% confidence interval, 0.43 to 0.88 per 1 mg/dl lower serum phosphate) and cardiovascular mortality (fully adjusted hazard ratio, 0.37; 95% confidence interval, 0.22 to 0.62) but not noncardiovascular mortality (fully adjusted hazard ratio, 1.33; 95% confidence interval, 0.9 to 1.96) or all-cause mortality (fully adjusted hazard ratio, 1.15; 95% confidence interval, 0.81 to 1.61).
Conclusions
Post-transplant hypophosphatemia develops early after transplantation. These data connect post-transplant hypophosphatemia with favorable long-term graft and patient outcomes.
Keywords: hypophosphatemia; graft survival; mortality; transplantation; chronic kidney disease-mineral and bone disorder; phosphate; Cardiovascular Diseases; Confidence Intervals; Follow-Up Studies; Humans; Hypophosphatemia; kidney transplantation; Longitudinal Studies; Phosphates; Proportional Hazards Models; Regression Analysis; Renal Insufficiency, Chronic; Risk
Introduction
CKD is characterized by progressively deregulated phosphate homeostasis reflected by increased circulating levels of the phosphaturic hormones fibroblast growth factor 23 (FGF23) and parathyroid hormone (PTH) and hyperphosphatemia (1,2). Emerging data have shown strong positive associations between plasma FGF23 concentrations and adverse cardiovascular outcomes, and FGF23 may contribute to the development of left ventricular hypertrophy in CKD (3–5). After kidney transplantation, FGF23 and PTH concentrations decline over a period of weeks to months (6). In the early post-transplant stage, the relatively high FGF23 and PTH concentrations in the context of a restored renal excretory capacity of phosphate may result in hypophosphatemia (7,8), which usually persists for weeks to months (9,10).
The clinical implications of post-transplant hypophosphatemia in terms of graft and patient survival are unclear. On the one hand, hypophosphatemia may reflect good graft function and thereby, be associated with prolonged graft survival (11) and favorable cardiovascular outcomes (12,13). On the other hand, post-transplant hypophosphatemia may reflect long-term exposure to high circulating FGF23 and PTH concentrations before transplantation, which may, at least in part, persist after transplantation (14,15). Such high FGF23 and PTH levels might contribute to an increased risk of graft failure and cardiovascular complications (16,17).
We investigated whether the development of post-transplant hypophosphatemia and the lowest serum phosphate concentration in the first year after transplantation are associated with death-censored graft failure as well as cardiovascular and all-cause mortality in a large cohort of renal transplant recipients.
Materials and Methods
Study Population
This single-center cohort consisted of 957 renal transplant recipients who underwent a first (solo) kidney transplantation and had a functioning graft at 1 year after transplantation. All transplantations were performed at the University Medical Center Groningen (Groningen, The Netherlands) or in a collaborating center in The Netherlands on behalf of the Dutch donor exchange program. From 1288 kidney transplantations performed between January of 1993 and February of 2008, 151 patients were excluded because of retransplantation, 65 patients were excluded because of simultaneous kidney/liver or kidney/pancreas transplantation, five patients were excluded because of missing serum phosphate data, and four were lost to follow-up. Finally, we excluded 106 patients with graft survival <1 year. After excluding these patients, 957 patients remained within the study cohort. The study was approved by the institutional ethical review board (METc 2014/077). All procedures were conducted in accordance with the declarations of Helsinki and Istanbul.
Transplantation and Follow-Up
Standard immunosuppression consisted of triple therapy with tacrolimus (Prograft or Avagraf; Astellas Pharma B.V., Leiden, The Netherlands; initial trough level of 8–12 ng/ml followed by 6–10 ng/ml [>2 months] and 4–6 ng/ml [>6 months]) or cyclosporin microemulsion (Neoral; Novartis Pharma B.V., Arnhem, The Netherlands; two times 4 mg/kg daily; initial trough level of 200–500 μg/L followed by 150–500 μg/L [>2 months] and 75–125 μg/L [>6 months]) in combination with mycophenolate mofetil (Cellcept; Roche B.V., Woerden, The Netherlands; 2 g daily or Myfortic, Novartis Pharma B.V.; 1440 mg daily) and prednisolone. Patients were followed up in our center initially weekly, were tapered to every 4–6 weeks during the first year, and later, had longer intervals up to once per year. Phosphate concentrations were assessed at each visit. Management of calcium/phosphate metabolism was not subject to a formal treatment protocol. Oral phosphate supplementation was given as potassium phosphate/sodium phosphate at the discretion of the treating nephrologist. Data on phosphate and vitamin D supplementation were collected before transplantation and at (or <4 weeks before) the lowest serum phosphate.
Study Outcomes
The primary outcomes were death-censored graft failure (defined as ESRD requiring dialysis or retransplantation), all-cause mortality, and cardiovascular mortality. The cause of death was obtained from death certificates coded by a physician according to the International Classification of Diseases, Ninth Revision (ICD-9) and were verified by reviewing the medical records. Cardiovascular mortality was defined as death from acute myocardial infarction (ICD-9 code 410), acute and subacute ischemic heart disease (ICD-9 code 411), coronary artery bypass grafting (ICD-9 code 414) or percutaneous transluminal coronary angioplasty, subarachnoid hemorrhage (ICD-9 code 430), intracerebral hemorrhage (ICD-9 code 431), other intracranial hemorrhage (ICD-9 code 432), occlusion or stenosis of the precerebral (ICD-9 code 433) or cerebral (ICD-9 code 434) arteries, or other vascular interventions, such as percutaneous transluminal angioplasty or bypass grafting of aorta and peripheral vessels.
Clinical and Biochemical Data
Data from all routinely determined biochemical measurements from all patients (real-life dataset) within the cohort were used. All routine measurements before March of 2006 were performed on the Merck Mega Analyzer (Merck, Darmstadt, Germany); measurements after this date were performed on the Roche Modular (Roche Ltd., Mannheim, Germany). From 1993 to 2006, PTH was measured using the PTH intact assay from Nichols Institute Diagnostics (San Juan Capistrano, CA). Since February of 2006, PTH has been analyzed using PTH intact assays using an Immulite 2500 (Siemens Healthcare Diagnostics, Deerfield, IL) and a Cobas e601 immunology analyzer (Roche Diagnostics, Mannheim, Germany). Measurements obtained before March of 2006 were converted according to the equations provided in Supplemental Table 1. The lowest available serum phosphate concentration during the first year after kidney transplantation for each patient was used in the analyses. Post-transplant hypophosphatemia was categorized as severe (lowest serum phosphate <1.55 mg/dl [<0.5 mmol/L]), mild (lowest serum phosphate =1.55–2.17 mg/dl [0.5–0.7 mmol/L]), or absent (lowest serum phosphate >2.17 mg/dl [>0.7 mmol/L]) according to the local reference value. Phosphate concentrations at fixed time points 6 and 12 months after transplantation were also collected. Renal function was estimated using the creatinine-based Chronic Kidney Disease Epidemiology Collaboration formula (18) using serum creatinine obtained on the same day as the lowest serum phosphate. Other biochemical parameters were collected on the same day as the lowest serum phosphate (±2 weeks). Clinical and transplant-related data were collected from our central hospital registry. Post-transplant diabetes mellitus was defined according to the American Diabetes Association guidelines: casual plasma glucose ≥200 mg/dl (≥11.1 mmol/L), fasting plasma ≥126 mg/dl (≥7.0 mmol/L), or use of antidiabetic medication (19). Rejection assessment was on the basis of histologic analysis by a pathologist and graded according to the Banff classification (20).
Statistical Analyses
Variable distribution was tested using histograms and probability plots. Normally distributed variables are presented as mean±SD, and non-normally distributed variables are presented as medians (first to third quartiles) unless indicated otherwise. Non-normally distributed variables were log transformed. Differences between hypophosphatemia categories were tested using one-way ANOVA for normally distributed variables, Kruskal–Wallis test for non-normally distributed variables, and Pearson chi-squared test for nominal variables (α=0.05).
To identify independent correlates of the lowest serum phosphate, backward linear regression was performed with initial inclusion of the following covariates: recipient age, sex, smoking status, etiology of kidney disease (glomerular disease, GN, tubular interstitial disease, polycystic kidney disease, dysplasia, renovascular disease, diabetes, and other), cytomegalovirus status, donor type, cold ischemia time, warm ischemia time, acute rejection, delayed graft function, proteinuria, PTH, calcium, albumin, eGFR, 24-hour urea excretion, immunosuppressive use (calcineurin inhibitor, mammalian target of rapamycin inhibitor, proliferation inhibitor, corticosteroids, and other), HLA A/B/DR mismatches, systolic BP, diastolic BP, pretransplant diabetes, pretransplant cardiovascular event, body mass index, donor cytomegalovirus status, donor age, and donor sex. Variables that were significantly associated with the lowest serum phosphate in the final backward regression model were considered independent correlates.
To investigate the association of hypophosphatemia severity with the study outcomes, univariable and multivariable Cox regression analyses were performed. Hypophosphatemia was analyzed as either a categorical (absent versus mild and absent versus severe) or continuous variable (per 1 mg/dl lower serum phosphate). Model 1 was adjusted for recipient age and sex. In model 2, we added eGFR given its potential influence on both the lowest serum phosphate and the risk of adverse graft and patient outcomes. Model 3 was additionally adjusted for nonimmunologic transplant-related factors (donor age and sex, donor type, cold ischemia time, delayed graft function, and proteinuria at time of lowest serum phosphate); model 4 was adjusted for immunologic transplant–related factors (total number of HLA mismatches, acute rejection, induction and maintenance immunosuppressive use, and panel of reactive antibodies), and model 5 was adjusted for potential confounders related to cardiovascular outcomes (dialysis vintage, smoking status, pretransplant diabetes mellitus, and cardiovascular history). Finally, model 6 was adjusted for additional factors that might influence serum phosphate levels: phosphate and vitamin D supplementation and corrected calcium concentrations (21). To visualize the hazard ratio (HR) for patients with hypophosphatemia, spline curves were made for all outcomes using the fully adjusted Cox regression models on the basis of a restricted cubic spline term with three knots. The lower limit of normal phosphate (2.17 mg/dl) was defined as the reference value.
We tested for effect modification by eGFR or timing of the lowest serum phosphate (days post-transplant) using multiplicative interaction terms. On the basis of missing data analysis (Supplemental Table 2), we used listwise deletion for our analyses and imputed data in additional analyses. Because of a relatively high number of unknown causes of death, we investigated whether these missing data could have influenced our model in an exploratory analysis by labeling all unknown causes of death as cardiovascular.
All statistical analyses were performed with SPSS software, version 22.0 for Windows (IBM, Armonk, NY); R, version 3.0.1 (R, Vienna, Austria; http://cran.r-project.org/), and Graphpad Prism 6 for Windows (GraphPad, San Diego, CA).
Results
Patient Characteristics and Correlates of Post-Transplant Hypophosphatemia
Baseline characteristics according to categories of hypophosphatemia (absent, mild, or severe) are shown in Table 1. A total number of 28,178 phosphate measurements were analyzed, with a median (first to third quartiles) of 27 (23–34) phosphate measurements per patient during the first year post-transplant. Patients who developed severe hypophosphatemia (<1.55 mg/dl; n=446; 47%) had a higher eGFR, were more often men, were less likely to have delayed graft function, and had shorter ischemia times compared with patients who did not develop hypophosphatemia. The lowest serum phosphate concentration was reached at 33 (21–51) days after transplantation. The time from transplantation to lowest serum phosphate was similar among hypophosphatemia categories. The intraindividual lowest serum phosphate concentrations were significantly lower than serum phosphate concentrations measured at 6 or 12 months post-transplantation (P<0.001) (Figure 1). Multivariable linear regression analysis revealed eGFR at the time of lowest phosphate as an independent correlate of the lowest serum phosphate (−0.14 mg/dl; 95% confidence interval [95% CI], −0.24 to −0.04 per 10 ml/min per 1.73 m2; P<0.01; model R2 =0.32).
Table 1.
Clinical characteristics of the cohort
| Variable | All Patients, n=957 | Absent Hypophosphatemia, n=136 | Mild Hypophosphatemia, n=375 | Severe Hypophosphatemia, n=446 | P Value |
|---|---|---|---|---|---|
| Lowest serum phosphate post-Tx, mg/dl | 1.58 [1.30–1.95] | 2.45 [2.26–2.73] | 1.80 [1.67–1.98] | 1.24 [1.05–1.42] | <0.001 |
| Age, yr | 49 [39–59] | 49 [36–58] | 50 [39–58] | 49 [39–59] | 0.40 |
| Men, n (%) | 557 (58) | 64 (47) | 221 (59) | 272 (61) | 0.02 |
| Smoking status, n (%) | |||||
| Current smoking | 48 (5) | 5 (4) | 13 (4) | 30 (7) | 0.40 |
| No smoking | 488 (51) | 65 (48) | 188 (50) | 235 (52) | |
| Unknown | 421 (44) | 66 (49) | 174 (46) | 181 (41) | |
| Etiology of kidney disease, % | |||||
| Primary glomerular disease | 24.6 | 26.8 | 24.9 | 20.9 | 0.44 |
| GN | 5.5 | 4.6 | 6.8 | 6.2 | |
| Tubulointerstitial nephritis/pyelonephritis | 10.2 | 10.8 | 9.0 | 14.0 | |
| Polycystic kidney disease | 18.4 | 20.2 | 18.4 | 16.3 | |
| Renovascular | 8.5 | 9.2 | 8.8 | 7.0 | |
| Diabetes | 4.1 | 3.7 | 4.7 | 4.7 | |
| Dysplasia | 1.8 | 1.4 | 2.5 | 1.6 | |
| Other/unknown | 24.1 | 23.4 | 24.9 | 29.5 | |
| Panel of reactive antibodies, % | 8.0 | 8.0 | 9.0 | 10.0 | 0.54 |
| Donor type: living/cadaveric, n | 220/737 | 28/108 | 64/311 | 128/318 | <0.001 |
| Cold ischemia time, h | 17.8 [10.1–23.0] | 18.2 [11.7–24.4] | 19.0 [13.3–23.8] | 16.0 [3.0–22.9] | <0.001 |
| Warm ischemia time, min | 40 [33–50] | 40 [35–52] | 40 [33–51] | 39 [32–45] | 0.02 |
| Delayed graft function, n (%) | 270 (28) | 50 (37) | 110 (29) | 110 (25) | 0.02 |
| Preemptive, n (%) | 31 (3.2) | 8 (5.9) | 11 (2.9) | 12 (2.7) | 0.14 |
| Dialysis vintage, mo | 39 [20–58] | 41 [18–62] | 40 [22–59] | 37 [20–57] | 0.56 |
| Serum calcium pre-Tx, mg/dl | 9.62 (1.04) | 9.62 (1.12) | 9.70 (1.12) | 9.94 (0.92) | 0.14 |
| Serum PTH pre-Tx, pg/ml | 160.3 [72.4–393.7] | 103.7 [61.1–212.2] | 122.6 [57.3–396.1] | 188.6 [88.2–393.7] | 0.04 |
| Acute rejection, n (%) | 342 (36) | 47 (35) | 135 (36) | 160 (36) | 0.95 |
| Time between Tx and lowest phosphate, d | 33 [21–51] | 35 [15–68] | 35 [22–53] | 32 [21–48] | 0.32 |
| eGFR (CKD-EPI),a ml/min per 1.73 m2 | 52 [39–66] | 41 [26–54] | 49 [38–60] | 58 [46–70] | <0.001 |
| Phosphate supplementation, n (%) | 67 (7.0) | 7 (5.2) | 18 (4.8) | 42 (9.4) | 0.06 |
| Vitamin D supplementation, n (%) | 176 (18.4) | 41 (30.1) | 57 (15.2) | 78 (17.5) | <0.001 |
| Vitamin D supplementation pre-Tx, n (%) | 399 (41.7) | 47 (34.6) | 149 (39.7) | 203 (45.5) | 0.25 |
| Hypophosphatemia symptoms,b n (%) | 5 (0.5) | 1 (0.74) | 3 (0.8) | 1 (0.22) | 0.44 |
| Serum calcium,a mg/dl | 9.22 (0.96) | 9.06 (1.04) | 9.38 (0.96) | 9.46 (0.96) | <0.001 |
| Corrected calcium,a mg/dl | 9.57 (0.82) | 9.32 (0.89) | 9.55 (0.78) | 9.67 (0.83) | <0.001 |
| Serum PTH,a pg/ml | 85.8 [61.5–137.8] | 69.5 [47.4–132.8] | 85.8 [70.0–129.5] | 91.3 [67.3–140.3] | <0.001 |
| Proteinuria,a g/24 h | 0.3 [0.2–0.6] | 0.2 [0.3–0.6] | 0.4 [0.2–0.7] | 0.2 [0.3–0.6] | 0.30 |
| Albumin,a g/dl | 3.8 [3.4–4.1] | 3.6 [3.4–4.0] | 3.8 [3.4–4.2] | 3.8 [3.4–4.1] | 0.65 |
| 24-h Urea excretion,a mg/dl | 2673 [2100–3302] | 2611 [2131–3108] | 2701 [2098–3342] | 2673 [2092–3325] | 0.22 |
| Medication use | |||||
| Cyclosporin use, % | 87.9 | 93.4 | 87.7 | 86.3 | 0.09 |
| Cyclosporin trough,a μg/L | 224 (165) | 209 (125) | 223 (144) | 230 (190) | 0.16 |
| Tacrolimus use, % | 7.4 | 2.9 | 7.5 | 8.7 | 0.08 |
| Tacrolimus trough,a μg/L | 11.1 (6.0) | 8.1 (4.9) | 11.6 (5.9) | 11.4 (6.2) | 0.56 |
| Proliferation inhibitor, % | 76.8 | 80.9 | 76.8 | 75.6 | 0.46 |
| mTOR inhibitor, % | 3.8 | 1.5 | 4.3 | 4.0 | 0.31 |
| Corticosteroids, % | 96.4 | 98.5 | 97.3 | 95.1 | 0.08 |
| Other, % | 23.4 | 26.5 | 24.3 | 21.7 | 0.46 |
| Induction therapy, n (%) | |||||
| Antithymocyte globulin | 87 (9) | 10 (7) | 38 (10) | 39 (9) | 0.59 |
| Anti–IL-2 mAb | 410 (43) | 62 (45) | 152 (41) | 196 (44) | 0.48 |
| Other/unknown | 460 (48) | 64 (47) | 185 (49) | 211 (47) | 0.82 |
| CMV-positive status | 376 (39.5) | 180 (40.5) | 141 (37.6) | 180 (40.5) | 0.74 |
| Pre-Tx diabetes mellitus (no/DM1/DM2), % | 94/2/4 | 93/2/5 | 94/3/3 | 94/2/4 | 0.82 |
| BMI at Tx, kg/m2 | 24.3 [22.0–27.2] | 23.7 [21.9–28.3] | 24.6 [22.0–27.1] | 24.3 [22.0–27.2] | 0.94 |
| Pre-Tx CV event, n (%) | 111(11.9) | 9 (6.9) | 46 (12.8) | 56 (12.6) | 0.16 |
| Systolic BP at 3 mo, mmHg | 140 [130–152] | 145 [130–160] | 140 [133–154] | 140 [130–150] | 0.18 |
| Diastolic BP at 3 mo, mmHg | 85 [80–90] | 82 [80–90] | 85 [80–90] | 85 [80–94] | 0.14 |
| Donor characteristics | |||||
| Age, yr | 47 [35–55] | 47 [38–54] | 47 [35–56] | 46 [33–54] | 0.65 |
| Men, n (%) | 477 (49.9) | 66 (48.5) | 197 (52.5) | 214 (48) | 0.41 |
| CMV-positive status, n (%) | 475 (49.9) | 76 (57.1) | 170 (45.3) | 229 (51.6) | 0.12 |
| HLA mismatches, valid % | |||||
| A (0/1/2) | 47/46/7 | 43/48/9 | 49/45/6 | 45/47/8 | 0.35 |
| B (0/1/2) | 40/47/1 | 37/51/13 | 43/44/14 | 37/50/13 | 0.53 |
| DR (0/1/2) | 52/45/2 | 54/44/2 | 56/42/2 | 48/48/3 | 0.12 |
Data are presented as mean (SD) or median [first to third quartiles] unless otherwise noted. Tx, transplantation; PTH, parathyroid hormone; CKD-EPI, Chronic Kidney Disease Epidemiology Collaboration; mTOR, mammalian target of rapamycin; CMV, cytomegalovirus; DM1, type 1 diabetes mellitus; DM2, type 2 diabetes mellitus; BMI, body mass index; CV, cardiovascular.
Measured at the time of lowest serum phosphate level.
Symptoms attributed to hypophosphatemia by treating nephrologist.
Figure 1.

Lowest serum phosphate are significantly lower than serum phosphate concentrations at 6 and 12 months after transplantation. Data presented as medians and interquartile ranges for (A) severe, (B) mild, and (C) absent hypophosphatemia. The overall lowest serum phosphate concentrations were significantly different from serum phosphate measured at 6 or 12 months (P<0.001 in all categories).
Post-Transplant Hypophosphatemia and Graft Failure
During a median follow-up of 9 (5–12) years after transplantation, 181 (19%) patients developed graft failure. Compared with no hypophosphatemia, both mild and severe hypophosphatemia were associated with a lower risk of death-censored graft failure. Also, when analyzing the lowest phosphate as a continuous variable, a more severe hypophosphatemia was associated with a lower risk of death-censored graft failure (fully adjusted HR per 1 mg/dl, 0.61; 95% CI, 0.43 to 0.88). This association remained significant after adjustment for potential confounders (Figure 2, Table 2). We found significant effect modification by eGFR (Pinteraction<0.001). On stratification, the association between hypophosphatemia category and death-censored graft failure was only significant in patients with an eGFR<40 ml/min per 1.73 m2 (Figure 3). There was no effect modification by time post-transplant (Pinteraction=0.19).
Figure 2.

Spline curves illustrating the association between the lowest serum phosphate levels and the risk of (A) graft failure, (B) all-cause mortality, (C) cardiovascular mortality, and (D) noncardiovascular mortality. Models are on the basis of a cubic spline term (restricted cubic spline) with three knots, with a serum phosphate level of 2.17 mg/dl as the reference value. The solid lines represent the fully adjusted hazard ratios (HRs) for graft failure (Cox regression model 6) and all-cause and cardiovascular mortality (Cox regression model 6). The gray areas represent the 95% confidence intervals of the HRs.
Table 2.
Cox regression analysis of hypophosphatemia versus graft failure risk
| Graft Failure (181 Events) | Hypophosphatemia Category, HR [95% CI] | Lowest Serum Phosphate per 1 mg/dl | |||
|---|---|---|---|---|---|
| Absent | Mild | Severe | HR [95% CI] | P Value | |
| Crude | Reference | 0.52 [0.36 to 0.77]a | 0.44 [0.30 to 0.64]b | 0.43 [0.33 to 0.56] | <0.001 |
| Model 1 | Reference | 0.55 [0.38 to 0.82]a | 0.46 [0.31 to 0.68]b | 0.46 [0.35 to 0.59] | <0.001 |
| Model 2 | Reference | 0.63 [0.42 to 0.92]c | 0.61 [0.41 to 0.91]c | 0.52 [0.39 to 0.68] | <0.001 |
| Model 3 | Reference | 0.82 [0.51 to 1.34] | 0.72 [0.43 to 1.19] | 0.56 [0.40 to 0.79] | 0.001 |
| Model 4 | Reference | 0.87 [0.51 to 1.48] | 0.72 [0.42 to 1.26] | 0.60 [0.42 to 0.85] | 0.004 |
| Model 5 | Reference | 0.79 [0.46 to 1.38] | 0.74 [0.42 to 1.29] | 0.65 [0.46 to 0.92] | 0.02 |
| Model 6 | Reference | 0.79 [0.45 to 1.38] | 0.70 [0.40 to 1.25] | 0.60 [0.42 to 0.86] | 0.007 |
Model 1: adjusted for recipient age and sex. Model 2: model 1 plus eGFR at the time of lowest phosphate. Model 3: model 2 plus donor age and sex, donor type, cold ischemia time, delayed graft function, and proteinuria (at the time of the lowest serum phosphate). Model 4: model 3 plus total number of HLA mismatches, acute rejection, induction and maintenance immunosuppressive use, and panel of reactive antibodies. Model 5: model 4 plus cardiovascular risk factors (dialysis vintage, smoking status, pretransplant diabetes mellitus, and cardiovascular history). Model 6: model 5 plus phosphate supplementation, vitamin D supplementation, vitamin D supplementation pretransplantation, and corrected calcium concentrations (before transplantation and at the time of the lowest serum phosphate). HR, hazard ratio; 95% CI, 95% confidence interval.
P<0.01.
P<0.001.
P<0.05.
Figure 3.

The risk of graft failure in crude Cox regression analysis according to eGFR above or below 40 ml/min per 1.73 m2. (A) In patients with an eGFR<40 ml/min per 1.73 m2, mild (hazard ratio [HR], 0.44; 95% confidence interval [95% CI], 0.26 to 0.75; P=0.003) and severe (HR, 0.38; 95% CI, 0.20 to 0.71; P=0.002) hypophosphatemia were significantly associated with the risk of graft failure compared with absent hypophosphatemia (70 events in this stratum). (B) In patients with eGFR≥40 ml/min per 1.73 m2, no significant association was observed for both mild (HR, 0.93; 95% CI, 0.48 to 1.81; P=0.84) and severe (HR, 0.85; 95% CI, 0.44 to 1.62; P=0.62) hypophosphatemia compared with absent hypophosphatemia (109 events in this stratum).
PTH data at the time of lowest serum phosphate were available for a subset of patients (n=193) (Supplemental Table 2). In a sensitivity analysis, we additionally adjusted for PTH using multiple imputation to handle missing data; this did not materially change the results (fully adjusted HR, 0.61; 95% CI, 0.41 to 0.88 per 1 mg/dl). Further adjustment for the number of phosphate measurements (fully adjusted HR, 0.62; 95% CI, 0.43 to 0.90 per 1 mg/dl) or the time between transplantation and lowest serum phosphate (fully adjusted HR, 0.59; 95% CI, 0.42 to 0.85 per 1 mg/dl) did not change the results; also, replacing eGFR by creatinine clearance (fully adjusted HR, 0.61; 95% CI, 0.43 to 0.87 per 1 mg/dl) did not change the results.
Post-Transplant Hypophosphatemia and Mortality
During follow-up, 295 (35%) patients died, of which 94 (32%) died from cardiovascular causes, 41 (14%) died of infectious causes, 40 (14%) died of malignancies, and 13 (4%) died of other causes. The cause of death was unknown in 107 (36%) patients. The development of hypophosphatemia after transplantation was not associated with all-cause mortality (Table 3). However, severe post-transplant hypophosphatemia was associated with a lower risk of cardiovascular mortality compared with no hypophosphatemia (fully adjusted HR, 2.70; 95% CI, 1.23 to 5.88; continuous HR per 1 mg/dl, 0.37; 95% CI, 0.22 to 0.62). The lowest serum phosphate was not associated with death by infectious disease (n=41; HR, 1.10; 95% CI, 0.60 to 2.04), malignancies (n=40; HR, 1.19; 95% CI, 0.62 to 2.27), other causes (n=13; HR, 1.15; 95% CI, 0.61 to 2.17), or unknown causes (n=107; HR, 1.09; 95% CI, 0.71 to 1.64).
Table 3.
Cox regression analysis of hypophosphatemia versus mortality risk
| Model | Hypophosphatemia Category, HR [95% CI] | Lowest Serum Phosphate per 1 mg/dl | |||
|---|---|---|---|---|---|
| Absent | Mild | Severe | HR [95% CI] | P Value | |
| All-cause mortality (295 events) | |||||
| Crude | Reference | 1.01 [0.69 to 1.47] | 0.97 [0.67 to 1.41] | 0.93 [0.74 to 1.19] | 0.59 |
| Model 1 | Reference | 0.96 [0.65 to 1.40] | 0.89 [0.61 to 1.30] | 0.85 [0.67 to 1.09] | 0.20 |
| Model 2 | Reference | 1.01 [0.69 to 1.48] | 1.02 [0.69 to 1.50] | 0.93 [0.71 to 1.20] | 0.69 |
| Model 3 | Reference | 1.14 [0.68 to 1.90] | 1.21 [0.72 to 2.03] | 0.92 [0.73 to 1.35] | 0.96 |
| Model 4 | Reference | 0.93 [0.55 to 1.58] | 0.98 [0.58 to 1.67] | 0.90 [0.66 to 1.23] | 0.53 |
| Model 5 | Reference | 1.02 [0.59 to 1.80] | 1.19 [0.68 to 2.08] | 1.08 [0.77 to 1.52] | 0.66 |
| Model 6 | Reference | 1.02 [0.60 to 1.72] | 1.18 [0.70 to 1.99] | 1.15 [0.82 to 1.63] | 0.42 |
| Cardiovascular mortality (94 events) | |||||
| Crude | Reference | 0.73 [0.42 to 1.29] | 0.58 [0.33 to 1.02] | 0.45 [0.28 to 0.63] | <0.001 |
| Model 1 | Reference | 0.65 [0.37 to 1.16] | 0.53 [0.30 to 0.94]a | 0.40 [0.29 to 0.56] | <0.001 |
| Model 2 | Reference | 0.65 [0.37 to 1.15] | 0.53 [0.29 to 0.97]a | 0.38 [0.27 to 0.55] | <0.001 |
| Model 3 | Reference | 0.64 [0.35 to 1.18] | 0.50 [0.26 to 0.95]a | 0.39 [0.25 to 0.60] | <0.001 |
| Model 4 | Reference | 0.62 [0.32 to 1.18] | 0.44 [0.22 to 0.88]a | 0.38 [0.24 to 0.59] | <0.001 |
| Model 5 | Reference | 0.44 [0.21 to 0.91]a | 0.36 [0.17 to 0.77]b | 0.39 [0.24 to 0.64] | <0.001 |
| Model 6 | Reference | 0.45 [0.21 to 0.95]a | 0.38 [0.17 to 0.84]a | 0.36 [0.21 to 0.61] | <0.001 |
| Noncardiovascular mortality (176 events) | |||||
| Crude | Reference | 0.85 [0.57 to 1.27] | 0.83 [0.56 to 1.23] | 0.91 [0.69 to 1.19] | 0.49 |
| Model 1 | Reference | 0.84 [0.56 to 1.25] | 0.80 [0.54 to 1.19] | 0.85 [0.65 to 1.12] | 0.25 |
| Model 2 | Reference | 0.93 [0.61 to 1.40] | 1.00 [0.66 to 1.52] | 1.01 [0.74 to 1.35] | 0.99 |
| Model 3 | Reference | 1.14 [0.68 to 1.90] | 1.21 [0.72 to 2.03] | 1.16 [0.82 to 1.64] | 0.40 |
| Model 4 | Reference | 0.93 [0.55 to 1.58] | 0.98 [0.58 to 1.67] | 1.04 [0.73 to 1.52] | 0.80 |
| Model 5 | Reference | 1.03 [0.59 to 1.80] | 1.19 [0.68 to 2.08] | 1.22 [0.85 to 1.79] | 0.28 |
| Model 6 | Reference | 1.04 [0.59 to 1.83] | 1.27 [0.71 to 2.25] | 1.32 [0.89 to 1.96] | 0.15 |
Model 1: adjusted for recipient age and sex. Model 2: model 1 plus eGFR at the time of the lowest phosphate. Model 3: model 2 plus donor age and sex, donor type, cold ischemia time, delayed graft function, and proteinuria (at the time of the lowest serum phosphate). Model 4: model 3 plus total number of HLA mismatches, acute rejection, induction and maintenance immunosuppressive use, and panel of reactive antibodies. Model 5: model 4 plus cardiovascular risk factors (dialysis vintage, smoking status, pretransplant diabetes mellitus, and cardiovascular history). Model 6: model 5 plus phosphate supplementation, vitamin D supplementation, vitamin D supplementation pretransplantation, and corrected calcium concentrations (before transplantation and at the time of the lowest serum phosphate). HR, hazard ratio; 95% CI, 95% confidence interval.
P<0.05.
P<0.01.
When assuming that all patients with unknown cause of death died due to cardiovascular disease, results were similar (fully adjusted HR, 0.49; 95% CI, 0.41 to 0.58 per 1 mg/dl). Further adjustment for serum PTH at the time of lowest phosphate (using imputed data; fully adjusted HR, 0.35; 95% CI, 0.2 to 0.59), number of phosphate measurements (fully adjusted HR, 0.50; 95% CI, 0.42 to 0.58), time between transplantation and lowest serum phosphate (fully adjusted HR, 0.47; 95% CI, 0.4 to 0.56), or replacing eGFR with creatinine clearance (fully adjusted HR, 0.52; 95% CI, 0.44 to 0.61) did not materially change the association between hypophosphatemia and cardiovascular mortality. No significant interaction with eGFR or time after transplantation was observed.
Discussion
Hypophosphatemia is very common after kidney transplantation: in our study, the incidence of hypophosphatemia (serum phosphate <2.17 mg/dl) was 86%. Moreover, we found that the occurrence of hypophosphatemia was associated with favorable graft and patient outcomes. We analyzed the lowest serum phosphate concentration reached within the first year after transplantation considering a large number of intraindividual phosphate measurements (n>28,000 real-life measurements). We found that patients who developed hypophosphatemia had a lower risk of graft failure compared with those who did not develop hypophosphatemia. Furthermore, post-transplant hypophosphatemia was associated with a lower risk of cardiovascular mortality but not all-cause mortality.
In line with our results, previous studies found that a lower serum phosphate concentration at 6 and 12 months after transplantation was associated with a lower risk of graft failure. These fixed time points may have been relatively late given our finding that 50% of patients develop their lowest serum phosphate concentration at approximately 1 month after transplantation. When considering all serum phosphate measurements within the first year after transplantation and selecting the lowest serum phosphate value at any given moment in the first year, we found that the incidence of severe hypophosphatemia was 47% in our cohort, which is in line with some (22) but not all (23) previous studies. We also show that the absence of hypophosphatemia or the occurrence of hyperphosphatemia was associated with a higher graft failure and cardiovascular mortality risk, in line with previous studies (11,24).
The development of hypophosphatemia was associated with a higher eGFR, in line with the concept that good graft function is accompanied by an increased renal capacity to excrete phosphate. Our findings do not support a previously postulated hypothesis that hypophosphatemia would contribute to nephrocalcinosis and premature graft failure (25). We hypothesize that hypophosphatemia is indicative of a restored renal capacity to filtrate phosphate to provide a tubular response to the high concentrations of phosphaturic hormones FGF23 and PTH by reducing sodium-phosphate cotransporters 2a and 2c (7). We cannot distinguish whether hypophosphatemia is a mere marker of a better prognosis or whether lower phosphate concentrations may actually protect graft function. For clinical purposes, our data raise the question whether the associations of hypophosphatemia with favorable graft and patient outcomes should have consequences for phosphate management after transplantation. However, proof for an etiologic role of low phosphate levels should precede clinical recommendations.
Interestingly, we found significant interaction by eGFR for the association between the severity of hypophosphatemia and the risk of death-censored graft failure. Patients with a lower eGFR (<40 ml/min per 1.73 m2) but a preserved capacity to excrete sufficient amounts of phosphate to induce hypophosphatemia apparently had better graft outcome than those with a low eGFR and no hypophosphatemia. Interestingly, the association between hypophosphatemia and graft survival remained materially unchanged after adjustment for eGFR, suggesting that factors other than the eGFR are also involved (26,27). Only 32% of the variation in the lowest phosphate concentration was explained by eGFR and PTH, further supporting the presence of one or more yet unidentified correlates. FGF23, not measured in this cohort, could at least partially further explain this variation.
The association between hypophosphatemia and lower cardiovascular mortality after transplantation can be reconciled with previous studies by our group and others. Early after successful kidney transplantation, when renal function is rapidly restored but FGF23 and PTH remain high, the resulting renal phosphate leak will lead to a subsequent decline in circulating FGF23 and PTH. This decline could be more efficient in those with hypophosphatemia than in those without hypophosphatemia. Because FGF23 has been implicated in cardiovascular disease (3–5) and associated with a higher cardiovascular mortality risk in renal transplant recipients (28,29), more efficient lowering of FGF23 may contribute to the lower risk of cardiovascular mortality in renal transplant recipients who developed hypophosphatemia in the first year after transplantation. The development of hypophosphatemia was associated with cardiovascular but not noncardiovascular mortality.
A limitation of our study is that we do not have data on FGF23 concentrations available, which precludes conclusions on the relationship between FGF23 and the severity of hypophosphatemia. Furthermore, data on PTH, a major regulator of serum phosphate levels, were available for a limited number of patients. However, on the basis of the available data and using multiple imputation to account for missing data, adjusting for PTH did not materially change the association between hypophosphatemia and patient or graft outcomes. Another limitation is the unknown cause of death in a relatively large proportion of patients. Cardiovascular disease was a cause of death in 32% of patients in our cohort, which is less common than previously observed (30). Probably, cardiovascular mortality is over-represented in the group of patients with an unknown cause of death. However, when we performed an exploratory analysis to find effects of the over-representation, the results were highly similar to the original analyses. Also, our patient population had a low incidence of diabetes mellitus and mainly consisted of whites, which may limit the generalizability of our findings. However, strengths of our study include the design, taking advantage of a large number of intraindividual measurements, the high external validity due to the use of real-life data, and the complete and long-term follow-up.
In conclusion, we found that patients who develop hypophosphatemia after kidney transplantation are at lower risk of graft failure and cardiovascular mortality compared with patients who do not develop hypophosphatemia. Future studies should address whether patients who do not develop hypophosphatemia require more intense monitoring for accelerated renal function loss or cardiovascular disease.
Disclosures
None.
Supplementary Material
Acknowledgments
S.J.L.B. is a principal investigator of the Transplant Lines Biobank and Data Repository of the University Medical Center Groningen.
M.H.d.B. is supported by Dutch Organization for Scientific Research Veni grant 016.146.014. This study was supported by Dutch Kidney Foundation consortium grant CP10.11 (to the NIGRAM Consortium).
The NIGRAM Consortium consists of the following principal investigators: Piet ter Wee (Vrije Universiteit University Medical Centre), Marc Vervloet (Vrije Universiteit University Medical Centre), René Bindels (Radboud University Medical Centre), Joost Hoenderop (Radboud University Medical Centre), G.N. (University Medical Centre Groningen), Jan-Luuk Hillebrands (University Medical Centre Groningen), and M.H.d.B. (University Medical Centre Groningen).
Footnotes
Published online ahead of print. Publication date available at www.cjasn.org.
This article contains supplemental material online at http://cjasn.asnjournals.org/lookup/suppl/doi:10.2215/CJN.10270916/-/DCSupplemental.
References
- 1.Larsson T, Nisbeth U, Ljunggren O, Jüppner H, Jonsson KB: Circulating concentration of FGF-23 increases as renal function declines in patients with chronic kidney disease, but does not change in response to variation in phosphate intake in healthy volunteers. Kidney Int 64: 2272–2279, 2003 [DOI] [PubMed] [Google Scholar]
- 2.Wolf M: Forging forward with 10 burning questions on FGF23 in kidney disease. J Am Soc Nephrol 21: 1427–1435, 2010 [DOI] [PubMed] [Google Scholar]
- 3.Faul C, Amaral AP, Oskouei B, Hu MC, Sloan A, Isakova T, Gutiérrez OM, Aguillon-Prada R, Lincoln J, Hare JM, Mundel P, Morales A, Scialla J, Fischer M, Soliman EZ, Chen J, Go AS, Rosas SE, Nessel L, Townsend RR, Feldman HI, St John Sutton M, Ojo A, Gadegbeku C, Di Marco GS, Reuter S, Kentrup D, Tiemann K, Brand M, Hill JA, Moe OW, Kuro-O M, Kusek JW, Keane MG, Wolf M: FGF23 induces left ventricular hypertrophy. J Clin Invest 121: 4393–4408, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Scialla JJ, Xie H, Rahman M, Anderson AH, Isakova T, Ojo A, Zhang X, Nessel L, Hamano T, Grunwald JE, Raj DS, Yang W, He J, Lash JP, Go AS, Kusek JW, Feldman H, Wolf M; Chronic Renal Insufficiency Cohort (CRIC) Study Investigators : Fibroblast growth factor-23 and cardiovascular events in CKD. J Am Soc Nephrol 25: 349–360, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hu MC, Shi M, Cho HJ, Adams-Huet B, Paek J, Hill K, Shelton J, Amaral AP, Faul C, Taniguchi M, Wolf M, Brand M, Takahashi M, Kuro-O M, Hill JA, Moe OW: Klotho and phosphate are modulators of pathologic uremic cardiac remodeling. J Am Soc Nephrol 26: 1290–1302, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Han SY, Hwang EA, Park SB, Kim HC, Kim HT: Elevated fibroblast growth factor 23 levels as a cause of early post-renal transplantation hypophosphatemia. Transplant Proc 44: 657–660, 2012 [DOI] [PubMed] [Google Scholar]
- 7.Shimada T, Hasegawa H, Yamazaki Y, Muto T, Hino R, Takeuchi Y, Fujita T, Nakahara K, Fukumoto S, Yamashita T: FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res 19: 429–435, 2004 [DOI] [PubMed] [Google Scholar]
- 8.Evenepoel P, Naesens M, Claes K, Kuypers D, Vanrenterghem Y: Tertiary ‘hyperphosphatoninism’ accentuates hypophosphatemia and suppresses calcitriol levels in renal transplant recipients. Am J Transplant 7: 1193–1200, 2007 [DOI] [PubMed] [Google Scholar]
- 9.Felsenfeld AJ, Gutman RA, Drezner M, Llach F: Hypophosphatemia in long-term renal transplant recipients: Effects on bone histology and 1,25-dihydroxycholecalciferol. Miner Electrolyte Metab 12: 333–341, 1986 [PubMed] [Google Scholar]
- 10.Baia LC, Heilberg IP, Navis G, de Borst MH; NIGRAM investigators : Phosphate and FGF-23 homeostasis after kidney transplantation. Nat Rev Nephrol 11: 656–666, 2015 [DOI] [PubMed] [Google Scholar]
- 11.Benavente D, Chue CD, Moore J, Addison C, Borrows R, Ferro CJ: Serum phosphate measured at 6 and 12 months after successful kidney transplant is independently associated with subsequent graft loss. Exp Clin Transplant 10: 119–124, 2012 [DOI] [PubMed] [Google Scholar]
- 12.Meier-Kriesche HU, Baliga R, Kaplan B: Decreased renal function is a strong risk factor for cardiovascular death after renal transplantation. Transplantation 75: 1291–1295, 2003 [DOI] [PubMed] [Google Scholar]
- 13.Briggs JD: Causes of death after renal transplantation. Nephrol Dial Transplant 16: 1545–1549, 2001 [DOI] [PubMed] [Google Scholar]
- 14.Kawarazaki H, Shibagaki Y, Fukumoto S, Kido R, Nakajima I, Fuchinoue S, Fujita T, Fukagawa M, Teraoka S: The relative role of fibroblast growth factor 23 and parathyroid hormone in predicting future hypophosphatemia and hypercalcemia after living donor kidney transplantation: A 1-year prospective observational study. Nephrol Dial Transplant 26: 2691–2695, 2011 [DOI] [PubMed] [Google Scholar]
- 15.Trombetti A, Richert L, Hadaya K, Graf JD, Herrmann FR, Ferrari SL, Martin PY, Rizzoli R: Early post-transplantation hypophosphatemia is associated with elevated FGF-23 levels. Eur J Endocrinol 164: 839–847, 2011 [DOI] [PubMed] [Google Scholar]
- 16.Evenepoel P: Recovery versus persistence of disordered mineral metabolism in kidney transplant recipients. Semin Nephrol 33: 191–203, 2013 [DOI] [PubMed] [Google Scholar]
- 17.Pihlstrøm H, Dahle DO, Mjøen G, Pilz S, März W, Abedini S, Holme I, Fellström B, Jardine AG, Holdaas H: Increased risk of all-cause mortality and renal graft loss in stable renal transplant recipients with hyperparathyroidism. Transplantation 99: 351–359, 2015 [DOI] [PubMed] [Google Scholar]
- 18.Levey AS, Stevens LA, Schmid CH, Zhang YL, Castro AF 3rd , Feldman HI, Kusek JW, Eggers P, Van Lente F, Greene T, Coresh J; CKD-EPI (Chronic Kidney Disease Epidemiology Collaboration) : A new equation to estimate glomerular filtration rate. Ann Intern Med 150: 604–612, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.American Diabetes Association : Diagnosis and classification of diabetes mellitus. Diabetes Care 31[Suppl 1]: S55–S60, 2008 [DOI] [PubMed] [Google Scholar]
- 20.Racusen LC, Solez K, Colvin RB, Bonsib SM, Castro MC, Cavallo T, Croker BP, Demetris AJ, Drachenberg CB, Fogo AB, Furness P, Gaber LW, Gibson IW, Glotz D, Goldberg JC, Grande J, Halloran PF, Hansen HE, Hartley B, Hayry PJ, Hill CM, Hoffman EO, Hunsicker LG, Lindblad AS, Yamaguchi Y: The Banff 97 working classification of renal allograft pathology. Kidney Int 55: 713–723, 1999 [DOI] [PubMed] [Google Scholar]
- 21.Keyzer CA, Riphagen IJ, Joosten MM, Navis G, Muller Kobold AC, Kema IP, Bakker SJL, de Borst MH; NIGRAM consortium : Associations of 25(OH) and 1,25(OH)2 vitamin D with long-term outcomes in stable renal transplant recipients. J Clin Endocrinol Metab 100: 81–89, 2015 [DOI] [PubMed] [Google Scholar]
- 22.Wolf M, Weir MR, Kopyt N, Mannon RB, Von Visger J, Deng H, Yue S, Vincenti F: A prospective cohort study of mineral metabolism after kidney transplantation. Transplantation 100: 184–193, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huber L, Naik M, Budde K: Frequency and long-term outcomes of post-transplant hypophosphatemia after kidney transplantation. Transpl Int 26: e94–e96, 2013 [DOI] [PubMed] [Google Scholar]
- 24.Jeon HJ, Kim YC, Park S, Kim CT, Ha J, Han DJ, Oh J, Lim CS, Jung IM, Ahn C, Kim YS, Lee JP, Kim YH: Association of serum phosphorus concentration with mortality and graft failure among kidney transplant recipients. Clin J Am Soc Nephrol 12: 653–662, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Evenepoel P, Lerut E, Naesens M, Bammens B, Claes K, Kuypers D, Vermeersch P, Meijers B, Van Damme B, Vanrenterghem Y: Localization, etiology and impact of calcium phosphate deposits in renal allografts. Am J Transplant 9: 2470–2478, 2009 [DOI] [PubMed] [Google Scholar]
- 26.Cirillo M, Ciacci C, De Santo NG: Age, renal tubular phosphate reabsorption, and serum phosphate levels in adults. N Engl J Med 359: 864–866, 2008 [DOI] [PubMed] [Google Scholar]
- 27.de Matos AC, Câmara NO, de Oliveira AF, Franco MF, Moura LA, Nishida S, Pereira AB, Pacheco-Silva A: Functional and morphologic evaluation of kidney proximal tubuli and correlation with renal allograft prognosis. Transpl Int 23: 493–499, 2010 [DOI] [PubMed] [Google Scholar]
- 28.Baia LC, Humalda JK, Vervloet MG, Navis G, Bakker SJ, de Borst MH; NIGRAM Consortium : Fibroblast growth factor 23 and cardiovascular mortality after kidney transplantation. Clin J Am Soc Nephrol 8: 1968–1978, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wolf M, Molnar MZ, Amaral AP, Czira ME, Rudas A, Ujszaszi A, Kiss I, Rosivall L, Kosa J, Lakatos P, Kovesdy CP, Mucsi I: Elevated fibroblast growth factor 23 is a risk factor for kidney transplant loss and mortality. J Am Soc Nephrol 22: 956–966, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Collins AJ, Foley RN, Herzog C, Chavers B, Gilbertson D, Herzog C, Ishani A, Johansen K, Kasiske B, Kutner N, Liu J, St Peter W, Ding S, Guo H, Kats A, Lamb K, Li S, Li S, Roberts T, Skeans M, Snyder J, Solid C, Thompson B, Weinhandl E, Xiong H, Yusuf A, Zaun D, Arko C, Chen SC, Daniels F, Ebben J, Frazier E, Hanzlik C, Johnson R, Sheets D, Wang X, Forrest B, Constantini E, Everson S, Eggers P, Agodoa L: US renal data system 2012 annual data report. Am J Kidney Dis 61[Suppl 1]: e1–e476, 2013 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.