

Abstract
Increased glucocorticoid receptor (GR) expression and activity following androgen blockade can contribute to castration-resistant prostate cancer (CRPC) progression. Therefore, we hypothesized that GR antagonism will have therapeutic benefit in CRPC. However, the FDA-approved nonselective, steroidal GR antagonist, mifepristone, lacks GR specificity, reducing its therapeutic potential. Here we report that two novel non-steroidal and highly selective GR modulators (SGRMs), CORT118335 and CORT108297, have the ability to block GR activity in prostate cancer (PC) and slow CRPC progression. In contrast to mifepristone, these novel SGRMs did not affect AR signaling, but potently inhibited GR transcriptional activity. Importantly, SGRMs decreased GR-mediated tumor cell viability following AR blockade. In vivo, SGRMs significantly inhibited CRPC progression in high GR-expressing, but not in low GR-expressing xenograft models. Transcriptome analysis following AR blockade and GR activation revealed that these SGRMs block GR-mediated proliferative gene expression pathways. Furthermore, GR-regulated proliferation-associated genes AKAP12, FKBP5, SGK1, CEBPD, and ZBTB16 are inhibited by CORT108297 treatment in vivo. Together, these data suggest that GR-selective non-steroidal SGRMs potently inhibit GR activity and PC growth despite AR pathway inhibition demonstrating the therapeutic potential of SGRMs in GR-expressing CRPC.
Keywords: prostate cancer, selective glucocorticoid receptor modulators (SGRMs), glucocorticoid receptor, androgen receptor, castrate-resistant
Introduction
Prostate cancer (PC) is the most commonly diagnosed and the second leading cause of cancer death in men in the United States (1). Pioneering work by Charles Huggins and colleagues over 70 years ago demonstrated that PC growth is driven by androgens (2). Since this discovery, androgen ablation therapies have been the primary treatment for metastatic PC; however, therapies targeting AR signaling inevitably fail because PC adapts with a variety of mechanisms bypassing androgen deprivation, thereby becoming “castration-resistant” (CRPC) (3). More recently, the second-generation AR antagonist enzalutamide was approved for the treatment of metastatic CRPC. Enzalutamide prolongs CRPC patient survival but PC resistance to potent AR pathway blockade is inevitable (4,5). Mechanisms of tumor resistance to androgen deprivation and AR antagonist therapy include AR-mediated progression driven by AR amplification or somatic mutations that allow AR activation by non-androgen ligands (6,7), as well as expression of constitutively active AR splice variants (e.g. AR-V7) (8). In addition, AR-independent bypass mechanisms of CRPC progression also exist, including activated oncogenic pathways such as PI3K, cMYC (9,10), and increased glucocorticoid receptor alpha (GRα) expression and activity (11,12), hereafter referred to as GR. Research from our group (11) and others (12) examining the role of GR in CRPC reveals that GR activity can indeed promote PC progression following AR blockade, suggesting that GR antagonism is a therapeutic strategy for CRPC (13).
The role of GR activity in PC is complex as GR-mediated functions evolve depending on AR activity (13). In fact, GR activation is growth inhibitory in PC cell lines that retain AR signaling (14,15), whereas with compromised AR signaling, GR activation promotes PC cell survival and proliferation (16). This is demonstrated by mifepristone inhibition of PC growth and survival in AR-negative, GR-expressing PC cell lines (17–19). Additionally, high dose synthetic glucocorticoids (GCs) have demonstrated palliative clinical benefits in metastatic PC patients (20,21). Importantly, GR expression increases in a significant portion of primary tumor samples from patients following androgen deprivation therapy compared to untreated patients (22,23). This upregulation coupled with our understanding of the pro-tumorigenic role of GR suggests an adaptive, cell survival function for GR activity in cancers being treated with androgen deprivation therapy or AR signaling blockade. The pro-tumorigenic GR function in PC that follows functional blockade of AR activity may be due to GR and AR homology, and overlapping AR/GR cistromes and target gene expression (24,25). Upregulated GR expression and transcriptional activity in AR-inhibited PC suggests that pro-tumorigenic gene activation may be acquired by GR in the setting of AR inhibition and tumor evolution. In support of this, we and others have shown that GR activation diminishes the anti-tumor efficacy of AR blockade and that simultaneously antagonizing GR and AR decreases PC cell proliferation in vitro and tumor growth in vivo relative to AR inhibition alone (11,12). Therefore, the development of molecules that specifically antagonize GR activity in advanced PC may provide clinical benefit by blocking the GR bypass mechanism for tumor growth.
The hypothesis that concomitant AR and GR antagonism can increase the time to endocrine therapy resistance is currently being tested in a Phase I/II clinical trial (NCT 02012296) using mifepristone (the only FDA-approved GR antagonist) in combination with the AR antagonist enzalutamide. However, mifepristone also modulates progesterone receptor (PR) and AR signaling, and alters the metabolism of other therapeutics through its potent effects on cytochrome P450 enzymatic activity (26). In addition, although mifepristone acts as a weak wild-type AR antagonist, occasional mutant AR proteins that evolve in metastatic PC may be activated by mifepristone (27). As such, the development of highly-selective GR modulators (i.e. SGRMs without AR or PR cross-reactivity) is an important step toward implementing GR antagonism as a targeted therapy in CRPC. SGRMs CORT118335 and CORT108297 (Table 1) are two non-steroidal highly specific GR ligands that have been developed based on potent functional GR antagonism without significant binding to other members of the nuclear hormone receptor family (28,29).
Table 1.
Structures and specificities of small molecule GR modulators
| Corcept Therapeutics compound |
Chemical structure | Nuclear hormone receptor specificity |
GR binding and functional Ki(nM) |
|---|---|---|---|
| Mifepristone |
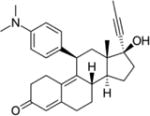
|
GR, PR, AR PR specificity is ~10x greater than for GR, and AR specificity is ~5x lower than for GR (26). | Binding: 0.4 Functional: 1.2 (26) |
| CORT118335 |
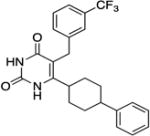
|
GR, MR GR specificity is ~8X greater than for MR (28). | Binding: 8.0 Functional: 24 (28) |
| CORT108297 |
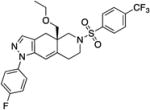
|
GR specific (26) | Binding:0.9 Functional: 6.8 (26) |
Nuclear hormone receptors specificities were previously determined by ligand binding assays (estrogen receptor-alpha, androgen receptor, and progesterone receptor) or reporter assays (mineralocorticoid receptor). Binding Ki was determined by [3H] dexamethasone displacement and antagonist functional Ki was determined by inhibition of dexamethasone-induced luciferase expression.
Here we demonstrate that unlike mifepristone, novel nonsteroidal SGRMs do not affect AR transcriptional activity. However, these SGRMs effectively reverse GR-specific gene expression and inhibit GR-mediated PC cell viability in the setting of enzalutamide. Furthermore, we report that SGRMs delay CRPC progression in the high-GR expressing CWR-22Rv1 xenograft model. PC cell transcriptome analysis following enzalutamide treatment reveals that subsequent SGRM treatment inhibits GR-mediated tumor cell proliferative and anti-apoptotic gene expression pathways, supporting their therapeutic potential in GR-expressing CRPC.
Material and Methods
Cell culture
LAPC4 and CWR-22Rv1 were a generous gift from Dr. John Isaacs (Johns Hopkins University) in 2009 and were regularly validated by DNA typing (most recently Arizona Research Labs 01/2017). Cell lines were routinely tested for mycoplasma contamination using the American Type Tissue Culture Universal Mycoplasma Detection Kit. LAPC4 cells were grown in IMDM (Hyclone) supplemented with 1% Penicillin/Streptomycin (Corning), L-Glutamine (Corning), 10% FBS (Atlanta Biologicals), and 1nM R1881 (Sigma-Aldrich). CWR-22Rv1 cells were grown in RPMI-1640 (Gibco) supplemented with 1% Penicillin/Streptomycin (Corning), L-Glutamine (Corning), 10% FBS (Atlanta Biologicals).
Cell treatments
Cells were plated in standard media described above and incubated overnight. Cells were washed with PBS (Hyclone) and placed into media containing charcoal stripped FBS (Atlanta Biologicals), 10% for LAPC4 or 1%/10% for CWR-22Rv1. Cells were treated for indicated times with media changes every other day with either vehicle control or specified treatment: 1nM R1881 (Sigma-Aldrich), 100nM dexamethasone (MP Biomedicals), 10µM enzalutamide (Selleck Chemicals or Medivation), 100nM mifepristone (Sigma-Aldrich), 1µM of Corcept Therapeutics compounds CORT118335, CORT108297. For all experiments, equimolar vehicle (ethanol +/−DMSO) was added to every sample for equal treatment periods.
Protein lysate preparation and immunoblotting
Cells were lysed in radio immunoprecipitation assay (RIPA) buffer and then sonicated. Xenograft tumor cell lysates were prepared similarly, with the additional step of motorized homogenization. Protein concentration was determined with the Pierce BCA Protein Assay Kit (Thermo Scientific) and lysates containing 100µg of protein were loaded onto gels, resolved, and transferred to nitrocellulose membranes (LI-COR). Membranes were blocked in 5% dry milk, incubated with indicated primary antibody overnight, and washed. Fluorescently labeled secondary antibodies (1:5,000, LI-COR) were incubated and washed. Detection was performed with the LI-COR Odyssey system. Antibodies and concentrations were as follows: anti-SGK1 (DB29, 1:100, Enzo Life Sciences) (30), anti-AR (N20, 1:2,000, Santa Cruz Biotechnology) or (441, 1:1,500, Santa Cruz Biotechnology), anti-GR (D8H2 XP, 1:1,000, Cell Signaling Technology), and anti-beta actin (AC-15, 1:10,000, Sigma-Aldrich).
Quantitative reverse transcription (qRT)-PCR
RNA was purified using the RNeasy Mini Kit (Qiagen). cDNA was synthesized with SuperScript III First-Strand Synthesis SuperMix (Invitrogen). mRNA transcript expression was quantified using PowerSYBR Green PCR Master Mix (Invitrogen) using primers for SGK1, KLK3, NR3C1(GR), GRβ, AKAP12, CEBPD, FKBP5, ZBTB16 and GAPDH (Supplemental Table 1). Melting curve analysis and efficiencies were determined for all primers with the requirement of a single peak for the melt curves and efficiencies between 0.90–1.0 (Supplemental Table 2). Expression was determined by the ΔΔCT method relative to controls. Experiments were performed in triplicate.
Trypan blue exclusion test for cell viability
Cells were plated and treated as above. At indicated days cells were washed, trypsinized, pelleted, and resuspended in media. Cells were then mixed 1:1 with trypan blue (Hyclone) and viable cells were counted in a blinded fashion. Three biological replicates were assayed per condition per time point and the mean of the biological replicates was reported.
IncuCyte proliferation assay
LAPC4 and CWR-22Rv1 cells were infected with IncuCyte Nuclight Green Lentivirus (Essen BioScience) and selected with puromycin. The resulting stable cell lines expressing nuclear GFP were plated in 96-well plates at 2.5×103 (CWR-22Rv1) or 5×103 (LAPC4) cells per well in 180µL of media supplemented with 10% CSS. Cells were incubated overnight in standard medium and the next day 20µL of 10x vehicle and/or drugs were added to cells at concentrations described above and plates were placed in the IncuCyte Zoom in vitro micro-imaging system (Essen Instruments). Four images (1.90×1.52 mm) in separate regions of each well were captured with a 10x objective at 4-hour intervals. Total numbers of cells (detected as GFP-positive objects) were calculated using IncuCyte Zoom software. Data was analyzed by one-way ANOVA to determine p-value.
Xenograft experiments
Animal studies were carried out in compliance with the U.S. Public Health Service Policy on Humane Care and Use of Laboratory Animals and approved by the University of Chicago Institutional and Animal Care and Use Committee. Male nude mice (8–10 weeks old, Envigo) were castrated and implanted with approximately 25mg testosterone (4-androsten-17β-OL-3-ONE, Steraloids) loaded into a 1cm “pellet” of silastic tubing to maintain consistent and equivalent levels of testosterone in all mice similar to the physiological concentration of testosterone in humans. Testosterone implants elevate and maintain testosterone levels to 530 ± 50 ng/dL (18.2 nM) which is similar to eugonadal adult human males (31). One week later, mice were then injected subcutaneously with 2.5×105 CWR-22Rv1 or 1×106 LAPC4 prostate cancer cells in 100µL (75% Matrigel (Corning) and 25% HBSS (Gibco)) subcutaneously in the flank. Testosterone pellets were removed (castration) when tumors reached a size of 250mm3. Mice began treatment one or two weeks following castration for CWR-22Rv1 or LAPC4, respectively. Mice received daily intraperitoneal injections of vehicle or 20mg/kg of indicated SGRM dissolved in ethanol and diluted 1:10 v/v in sesame oil (Sigma). Treatment was continued until an endpoint defined as tumor doubling relative to size at treatment initiation. Time to endpoint Kaplan-Meier curves were generated using GraphPad Prism and were compared using the Grehan-Breslow-Wilcoxon test to determine statistical significance. Mice were sacrificed at of castration, treatment initiation, one week of treatment, and the endpoint of tumor doubling. Tumors were harvested and frozen for protein analysis, stored in RNAlater (Qiagen) for RNA extraction, and formalin fixed for subsequent paraffin embedding.
PSA quantification
Cells (5×105) were seeded in 12-well plates and treated for three days as described above. Supernatants were harvested and submitted to the Clinical Chemistry Core (The University of Chicago) for analysis.
Immunohistochemistry
Slides were deparaffinized and processed through rehydration steps, then immersed in citrate pH 8 buffer (Diagnostic Biosystems) and heated in a steamer above 97°C for 3 minutes. The slides were cooled to room temperature for 20 minutes, rinsed in tap water for 5 minutes, transferred to TBS for 5 minutes, and loaded into a Leica Bond RX for automated staining. Briefly, slides were incubated with primary antibody for 1 hour, anti-AR (N20, 1:200, Santa Cruz Biotechnology) or anti-GR (D8H2 XP, 1:200, Cell Signaling Technology), incubated with Envision+anti-rabbit system (DAKO) for 30 minutes, followed by DAB+chromogen (DAKO) for 5 minutes. Tissue sections were immersed in Bond Polymer Refine Detection (Leica Microsystems) for 5 minutes for hematoxylin counterstaining. Slides were removed from the Leica Bond RX and mounted with cover glasses. Slides were digitized using the Aperio ScanScope XT and images were captured using Aperio ImageScope v12.3.2.5030 (Leica Biosystems).
Immunohistochemical scoring
Tissues were analyzed in a blinded fashion by a trained pathologist (RL). GR and AR-stained slides were scored by assessing both the percentage of positively stained cells and the intensity of the staining. GR slides were assessed as 0–2%, 2–10%, 10–25%, 25–50%, 50–75%, or 75–100% positivity. AR stained slides were assessed in a similar manner (0–25%, 25–50%, 50–75%, 75–100%). The results were quantified by taking the mean of the percentage positivity score (i.e. a slide in the 25–50% positive category received a mean positive score of 37.5) and then multiplied by the predominant intensity.
RNA-sequencing and analysis
Total RNA was isolated using the ‘RNeasy Mini’ kit (Qiagen) following the manufacturer’s instructions. Quality of extracted RNA was evaluated using a 2200 TapeStation system (Agilent). Samples with a RIN score ≥ 7 were selected for making libraries. RNA sequencing (RNA-seq) libraries were built using the ‘Stranded mRNA-Seq’ kit for Illumina platforms (KAPA) with OligodT magnetic beads to enrich the samples for informative mRNA species. Libraries were run on a 2200 TapeStation system (Agilent) to confirm fragment size. Libraries were then quantified by qPCR using the ‘Library Quantification’ kit (KAPA). Sequencing was done on a HiSeq 2000 sequencing system (Illumina) in a 100bp, paired-end run. The quality of raw reads was accessed by FastQC (v0.11.4). Sequencing data uploaded into public database (GEO Accession #: GSE97204). All reads were mapped to the human genome assembly (NCBI build 19) using STAR (v2.5.1b). Alignment metrics were collected by Picard tools (v2.8.1) and RSeQC (v2.6.4). Transcripts were assembled from the aligned reads using Cufflinks and combined with known gene annotation. The expression level of transcripts was quantified using FPKM (Fragments Per Kilobase of transcript per Million mapped reads)-based and read count-based methods. Transcript expression was normalized across samples. Differentially expressed genes (DEGs) and isoforms were detected using Cuffquant-Cuffnorm-Cuffdiff suite (FPKM-based method) and featureCounts-DESeq2 or EdgeR (read count-based method). Transcripts were further filtered by fold change ≥ 1.5 Genes detected by at least more than one method were collected to create a high-confidence DEG lists. Biological insights from candidate gene lists were gained by performing gene set enrichment analysis (GESA) and Ingenuity Pathways Analysis (IPA) (Ingenuity® Systems) to identify functional categories or pathways that are significantly altered under condition. We completed a comparative analysis for the treatments noted and examined the diseases and functions activation z-scores calculated in IPA. We focused on cellular functions that were relevant to the studies at hand (cell proliferation, survival, apoptosis). Heat maps were generated from IPA activation Z-scores using Morpheus (https://software.broadinstitute.org/morpheus/).
Results
Prostate cancer GR expression and function vary dependent on AR signaling context
We first examined glucocorticoid receptor (GR) activity in PC cells with various levels of androgen sensitivity. LAPC4 cells depend on exogenous androgen for growth, and CWR-22Rv1 cells do not require androgen for growth but are androgen-responsive. The LAPC4 cell line was derived from a human tumor before androgen-deprivation therapy and expresses wild-type AR and minimal GR, although GR expression increases following AR blockade (11,32). CWR-22Rv1 cells are derived from CRPC and express both wild-type AR and AR with an H874Y mutation in the ligand binding domain (LBD) allowing activation by non-androgen hormones (6), and express constitutively active AR splice variants (e.g. AR-V7) (8). The CRPC CWR-22Rv1 cells have high basal GRα expression in comparison to LAPC4, which remains elevated with enzalutamide treatment (Fig. S1) (11). Of note, GRβ expression is low (similar to LNCaP negative control) in both cell lines, with or without AR modulation (Fig. S1). These two PC cell lines have phenotypes at both ends of the AR sensitivity spectrum, express variable GR, and appropriately represent human PC biology.
GR and AR steady-state protein levels were assessed in both cell lines following 3 days of treatment with AR agonist (R1881, 1nM) (33) +/− GR agonist (dexamethasone, 100nM). Consistent with previous reports, in LAPC4 cells, AR expression increased with R1881 treatment and remained elevated with the addition of dexamethasone (Fig. 1A); LAPC4 cells had very low GR expression with R1881 +/− dexamethasone treatment but GR expression increased following three days of androgen deprivation in the vehicle alone condition (Fig. 1A). In contrast, CWR-22Rv1 cells had consistently high steady-state levels of full length and splice variant AR +/− R1881 and +/− dexamethasone treatment; interestingly, independent of AR ligand presence they have high basal GR expression, which is not impacted by dexamethasone (Fig. 1A). AR and GR transcriptional activity was then assessed by target gene qRT-PCR. In LAPC4 cells, which express GR at very low levels, activating AR with R1881 induces SGK1 and KLK3 (2.7-fold and 8.9-fold relative to vehicle respectively, p<0.05), while there is no additional induction of these genes following GR activation by dexamethasone. In contrast, in high-GR-expressing CWR-22Rv1 cells SGK1 is not induced by R1881 alone, but is dramatically induced (~150-fold) by GR activation with dexamethasone, while KLK3 (PSA) is induced 6-fold by R1881 alone and even further (11.5-fold) with dexamethasone treatment (Fig. 1B; p<0.05 compared to R1881 alone). Consistent with RNA induction, SGK1 protein expression and secreted PSA were induced with similar patterns in both cell lines (Fig. 1C). We then examined how GR activation impacts AR agonized PC cell viability. As anticipated, R1881 increased viable LAPC4 cell numbers in comparison to vehicle treated cells by 3.3-fold, while adding dexamethasone had little effect in these cells. In contrast, CWR-22Rv1 total cell number was not significantly increased by exogenous androgen; however, GR activation with dexamethasone decreased cell growth by 48% (Fig. 1D, p<0.05). As described previously, GR activation in PC cells can be growth inhibitory when AR signaling is intact (11,13). These data indicate that GR activation, in the context of highly active AR, can be growth inhibitory. This supports the notion that GR function in PC is dependent on both its level of expression and the context of AR activity.
Figure 1. Variable GR expression and transcriptional activity in PC cells is dependent on AR function.

For A-C LAPC4 and CWR-22Rv1 cells were treated with vehicle (Veh), R1881 (R, 1nM) +/− dexamethasone (D, 100nM) for three days. A. Cell lysates were analyzed by Western blot for AR (full length (fl) and AR-V7 splice variant are indicated), GR, and β-actin. B. RNA was isolated and SGK1 and KLK3 mRNA levels were quantified by qRT-PCR. C. Supernatants were analyzed by ELISA to measure PSA (top panels) and cell lysates were analyzed by Western blot for SGK1, and β-actin (lower panels). D. Cells were treated as in A for 10 days and assessed for the total number of viable cells by trypan blue exclusion assay. Data is representative of 3 independent experiments. Error bars represent SEM. Statistical significance was determined by Student’s t-test, *p<0.05, **p<0.01.
Novel selective GR modulators (SGRMs) antagonize CRPC-associated GR activity concurrent with AR blockade
As a first step to studying the newly available SGRMs, we tested their potential for off-target activity with respect to AR function. LAPC4 cells expressing wild-type AR and no appreciable GR under normal growth conditions were examined. LAPC4 cells were treated for 3 days with the AR agonist (R1881, 1nM), with or without the AR antagonist enzalutamide (10µM), mifepristone (100nM), or SGRMs (1µM), and then cells were analyzed for AR-driven target gene expression and proliferation. The putative AR/GR target genes SGK1 and KLK3 were selected for their established roles in CRPC; SGK1 independently confers castration resistant progression (11,34) and KLK3/PSA is a commonly used serum biomarker associated with CRPC progression. As expected, analysis of mRNA expression of SGK1 and KLK3 demonstrated that AR agonist (R1881) increased expression of both genes (3.5-fold and 36-fold, respectively) and enzalutamide completely blocked this upregulation (p<0.01, Fig. 2A). The non-specific GR antagonist mifepristone, also inhibited AR-induced expression of SGK1 and KLK3 (99% and 37%, respectively, p<0.05) (Fig. 2A). In contrast, treatment with CORT118335 or CORT108297 had no impact on AR-induced gene expression either as single agents or in combination with R1881 (Fig. 2A, S2A). Analysis of protein products SGK1 and PSA yielded similar results (Fig. 2B). These data suggest much greater GR specificity with the new SGRMs compared to mifepristone.
Figure 2. Novel GR-selective modulators do not impact AR function.

A. LAPC4 cells were treated with vehicle (Veh) or R1881 (R, 1nM) +/− enzalutamide (E, 10µM), mifepristone (Mif, 100nM), CORT108297 (297, 1µM), or CORT118335 (335, 1µM) for three days. RNA was isolated and SGK1 and KLK3 mRNA levels were quantified by qRT-PCR. B. Cells were treated as in A, and cell lysates were analyzed by Western blot for AR, SGK1, and β-actin (left panel) and supernatant was analyzed by ELISA to measure PSA (right panel). C. Cells were treated as in A for ten days and assessed for the total number of viable cells by trypan blue exclusion assay. Data are representative of 3 independent experiments. Error bars represent SEM. Statistical significance was determined by Student’s t-test, *p<0.05, **p<0.01.
We next analyzed the effect of SGRM treatment on AR-driven proliferation. LAPC4 cells treated with R1881 demonstrated increased cell number compared to vehicle-treated control cells (4.1-fold) and enzalutamide fully antagonized this increase (Fig. 2C). Likewise, mifepristone blocked the AR-induced increase in viable cell numbers. However, SGRM treatment did not impact AR-driven cell growth (Fig. 2C, S2B). One outcome of enzalutamide binding to AR is decreased total AR protein levels. Interestingly, mifepristone similarly decreased AR protein level, while treatment with SGRMs had no effect on steady-state AR protein levels (Fig. 2B). Overall, these data indicate that SGRMs, in contrast to mifepristone, do not associate with or alter the function of AR and are therefore appropriate for testing the specific role of GR antagonism in prostate cancer.
To assess how SGRM treatment affects GR activity in combination with AR inhibition, LAPC4 and CWR-22Rv1 cells were treated with combinations of R1881 and enzalutamide +/− dexamethasone, and +/− SGRMs. As anticipated, enzalutamide increased GR expression in LAPC4 cells (Fig. 3A, 1A) due to release of AR-mediated repression of the NR3C1/GR gene (35) (Fig. 3A). In contrast to AR, where agonist ligand binding stabilizes and increases AR protein levels, GR steady-state protein levels are down-regulated following agonist binding as a result of ubiquitin modification and proteasomal-degradation and/or decreased GR mRNA levels (11,36,37). Interestingly, SGRM treatment, especially CORT108297 mitigated the decreases in GR steady-state protein levels observed with glucocorticoid treatment, suggesting functional antagonism of dexamethasone binding to GR LBD (Fig. 3A). CWR-22Rv1 GR levels were not changed by AR antagonism (Fig. 3A), and as was observed in LAPC4 cells CORT108297 appeared to stabilize GR levels (Fig. 3A).
Figure 3. SGRMs block GR activity subsequent to AR blockade with enzalutamide.

For A-C LAPC4 and CWR-22Rv1 cells were treated with vehicle (Veh), or R1881 (R, 1nM) and enzalutamide (E, 10µM), +/− dexamethasone (D, 100nM), +/− CORT108297 (297, 1µM), or CORT118335 (335, 1µM) for three days. A. Cell lysates were utilized for Western blot analysis for AR (full length (fl) and AR-V7 splice variant are indicated), GR, SGK1, and β-actin. B. RNA was isolated and SGK1 and KLK3 mRNA levels were quantified by qRT-PCR. C. Supernatants were analyzed by ELISA to measure PSA. Data are representative of 3 independent experiments. Error bars represent SEM. Statistical significance was determined by Student’s t-test, *p<0.05, **p<0.01.
We next examined the effect of SGRMs on GR-induced gene expression. In LAPC4 cells, co-treatment with dexamethasone induced steady-state SGK1 expression 1.7-fold compared to R1881/enzalutamide (RE) treatment alone (Fig. 3B). Addition of CORT118335 (1µM) inhibited dexamethasone-induced SGK1 expression 50% while CORT108297 completely blocked the dexamethasone-mediated SGK1 increase (p<0.05) (Fig. 3B). KLK3 expression was increased 2.5-fold by dexamethasone compared to treatment with RE. Both CORT108297 and CORT118335 antagonized dexamethasone-induced KLK3 expression (by 48% and 60%, respectively, p<0.05) (Fig. 3B). Following 3 days of dexamethasone +/− SGRMs in CWR-22Rv1 cells, SGK1 gene expression was dramatically induced by ~100-fold compared to RE-treated cells and this induction was completely abrogated by both CORT108297 and CORT118335 (p<0.01, Fig. 3B). KLK3 was also induced (7.5-fold) by dexamethasone compared to RE in CWR-22Rv1 cells; CORT108297 and CORT118335 inhibited this induction by 70% and 75%, respectively (p<0.01, Fig. 3B). GR activation (dexamethasone alone) induced both genes compared to vehicle in both cell lines, which was antagonized by SGRMs (Fig. S3A). SGRM thus antagonized steady state GR target gene expression in both lines. To determine the impact of SGRMs on GR-mediated transcription kinetics, rather than steady state expression, cells were treated with RE for 3 days then pulsed with dexamethasone +/− SGRM for 2 or 6 hours. The SGRMs antagonized SGK1 at both time-points and KLK3 at 6 hours (Fig. S3B).
To validate day 3 steady-state gene expression results as resulting in corresponding changes in protein, we examined cell lysate SGK1 and secreted PSA from both cell lines treated as above with dexamethasone +/− SGRMs for 3 days. Similar to the RNA steady-state PCR results, SGK1 and KLK3 protein levels increased following dexamethasone treatment (compared to RE) and subsequently decreased following treatment with either CORT108297 or CORT118335 (Fig. 3C). Thus, in both cell lines these SGRMs demonstrate selective GR antagonism following GR activation.
SGRMs inhibit GR-mediated prostate cancer cell viability following AR blockade with enzalutamide
To further determine whether these SGRMs may be useful adjuncts to CRPC therapeutics, we next asked whether GR antagonism could decrease cell viability in AR-antagonized LAPC4 and CWR-22Rv1 cells. Cell lines were treated for 7–10 days with R1881, R1881/enzalutamide (RE), RE/dexamethasone (RED) or RED+ an individual SGRM. Cell numbers were assessed every 4 hours using automated live cell imaging (IncuCyte). As anticipated (11), GR activation with concomitant AR blockade (RE) increased cell number in both cells lines (although to a greater extent in the higher GR-expressing CWR-22Rv1s). It should be also noted that, despite the presence of AR splice-variants lacking the AR LBD, enzalutamide treatment still decreased cell proliferation, perhaps due to full-length and splice variant dimerization (11,38). In both cell lines, adding SGRM treatment to RED significantly decreased cancer cell proliferation (Fig. 4A). Although there was a decrease in viable cells with SGRM treatment in both cell lines, the impact of SGRM was more pronounced in the CWR-22Rv1 cell line. Using trypan blue exclusion assay to validate these data, there was a 41% and 42% decrease in cell viability by CORT118335 (p=0.04) and CORT108297 (p=0.05) respectively at 10 days compared to the RED condition, versus a 20% and 26% reduction for CORT118335 (p=0.003) and CORT108297 (p=0.002) treatment in LAPC4 cells (Fig. S4). As predicted, SGRM treatment decreased GR-mediated cancer cell proliferation when AR was blocked, most notably in the highly GR expressing cells.
Figure 4. SGRMs inhibit PC growth following AR blockade.

A. Cells were treated as in Figure 1 and 3 then assessed for the total number of viable cells by digital counting of nuclei every 4 hours over 7 or 10 days. Error bars represent SEM. Statistical significance was determined by one-way ANOVA, **p<0.01. B. Mice were castrated and implanted with testosterone pellets. One week later, mice were injected subcutaneously with LAPC4 or CWR-22Rv1 cells. Following the establishment of primary tumors, mice were castrated and following 7 or 14 days of recovery, and began treatments of vehicle (Veh), 20mg/kg CORT108297 (297), or CORT118335 (335). C. Kaplan-Meier survival curves are depicted showing time to CRPC development defined as doubling of tumor volume. *Denotes p<0.05 with Log-rank test. D. LAPC4 and CWR-22Rv1 tumors were dissected from mice prior to castration (UNTR), and at the endpoint (tumor doubling from size at castration) and formalin-fixed for immunohistochemistry. Serial sections of tumor tissue were labeled with antibodies for GR and AR. Representative images are shown.
SGRMs inhibit castrate-resistant tumor growth
Following the conclusion that SGRMs antagonize GR-mediated target gene expression and concomitantly decrease cell proliferation in the context of AR antagonism, we next asked whether SGRMs could slow CRPC progression in vivo. Male mice were first castrated and then implanted with testosterone pellets to maintain equivalent levels of testosterone in all mice which are in the same range as physiological testosterone concentrations in human male subjects (31). Mice were then injected subcutaneously with CWR-22Rv1 or LAPC4 cells and when tumors reached 250mm3, testosterone pellets were removed to mimic castration in PC patients. After one week (CWR-22Rv1) or two weeks (LAPC4) following castration/pellet removal, mice began daily intraperitoneal injections of vehicle, 20 mg/kg CORT108297 or CORT118335. Tumor size was followed until mice developed CRPC, which was defined as tumor size doubling from SGRM treatment initiation post castration (Fig. 4B). Of note, there was no evidence of drug-related toxicity (e.g. weight loss or lethargy) in mice receiving daily SGRM treatment compared to vehicle (Fig. S5). In mice with LAPC4 xenografts, neither CORT118335 nor CORT108297 significantly prolonged time to CRPC. In mice with CWR-22Rv1 tumors, both CORT118335 (hazard ratio, HR=0.45, p<0.05) and CORT108297 (HR=0.38, p<0.05) treatment resulted in a near doubling of the median time to CRPC compared to vehicle-treated mice (17.5, 21, 11.5 days for CORT118335, CORT108297, and vehicle, respectively; Fig. 4C, Fig. S6).
Given the effectiveness of SGRM treatment in delaying CRPC growth in CWR-22Rv1 compared to LAPC4 tumors, we hypothesized that the SGRMs were less effective in LAPC4 tumor progression because they have less GR expression. To evaluate GR and AR protein expression in LAPC4 and CWR-22Rv1 xenografts, a subset of mice were sacrificed at the time of castration, after 7 days of SGRM treatment, and at CRPC progression (endpoint) and analyzed by IHC and Western blot (Fig. 4D, Fig. S7). Of note, the xenografts were comprised of 95–100% tumor cells. Prior to castration, LAPC4 xenografts had very few GR expressing cells and near ubiquitous tumor cell AR expression (Fig. 4D, Fig. S7), consistent with cultured cells evaluated by Western blot analysis (Fig. 1). GR levels increased slightly and involved focal pockets of high GR-expressing cells representing ~10–20% of the tumor cells in post-castration mice. Notably, AR dramatically decreased within LAPC4 xenografts following castration in both vehicle- and SGRM-treated mice (Fig. 4D, Fig. S7). This is similar to the effect of enzalutamide on these cells in vitro. Subsequent SGRM treatment did not increase AR or GR expression in LAPC4 xenografts (Fig. 4D, Fig. S7). In contrast to LAPC4, CWR-22Rv1 tumors highly express GR and AR prior to castration, and both remained high in castrated mice. SGRM treatment did not change AR or GR levels in CWR-22Rv1 xenografts (Fig. 4D, Fig. S7). As described previously (11), prior to castration, GR expression within CWR-22Rv1 xenografts appeared predominantly cytoplasmic, whereas it was mainly nuclear post castration. This suggests that although GR protein levels do not change with inhibition of AR in CWR-22Rv1 xenografts, GR nuclear localization and therefore function may increase. In conclusion, both SGRMs delayed time to CRPC in the high GR-expressing CWR-22Rv1 xenografts but not in the low GR-expressing LAPC4 xenografts, suggesting that SGRM efficacy may be dependent on high endogenous levels of GR expression within CRPC tumors.
SGRMs inhibit GR-mediated pro-proliferative gene expression pathways following AR blockade
The ability of SGRMs to antagonize dexamethasone-mediated GR target gene expression, including the pro-survival gene SGK1 (39), and counteract GR-driven cell survival following AR inhibition both in vitro and in vivo, suggests that GR antagonism with SGRMs concurrently with enzalutamide may promote apoptosis. Alternatively, the GR-mediated impact on cell number may be due to changes in cell proliferation. To better understand the mechanisms underlying SGRM inhibition of CRPC growth, we examined SGRM-mediated global gene expression. Cells were treated with R1881 and enzalutamide +/− dexamethasone +/− CORT118335 or CORT108297. Whole-transcriptome sequencing (RNA-seq) followed by Ingenuity Pathway Analysis (IPA) was performed to assess cell growth pathways regulated by SGRM treatment.
To validate the RNA-seq data, we followed KLK3 and SGK1 mRNA levels throughout the treatment groups for both LAPC4 and CWR-22Rv1 cells. Gene expression changes were similar in pattern to qRT-PCR data shown in Figures 1B and 2A, i.e. SGRMs reversed GR-mediated induction of both genes (Fig. S8A). Global analysis of mRNA expression in LAPC4 cells during concurrent AR-blockade revealed that dexamethasone upregulated genes associated with several proliferation-associated pathways and downregulated genes involved in apoptotic pathways (p<0.05). The addition of SGRMs reversed these dexamethasone-mediated pathway activations (Fig. 5, p<0.05). In CWR-22Rv1 cells, dexamethasone also led to activation of proliferation pathways, which were to a large extent reversed with SGRMs. The highly relevant Proliferation of prostate cancer cell lines and Cell proliferation of tumor cell lines IPA pathway activation scores were reduced following SGRM treatment in both cell lines. Paradoxically, SGRMs treatment did not induce apoptosis pathways in cells.
Figure 5. SGRMs inhibit GR-activated PC cell proliferation pathways.

A. LAPC4 and CWR-22Rv1 cells were treated with R1881 (R, 1nM) and enzalutamide (E, 10µM) for 3 days, then dexamethasone (D, 100nM) +/− CORT108297 (297, 1µM), or CORT118335 (335, 1µM) for 6 hours. RNA-seq data was analyzed by Ingenuity Pathway Analysis (IPA). Proliferation/growth and apoptosis pathways are shown. Pathways that are significantly altered (p<0.05) are shown as activated (red) or repressed (blue). Mice treated as in Fig. 4 were euthanized following 7 days of treatment (Veh n=6; 297 n=9) B or at tumor doubling C (Veh n=6; bottom quartile n=4; top quartile n=4), the RNA was isolated from dissected tumors and analyzed by qRT-PCR. Error bars represent SEM. Statistical significance was determined by Student’s t-test, *p<0.05, **p<0.01. D. Schematic showing SGRMs competitively binds to GR to block GR-mediated PC cell proliferation and PC progression subsequent to AR-blockade.
To determine if gene expression changes revealed by IPA analysis reflects gene expression in vivo, xenografts were analyzed for expression of Proliferation pathway genes AKAP12, FKBP5, SGK1, CEBPD, ZBTB16 all of which were dexamethasone-induced and reversed by both SGRMs (Fig. S8B). Following 7 days of CORT108297 treatment in vivo, expression of these pro-proliferation genes was significantly reduced compared to vehicle-treated tumors (Fig. 5B). It was notable that within the CWR-22Rv1 in vivo experiments, there was heterogeneity of tumor progression kinetics of treated mice. To test the hypothesis that slower tumor growth was associated with increased SGRM-mediated inhibition of proliferation genes, we examined mRNA expression from the top and bottom quartiles of growth rate for tumors from SGRM-treated endpoint xenografts. As hypothesized, SGRM-treated slow growing tumors had lower expression of the majority of the proliferation genes tested compared to fast growing tumors (Fig. 5C).
In summary, these data suggest that SGRM treatment in the setting of AR inhibition diminishes GR-mediated PC proliferation and associated gene expression suggesting that high GR-expressing CRPC patients may benefit from SGRM co-treatment with an AR antagonist (Fig. 5D).
Discussion
There are currently no FDA-approved therapies specific to enzalutamide-resistant CRPC. Therefore, defining and targeting resistance pathways is vital to reduce death from PC. A subset of CRPC patients develops tumors with high GR expression, correlating with a poor response to enzalutamide (12) and suggesting that GR could be a therapeutic target in this context. Importantly, GR activation can drive PC progression in the presence of androgen ablation, as demonstrated by pre-clinical models (11,12). The FDA-approved non-selective GR mixed antagonist mifepristone was shown previously to block GR-mediated PC growth (11,27). Although mifepristone is an effective GR antagonist, it can potentially activate AR [depending on AR ligand binding site mutations and dose (27)], thereby reducing its therapeutic utility in CRPC. Due to the potential effectiveness of GR antagonism for CRPC treatment and the non-specific activity of mifepristone, there is an immediate need for the development of highly specific GR modulators with principally antagonist activity.
We examined two structurally distinct and yet highly selective SGRMs with principally GR antagonistic activity for this pre-clinical study: CORT118335 and CORT108297. For in vitro experiments concentrations were based on human PK studies with mifepristone (40,41), and on previous studies utilizing CORT118335 (42). For in vivo experiments, we tested the SGRMs at concentrations based on mifepristone use in previous mouse studies (11,40,43). At these doses, the SGRMs block dexamethasone-induced, GR-mediated mRNA and protein expression and inhibit viability in PC cells that are concurrently treated with enzalutamide. Interestingly, the CWR-22Rv1 castrate-resistant PC cells, which have relatively high GR expression, showed increased responsiveness to SGRMs compared to lower GR expressing LAPC4 cells. These data suggest that PC cells with high GR expression may be particularly sensitive to SGRM treatment during AR pathway inhibition.
The potential mechanisms underlying GR-mediated tumor progression in the context of AR inhibition were examined using global RNA sequencing. IPA analysis of gene expression following AR blockade with enzalutamide and concomitant GR activation in PC cells demonstrates that SGRMs predominantly inhibit the expression of genes associated with proliferation pathways. The expression of key genes within these pathways was examined within xenografts, and importantly, inhibition of expression correlated with SGRM efficacy. Future efforts will focus on these and potentially other downstream mediators of cell proliferation inhibited by SGRMs. Because GR and AR can associate with highly homologous DNA elements, work is needed to understand how chromatin is remodeled in the context of SGRM treatment. This will provide further insights into SGRM antagonism of GR bypass mechanisms in AR-blocked cancers.
The strengths of this investigation were its inclusion of human PC cell lines with varied AR sensitivity, robust in vitro and in vivo interrogation of SGRM function and global assessment of SGRM effect on mRNA expression. In addition, the demonstration that two novel selective GR modulators of distinct non-steroidal structures both potently antagonize GR activation to inhibit PC growth supports the hypothesis that SGRM antagonism of GR gene expression pathways that bypass AR blockade in CRPC should be explored further. One limitation of this study is the lack of enzalutamide treatment in vivo. Enzalutamide is an inducer of CYP3A4, a P450 isozyme that commonly metabolizes drugs and thus could potentially impact the pharmacokinetics (PK) of co-administered therapies such as SGRMs (44). The SGRMs could also affect enzalutamide PK. Therefore, for this study, the use of castration allowed for in vivo testing of the SGRMs without concern over relative drug concentrations, while maintaining the ability to test the hypothesis that SGRMs could potentiate the efficacy of AR pathway modulation. Furthermore, murine castration models cause near-complete AR ligand depletion, analogous to the FDA-approved abiraterone acetate treatment (31). The models used human PC cells, which necessitated an immunocompromised host for in vivo work. Thus, the effect of these SGRMs on metastatic disease within an immune-competent microenvironment is not known. Because GR activation by synthetic glucocorticoids can cause lymphocyte apoptosis and depletion, it is conceivable that SGRMs could increase lymphocyte activity in the tumor microenvironment thereby facilitating tumor regression in the immunocompetent host (45).
CWR-22Rv1 cells undergo decreased cell proliferation with single-agent dexamethasone activation of GR, yet are sensitive to GR blockade with SGRMs when AR is also blocked with either enzalutamide or castration, supporting an AR context dependency for GR activity as is noted in breast cancer (13). This anti-tumorigenic effect of GR blockade occurs despite expression of constitutively active AR splice variants, which are associated with diminished efficacy of AR targeted therapies (46,47). The finding that GR antagonism is effective despite the presence of these AR variants is provocative and suggests that GR antagonism may block pathways independent of AR or may directly or indirectly mitigate pro-tumorigenic AR splice variant function. Based on this finding, further studies of a potential interaction between AR splice variants (e.g. AR-V7) and GR are underway.
Although a previous single agent clinical trial of mifepristone intended as an AR antagonist for CRPC failed to show objective clinical responses, this failure was attributed to secondary increased androgen production (48). Thus, we hypothesize here that even highly specific GR antagonist treatment must be administered in conjunction with potent AR blockade to inhibit PC progression. A clinical trial of a mifepristone with enzalutamide is enrolling (NCT02012296) and a trial of novel SGRM administered with enzalutamide is under development; both include careful serial PK assessment to characterize any potential drug-drug interaction. The trials will examine patients’ GR expression in circulating tumor cells (CTCs) to test the hypothesis that relatively high CTC GR expression will predict the efficacy of GR antagonism. There is likely to be heterogeneity of GR expression and activity, therefore repeated assessment of PC CTC GR expression throughout the study will be valuable. Based on the proliferation IPA and the correlation of decreased proliferation gene expression with delayed in vivo progression, examination of a proliferation gene signature in biospecimens may be an additional pharmacodynamic biomarker associated with SGRM efficacy. Although the addition of SGRM treatment is predicted to delay CRPC progression compared to enzalutamide alone, our xenograft data suggest the eventual emergence of resistance to dual AR/GR blockade. Nevertheless, delineating the bypass mechanisms employed by GR in CRPC is a critical step in overcoming resistance to endocrine therapy.
Supplementary Material
Acknowledgments
The authors would like to thank Friends Against Cancer for their philanthropic support. We would like to thank David Hosfield and Geoffrey Greene for generous supply of the Nu-GFP labeled cell lines. The RNA sequencing was performed by the Functional Genomics Core Facility at the University of Chicago supported in part by the University of Chicago Medicine Comprehensive Cancer Center Cancer Center support grant (P30CA014599) under the direction of Pieter Faber. The Center for Research Informatics, under the direction of Jorge Andrade, is funded by the Biological Sciences Division at the University of Chicago with additional funding provided by the Institute for Translational Medicine, CTSA grant number UL1 TR000430 from the National Institutes of Health. Dr. Andrade also contributed to the RNA-sequencing bioinformatics pipeline utilized for this study. Authors CVO and LW are supported by Cancer Biology Training Grant T32 CA 009594.
Financial support: Movember-Prostate Cancer Foundation Challenge Award, Cancer Research Foundation, NIH SPORE 1P50CA180995
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66(1):7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 2.Huggins C, Stevens RJ, Hodges C. Studies on prostatic cancer. II. The effects of castration on advanced carcinoma of the prostate gland. Archives of Surgery. 1941;43:209–23. Archives of Surgery 1941;43:209-23. [Google Scholar]
- 3.Vogelzang N. Comprehensive textbook of genitourinary oncology. Philadelphia: Lippincott Williams & Wilkins; 2006. xxix, 907 p., 8 p. of plates p. [Google Scholar]
- 4.Scher HI, Fizazi K, Saad F, Taplin M-E, Sternberg CN, Miller K, et al. Effect of MDV3100, an androgen receptor signaling inhibitor (ARSI), on overall survival in patients with prostate cancer postdocetaxel: Results from the phase III AFFIRM study. ASCO Meeting Abstracts. 2012;30(5_suppl) LBA1. [Google Scholar]
- 5.Kregel S, Chen JL, Tom W, Krishnan V, Kach J, Brechka H, et al. Acquired resistance to the second-generation androgen receptor antagonist enzalutamide in castration-resistant prostate cancer. Oncotarget. 2016 doi: 10.18632/oncotarget.8456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tan J, Sharief Y, Hamil KG, Gregory CW, Zang DY, Sar M, et al. Dehydroepiandrosterone activates mutant androgen receptors expressed in the androgen-dependent human prostate cancer xenograft CWR22 and LNCaP cells. Mol Endocrinol. 1997;11(4):450–9. doi: 10.1210/mend.11.4.9906. [DOI] [PubMed] [Google Scholar]
- 7.Romanel A, Gasi Tandefelt D, Conteduca V, Jayaram A, Casiraghi N, Wetterskog D, et al. Plasma AR and abiraterone-resistant prostate cancer. Sci Transl Med. 2015;7(312):312re10. doi: 10.1126/scitranslmed.aac9511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hu R, Dunn TA, Wei S, Isharwal S, Veltri RW, Humphreys E, et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 2009;69(1):16–22. doi: 10.1158/0008-5472.CAN-08-2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carver BS, Chapinski C, Wongvipat J, Hieronymus H, Chen Y, Chandarlapaty S, et al. Reciprocal Feedback Regulation of PI3K and Androgen Receptor Signaling in PTEN-Deficient Prostate Cancer. Cancer Cell. 2011;19(5):575–86. doi: 10.1016/j.ccr.2011.04.008. doi S1535-6108(11)00155-3 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beltran H, Prandi D, Mosquera JM, Benelli M, Puca L, Cyrta J, et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat Med. 2016;22(3):298–305. doi: 10.1038/nm.4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Isikbay M, Otto K, Kregel S, Kach J, Cai Y, Vander Griend DJ, et al. Glucocorticoid receptor activity contributes to resistance to androgen-targeted therapy in prostate cancer. Horm Cancer. 2014;5(2):72–89. doi: 10.1007/s12672-014-0173-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arora Vivek K, Schenkein E, Murali R, Subudhi Sumit K, Wongvipat J, Balbas Minna D, et al. Glucocorticoid Receptor Confers Resistance to Antiandrogens by Bypassing Androgen Receptor Blockade. Cell. 2013;155(6):1309–1322. doi: 10.1016/j.cell.2013.11.012. doi http://dx.doi.org/10.1016/j.cell.2013.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kach J, Conzen SD, Szmulewitz RZ. Targeting the glucocorticoid receptor in breast and prostate cancers. Science translational medicine. 2015;7(305):305ps19. doi: 10.1126/scitranslmed.aac7531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nishimura K, Nonomura N, Satoh E, Harada Y, Nakayama M, Tokizane T, et al. Potential mechanism for the effects of dexamethasone on growth of androgen-independent prostate cancer. J Natl Cancer Inst. 2001;93(22):1739–46. doi: 10.1093/jnci/93.22.1739. [DOI] [PubMed] [Google Scholar]
- 15.Smith RG, Syms AJ, Nag A, Lerner S, Norris JS. Mechanism of the glucocorticoid regulation of growth of the androgen-sensitive prostate-derived R3327H-G8-A1 tumor cell line. J Biol Chem. 1985;260(23):12454–63. [PubMed] [Google Scholar]
- 16.Yan TZ, Jin FS, Xie LP, Li LC. Relationship between glucocorticoid receptor signal pathway and androgen-independent prostate cancer. Urol Int. 2008;81(2):228–33. doi: 10.1159/000144067. [DOI] [PubMed] [Google Scholar]
- 17.Tieszen CR, Goyeneche AA, Brandhagen BN, Ortbahn CT, Telleria CM. Antiprogestin mifepristone inhibits the growth of cancer cells of reproductive and non-reproductive origin regardless of progesterone receptor expression. BMC Cancer. 2011;11:207. doi: 10.1186/1471-2407-11-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lin MF, Kawachi MH, Stallcup MR, Grunberg SM, Lin FF. Growth inhibition of androgen-insensitive human prostate carcinoma cells by a 19-norsteroid derivative agent, mifepristone. Prostate. 1995;26(4):194–204. doi: 10.1002/pros.2990260405. [DOI] [PubMed] [Google Scholar]
- 19.Gabaglia CR, DeLaney A, Gee J, Halder R, Graham FL, Gauldie J, et al. Treatment combining RU486 and Ad5IL-12 vector attenuates the growth of experimentally formed prostate tumors and induces changes in the sentinel lymph nodes of mice. J Transl Med. 2010;8:98. doi: 10.1186/1479-5876-8-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tannock I, Gospodarowicz M, Meakin W, Panzarella T, Stewart L, Rider W. Treatment of metastatic prostatic cancer with low-dose prednisone: evaluation of pain and quality of life as pragmatic indices of response. J Clin Oncol. 1989;7(5):590–7. doi: 10.1200/JCO.1989.7.5.590. [DOI] [PubMed] [Google Scholar]
- 21.Fakih M, Johnson CS, Trump DL. Glucocorticoids and treatment of prostate cancer: a preclinical and clinical review. Urology. 2002;60(4):553–61. doi: 10.1016/s0090-4295(02)01741-7. doi S0090429502017417 [pii] [DOI] [PubMed] [Google Scholar]
- 22.Szmulewitz RZ, Clark R, Lotan T, Otto K, Taylor Veneris J, Macleod K, et al. MKK4 suppresses metastatic colonization by multiple highly metastatic prostate cancer cell lines through a transient impairment in cell cycle progression. Int J Cancer. 2012;130(3):509–20. doi: 10.1002/ijc.26005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yemelyanov A, Bhalla P, Yang X, Ugolkov A, Iwadate K, Karseladze A, et al. Differential targeting of androgen and glucocorticoid receptors induces ER stress and apoptosis in prostate cancer cells: a novel therapeutic modality. Cell Cycle. 2012;11(2):395–406. doi: 10.4161/cc.11.2.18945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bolton EC, So AY, Chaivorapol C, Haqq CM, Li H, Yamamoto KR. Cell- and gene-specific regulation of primary target genes by the androgen receptor. Genes Dev. 2007;21(16):2005–17. doi: 10.1101/gad.1564207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cleutjens CB, Steketee K, van Eekelen CC, van der Korput JA, Brinkmann AO, Trapman J. Both androgen receptor and glucocorticoid receptor are able to induce prostate-specific antigen expression, but differ in their growth-stimulating properties of LNCaP cells. Endocrinology. 1997;138(12):5293–300. doi: 10.1210/endo.138.12.5564. [DOI] [PubMed] [Google Scholar]
- 26.Clark RD. Glucocorticoid receptor antagonists. Curr Top Med Chem. 2008;8(9):813–38. doi: 10.2174/156802608784535011. [DOI] [PubMed] [Google Scholar]
- 27.Song LN, Coghlan M, Gelmann EP. Antiandrogen effects of mifepristone on coactivator and corepressor interactions with the androgen receptor. Mol Endocrinol. 2004;18(1):70–85. doi: 10.1210/me.2003-0189. [DOI] [PubMed] [Google Scholar]
- 28.Clark RD, Ray NC, Williams K, Blaney P, Ward S, Crackett PH, et al. 1H-Pyrazolo[3,4-g]hexahydro-isoquinolines as selective glucocorticoid receptor antagonists with high functional activity. Bioorg Med Chem Lett. 2008;18(4):1312–7. doi: 10.1016/j.bmcl.2008.01.027. [DOI] [PubMed] [Google Scholar]
- 29.Hunt HJ, Ray NC, Hynd G, Sutton J, Sajad M, O’Connor E, et al. Discovery of a novel non-steroidal GR antagonist with in vivo efficacy in the olanzapine-induced weight gain model in the rat. Bioorg Med Chem Lett. 2012;22(24):7376–80. doi: 10.1016/j.bmcl.2012.10.074. [DOI] [PubMed] [Google Scholar]
- 30.Wu W, Chaudhuri S, Brickley DR, Pang D, Karrison T, Conzen SD. Microarray analysis reveals glucocorticoid-regulated survival genes that are associated with inhibition of apoptosis in breast epithelial cells. Cancer Res. 2004;64(5):1757–64. doi: 10.1158/0008-5472.can-03-2546. [DOI] [PubMed] [Google Scholar]
- 31.Michiel Sedelaar JP, Dalrymple SS, Isaacs JT. Of mice and men--warning: intact versus castrated adult male mice as xenograft hosts are equivalent to hypogonadal versus abiraterone treated aging human males, respectively. Prostate. 2013;73(12):1316–25. doi: 10.1002/pros.22677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Klein KA, Reiter RE, Redula J, Moradi H, Zhu XL, Brothman AR, et al. Progression of metastatic human prostate cancer to androgen independence in immunodeficient SCID mice. Nat Med. 1997;3(4):402–8. doi: 10.1038/nm0497-402. [DOI] [PubMed] [Google Scholar]
- 33.Schilling K, Liao S. The use of radioactive 7 alpha, 17 alpha-dimethyl-19-nortestosterone (mibolerone) in the assay of androgen receptors. Prostate. 1984;5(6):581–8. doi: 10.1002/pros.2990050603. [DOI] [PubMed] [Google Scholar]
- 34.Sherk AB, Frigo DE, Schnackenberg CG, Bray JD, Laping NJ, Trizna W, et al. Development of a small-molecule serum- and glucocorticoid-regulated kinase-1 antagonist and its evaluation as a prostate cancer therapeutic. Cancer Res. 2008;68(18):7475–83. doi: 10.1158/0008-5472.CAN-08-1047. doi 68/18/7475 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xie N, Cheng H, Lin D, Liu L, Yang O, Jia L, et al. The expression of glucocorticoid receptor is negatively regulated by active androgen receptor signaling in prostate tumors. Int J Cancer. 2015;136(4):E27–38. doi: 10.1002/ijc.29147. [DOI] [PubMed] [Google Scholar]
- 36.Wallace AD, Cao Y, Chandramouleeswaran S, Cidlowski JA. Lysine 419 targets human glucocorticoid receptor for proteasomal degradation. Steroids. 2010;75(12):1016–23. doi: 10.1016/j.steroids.2010.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vedeckis WV, Ali M, Allen HR. Regulation of glucocorticoid receptor protein and mRNA levels. Cancer Res. 1989;49(8 Suppl):2295s–302s. [PubMed] [Google Scholar]
- 38.Watson PA, Chen YF, Balbas MD, Wongvipat J, Socci ND, Viale A, et al. Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proc Natl Acad Sci U S A. 2010;107(39):16759–65. doi: 10.1073/pnas.1012443107. doi 1012443107 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Szmulewitz RZ, Chung E, Al-Ahmadie H, Daniel S, Kocherginsky M, Razmaria A, et al. Serum/glucocorticoid-regulated kinase 1 expression in primary human prostate cancers. Prostate. 2012;72(2):157–64. doi: 10.1002/pros.21416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Skor MN, Wonder EL, Kocherginsky M, Goyal A, Hall BA, Cai Y, et al. Glucocorticoid receptor antagonism as a novel therapy for triple-negative breast cancer. Clin Cancer Res. 2013;19(22):6163–72. doi: 10.1158/1078-0432.CCR-12-3826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Heikinheimo O, Kekkonen R, Lahteenmaki P. The pharmacokinetics of mifepristone in humans reveal insights into differential mechanisms of antiprogestin action. Contraception. 2003;68(6):421–6. doi: 10.1016/s0010-7824(03)00077-5. [DOI] [PubMed] [Google Scholar]
- 42.Mammi C, Marzolla V, Armani A, Feraco A, Antelmi A, Maslak E, et al. A novel combined glucocorticoid-mineralocorticoid receptor selective modulator markedly prevents weight gain and fat mass expansion in mice fed a high-fat diet. Int J Obes (Lond) 2016;40(6):964–72. doi: 10.1038/ijo.2016.13. [DOI] [PubMed] [Google Scholar]
- 43.Masson MJ, Collins LA, Carpenter LD, Graf ML, Ryan PM, Bourdi M, et al. Pathologic role of stressed-induced glucocorticoids in drug-induced liver injury in mice. Biochem Biophys Res Commun. 2010;397(3):453–8. doi: 10.1016/j.bbrc.2010.05.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gibbons JA, de Vries M, Krauwinkel W, Ohtsu Y, Noukens J, van der Walt JS, et al. Pharmacokinetic Drug Interaction Studies with Enzalutamide. Clin Pharmacokinet. 2015;54(10):1057–69. doi: 10.1007/s40262-015-0283-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang D, Muller N, McPherson KG, Reichardt HM. Glucocorticoids engage different signal transduction pathways to induce apoptosis in thymocytes and mature T cells. J Immunol. 2006;176(3):1695–702. doi: 10.4049/jimmunol.176.3.1695. [DOI] [PubMed] [Google Scholar]
- 46.Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, Roeser JC, et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med. 2014;371(11):1028–38. doi: 10.1056/NEJMoa1315815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Scher HI, Lu D, Schreiber NA, Louw J, Graf RP, Vargas HA, et al. Association of AR-V7 on Circulating Tumor Cells as a Treatment-Specific Biomarker With Outcomes and Survival in Castration-Resistant Prostate Cancer. JAMA oncology. 2016;2(11):1441–9. doi: 10.1001/jamaoncol.2016.1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Taplin ME, Manola J, Oh WK, Kantoff PW, Bubley GJ, Smith M, et al. A phase II study of mifepristone (RU-486) in castration-resistant prostate cancer, with a correlative assessment of androgen-related hormones. BJU Int. 2008;101(9):1084–9. doi: 10.1111/j.1464-410X.2008.07509.x. doi BJU7509 [pii] [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.