

Abstract
Targeting KRAS and MYC has been a tremendous challenge in cancer drug development. Genetic studies in mouse models have validated the efficacy of silencing expression of both KRAS and MYC in mutant KRAS driven tumors. We investigated the therapeutic potential of a new oligonucleotide-mediated gene silencing technology (U1 Adaptor) targeting KRAS and MYC in pancreatic cancer. Nanoparticles in complex with anti-KRAS U1 Adaptors (U1-KRAS) showed remarkable inhibition of KRAS in different human pancreatic cancer cell lines in vitro and in vivo. As a nanoparticle-free approach is far easier to develop into a drug, we refined the formulation of U1 Adaptors by conjugating them to tumor targeting peptides (iRGD and cRGD). Peptides coupled to fluorescently tagged U1 Adaptors showed selective tumor localization in vivo. Efficacy experiments in pancreatic cancer xenograft models showed highly potent (>90%) anti-tumor activity of both iRGD and (cRGD)2-KRAS Adaptors. U1 Adaptors targeting MYC inhibited pancreatic cancer cell proliferation caused apoptosis in vitro (40–70%) and tumor regressions in vivo. Comparison of iRGD conjugated U1 KRAS and U1 MYC Adaptors in vivo revealed a significantly greater degree of cleaved caspase-3 staining and decreased Ki67 staining as compared with controls. There was no significant difference in efficacy between the U1 KRAS and U1 MYC Adaptor groups. Our results validate the value in targeting both KRAS and MYC in pancreatic cancer therapeutics and provide evidence that the U1 Adaptor technology can be successfully translated using a nanoparticle free delivery system to target two undruggable genes in cancer.
Keywords: U1 Adaptor, gene silencing, KRAS, MYC, pancreatic cancer
Introduction
One of the most significant unmet needs in cancer therapeutics is to effectively target both the KRAS and MYC oncogenes. Genetic studies in murine tumor models of both lung and pancreatic cancer have validated the therapeutic potential of silencing mutant KRAS and MYC (1–4). This unmet need is particularly conspicuous in pancreatic ductal adenocarcinoma (PDAC) where KRAS mutations have been found to be has high as 93% in cohorts of patient samples (5). MYC deregulation in PDAC occurs as a consequence of mutant KRAS and inhibition of it results in regression of both tumor and stromal compartments (3,4). PDAC, the fourth leading cause of cancer related death in the United States, is plagued by a lack of effective systemic therapy highlighting the need for novel therapeutics in this disease.
U1 Adaptor is a new generation gene silencing technology that represents a novel therapeutic platform for targeted suppression of any oncogene (6,7). The mechanism of action is based on targeted interference with pre-mRNA processing inside the cell nucleus. Pre-mRNAs of nearly all protein-coding genes require cleavage within the terminal exon and addition of a polyadenosine (polyA) tail to become mature, functional mRNA. PolyA tail addition is a critical step in mRNA maturation, and its failure results in rapid degradation of nascent message by nucleases. U1 Adaptors are named for the U1 small nuclear ribonucleoprotein (U1 snRNP) complex, which mediates their gene silencing action. U1 snRNP is best known as a premRNA splicing factor, but also normally acts to silence certain genes by suppressing polyadenylation. When U1 snRNP is able to bind stably within the terminal exon of a pre-mRNA, it directly inhibits polyA polymerase and so prevents addition of a polyA tail. U1 Adaptors are synthetic oligonucleotides that enable the U1 snRNP complex to stably bind within the terminal exon of any chosen pre-mRNA through extended base pairing, thereby interfering with polyA tail addition and causing the pre-mRNA to be degraded inside the nucleus (Fig. 1A).
Figure 1. Design, selection, and validation of U1 Adaptors targeting KRAS in human pancreatic cancer cells.

A, The U1 Adaptor has two sequence specific domains that tether the RNA component of the U1 snRNP complex to the target pre-mRNA, blocking the addition of the poly-A tail and selectively triggering pre-mRNA destruction. B, Eight U1 Adaptors targeting human KRAS were screened in MIA PaCa-2 cells, demonstrating a range of mRNA knockdown. The most effective KRAS Adaptors were KRAS1, 2 and 3, within the terminal exon. C, Validation of human KRAS1B Adaptor (LNA modified) in MIA PaCa-2 and PANC-1 cells after 72-hour transfection. A control adaptor with a scrambled target domain and siRNA to KRAS serve as negative/positive controls (P<0.0001). Western blots for KRAS (top) and actin (bottom) 72 hours following transfection of control and KRAS1B Adaptors.
A major factor complicating the translation of therapeutic gene silencing technologies is tumor delivery. Passage of oligonucleotides through cell membranes is hampered by charge repulsion, as both display net negative charges. Moreover, the densely fibrotic stroma of PDAC presents problems for drug delivery that impede effective chemotherapy. To address the problem of charge repulsion, we and others have formulated oligonucleotides for delivery in vivo as non-covalent complexes with positively charged polypropyleneimine (PPI) and polyamidoamine (PAMAM) dendrimer nanocarriers (8). To make delivery more selective for tumor cells, we and others have used tumor-targeting RGD peptides covalently linked to dendrimer nanocarriers. RGD peptides are synthetic oligopeptides that include an Arginine–Glycine-Aspartate (RGD) motif, which is bound by several cell-surface integrin receptors, notably the αvβ5 heterodimer which is highly expressed on tumor vasculature (9). One RGD peptide that is particulary well suited for delivery in PDAC is the “internalizing RGD” (iRGD), a modified RGD peptide that is bound by αvβ5 integrin, but undergoes cleavage at the cell surface, followed by secondary binding to neuropilin (NPR-1), a receptor that triggers permeabilization of tumor endothelium and internalization by cells (10,11). The limitations of using dendrimer based systems are: 1) toxicity at high doses, 2) the solubility of oligonucleotide:dendrimer complexes is significantly less than that of either component alone which limits effective dosing, and 3) commercial preparation of dendrimers lack a source for cGMP-compliant synthesis at scale. Here, we demonstrate for the first time that U1 Adaptor covalently linked to tumor targeting peptides (dendrimer free system) can be employed to therapeutically target both KRAS and MYC in pancreatic cancer.
Materials and Methods
Cell culture and transfection
Cell lines were maintained in 5%CO2/95% air at 37°C. PDAC cells were purchased from ATCC in February 2015. Experiments were completed with early passage number cell lines. Cell lines were tested and authenticated by ATCC. The cell lines MIA PaCa-2, PANC-1, and IMR-90 cells were maintained in DMEM with 10% fetal bovine serum (FBS). BxPC-3 cells were maintained in RPMI with 10% FBS. Capan-1 was maintained in IMDM with 10% FBS.
U1 Adaptor oligonucleotides were designed using a proprietary computational target site algorithm developed by Silagene to select target sequences within the terminal exon and 3′UTR. MIA PaCa-2 cells were screened using eight different KRAS U1 Adaptors. PAMAM G5 (150 nM) was used as a control. All transfections used 10 nM of adaptor or siRNA in complex with RNAiMax (FisherSci) or PAMAM. After 72 hours, cells were collected for expression analysis. The most effective sequences were modified using locked nucleic acid (LNA) 2′OMe chemistry. A U1 Adaptor designed with a scrambled target domain sequence and a wild type U1 domain served as a negative control.
Cell growth inhibition assay
Cells (5,000) were plated in 96-well plates in triplicate and transfected on Day 0 using RNAiMax with 10 nM of Control adaptor, U1 KRAS1B, U1 KRAS1B mutant, U1 MYC2, U1 MYC3, MYC siRNA (Santa Cruz), or KRAS siRNA SMARTpool (Dharmacon). On Day 3, cells were harvested for viability using the ViaCount reagent on the Guava (Millipore)or with CCK-SK (Dojindo) reagent according to manufacturer’s protocol. For unharvested cells, the old medium was replaced and transfected again. The experiment was completed after 3 treatments.
RNA extraction and RT-quantitative real-time PCR (RT-qPCR)
RNA was extracted using RNeasy kit (Qiagen) and gene expression level was measured by quantitative RT-qPCR using TaqMan gene expression assays (Life Technologies/Applied BioSystems). The gene expression level was normalized with β-actin and the average presented with standard deviation.
Western blot
Western blotting of cell and tumor lysates was performed as previously described (12). Antibodies used were as follows: human KRAS (Thermofisher Scientific (PA5-27234)), pERK1/2 (Cell Signaling, #4370), pAKT (Cell Signaling #9018), ERK1/2 (Cell Signaling), AKT (Cell Signaling #9272), MYC (R&D Systems, clone 9E10), and actin (Santa Cruz Biotechnology).
Apoptosis assay
Annexin V staining was completed according to the manufacture’s instructions using the Nexin reagent for Guava (EMD Millipore). Similar to the transfection experiments, the MIA Paca-2 cells were cultured in a 12-well plate, followed by treatment with U1 Adaptors at 10 nM using RNAiMax as a transfection reagent for 72 hours.
RGD peptide-U1 Adaptor Conjugate Preparation and Purification
A variation of CLICK conjugation chemistry was used to prepare peptide (cRGD)2 or iRGD-U1 Adaptor conjugates as follows: a solution containing the following was degassed with argon: 0.1 mM U1 Adaptor with a 5′-end Hexynyl group, 0.2 M Triethylamine acetate, 50% DMSO, 0.3 mM Azido-peptide and 0.5 mM Ascorbic acid. Conjugation was initiated by addition of 1/20th volume of 10 mM CuSO4-TBTA solution and the reaction proceeded overnight with gentle agitation at room temperature. The reaction was halted by addition of 1/50th volume of 0.5 M EDTA, pH 8.0 followed by desalting over a Zeba spin column that also removed unreacted Azido-peptide. In some cases a higher stoichiometric ratio of Azido-peptide was used.
Purity of samples was tested by HPLC of 15 μg Hex-KRAS2 unconjugated U1 Adaptor versus 15 μg of conjugated (cRGD)2-KRAS2 peptide. HPLC was done on a Hypersil ODS C18 column (Buffer A = 0.1 M TEAA pH 7.0; Buffer B= 100% acetonitrile). Conjugation was further tested for purity and efficiency by subjecting 700 ng of peptide-adaptor conjugate to 8% PAGE-UREA-TBE gel electrophoresis followed by methylene blue staining.
Animal studies
All animal protocols were approved by the Rutgers Biomedical and Health Sciences Animal Care and Use Committee. Five to six week old female athymic nude mice (NCR nu/nu) were purchased from Taconic Biosciences. KPC mice were obtained from the NCI mouse repository. For xenograft studies, 5 × 106 pancreatic cells were injected subcutaneously into both flanks of each nude mouse after the induction of anesthesia with isofluorane. Treatment began when xenograft tumors were either just palpable (10–20 mm3) or larger (40–50 mm3 and in one case 100 mm3). Mice were randomly divided and assigned to their respective treatment groups (9–10 mice/group). Tumor volumes (V) were calculated from caliper measurments by length (L) and width (W) by using the formula: V= 0.5xLxW2. Treatment was twice a week by tail vein injection until the tumors in the vehicle control group (buffer solution) reached ~1.5 cm3.
For the biodistribution studies of the Peptide-Adaptor conjugates in the KPC mouse model (Pdx-1cre/+; KRASG12D/+; p53R172H/+) (13), Cy3-labeled KRAS3 Adaptor was conjugated to either no peptide, iRGD or (cRGD)2. Mice were randomly selected into each treatment group (3 mice/group). When the pancreatic tumors reached 50 mm3 (by ultrasound), the mice were treated by a single tail vein injection (30 μg) and sacrificed after 24 hours when organs were harvested for imaging. The IVIS Spectrum imaging system (Perkin-Elmer) was used to detect Cy3-labeled adaptors, and the total flux (p/s) was calculated based upon a region of interest that remained a constant size per mouse.
For the pharmacodynamics experiment, PANC-1 xenografts were established as before. Mice were treated with vehicle (HEPES buffer), iRGD-Control, iRGD-KRAS3, or iRGD-MYC2 at 30 μg per dose, for a total of 3 treatments (1.5 weeks). Mice were sacrificed the day following the last injection and tumors processed for immunostaining.
Statistical Analysis
Mouse numbers were determined with assistance from the Biometrics Facility Core at the Cancer Institute of New Jersey. Statistical analyses were carried out using GraphPad Prism V software. Tumor volumes for each group were compared using student’s t-test (for comparisons of two groups) and analysis of variance (for multiple group comparisons, 1 way ANOVA). p <0.05 was considered significant.
Immunofluorescence
Immunoflourescence of tissue sections was performed as previously described (12). Antibodies to Ki67 (D3B5, Cell Signaling) and cleaved caspase 3 (D3E9, Cell Signaling) were diluted into 10% goat serum and incubated on the sections at 4°C overnight. Images were acquired using a fluorescence microscope with CCD camera (Keyence). Cells with positive staining were scored in at least 3 separate fields at 200X and reported as mean ± SD, n=3. Scale bars were 50 μm in length.
Results
Design and validation of U1-Adaptor targeting human KRAS
U1 Adaptor targeting KRAS was designed using a proprietary computational Target Site Algorithm developed by Silagene to select target sequences within the terminal exon and 3′UTR. Eight anti-KRAS U1 Adaptors (Supp. Table) were screened through transfection of an established human pancreatic cancer cell line (MIA PaCa-2 KRASG12C) and measurement of mRNA levels by RT-qPCR. The cells were treated once, and the mRNA levels measured after 72 hours. An anti-KRAS siRNA was used as a control. The eight U1 Adaptors demonstrated a range of mRNA knockdown from 0.24 (U1-KRAS3) to 1.17 (U1 KRAS8) compared to the untreated control, with three U1 Adaptor equal to or less than 0.35. The siKRAS was 0.23 compared to the untreated control (Fig. 1B). For further validation, a modified KRAS Adaptor (1B) containing five locked nucleic acid (LNA) modifications was evaluated in MIA PaCa-2 cells since LNAs are able to provide a “boost”, albeit unpredictable, in silencing activity (7). This was compared to siRNA, but also a control U1 Adaptor, which had a scrambled target domain (TD) (Supp. Table). There was a slight decrease in the mRNA using the control U1 Adaptor compared with the transfection control while that of the U1 KRAS1B Adaptor showed significant knockdown of both the mRNA and protein level (Fig. 1C, left). A 60% reduction in KRAS mRNA was observed with treatment in U1 KRAS1B in another established human pancreatic cancer cell line, Panc1 (KRASG12D mutation) compared to the controls. In both of these cell lines, the anti-KRAS siRNA showed a greater degree of KRAS mRNA knockdown (Fig. 1C, right).
U1 Adaptors targeting KRAS inhibit pancreatic cancer cell proliferation in vitro and in vivo
We then examined if these U1 Adaptors targeting KRAS could inhibit pancreatic cancer cell proliferation. We transfected U1 KRAS1B into MIA PaCa-2 (Fig. 2A left) and PANC-1 (Fig. 2A middle) cells every 72 hours over 9 days, and observed potent inhibition of cell proliferation in both cell lines when compared to an RNAiMAX transfection control and Control U1 Adaptor with a scrambled TD. There was no difference between the U1-KRAS1B and siKRAS groups. We further evaluated cell growth inhibition in another established human pancreatic cancer cell line containing another commonly found missense mutation in pancreatic cancer (Capan-1, KRASG12V) and observed, by day 14, a significant inhibition of cell growth as compared with the untreated and control U1 Adaptor with a scrambled TD that was very similar in magnitude to the siKRAS (Supp. Fig. 1A).
Figure 2. U1 Adaptors targeting KRAS are effective in vitro and in vivo.

A, Cell growth inhibition assay of human pancreatic cancer cell lines MIA PaCa-2, PANC-1, and BxPC-3, after transfection with U1 KRAS1B every 72 hours over 9 days compared to U1 Control adaptor and siKRAS. B, (Top) Schematic of the U1 KRAS1B mutant adaptor. Nucleotides underlined in the KRAS domain have been LNA-modified. Red lowercase nucleotides represent the mutated nucleotides of the U1 domain. (Bottom) Apoptosis assay using Annexin V staining following transfection of MIA PaCa-2 cells with 10 μM U1 Control, U1 KRAS1B, U1 KRAS1B mutant, or siKRAS. after 72 hours (P=0.0001), with RNAiMAX serving as an additional negative control. C, Western blots for KRAS, MYC, p-AKT, AKT, p-ERK, and ERK, 72 hours following transfection of MIA PaCa-2 cells as in B. Actin serves as a loading control. D, Schematic of the peptide (cRGD)-dendrimer (PPI) linked to the U1 KRAS3 adaptor (Top) used for in vivo delivery of the KRAS adaptor. (Bottom) MIA PaCa-2 xenograft mice (n=9) were treated with cRGD-PAMAM-KRAS3 complex (2 μg KRAS3 Adaptor, 7.5 μg cRGD-PAMAM complex per dose) by tail vein injection twice weekly starting when tumors reached 40–50mm3. 68% growth inhibition observed when compared with vehicle control (p=0.0002).
KRAS U1 Adaptors are not specific to mutant KRAS, which raises two questions: 1) are they effective in pancreatic cell lines with wild type KRAS and 2) would they be toxic to non-cancer cell proliferation? To answer the first question, we tested U1 KRAS1B in the established human pancreatic cancer cell line BxPC3 (KRAS wild type). We found that by day 10 there was a mild degree of growth inhibition as compared to the untreated control and this magnitude was similarly seen for the siKRAS indicating that this cell line is much less dependent on KRAS signaling for proliferation (Fig. 2A, right). To determine if the U1 Adaptors would inhibit the growth of a non-cancer cell line we evaluated the human lung fibroblast line (IMR-90) and found that by day 12 there was no significant difference in proliferation between U1 KRAS1B, siKRAS, and the U1 Control groups (p=0.894, 1 way ANOVA) (Supp. Fig. 1B). Knockdown of U1 KRAS1B and siKRAS KRAS was confirmed by Western blot (Supp. Fig. 1B).
It has been previously shown that a marker of KRAS dependency or “oncogene addiction” is the induction of apoptosis upon KRAS knockdown (14). We next performed an apoptotic assay using Annexin V staining. We found that by day 3, there was a nearly four-fold greater percentage of apoptotic cells for the U1 KRAS1B treated apoptotic cells than the untreated control and a control U1 Adaptor with a mutant TD. As an additional control we included a U1 Adaptor that contains the same KRAS targeting sequence as the U1 KRAS1B Adaptor, but the U1 domain sequence has a two nucleotide mutation (Fig. 2B, top). We found that this mutant adaptor (U1 KRAS1B mutant) failed to significantly induce apoptosis in the MIA PaCa-2 cells compared with the untreated control indicating the specificity of the U1 Adaptor mechanism for this effect (Fig. 2B, bottom).
We then examined the MIA PaCa-2 cells for evidence of inhibition of molecules downstream of KRAS signaling such as ERK, AKT and MYC in U1 Adaptor treated cells. We found a significant reduction in overall MYC, phosphorylated ERK and phosphorylated AKT as compared to the untreated control (Fig. 2C). This magnitude was greater than or equal to that of the siKRAS control, with a greater reduction in AKT activation compared to ERK. Note the lack of inhibition of these markers in either the U1 Control or the U1 KRAS1B mutant, indicating the specificity for KRAS signaling inhibition. For further validation, we evaluated PANC-1 for these biochemical markers of KRAS signaling inhibition and found similar effects as in the MIA PaCa-2 cells with an inhibition of MYC, phosphorylated AKT for the U1 KRAS1B and siKRAS treated cells that is lost in the KRAS-1B mutant (Supp. Fig. 1C).
To determine if the KRAS U1 Adaptors could inhibit pancreatic xenograft tumor growth, we evaluated the U1 KRAS3 adaptor using our previously validated formulation containing PAMAM dendrimers covalently linked to the cRGD peptide (7) (Fig. 2D, top). Twice-weekly intravenous injection of the cRGD-PAMAM-KRAS3 adaptor led to a significant growth inhibition of MIA PaCa-2 xenograft tumors (68% inhibition, KRAS3) (Fig. 2D, bottom). This provides evidence that KRAS U1 adaptors inhibit pancreatic tumor growth in vivo.
KRAS U1Adaptors Directly Conjugated to Tumor Targeting Peptides Inhibit Tumor Growth
In an effort to pursue a PAMAM dendrimer-free system of delivery, we directly conjugated the U1 Adaptor to a tumor targeting peptide. After in vitro and in vivo efficacy testing of several conjugation methods we settled on copper-based CLICK chemistry as it provided a higher conjugation efficiency, precluding the need for vast excess of peptide and HPLC purification (Fig. 3A, Sup. Fig 2). cRGD is amenable to monomeric and dimeric coupling whereas iRGD is amenable only to monomeric coupling due to internal cysteines not found in cRGD, (Fig. 3A).
Figure 3. U1 Adaptors directly conjugated to tumor targeting peptides inhibit tumor growth.

A, Schematic of copper CLICK peptide-U1 Adaptor conjugation (top). Purity of the reaction was tested by gel electrophoreses on an 8% TBE-Urea gel (bottom). B, (left) Biodistribution study of Cy3 labeled RGD-Adaptor conjugates in the KPC mouse model. 24 hours after treatment, mice (n=3) were sacrificed, and tumors and organs were harvested and imaged using IVIS Spectrum. Bioluminescence was measured in total flux (photons/second) (p=0.01) (right) Statistical significance was calculated by student’s t-test. C, In vivo efficacy study using the best formulation of (cRGD)2-KRAS3 U1 Adaptor (30 μg) in MIA PaCa-2 xenografts. Nude mice (n=10, Vehicle, n=9 (cRGD)2-KRAS3) were treated by tail vein injection twice weekly once tumors reached 20 mm3. There was greater than 90% growth inhibition observed in both treatment groups when compared with the vehicle control (p=0.0001) (left) Statistical significance was calculated by student’s t-test. Waterfall plot showing individual MIA PaCa-2 xenograft tumor response to treatment (right). Sizes were measured at time of treatment up to 29 days. Note that the (cRGD)2-KRAS3-treated (bottom) group had tumor stasis (7/9) or regression (2/9) in individual mice.
Given the iRGD mechanism of delivery not only binds to tumor endothelium, but also results in greater tissue penetration through a NP-1 mediated vascular permeablization (10), we reasoned that iRGD would be particularly suitable for delivery to PDAC of the KRAS U1 Adaptors considering that a hallmark of PDAC is a fibrous stroma that is a barrier to systemic agents (15,16). We evaluated both iRGD and cRGD peptides for PDAC delivery by conjugating each peptide to the 5′ end of a Cy3-labelled U1 Adaptor using the autochthonous KPC genetically engineered mouse model, which has been shown to reproduce key features of the stromal compartment of human PDAC (13). Twenty-four hours after injection, we sacrificed the mice and examined in vivo fluorescence of the brain, heart, lung, liver, kidney, spleen and tumor. We found the highest levels of fluorescence accumulated in the brain, kidney and tumor with very little fluorescence detected in the heart, lung, liver or spleen (Fig. 3B, left). Interestingly, the control (no peptide) Cy3-U1 Adaptor also highly localized to the brain and kidney indicating that the uptake of the iRGD and cRGD conjugated adaptors in the brain is not peptide related. Alternatively, accumulation of fluorescence in the tumor did demonstrate a significant increase of fluorescence in the peptide conjugated tumor tissue as compared with the non-peptide conjugated control, p=0.01 (Fig. 3B, right). We did not see a significant difference between the cRGD and iRGD peptide conjugated U1 Adaptor groups.
We then sought to compare the formulation of the iRGD-KRAS-3 Adaptor with the cRGD-PAMAM dendrimer for in vivo antitumor activity. Again we used the MIA PaCa-2 xenograft assay and administered a dose of 10 μg intravenously twice weekly. We found that both the PAMAM-cRGD:KRAS-3 complex and iRGD-KRAS-3 conjugate potently inhibited tumor growth essentially causing stasis of tumor growth (92 and 93% growth inhibition by day 39) (Supp. Fig. 3A). This provides evidence that KRAS U1 Adaptors directly conjugated to tumor targeting peptides demonstrate potent antitumor effects that are equivalent to the dendrimer-based system. We evaluated the iRGD-KRAS-3 adaptor to the (cRGD)2-KRAS-3 in the same Mia PaCa-2 xenograft assay and found that by Day 39 there was significantly greater tumor growth inhibition with iRGD-KRAS-3, p<0.05 (Supp. Fig. 3B).
Optimizing the KRAS U1-Adaptor-tumor targeting peptide formulation
To optimize the KRAS U1 adaptor formulation, we evaluated a number of variables all using the MIA PaCa-2 xenograft assay as the readout for activity. We first examined a KRAS adaptor with a longer targeting KRAS domain (24 nucleotides versus 20) (Supp. Fig. 3C) as we had observed longer targeting domains can in some cases enhance silencing activity (6). We found that there was no significant difference between the KRAS Adaptors using a targeting domain of 20 nucleotides versus 24 (63% versus 74% tumor growth inhibition respectively).
As stated earlier, PAMAM dendrimer:U1 Adaptor formulations have limited utility due to precipitation at higher doses (just 3-fold higher concentrations result in extensive precipitation). This problem is obviated with a targeting peptide conjugated system that is dendrimer-free, allowing us to escalate the dose of the peptide conjugated U1 Adaptor. We then performed a dose escalation experiment by testing three doses of the (cRGD)2-KRAS2 conjugate formulation at 10, 30, and 90 μg as well as the (cRGD)2-KRAS3 at 30 μg. We found that there was no significant difference between the 10 and 30 μg doses of the (cRGD)2-KRAS-2 conjugate (75% growth inhibition compared to Vehicle, p<0.0001, student’s t test), by day 29. Interestingly the 90 μg dose had a decreased antitumor effect with only 61% growth inhibition, but still significantly different than the Vehicle (p<0.002, student’s t test) (Supp. Fig. 3D). The (cRGD)2-KRAS3 formulation performed the best, producing 90% growth inhibition (p<0.0001, student’s t test). To validate this result, we used this formulation to perform an efficacy experiment using the MIA PaCa-2 xenograft assay but starting treatments when tumors were larger (100 mm3) and found these treated mice exhibited near stasis in tumor growth (Fig. 3C, left). In fact when we examined the effect on tumor growth in individual mice, we found regressions in 7/9 mice and complete responses in 2/9 mice (Fig. 3C, right).
U1 Adaptors targeting MYC in mutant KRAS driven pancreatic cells are effective both in vitro and in vivo
MYC is a key downstream effector of mutant KRAS. Silencing MYC in a KRAS-driven lung cancer mouse model has been shown to be highly therapeutic (3). Given that MYC remains an undruggable target, we designed U1 Adaptors to target MYC not only to determine feasibility but also to compare the antitumor effect to that of KRAS in the same pancreatic tumor model.
Screening a panel of anti-MYC U1 Adaptors in vitro identified three with superior silencing activity (Supp. Table). We validated these adaptors in the MIA PaCa-2 cells by Western blot and found potent MYC inhibition (Fig. 4A). We performed a cell growth inhibition assay and found that all three MYC U1 adaptors potently inhibited growth (Fig. 4B). We also examined the percent of apoptotic cells induced by U1-MYC adaptors and found that all three adaptors significantly induced apoptosis (40–70%) in MIA PaCa-2 pancreatic cancer cells in comparison to the untreated and U1 Control adaptor groups (Figure 4C).
Figure 4. U1 Adaptors targeting MYC in mutant KRAS driven pancreatic cells are effective both in vitro and in vivo.
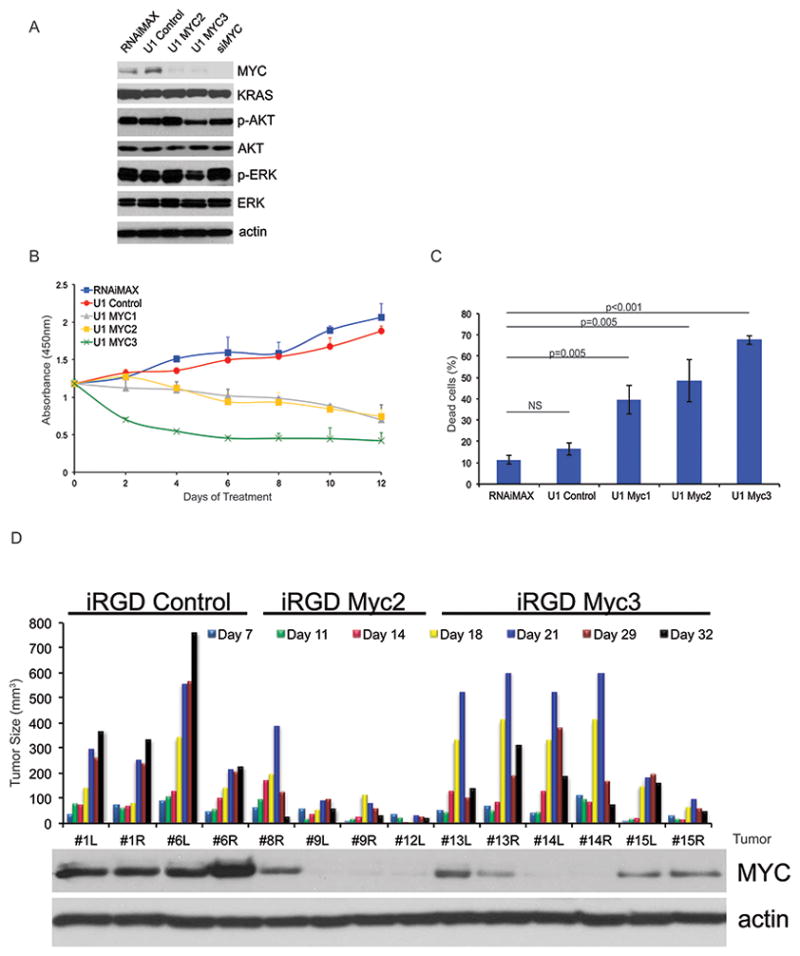
A, Western blotting of MIA PaCa-2 cells transfected with 10 μM U1 Control, U1 MYC1, U1 MYC2, U1 MYC3, and siMYC after 72 hours of treatment. B, Cell proliferation assay of MIA PaCa-2 cells treated as in A, except for every 72 hours over 9 days. C, Annexin V assay of MIA PaCa-2 cells treated as in A, except cells were harvested on Day 6 following 2 transfections 72 hours apart. Statistical significance was calculated by student’s t-test. D, Nude mice harboring MIA PaCa-2 xenograft tumors were treated 2 times a week for a total of 32 days with 30 μg iRGD-Control (n=4), iRGD-MYC2 (n=4), and iRGD-MYC3 adaptors (n=6). Waterfall plot of tumor size with the correlate protein expression was measured for treated tumors. Western blotting using antibodies for actin and MYC were completed on tumor cell lysates from iRGD-Control, iRGD-MYC2, and iRGD-MYC3 tumors at the end of treatment. Representative images are shown. NS, not significant.
To evaluate the MYC U1 Adaptor in vivo, we synthesized iRGD-MYC2 and MYC3 adaptor conjugates using the same chemistry used in the iRGD-KRAS. We tested these formulations using a 10 μg dose administered in the MIA PaCa-2 xenograft assay. We observed significant antitumor activity in both the iRGD-MYC2 and iRGD-MYC3 U1 Adaptor formulations with regressions in 4/4 iRGD-MYC2 mice and 5/6 iRGD-MYC3 mice (Fig. 4D). Interestingly in both of these treatment groups regressions were typically seen after day 18. Western blot for MYC in the tumor tissue of the mice in these three treatment groups indicated inhibition of MYC with levels corresponding to the degree of tumor growth inhibition (Fig. 4D).
Targeting KRAS versus MYC in Pancreatic Xenograft Tumors
Genetic experiments in mouse models of pancreatic and lung cancer have shown that inhibition of either KRAS or MYC signaling in mutant KRAS driven tumors is therapeutic. We sought to validate this using U1 adaptors targeting either KRAS or MYC in the same tumor model. We first performed a pharmacodynamic experiment in which mice bearing PANC-1 xenografts were treated with either the iRGD-KRAS or iRGD-MYC adaptor along with the iRGD-Control adaptor. Western blot data in Figure 5A confirmed inhibition of protein expression for both KRAS (left) and MYC (right). We then compared both cell proliferation and apoptosis in the tumors using Ki67 and cleaved caspase-3 (CC3) immunofluorescence, respectively. We observed a significantly greater amount of CC3 staining in the U1-KRAS and U1-MYC adaptor-treated groups as compared to the buffer-treated and iRGD-Control groups (Fig. 5B, p<0.007). Interestingly the U1 MYC group displayed a greater number of CC3 stained cells as compared with the U1 KRAS group (Fig. 5B, right) that was significant (p=0.048, student’s t test). For Ki67 staining, we found that the two U1Adaptor treated groups had significantly decreased staining as compared to the controls (Fig. 5C, p<0.001). We also evaluated the effects of both KRAS and MYC suppression on stromal collagen by Masson’s trichrome staining at day 9 and 29 after treatment and observed a reduction in collagen in the MYC U1 Adaptor treated tumors at day 29, which was not seen in the KRAS or control treated tumors (Supp. Fig. 4A). There was no difference in either KRAS or MYC treated tumors at day 9. We then compared the U1 KRAS and U1 MYC groups for efficacy in the PANC-1 xenograft tumor assay. We found that both the iRGD-KRAS and MYC formulations caused tumor regressions that were not significantly different, although they were when compared to controls (p<0.001). Note the iRGD-Control U1 Adaptor group did not have any antitumor activity compared to the untreated control (Fig. 5D). This demonstrates that in a head-to-head comparison of the KRAS and MYC adaptors, both were equally efficacious.
Figure 5. Targeting KRAS versus MYC in Pancreatic Xenograft Tumors.

A, Nude mice harboring PANC-1 xenograft tumors were treated 3 times with 30 μg iRGD-Control (n=2), iRGD-KRAS3 (n=4), or iRGD-MYC2 (n=4) adaptors. Tumor protein lysates were probed for KRAS (left) and MYC (right) expression. Actin served as a loading control. B, Cleaved caspase 3 and C, Ki67 immunofluorescence staining of tumors treated as in A. Three fields at 200X were quantitated for total cell number (DAPI, blue) and FITC-positive cells (B, cleaved caspase 3, green; C, Ki67, green). Statistical significance was calculated by student’s t-test. D, Nude mice harboring PANC-1 xenograft tumors were treated 5 times over 17 days with HEPES buffer or 30 μg iRGD-Control (n=7), iRGD-KRAS3 (n=7), or iRGD-MYC2 (n=7) adaptors. Tumor size was measured and plotted at each treatment day. Statistical significance was calculated by student’s t-test. Scale bar=50 μm
Discussion
Pancreatic cancer is plagued by a lack of effective systemic chemotherapy. One reason for this is the desmoplastic stromal compartment of the tumor, which serves as a barrier to the delivery of therapeutic agents (15,16). Recently, this aspect of its biology has been linked to signaling from the KRAS-MYC oncogenic axis as withdrawal of either oncogenic KRAS or MYC leads to a general dissolution of the stromal compartment (1–3). This further substantiates the significance of finding novel therapeutics to target these genes in PDAC. Our results with Masson’s trichrome staining revealed conflicting results between KRAS and MYC Adaptor treated tumors with a decrease seen in the MYC group and no change in the KRAS group. We are hesitant to draw any conclusions from this because nude mouse xenograft models do not effectively recapitulate the tumor microenvironment (TME), which is one their limitations. It is possible that the efficacy of the U1 Adaptor is overestimated in these models due to the lack of a stromal barrier. The mouse autochthonous model (KPC) is a better model to evaluate efficacy because it does effectively recapitulate the human TME; however, we could not perform these experiments as our efforts to silence murine KRAS were unsuccessful.
Genetic experiments using mouse models of cancer have demonstrated the therapeutic value in silencing both KRAS and MYC in pancreatic and lung cancers driven by mutant KRAS; however, these results have yet to be validated using developmental therapeutics. This is primarily due to the fact that KRAS and MYC have thus far proven refractory to targeting using small molecules. In translating U1 Adaptors to target KRAS and MYC, we provide further pre-clinical evidence of the therapeutic value in targeting KRAS and the first evidence for targeting MYC in pancreatic cancer. It remains a valid question if inhibiting these genes systemically will lead to toxicity. Given that KRAS is upstream of MYC and activates multiple signaling pathways such as PI3K and RAL-GDS, we would speculate that the efficacy of the combination would be increased. We have shown that the adaptors inhibit non-cancer cell proliferation slightly at doses that potently inhibit pancreatic cancer cell proliferation, suggesting there may be a therapeutic window. Moreover, Soucek et al. have demonstrated that the Omomyc mice can tolerate chronic inhibition of MYC (3).
Gene silencing as a therapeutic modality has long generated excitement with promises for rapid translation into the clinic. Excitement faded with the realization that oligonucleotide drug candidates lack certain key pharmacological properties most notably targeted delivery to specific cell and organ types, short half-lives and unwanted toxicity and immune responses. Over the years improvements have been made, mostly through systematic changes in the phosphate and ribose groups that comprise the backbone of the oligonucleotide. While such changes appear minor, they greatly increase stability and reduce immunogenicity and overall toxicity. With a few notable exceptions (GalNac/liver) targeted delivery remains a significant challenge resulting in reliance on nanoparticle-based delivery that brings its own challenges with toxicity and stability inherent to a two-component drug system. Recently siRNA technology was used to silence KRAS in murine models of lung and colon cancer that required coupling with lipid-based nanoparticles (17). Alternatively a biodegradable polymeric matrix (LODER) was developed to provide a slow release of siRNA to KRAS in a murine pancreatic cancer model (18). This delivery system showed efficacy, but needs to be injected intratumorally and would not be a useful systemic agent. While cell growth inhibition was similar between the U1 Adaptor and siRNA to KRAS, the U1 Adaptor offers efficacy when conjugated to cell-targeting peptides such as the RGD family, while relying solely on the relatively non-toxic and stable 2′-O-methyl backbone chemistry and eliminating the need for a nanoparticle. These features give the U1 Adaptor the possibility to become first in class as a gene silencing anti-cancer modality.
As with any gene silencing technology, an effective delivery system is required. Here we provide evidence that tumor-targeting peptides directly conjugated to the U1 Adaptor can be used as an effective delivery system, which greatly simplifies GMP manufacturing and testing in humans. Interestingly we did not observe a difference in fluorescence between the iRGD and cRGD-Cy3 labeled peptides in the KPC model as we had hypothesized, suggesting neither peptide is superior. However, a robust comparison of the two peptides was not the aim of this study. We did however observe a difference in tumor growth inhibition in favor of the iRGD.
While these experiments certainly validate the therapeutic potential of U1 Adaptor, there are still several important knowledge gaps that remain to effectively translate this technology to the clinic. The first is a thorough understanding of the pharmacokinetics and pharmacodynamics (PK/PD) of the U1 Adaptor, which is needed to optimize dose and schedule. We chose twice weekly intravenous treatments both for convenience and because this worked well in our 2013 proof of concept that relied on a nanoparticle delivery system (8). The second is the optimal dose. Conjugating tumor targeting peptides allowed us to perform a dose escalation experiment in which we found that a dose of 90 μg was not more effective than 10 or 30 μg. This would need to be examined using pharmacodynamic assays as well as with other target genes to determine if this is specific to KRAS or is more general to U1 Adaptors.
The third is the design of the nucleic acid chemistry. While LNA modifications should in principle increase in vivo potency, we have avoided extensive testing of LNA-U1 Adaptors in vivo due to their unpredictable behavior and known toxicities at higher doses (unpublished observations). That said LNA modification might be necessary for cases where deep silencing of the target gene is necessary to achieve an efficacious response.
What is also unclear is the duration of efficacy with U1 Adaptors. As with any targeted agent, the development of resistance is a factor that must be considered. U1-Adaptor formulations currently require intravenous dosing, which does limit the ability to conduct lengthy treatment experiments in mice. Future studies will focus on these knowledge gaps to effectively translate the U1 Adaptor technology therapeutically.
Supplementary Material
Acknowledgments
Grant Support: This work was supported by the following grants: R21CA191622 (to S.I.G. and D.R.C.), QED Proof of Concept (University City Science Center) (to S.I.G), K08CA172676 and the Breast Cancer Research Foundation to D.R.C.
We would like to thank Dr. Erkki Ruoslahti for providing us with the iRGD peptide and Dr. Joseph Bertino for sharing results of an in vitro screen to identify anti-MYC U1 Adaptors.
Footnotes
Conflict of Interest Statement:
Drs. Gunderson and Goraczniak are inventors of the U1 Adaptor technology and have an equity stake in Silagene Inc. that has licensed the technology from Rutgers University
The following authors declare no potential conflicts of interest: ATT, CD, LY, XY, SK, EH, DRC
References
- 1.Collins MA, Bednar F, Zhang Y, Brisset JC, Galban S, Galban CJ, et al. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J Clin Invest. 2012;122(2):639–53. doi: 10.1172/JCI59227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell. 2012;149(3):656–70. doi: 10.1016/j.cell.2012.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Soucek L, Whitfield JR, Sodir NM, Masso-Valles D, Serrano E, Karnezis AN, et al. Inhibition of Myc family proteins eradicates KRas-driven lung cancer in mice. Genes Dev. 2013;27(5):504–13. doi: 10.1101/gad.205542.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Soucek L, Whitfield J, Martins CP, Finch AJ, Murphy DJ, Sodir NM, et al. Modelling Myc inhibition as a cancer therapy. Nature. 2008;455(7213):679–83. doi: 10.1038/nature07260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Biankin AV, Waddell N, Kassahn KS, Gingras MC, Muthuswamy LB, Johns AL, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012;491(7424):399–405. doi: 10.1038/nature11547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goraczniak R, Behlke MA, Gunderson SI. Gene silencing by synthetic U1 adaptors. Nat Biotechnol. 2009;27(3):257–63. doi: 10.1038/nbt.1525. [DOI] [PubMed] [Google Scholar]
- 7.Goraczniak R, Wall BA, Behlke MA, Lennox KA, Ho ES, Zaphiros NH, et al. U1 Adaptor Oligonucleotides Targeting BCL2 and GRM1 Suppress Growth of Human Melanoma Xenografts In Vivo. Mol Ther Nucleic Acids. 2013;2:e92. doi: 10.1038/mtna.2013.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weirauch U, Grunweller A, Cuellar L, Hartmann RK, Aigner A. U1 adaptors for the therapeutic knockdown of the oncogene pim-1 kinase in glioblastoma. Nucleic Acid Ther. 2013;23(4):264–72. doi: 10.1089/nat.2012.0407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen K, Chen X. Integrin targeted delivery of chemotherapeutics. Theranostics. 2011;1:189–200. doi: 10.7150/thno/v01p0189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sugahara KN, Teesalu T, Karmali PP, Kotamraju VR, Agemy L, Girard OM, et al. Tissue-penetrating delivery of compounds and nanoparticles into tumors. Cancer Cell. 2009;16(6):510–20. doi: 10.1016/j.ccr.2009.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sugahara KN, Teesalu T, Karmali PP, Kotamraju VR, Agemy L, Greenwald DR, et al. Coadministration of a tumor-penetrating peptide enhances the efficacy of cancer drugs. Science. 2010;328(5981):1031–5. doi: 10.1126/science.1183057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu X, Vazquez A, Levine AJ, Carpizo DR. Allele-specific p53 mutant reactivation. Cancer cell. 2012;21(5):614–25. doi: 10.1016/j.ccr.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7(5):469–83. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 14.Singh A, Greninger P, Rhodes D, Koopman L, Violette S, Bardeesy N, et al. A gene expression signature associated with “K-Ras addiction” reveals regulators of EMT and tumor cell survival. Cancer Cell. 2009;15(6):489–500. doi: 10.1016/j.ccr.2009.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324(5933):1457–61. doi: 10.1126/science.1171362. 1171362 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21(3):418–29. doi: 10.1016/j.ccr.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pecot CV, Wu SY, Bellister S, Filant J, Rupaimoole R, Hisamatsu T, et al. Therapeutic silencing of KRAS using systemically delivered siRNAs. Mol Cancer Ther. 2014;13(12):2876–85. doi: 10.1158/1535-7163.MCT-14-0074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zorde Khvalevsky E, Gabai R, Rachmut IH, Horwitz E, Brunschwig Z, Orbach A, et al. Mutant KRAS is a druggable target for pancreatic cancer. Proc Natl Acad Sci U S A. 2013;110(51):20723–8. doi: 10.1073/pnas.1314307110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.