

Abstract
Rationale
Acute pulmonary oxygen sensing is essential to avoid life-threatening hypoxemia via hypoxic pulmonary vasoconstriction (HPV) which matches perfusion to ventilation. Hypoxia-induced mitochondrial superoxide release has been suggested as critical step in the signaling pathway underlying HPV. However, the identity of the primary oxygen sensor and mechanism of superoxide release in acute hypoxia, as well as its relevance for chronic pulmonary oxygen sensing remains unresolved.
Objectives
To investigate the role of the pulmonary specific isoform 2 of subunit 4 of mitochondrial complex IV (Cox4i2) and the subsequent mediators superoxide and hydrogen peroxide for pulmonary oxygen sensing and signaling.
Methods and Results
Isolated ventilated and perfused lungs from Cox4i2−/− mice lacked acute HPV. In parallel, pulmonary arterial smooth muscle cells (PASMCs) from Cox4i2−/− mice showed no hypoxia-induced increase of intracellular calcium. Hypoxia-induced superoxide release which was detected by electron spin resonance spectroscopy in wild type (WT) PASMCs was absent in Cox4i2−/− PASMCs and was dependent on cysteine residues of Cox4i2. HPV could be inhibited by mitochondrial superoxide inhibitors proving functional relevance of superoxide release for HPV. Mitochondrial hyperpolarization, which can promote mitochondrial superoxide release, was detected during acute hypoxia in WT but not Cox4i2−/− PASMCs. Downstream signaling determined by patch clamp measurements showed decreased hypoxia-induced cellular membrane depolarization in Cox4i2−/− PASMCs compared to WT PASMCs, which could be normalized by application of hydrogen peroxide. In contrast, chronic hypoxia-induced pulmonary hypertension and pulmonary vascular remodeling were not or only slightly affected by Cox4i2 deficiency, respectively.
Conclusion
Cox4i2 is essential for acute but not chronic pulmonary oxygen sensing by triggering mitochondrial hyperpolarization and release of mitochondrial superoxide which, after conversion to hydrogen peroxide, contributes to cellular membrane depolarization and HPV. These findings provide a new model for oxygen sensing processes in the lung and possibly also in other organs.
Keywords: Cytochrome c oxidase subunit 4 isoform 2, hypoxic pulmonary vasoconstriction, pulmonary hypertension, reactive oxygen species, hypoxia
Subject Terms: Cell Signaling/Signal Transduction, Oxidant Stress, Pulmonary Biology, Vascular Disease, Pulmonary Hypertension
INTRODUCTION
Although oxygen sensing is essential for aerobic life, the molecular nature, particularly of acute oxygen sensing processes in specific organs is not fully understood. One of the best characterized systems for long-term adaptation at the transcriptional level is through stabilization of HIF-1α (1). However, acute oxygen sensing mechanisms are less well defined, such as in the pulmonary vasculature that reacts to alveolar hypoxia within seconds by hypoxic pulmonary vasoconstriction (HPV) (2, 3). Acute HPV (occurring within seconds to minutes) diverts blood from poorly to well oxygenated alveoli of the lung and thereby prevents shunt flow and arterial hypoxemia on a breath-to-breath basis. This principle was first described by von Euler and Liljestrand in 1946 (4). Exposure to prolonged hypoxia results in sustained HPV (lasting minutes to hours) and leads to the activation of chronic mechanisms resulting in pathologic remodeling of the pulmonary vasculature and development of pulmonary hypertension (PH) (2, 3). Decreased HPV, e.g., during anesthesia, pneumonia, the adult respiratory distress syndrome, septic events or liver failure can result in shunting and life threatening hypoxemia, whereas exaggerated HPV in chronic lung disease or at high altitude contributes to PH (2, 3), which can culminate in right ventricular (RV) failure. In the newborn, prolonged HPV can also contribute to persistent PH, a disease in which the postnatal vasodilatory mechanism, which usually results in low pulmonary vascular resistance in adults, is disturbed (5).
Since the systemic vasculature dilates in response to hypoxia, a unique oxygen sensing mechanism has been suggested for the pulmonary vasculature. Acute and chronic oxygen sensing mechanisms of HPV and PH have been shown to be located in pulmonary arterial smooth muscle cells (PASMCs). PASMCs can react to acute hypoxic exposure by increasing intracellular calcium concentrations following smooth muscle contraction, and by induction of proliferation when exposed to chronic hypoxia, even when isolated. Despite decades of research the acute oxygen sensor in the lung remains unknown. For acute HPV both increased or decreased release of reactive oxygen species (ROS) originating from mitochondrial electron transport chain (ETC) complexes I and/or III have been suggested to initiate the oxygen sensing pathway. The change in ROS release is proposed to then activate sarcoplasmic and/or plasmalemmal ion channels, leading to intracellular calcium increase in PASMCs, triggering vasoconstriction (2, 3, 6). Evidence has accumulated in recent years that triggering acute HPV results from increased superoxide concentration in PASMCs, specifically released into the mitochondrial intermembrane space and originating from complex III of the mitochondrial electron transport chain (ETC) (7, 8). However, the mechanism for such an increase of superoxide production in hypoxia remains unclear. Given the exponential relationship of the mitochondrial membrane potential (ΔΨm) and superoxide production at the outer ubiquinol binding site of complex III (9, 10), which results in a release of superoxide into the intermembrane space, an integrated model of HPV likely involves ΔΨm hyperpolarization as an early upstream signal followed by ROS production at complex III. Indeed, we and others previously showed that HPV is associated with increased mitochondrial ROS production as well as ΔΨm hyperpolarization (11, 12). Similar to acute HPV, for chronic processes and development of PH either an increase (13) or decrease (14) of mitochondrial ROS have been suggested to contribute to PASMC proliferation and pulmonary vascular remodeling, e.g., by activation of transcription factors, such as HIF-1α (13–16).
Although ROS are preferentially released from mitochondrial respiratory chain complexes I and III (17), mitochondrial complex IV (or cytochrome c oxidase, COX) catalyzes the final step of the electron transport chain (ETC) and should be particularly susceptible to hypoxia since it converts oxygen to water. The isoform 2 of COX subunit 4 (Cox4i2) was suggested to increase COX activity (18, 19), thereby promoting ETC activity, ΔΨm, and ROS production. Cox4i2 is preferentially expressed in the adult mammalian lung, specifically in smooth muscle cells and to a lesser degree also in the fetal lung (20, 21). We show here that Cox4i2 is an essential component in the oxygen sensing process of the pulmonary vasculature by promoting ΔΨm hyperpolarization and ROS production during hypoxia.
METHODS
Additional information is provided in Supplemental Methods.
Animals
All animal experiments were approved by the Institutional Animal Investigation Care and Use Committee or appropiate governmental committee. Cox4i2−/− mice were generated as described (18).
Hemodynamic measurements in isolated, perfused, and ventilated mouse lungs
Lungs of either WT or Cox4i2−/− mice were isolated, perfused and ventilated as described previously (22). For arterial pO2 measurement and airway fluid load see Supplemental Methods.
Mouse PASMC isolation and human PASMCs
Mouse PASMCs were isolated from precapillary pulmonary arterial vessels as described previously (22). Human PASMCs were purchased from PromoCell (Heidelberg, Germany).
Calcium measurement
The fluorescent dye Fura 2-AM (Sigma-Aldrich, Munich, Germany) was used for detection of changes in intracellular Ca2+ concentration ([Ca2+]i) in PASMCs (passage 0) isolated from WT and Cox4i2−/− mice. Hypoxia was induced by application of hypoxic (1% O2, balanced with N2) HEPES buffer at minute 2. All gas concentrations in % are given for normobaric conditions.
Lentivirus production and transduction of primary PASMCs
Full length Cox4i1, or wild type or mutated Cox4i2 were subcloned into the pWPXL plasmid (Addgene, Boston, USA) and packaged with a second-generation lentivirus transduction system with pMD2.G as the envelope and psPAX2 as a packing vector (Addgene, Boston, USA).
Stable expression of Cox4i2 and Cox4i1 in CMT 167 cells
Mouse cDNA of Cox4i1, wild-type, or of mutated Cox4i2 were subcloned in the pCI-neo plasmid. CMT 167 (mouse lung carcinoma cell line) cells were transfected with the corresponding plasmids using TurboFectin 8.0 transfection reagent.
Measurement of superoxide by electron spin resonance spectroscopy
Intracellular and extracellular ROS concentration was measured using an EMXmicro Electron Spin Resonance (ESR) spectrometer (Bruker Biospin GmbH, Rheinstetten, Germany) using 0.5 mM of the spin probe CMH (1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethylpyrrolidine; Noxygen, Elzach, Germany). The superoxide portion of ROS was determined by subtracting the ESR signal of the sample with polyethylene-glycol conjugated superoxide dismutase (pSOD) from the sample incubated for 90 min without 45 U/ml pSOD in ESR-Krebs HEPES buffer (23, 24). Hypoxia was applied by incubating the cells in a hypoxic chamber (1% O2, 5min).
Patch clamp recordings of cellular membrane potential and KV-channel currents
Membrane potential was recorded under current clamp conditions (I = 0) in whole cell configuration using an EPC10 USB single amplifier (HEKA, Lambrecht, Germany), whereas whole cell Kv-currents were measured in voltage clamp mode using a standardized depolarizing pulse protocol. Acute hypoxia was applied by switching from normoxic (bubbled with a gas mixture of 21 % O2, 5.3 % CO2, rest N2) to hypoxic (1 % O2, 5.3 % CO2, rest N2) bath solution and the pO2 near the cell was recorded by an optical needle-type oxygen sensor (Firesting, Pyro Science, Aachen, Germany).
Mitochondrial membrane potential and cytosolic hydrogen peroxide
Mitochondrial membrane potential was investigated by fluorescent microscopy using JC-1 (5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide) as described previously (11). Acute hypoxia was prompted by switching from normoxic perfusion buffer to hypoxic buffer (1% O2). For intracellular H2O2 detection, the cytosolic HyPer sensor (Evrogen, Moscow, Russian Federation) subcloned into the pWPXL plasmid (distributed by Addgene, Boston, USA) was used.
High resolution respirometry
Oxygen consumption rate was determined at 37°C using an Oxygraph-2k (Oroboros Instruments, Innsbruck, Austria).
Quantification of hypoxia-induced pulmonary hypertension by in vivo hemodynamics, right ventricular morphometry, vascular remodelling, and echocardiography
Mice were exposed to normobaric hypoxia (10% O2) for 4 weeks. Quantification of pulmonary hypertension was performed as described previously (22).
Measurement of transthoracic echocardiography was performed with the Vevo2100 high-resolution imaging system equipped with a 40-MHz transducer (VisualSonics, Toronto, Canada) (25).
Proliferation of PASMCs was determined by BrdU assay (cell proliferation ELISA, Roche, Basel, Switzerland).
Statistics
Normal distribution of the sample sets was determined by Shapiro Wilk normality test. For sample sets with Gaussian distribution, student’s two-tailed t-test or 2-way-ANOVA were used. For the sample sets with a non-Gaussian distribution, Mann-Whitney-test or Kruskal-Wallis-test was used. Multiple comparisons, using the same group in more than one analysis or hypothesis, were adjusted using Bonferroni correction. All analyses were considered statistically significant at p<0.05. Statistical analysis was performed using Prism 6 (GraphPad Software Inc., San Diego, USA).
RESULTS
Cox4i2−/− mice lack HPV and hypoxia-induced calcium increase in PASMCs
We generated Cox4i2 knockout (Cox4i2−/−) mice (18) and investigated HPV in isolated ventilated and perfused mouse lungs from wildtype (WT) and Cox4i2−/− mice by measuring the increase of pulmonary arterial pressure (∆PAP) during hypoxic ventilation (1% O2, 5% CO2, 94% N2, Figure 1A) Exposure of isolated lungs to acute hypoxia for 10 min resulted in an increase of PAP in lungs from WT, but not Cox4i2−/− mice (Figure 1B, D). Under normoxic ventilation the course of PAP did not differ between knockout and WT mice (Figure 1C). While acute HPV (10 min, 1% O2) was absent in Cox4i2−/− lungs, the hypoxia-independent vasoconstriction artificially induced by application of potassium-chloride (KCl) was unchanged (Figure 1D), indicating that the contractile apparatus of the lung vasculature is intact in Cox4i2−/− lungs. These results show that Cox4i2 is essential for acute HPV. Furthermore, the response to prolonged hypoxia was blunted in Cox4i2−/− lungs (Figure 1B). Accordingly, the drop of the arterial pO2 after an intratracheal fluid challenge of anesthetized mice to induce local alveolar hypoventilation was higher in Cox4i2−/− mice compared to WT mice (Figure 1E), representing disturbed ventilation-perfusion matching in Cox4i2−/− mice. Expression analysis showed that Cox4i2 protein is expressed in the pulmonary artery, preferentially in PASMCs (Figure 1F). As PASMCs have been identified as the sensor and effector site of HPV, (3) and Cox4i2 protein was found to be expressed predominantly in this cell type (Figure 1F), we next investigated PASMCs isolated from precapillary arteries. Cox4i2−/− PASMCs largely lacked the hypoxia-induced calcium increase observed in WT PASMCs (Figure 1G). The lack of HPV did not affect the number of offsprings per litter (Online Figure IA) or baseline arterial oxygen concentration (Figure 1E). Lungs of Cox4i2−/− mice aged 2–3 months did not show histological alterations compared to WT mice (Online Figure IB).
Figure 1. Acute HPV is absent in Cox4i2-deficient (Cox4i2−/−) mice.
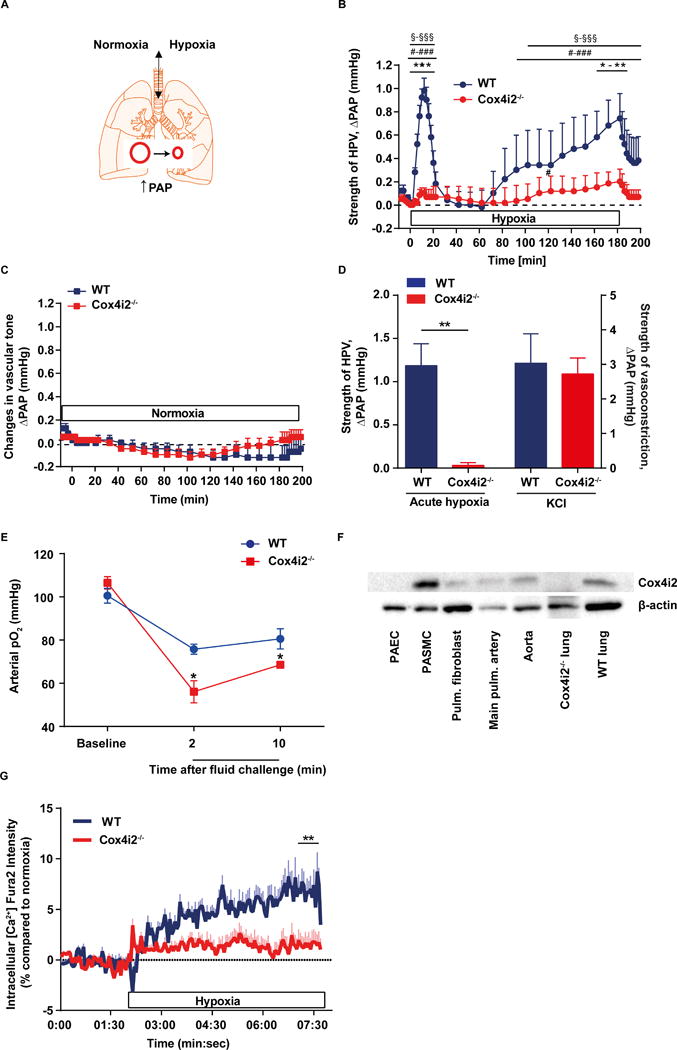
(A) Schematic of isolated mouse lungs ventilated with normoxic or hypoxic gas. Hypoxic ventilation results in pulmonary vasoconstriction detected as an increase in pulmonary arterial pressure (PAP). (B) Time course of the strength of hypoxic pulmonary vasoconstriction (HPV) in isolated, buffer-perfused, and ventilated mouse lungs during 180 min of hypoxic ventilation (1% O2). Changes in pulmonary arterial pressure (∆PAP) are shown for lungs from WT (n=5) and Cox4i2−/− (n=6) mice. *p<0.05, **p<0.01, ***p<0.001 for comparison between WT and Cox4i2−/− mice: §p<0.05, §§§p<0.001 for comparison between hypoxic and normoxic experiments (data from Figure 1c), and #p<0.05, ###p<0.001 for comparison between hypoxic and normoxic ventilation at time point 0 min. Data were analyzed by 2-way-ANOVA. (C) Time course of ∆PAP during 180 min of normoxic ventilation (n=4, each group). (D) Specificity of Cox4i2 for the hypoxia-induced vasoconstrictor response. Lungs of WT and Cox4i2−/− mice (n=5 each) were challenged either with hypoxic ventilation (1% O2, 10 min, acute hypoxia) or with infusion of potassium chloride (KCl) into the pulmonary artery. **p<0.01 (by two-tailed Student’s t-test). (E) Arterial pO2 in anesthetized WT and Cox4i2−/− mice after regional ventilatory disturbance by tracheal instillation of 25 μl of saline at time point zero. *p<0.05 comparing WT and Cox4i2−/− mice. Data are from n = 4 mice per group (F) Expression of Cox4i2 in mouse lung tissue. β-actin was used as a loading control. PAEC: pulmonary arterial endothelial cell, PASMC: pulmonary arterial smooth muscle cell, Pulm.: pulmonary. (G) Intracellular calcium increase in PASMCs upon exposure to hypoxic medium compared to normoxic medium measured by the increase in fura-2 intensity. The horizontal bar indicates presence of hypoxic medium. Data are from n=50 (WT) and n=52 (Cox4i2−/−) cells, from 5 individual experiments per group isolated from 3 mice per group. * significant difference (p<0.05) between WT and Cox4i2−/− analyzed by two-tailed Mann-Whitney-test comparing averaged values from minute 7 to 8.
All data are given as mean+SEM.
Hypoxia-induced superoxide increase is absent in Cox4i2−/− PASMCs and depends on Cysteine 109 of Cox4i2
In order to address the underlying signaling mechanisms in more detail, we measured superoxide release by ESR of PASMCs exposed to acute hypoxia (5 min, 1% O2) and immediately frozen in the hypoxic atmosphere to avoid re-exposure to normoxia. Superoxide concentration markedly increased in hypoxic WT PASMCs (Figure 2A, C). This increase was absent in Cox4i2−/− PASMCs (Figure 2A, C) and similarly in PASMCs treated with Cox4i2-specific siRNA (Figure 2B, S3A). Mitochondrial superoxide release during hypoxia is necessary for HPV, as the mitochondrial superoxide dismutase (SOD) mimetic MitoTempol, which converts superoxide to hydrogen peroxide, but has also been shown to act as unspecific mitochondrial antioxidant (26), inhibited HPV in intact lungs (Figure 2D), but 1) only partially inhibited KCl induced vasoconstriction (Figure 2E) and 2) did not affect pulmonary vasoconstriction induced by the thromboxane mimetic U46619 (Online Figure IIA). Moreover, the novel inhibitor of mitochondrial superoxide release from complex III (27), S3QEL2, inhibited HPV (Figure 2F), but not KCl (Figure 2G) or U46619 induced vasoconstriction (Online Figure IIB). Similarly the mitochondria-targeted antioxidant SkQ1 inhibited HPV, but not U46619-induced vasoconstriction (Online Figure IIC,D). The specific role of Cox4i2 was shown by re-introduction of Cox4i2 in Cox4i2−/− PASMCs, which re-established the hypoxia-induced superoxide increase, whereas Cox4i1 transfection was ineffective (Figure 2H). In contrast to Cox4i1, Cox4i2 contains several cysteine residues that are proposed to form disulfide bonds and modulate allosteric ATP-inhibition of COX activity (20); these may in turn affect ΔΨm and thus subsequent ROS production. We thus introduced a mutated Cox4i2 plasmid, in which the conserved cysteine residue 109 was exchanged with serine (C109S) or alanine (C109A) into Cox4i2−/− cells. In contrast to WT Cox4i2 overexpression, overexpression of such Cox4i2 mutants in Cox4i2−/− PASMCs showed little or no increase of superoxide after hypoxic exposure (Figure 2H), indicating functional importance of the C109 residue. In order to exclude PASMC-specific hypoxia-induced signaling pathways upstream of Cox4i2, we expressed Cox4i2 in a completely different cell type, choosing a mouse lung carcinoma cell line (CMT cells). We detected increased superoxide production during hypoxia in Cox4i2 but not in Cox4i1 expressing CMT cells (Figure 2I). Mutation of cysteine-109 (Figure 2I) and the other cysteine residues (Online Figure IVA) did abolish hypoxia-induced superoxide increase in CMT cells. Lack of hypoxia-induced superoxide production was not due to failed expression or increased degradation of proteins because Western blot analysis showed the mutated Cox4i2 was expressed in the cells (Online Figure III B, C) or mitochondria of CMT cells (Online Figure IV B) Superfusion of PASMCs with hypoxic medium caused an increase in cytosolic H2O2 concentration in WT PASMCs (Figure 2J), but not in Cox4i2−/− PASMCs (Figure 2K). In order to investigate, if decreased activity of the SOD could cause a decrease in H2O2 concentration, we tested SOD expression and activity. Although mRNA expression of SOD1, which is located in the mitochondrial intermembrane space and cytosol, was higher in Cox4i2−/− PASMCs (Online Figure VA), we could not detect differences in SOD1 protein expression and SOD activity (Online Figure VB, C).
Figure 2. Cox4i2 is required for hypoxia-induced ROS increase to trigger HPV.
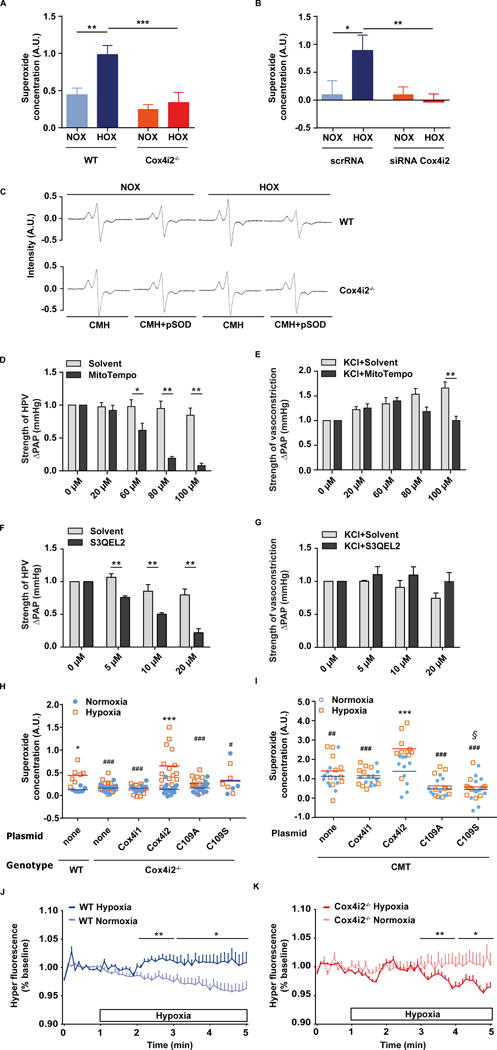
(A, B) Superoxide concentration in PASMC isolated from WT (n=7) and Cox4i2−/− mice (n=8) (A) or WT PASMC after transfection with scrambled (scr) siRNA (normoxia: n=9, hypoxia: n=8) or siRNA against Cox4i2 (normoxia: n=8, hypoxia: n=9) (B) during exposure to normoxia (room air) or hypoxia (1% O2, 5 min). Downregulation of Cox4i2 by siRNA was confirmed by Western Blot (Online Figure IA). AU: arbitrary units. *p<0.05, **p<0.01, ***p<0.001 determined by 2-way-ANOVA with Bonferroni posthoc test. Data are from 3 independent experiments from PASMC of one mouse per sample. (C) Representative ESR spectra of PASMC from WT and Cox4i2−/− mice. Superoxide concentration (A, B) was calculated as the portion of the 1-hydroxy-3-methoxycarbonyl-2,2,5, 5-tetramethylpyrrolidine (CMH) signal that was inhibited by the pegylated superoxide-dismutase (pSOD). (D–G) Strength of hypoxic pulmonary vasoconstriction (HPV, n=6 isolated lungs each group) (D,F) and potassium chloride (KCl) induced pulmonary vasoconstriction (n=5/8 isolated lungs for the solvent/MitoTempo group, n=4/4 isolated lungs for the solvent/S3QEL2 group) (E,G), determined as an increase of pulmonary arterial pressure (∆PAP) in presence (MitoTempo/S3QEL2 group) and absence (solvent group) of MitoTempo/S3QEL2 in WT mice. MitoTempo or S3QEL2 was applied in increasing concentrations in the same lung 5 min before each repetitive maneuver of hypoxic ventilation or KCl application, respectively, and compared to the respective increase of PAP in presence of the solvent only. *p<0.05, **p<0.01 analyzed by two-tailed Mann-Whitney test. (H) Intracellular superoxide concentration during normoxic and hypoxic exposure of PASMCs isolated from WT and Cox4i2−/− mice. COX4i2−/− cells were transfected with different plasmids encoding Cox4i1, Cox4i2, and Cox4i2 mutants in which the cysteine residue at position 109 was replaced with alanine (C109A) or serine (C109S). WT none: n=7 per group, Cox4i2−/− none: n=14 (normoxia)/n=15 (hypoxia), Cox4i1 plasmid: n=10 per group, Cox4i2 plasmid: n=16 (normoxia)/n=15 (hypoxia), C109A plasmid: n=15 (normoxia)/n=14 (hypoxia), C109S plasmid: n=4 (normoxia)/n=5 (hypoxia). Data are from 2–3 independent experiments with PASMC from one mouse per sample. *p<0.05, ***p<0.001 comparing the normoxic and hypoxic groups, #p<0.05, ###p<0.001 compared to the hypoxic Cox4i2 group (analysis by 2-way-ANOVA with Bonferroni posthoc test). (I) Intracellular superoxide concentration during hypoxic exposure of a mouse lung carcinoma cell line (CMT). CMT cells were transfected as stated in (F). n=10 per group for empty cells (none), n=10 (normoxia) or 9 (hypoxia) for cells transfected with Cox4i1, n=10 per group for cells transfected with Cox4i2 and C109A, n=13 (normoxia) or 11 (hypoxia) for cells transfected with C109S. Data are from 4 independent experiments with PASMC isolated from one mouse per sample. ***p<0.001 comparing the normoxic and hypoxic group, ##, p<0.05, ###p<0.001 compared to the hypoxic Cox4i2 group, §p<0.05 compared to the normoxic Cox4i2 group (analysis by 2-way-ANOVA with Bonferroni posthoc test). Lack of hypoxia-induced superoxide production was not due to failing expression or increased degradation of proteins because Western blot analysis showed the mutated Cox4i2 was expressed in the cells (Online Figure III3B,C). (J, K) Cytosolic hydrogen peroxide concentration determined as fluorescence intensity of the HyPer dye, given as percent (%) of the normoxia in PASMCs from WT (A) or Cox4i2−/− (B) mice. The horizontal bar indicates the presence of hypoxic medium for the hypoxic group. Data are from n=24/23 cells (hypoxia: WT/Cox4i2−/−) or n=14/21 cells (normoxia: WT/Cox4i2−/−) from at least three individual PASMC isolations. *, ** significant difference (p<0.05 or p<0.01) between normoxia and hypoxia analyzed by two-tailed Mann-Whitney-test comparing values averaged over one minute.
Data in A, B, D, E are given as mean+SEM. Data in F, G are presented as mean and individual values.
Hypoxia-induced cellular membrane depolarization is attenuated in Cox4i2−/− PASMCs
In order to determine the downstream signaling effects of ROS in PASMC, we performed patch clamp experiments to measure cellular membrane potential. We found that WT PASMCs exhibited cellular membrane depolarization upon exposure to acute hypoxia (Figure 3A, B), whereas this depolarization was blunted in Cox4i2−/− PASMCs (Figure 3C, D). In contrast to WT PASMCs, addition of H2O2, could further depolarize cellular membrane potential in Cox4i2−/− PASMCs to the level reached in WT PASMCs under hypoxia (Figure 3B, D; Online Figure VIA–E). Furthermore, basal membrane potential in Cox4i2−/− PASMCs was hyperpolarized compared WT PASMC (Figure 3C, D, Online Figure IVF). Application of S3QEL2 to inhibit mitochondrial superoxide release attenuated the hypoxia-induced depolarization in WT PASMCs (Figure 3E–H). Next, we investigated the effect of acute hypoxia on Kv channel closing, which has been shown to play an important role in hypoxia-induced membrane depolarization in PASMCs (6). Hypoxia caused an inhibition of Kv channels in WT and to a lesser degree in Cox4i2−/− PASMCs (Figure 3I–L). Application of H2O2 decreased Kv channel current density to the same level as in WT PASMCs (Figure 3I–L). Accordingly, application of H2O2 caused Kv channel inhibition and cellular membrane depolarization in WT PASMCs under normoxia (Figure 3M–P). Consequently, we could not find differences in Kv channel expression as an underlying reason for the described differences (Online Figure VI).
Figure 3. Hypoxia-induced cellular membrane depolarization is decreased in Cox4i2−/− PASMCs and dependent on reactive oxygen species.
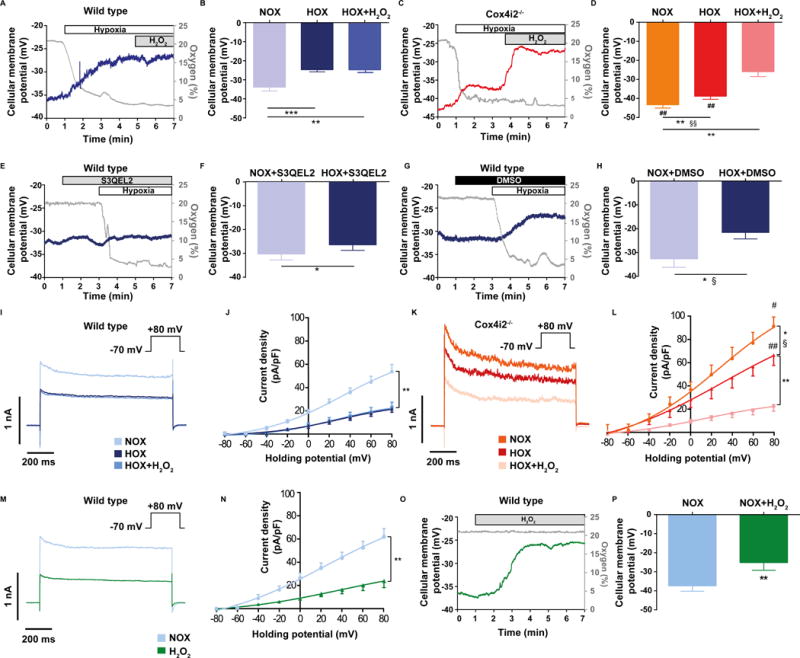
(A–D) Hypoxia-induced depolarization in WT (A, B) and Cox4i2−/− PASMCs (C, D). Representative tracings of patch clamp measurements in whole-cell current clamp-mode to determine the cellular (plasma) membrane potential during acute hypoxia in WT (A) and Cox4i2−/− PASMCs (C). The gray tracings show the oxygen concentration in %, the blue and red tracing the cellular membrane potential in WT (A) and Cox4i2−/− PASMCs (C), respectively. Summary of cellular membrane potential during normoxia (NOX), acute hypoxia (HOX) and acute hypoxia after application of hydrogen peroxide (HOX+H2O2, resulting in a concentration of 124 nM H2O2 in the bath solution) in WT (B) and Cox4i2−/− PASMCs (D). n=7 for WT, and n=6 for Cox4i2−/− PASMCs from 3 individual cell isolations, respectively. *p<0.05, **p<0.01 compared to respective NOX value. #p<0.05, ##p<0.05 compared to WT. §§p<0.01 comparing the differences of normoxic and hypoxic measurements between the WT and Cox4i2−/− group. (E–H) Hypoxia-induced depolarization in presence of the mitochondrial superoxide scavenger S3QEL2 (E, F) and the control solvent DMSO (G, H) in WT PASMCs. Representative tracings of hypoxic depolarization in presence of S3QEL2 (E) and DMSO (G). The gray tracings show the oxygen concentration in %, the blue tracings the cellular membrane potential. (F, H) Cellular membrane potential during normoxia (NOX) and acute hypoxia (HOX) in presence of S3QEL2 (F) and DMSO (H). n=6 for S3QEL2, and n=5 for DMSO from 3 and 2 individual cell isolations, respectively. *p<0.05 compared to respective NOX group, §p<0.05 comparing the differences of normoxic and hypoxic measurements between the S3QEL2 and DMSO group. (I–L) Whole-cell Kv channel activity determined as Kv current density in WT PASMCs (I, J) and Cox4i2−/− PASMCs (K, L) during normoxia (NOX) and after exposure to hypoxia (HOX) in absence and presence of hydrogen peroxide (HOX+ H2O2, resulting in a concentration of 124 nM H2O2 in the measurement buffer). Currents were evoked from a holding potential of −70 mV in incremental depolarizing 20-mV steps from −80 mV to + 80 mV and shown as raw data traces for the +80 mV step (I, K, traces overlayed for comparison) and as averaged whole-cell I–V relationship (J, L) from n=8 for WT and n=6 for Cox4i2−/− PASMCs from at least 3 independent cell isolations. *p<0.05, **p<0.01 compared to normoxia, #p<0.05, ##p<0.01 compared to WT hypoxia. §p<0.05 comparing the differences of normoxic and hypoxic measurements between the WT and Cox4i2−/− group. (M–P) Kv channel activity (M, O) and cellular membrane depolarization (N, P) after application of H2O2 (final concentration 124 μM) in WT PASMCs during normoxia. n=5 PASMCs each group from 3 independent cell isolations, **p<0.01 determined by t-test. (I–L) Kv channel activity determined as Kv current density in WT PASMCs (I, K) and Cox4i2−/− PASMCs (J, L) during normoxia (NOX) and hypoxia (HOX) in absence and presence of hydrogen peroxide (HOX+ H2O2, resulting in a concentration of 124 nM H2O2 in the measurement buffer). n=8 for WT and n=6 for Cox4i2−/− PASMCs from at least 3 independent cell isolations. *p<0.05, **p<0.01 compared to normoxia, #p<0.05, ##p<0.01 compared to WT hypoxia. §p<0.05 comparing the differences of normoxic and hypoxic measurements between the WT and Cox4i2−/− group. (M–P) Kv channel activity (M, O) and cellular membrane depolarization (N, P) after application of H2O2 (final concentration 124 μM) in WT PASMCs during normoxia. n=5 PASMCs each group from 3 independent cell isolations, **p<0.01 determined by t-test.
Hypoxia-induced mitochondrial hyperpolarization is absent in Cox4i2−/− PASMCs
As mitochondrial hyperpolarization was shown to increase mitochondrial ROS production (9, 10), we next focused on ΔΨm, as a possible mechanism by which Cox4i2 causes hypoxia-induced superoxide release. We confirmed the dependency of mitochondrial superoxide release on ΔΨm in PASMCs and showed that increasing ΔΨm with oligomycin increased the superoxide concentration of PASMCs, whereas decreasing ΔΨm by FCCP decreased superoxide concentration (Online Figure VIII8G). With regard to the hypoxic response, we detected an increase in ΔΨm in WT PASMCs when superfused with hypoxic medium (Figure 4A, Online Figure VIIIA, D) that was absent in Cox4i2−/− PASMCs (Figure 4B, Online Figure VIIIB, C). In presence of MitoTempol or S3QEL2 we could still detect the hypoxia-induced increase of ΔΨm in WT cells (Figure 4C, Online Figure VIIIF). Furthermore, during acute hypoxia mitochondrial matrix pH was increased in WT, but not in Cox4i2−/− PASMCs, which indicates fewer protons on the matrix side of the inner mitochondrial membrane thereby possibly promoting the ΔpH contributing to ΔΨm (Online Figure VIII8H). However, we could not detect significant differences in respiration when comparing WT and Cox4i2−/− PASMCs (Figure 4D, S9B). CMT cells overexpressing Cox4i2 respired less when maximally stimulated with FCCP compared to Cox4i1-overexpressing cells (Online Figure XIA). In permeabilized PASMCs and CMT cells, respiration under saturating conditions of different substrates was similar (Online Figure XIB,C). When decreasing the pO2 from normoxia (18% O2) to hypoxia (4% O2), respiration decreased in both WT and Cox4i2−/− PASMCs to a similar degree (Figure 4E). Moreover, investigating the respiration at more severe hypoxia (O2<3%) revealed a similar pO2-dependent respiration in WT and Cox4i2−/− PASMCs (Figure 4F).
Figure 4. Mitochondrial membrane potential, but not respiration is decreased in Cox4i2−/− PASMCs in acute hypoxia.
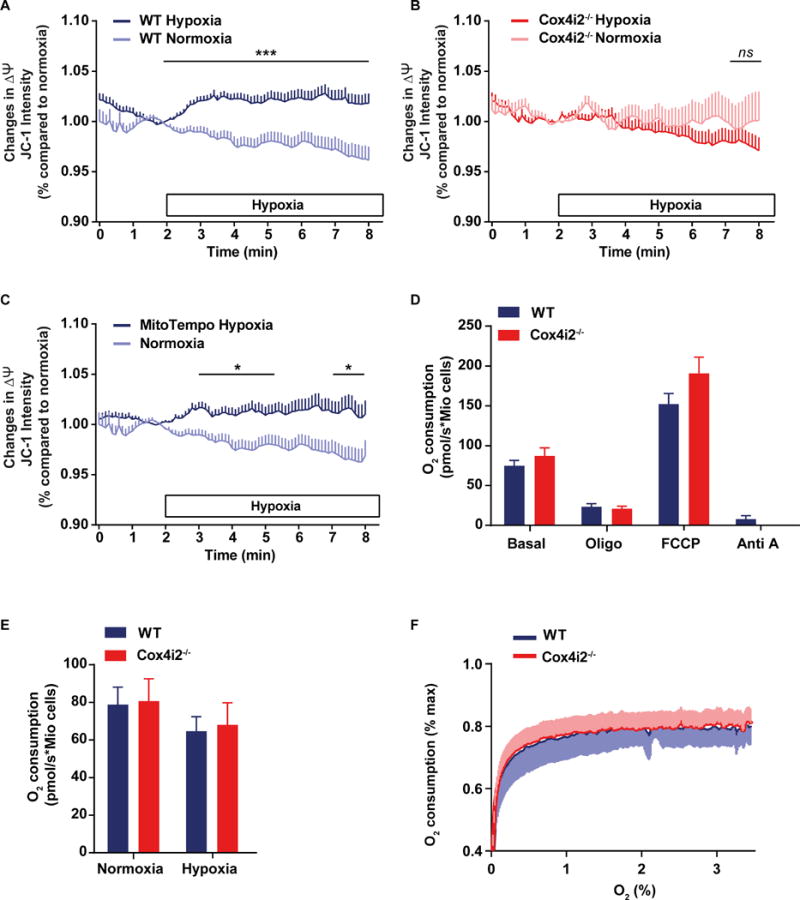
(A–C) Mitochondrial membrane potential determined as fluorescence intensity of red/green ratio of JC-1 staining, given as percent (%) of the normoxic value at minute 2 in PASMCs from WT (A) or Cox4i2−/− (B) mice, and in WT PASMCs in absence and presence of MitoTempo (C). The horizontal bar indicates the presence of hypoxic medium for the hypoxic group. Data are from n=180/207 cells (hypoxia: WT/Cox4i2−/−) or n=107/95 cells (normoxia: WT/Cox4i2−/−) from at least three individual PASMC isolations for Fig. A and B and n=31 cells from one PASMC isolation for the MitoTempo group in Figure 3C. The normoxic group in Figure 3C is the same as in Figure 3A. *, *** significant difference (p<0.05 or p<0.001) between normoxia and hypoxia analyzed by two-tailed Mann-Whitney-test comparing values averaged over one minute. (D) Respiration rate of intact PASMCs isolated from WT or Cox4i2−/− mice displayed as O2 consumption in pmol/s per million (Mio) cells under unstimulated conditions (basal) and in presence of oligomycin (oligo), FCCP, and antimycin A (Anti A). Data are from n=6 (WT) or n=8 (Cox4i2−/−) experiments containing 100,000 to 300,000 cells isolated from one mouse per experiment.
(E) Respiration in intact PASMCs isolated from WT or Cox4i2−/− mice under unstimulated conditions in normoxia (O2=18%) and hypoxia (O2=4%). Data are from n=9 each group containing 100,000 to 300,000 cells isolated from one mouse per experiment. (F) Oxygen-dependent respiration rate in intact PASMCs isolated from WT or Cox4i2−/− mice, expressed as percent (%) O2 consumption at normoxia (O2=18%). Data are from n=7 experiments, each group containing 100,000 to 300,000 cells isolated from one mouse per experiment.
All data are given as mean+SEM or −SEM.
Hypoxia-induced depolarization in human PASMCs is attenuated by Cox4i2 knockdown
To assess the relevance of our findings for the human system, we showed that Cox4i2 protein is expressed in lung homogenate and specifically pulmonary arterial vessels of human lungs (Online Figure XA). We could also detect Cox4i2 mRNA specifically in the media of human pulmonary arterial vessels and cultivated human PASMCs (Online Figure XB). Downregulation of Cox4i2 by siRNA (Online Figure XC) decreased the hypoxia-induced depolarization in human PASMCs (Figure 5).
Figure 5. Hypoxia-induced cellular membrane depolarization in human PASMCs.
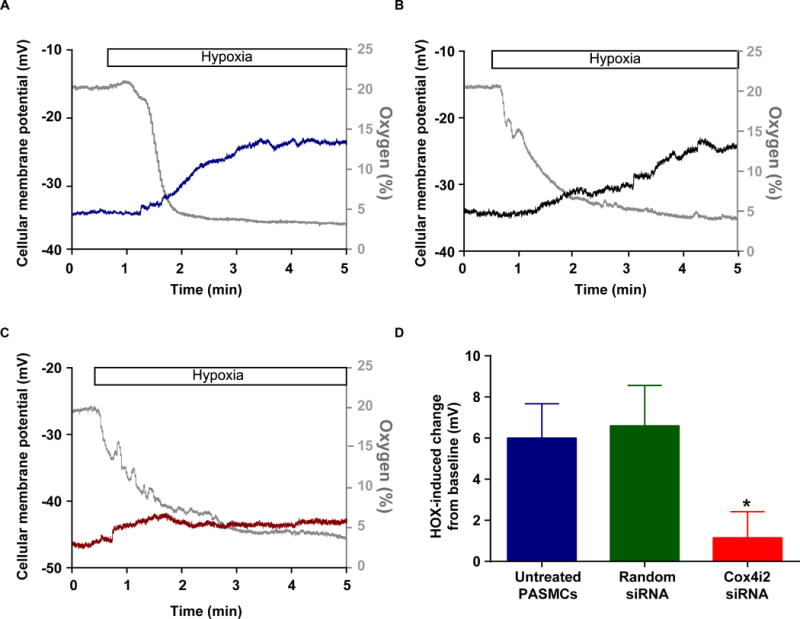
A–C) Representative whole-cell tracings of patch clamp measurements to determine the cellular membrane potential during acute hypoxia in human PASMCs (A), human PASMCs transfected with unspecific siRNA (Random siRNA, B), and human PASMCs transfected with siRNA for Cox4i2 (C). The gray tracings show the oxygen concentration in %, the blue, green and red tracing the cellular membrane potential. (D) Comparison of hypoxia-induced cellular membrane depolarization in human PASMCs after downregulation of Cox4i2.
Chronic hypoxia-induced PH is only slightly influenced by Cox4i2 deficiency
We investigated whether Cox4i2 is also essential for chronic pulmonary vascular oxygen sensing. After chronic exposure of mice to a hypoxic atmosphere (10% O2, 4 weeks) both WT and Cox4i2−/− developed PH to a similar degree. Although right ventricular pressure was higher in Cox4i2−/− mice, the hypoxia induced increase was not different between the strains (Figure 6A). Both strains developed right heart hypertrophy (Figure 6B). Pulmonary vascular remodeling was slightly lower in Cox4i2−/− mice, characterized by a higher portion of non muscularized vessels after chronic hypoxic exposure (Figure 6C). Cardiac index was decreased after hypoxic exposure, but not differently in the two strains (Figure 6D). In parallel HIF-1α stabilization was increased to a similar level in PASMCs after exposure to hypoxia (Figure 6E). PDGF-induced proliferation during hypoxia was similar in WT and Cox4i2−/− PASMCs, with a tendency for lower proliferation in Cox4i2−/− PASMCs (Figure 6F). After exposure of mice to chronic hypoxia (4 weeks, 10%O2), acute hypoxic response was also absent in isolated lungs of Cox4i2−/− mice (Online Figure XI).
Figure 6. Hypoxia-induced pulmonary hypertension in WT and Cox4i2-deficient mice after exposure to chronic normobaric hypoxia (10% O2) for 28 days.
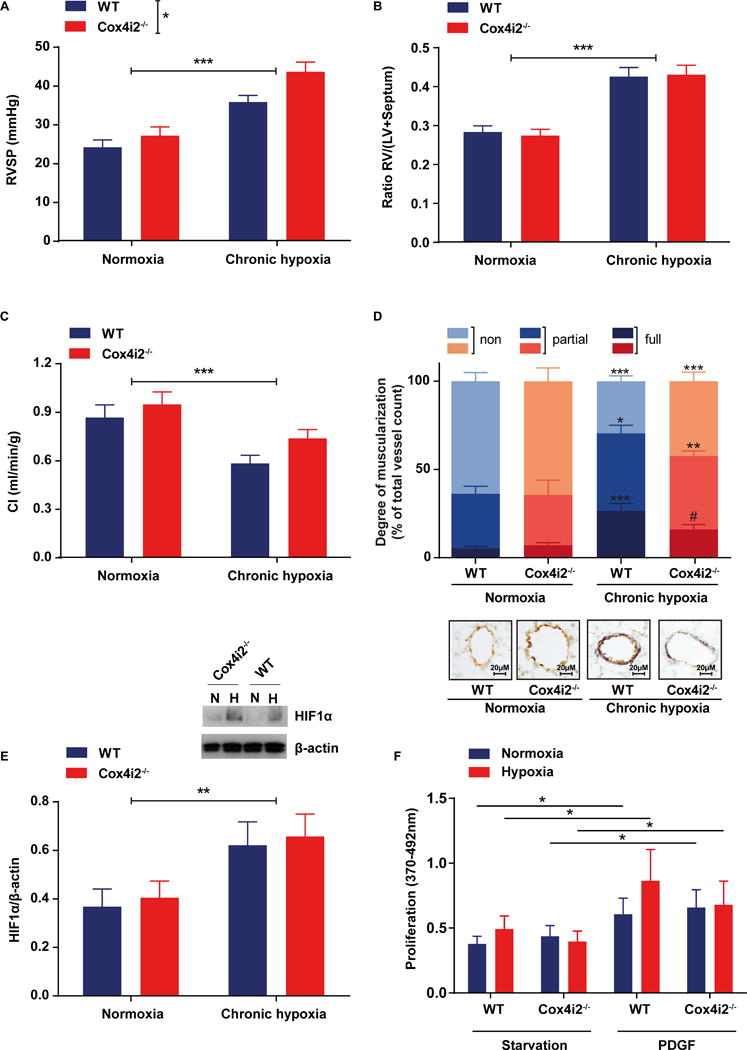
(A) Right ventricular systolic pressure (RVSP) quantified in vivo in WT and Cox4i2−/− mice after exposure to normoxia (n = 10 each group) and chronic hypoxia (WT, n=8, Cox4i2−/−, n=6). RVSP was significantly increased after hypoxia and higher in Cox4i2−/− mice (*indicates significant difference [p<0.01] between genotypes), but the hypoxia-induced increase was not higher in Cox4i2−/− mice compared to WT mice according to the interaction analysis by 2-way-ANOVA. (B) Ratios of the right ventricle (RV) of the heart and the left ventricle plus septum (LV+S). Data are from animals either exposed to normoxia (n=10, each group) or chronic hypoxia (WT, n=10, Cox4i2−/−, n=9). (C) Cardiac index (CI) quantified by echocardiography in WT and Cox4i2−/− mice exposed to normoxia (WT, n=10, Cox4i2−/−, n=8) or chronic hypoxia (WT, n=10, Cox4i2−/−, n=9).
For statistical analysis of data from figure a, b, and c 2-way-ANOVA with Bonferroni posthoc test was used. *** indicates significant difference (p<0.001) between normoxic or hypoxic treatment, while Bonferroni posthoc test indicated no significant differences between single groups.
(D) Vascular remodeling quantified as the degree of muscularization of small (20–70 μm diameter) pulmonary arterial vessels. Vessels were categorized as nonmuscularized, partially muscularized, or fully muscularized after immunostaining against α-smooth-muscle actin (purple) for detection of PASMC and von-Willebrand (brown) for discrimination of endothelium. Eighty-five vessels from each individual lung (WT, n=8, normoxia, n=9, hypoxia, Cox4i2−/−, n=6, hypoxia n=8) were analyzed. *** indicates significant difference (p<0.001) between respective normoxic group. # indicate significant difference between WT and Cox4i2−/−. Data were analyzed by 2-way-ANOVA with Bonferroni posthoc test. (E) HIF-1α expression in PASMCs isolated from WT and Cox4i2−/− normalized to β-actin expression after exposure to normoxia or hypoxia (36 h, 1% O2). n=11 per group from 4 independent experiments and one mouse per sample. ** indicates significant difference (p<0.01) between hypoxic and normoxic treatment. (F) Proliferation determined as EdU incorporation, measured by fluorescence intensity of WT and Cox4i2−/− PASMCs in the presence of hypoxia and platelet derived growth factor (PDGF). n=6–7 each group. *p<0.05, comparing the respective starved and PDGF-treated group.
All data are given as mean+SEM.
DISCUSSION
Our study reveals an essential mechanism for acute pulmonary oxygen sensing leading to HPV and uncovers a novel component for hypoxia-induced mitochondrial superoxide release in acute hypoxia. We found that the mitochondrial complex IV subunit 4 isoform 2 initiates acute HPV by mitochondrial hyperpolarization, which promotes the superoxide release preferentially at complex III of the ETC, previously suggested as an essential step of HPV (8, 13). Subsequently, ROS lead to cellular membrane depolarization including inhibition of Kv channels, cellular calcium entry and HPV. Moreover, our data emphasize that different signaling mechanisms underlie acute and chronic oxygen sensing. This conclusion is based on the findings that 1) Cox4i2−/− lungs lack acute HPV, and Cox4i2−/− PASMCs show no hypoxia-induced increase in intracellular calcium, superoxide and mitochondrial membrane potential; 2) decreased hypoxia-induced cellular membrane depolarization and Kv channel inhibition in Cox4i2−/− PASMCs can be enhanced to the WT level by H2O2 application, while HPV can be inhibited by two different mitochondrial superoxide scavengers; and 3) chronic hypoxia-induced PH is only slightly affected by Cox4i2 deficiency and HIF1α stabilization is not different in the genotypes. Thus our data suggests that complex IV acts as primary oxygen sensor and regulates downstream superoxide release by complex III. The lung specific isoform 2 of subunit 4 is essential for hypoxia-induced, ROS-dependent cellular membrane depolarization and HPV. Accordingly, a lack of Cox4i2 results in a lower arterial oxygenation during induction of localized hypoventilation, which is explained by ventilation-perfusion mismatch due to the lack of HPV. These mechanisms may be relevant also in humans, as we showed that Cox4i2 is relevant for hypoxia-induced cellular membrane depolarization in human PASMCs. Whereas Cox4i2 activity may be involved in increased shunting under conditions of local hypoventilation in the adult (e.g. pneumonia) or premature infants, physiological in utero vasoconstriction could presumably be maintained by a variety of Cox4i2-independent mechanisms (28).
The search for the oxygen sensor and its sensing mechanism has been hampered in the past by controversial findings with regard to whether an increase or decrease of ROS trigger HPV. These discrepant findings may be attributed to localized and time-dependent regulation of hypoxic ROS release, as well as by varying experimental protocols and limitations of fluorescent sensors, which are prone to autoxidation (29). By using ESR measurements in samples that were frozen in the hypoxic environment, we excluded possible artifacts due to re-oxygenation and autoxidation of the probe. Doing so, we found increased release of superoxide dependent on Cox4i2 during acute hypoxia in PASMCs. Cox4i2 triggered superoxide release was independent of upstream components specific to PASMCs, as increased hypoxia-induced superoxide production was detected in Cox4i2 but not in Cox4i1 overexpressing CMT cells. Next, we sought to identify specific properties of Cox4i2 predisposing this subunit as oxygen sensor. The HPV oxygen sensor has to act quickly and in a fully reversible manner. Cysteine residues are established redox sensitive residues, which can adopt multiple reversible modifications (30), such as the formation of an inter- or intra-molecular disulfide bond. We noticed that in contrast to Cox4i1, which does not contain any cysteines, mammalian Cox4i2 contains three such residues. One of them, cysteine 109 (C109), is located in the inner mitochondrial membrane, at a position within the highest oxygen gradient (20). Indeed, hypoxia-induced superoxide release was dependent on the presence of C109, as overexpression of the WT form of Cox4i2 could rescue superoxide release in Cox4i2−/− PASMCs and CMT cells, but not variants of Cox4i2 in which C109 was mutated. However, mutation of the other two cysteines also abolished superoxide release, suggesting a possible role in oxygen sensing. The latter two cysteines are located in the matrix domain of COX subunit 4, near the key regulatory ADP/ATP binding site (20). Thus, given their unique location, modification of any of the three cysteines could lead to changes in COX function. As cysteine bond formation can either be achieved by redox dependent modifications, or by oxygen-dependent enzymatic modifications shown to occur in the endoplasmic reticulum during post-translational modifications (31), we hypothesize that a decreased disulfide bond formation may occur during hypoxia, thereby modifying the function of Cox4i2 and subsequently causing ROS production in PASMCs. However, we cannot exclude that indirect effects, like decreased electron flux through COX in hypoxia or alterations in the ATP/ADP ratio, act as a trigger for Cox4i2 dependent hypoxic signaling. These hypotheses should be investigated in future experiments.
Next, we focused on downstream signaling mechanisms of increased mitochondrial superoxide release. First, we demonstrated that mitochondrial superoxide release is essential for HPV, as inhibition of mitochondrial superoxide with MitoTempo and S3QEL2specifically inhibited HPV (although there was some minor effect of MitoTempo on U46619- and potassium-induced vasoconstriction, suggesting some effects on hypoxia-independent pathways). Although MitoTempo is used as a SOD mimetic, which should decrease superoxide, and increase H2O2 concentration, there is evidence that MitoTempo rather acts as unspecific antioxidant, possibly decreasing both ROS types, as a decrease of H2O2 was measured during MitoTempo application and it has been shown that the similar substance MitoTempol is intracellularly converted to MitoTempol-H, which has antioxidant but not SOD-mimetic properties (26, 32). Thus, in order to confirm that indeed inhibition of mitochondrial superoxide release and not increased H2O2 mediates HPV, we applied S3QEL, which has been shown to inhibit superoxide release from mitochondrial complex III (27). This agent inhibited HPV, again specifically compared to U46619-, and potassium-induced vasoconstriction. Moreover, hypoxia induced an increase in cytosolic H2O2 concentration in WT, but not Cox4i2−/− PASMCs. Therefore, superoxide generated under hypoxia can be converted to H2O2 which is likely acting as the mediator for HPV instead of the less diffusible and unstable superoxide molecule (29). Subsequently, H2O2 or other ROS can interact with ion channels that lead to a release of calcium from intracellular stores and its influx through the plasma membrane by cellular membrane depolarization (3) that has been shown to be essential for HPV (22). We found that Cox4i2−/− PASMCs showed hyperpolarized basal cellular membrane potential and less hypoxia-induced depolarization than WT PASMCs. Hypoxic membrane potential reached a value of −25±1 mV in WT PASMCs, sufficient to activate voltage dependent calcium channels, such as L-type and T-type calcium channels, which can cause intracellular calcium increases leading to HPV (6, 33). In contrast, the cellular membrane potential of Cox4i2−/− PASMCs depolarized only up to −39±2 mV not reaching the threshold for activation of voltage dependent calcium channels, which has been shown to be at a membrane potential between −40 and −30 mV in mouse PASMCs (34). However, after addition of H2O2 the cellular membrane potential in Cox4i2−/− PASMCs during hypoxia depolarized to the level of WT PASMCs under hypoxia. These findings indicate that by substitution of the ROS portion, which is missing in the Cox4i2−/− PASMCs, levels of membrane depolarization were reached that are sufficient to activate cellular calcium inflow. Along these lines, hypoxia-induced Kv channel inhibition, which plays an important role in HPV (3, 6) was decreased in Cox4i2−/− PASMCs and could be restored to WT level by application of H2O2. Our data are in accordance with an investigation showing that application of a membrane-permeable form of H2O2, t-butyl hydroperoxide, inhibited Kv-currents and induced a contractile response in rat pulmonary arteries (35). Moreover, in a study investigating oxygen sensing in the carotid body, application of H2O2 showed effects on potassium channel properties that mimicked hypoxia (36). However, in contrast to our data it has been shown that antioxidants (e.g. DTT, duroquinone) can inhibit Kv-channels or potassium currents in PASMCs and that oxidants (e.g. DTBNP, diamide) can activate them, leading to vasodilation (37–40). Such discrepancies may be explained by different effects of oxidizing/reducing agents and H2O2 in the different studies as well as the site of the pulmonary vasculature (microvessels versus larger vessels) from which the PASMCs were derived. Further studies are needed to clarify these discrepancies.
Interestingly a portion of the hypoxia-induced Kv channel inhibition was independent of Cox4i2. We thus conclude that Cox4i2 is an essential oxygen sensor for HPV induction and that HPV is a multifactorial event which might be caused by a combination of 1) Cox4i2-derived H2O2 generation, 2) primary oxygen sensing properties of the Kv channel itself (41) and/or 3) ROS originating from non-mitochondrial sources. Moreover, Cox4i2 presence sensitizes PASMCs for the HPV response by normoxic inhibition of Kv channels and slight depolarization of the cellular membrane potential. Speculatively, a different and very localized superoxide release from mitochondria close to Kv channels (42) might play a role, as we could detect a tendency for decreased superoxide concentration in Cox4i2−/− PASMCs under normoxia.
To address the mechanism by which Cox4i2 triggered superoxide release, we considered that a subtle inhibition of mitochondrial respiration during hypoxia, when the terminal substrate oxygen becomes limiting, could result in a shift toward higher reduction rates of the upstream ETC components, resulting in increased lifetime of the ubisemiquinone radical at the outer ubiquinol binding site of complex III (13). In line with this reasoning, inhibition of complex IV by cyanide or nitric oxide could increase mitochondrial ROS production at complex III in non-pulmonary cells (43, 44). Indeed, a small inhibition of respiration could be detected in the hypoxia sensitive range of PASMCs in the current study and in previous investigations (11). However, it has been challenged whether inhibition of complex IV is sufficient for ROS production at complex III (10, 45, 46). Nevertheless, ROS production at complexes I and III can be promoted by increased ΔΨm (47), which we and others previously found in PASMCs during perfusion with hypoxic medium (11, 12). Along these lines, we now detected that the increase in ΔΨm in WT PASMCs was absent in Cox4i2−/− PASMCs. In presence of the superoxide scavengers MitoTempol and S3QEL2 we could still detect the hypoxia-induced increase of ΔΨm in WT cells, suggesting that alterations of ΔΨm are independent and apparently not downstream of mitochondrial ROS release. Our findings are in line with previous reports showing that Cox4i2 increases ROS production in astrocytes (48, 49). To further investigate the underlying mechanism for the increase in ΔΨm we found increased mitochondrial matrix pH, suggesting an increase of the proton gradient ΔpH contributing to increased ΔΨm. Furthermore, alkalization of the mitochondrial matrix has also been shown to promote superoxide release of complex III (50). In search for the mechanism of increased ΔΨm, we tested, wether increased respiration, quantified by measurement of oxygen consumption, caused mitochondrial hyperpolarization. In this regard, it was suggested by previous studies that Cox4i2 can enhance mitochondrial respiration by increasing complex IV activity thereby increasing proton pumping of the respiratory chain complexes and ΔΨm. (18, 51). However, our own measurements showed decreased respiration in mild hypoxia in both WT and Cox4i2−/− PASMCs. Moreover, we could not detect increased respiration in WT PASMCs compared to Cox4i2−/− PASMCs and in Cox4i2 overexpressing CMT cells compared to Cox4i1-overexpressing cells, neither in normoxia nor in hypoxia. These data are consistent with our previous measurements, showing similar respiration rates in Cox4i2−/− and WT mitochondria (18), and with a further study employing Cox4i2 knockdown in cell lines when cultivated under normoxia (52). Thus, increased complex IV activity resulting in increased respiration and proton pumping does not seem to cause mitochondrial hyperpolarization in acute hypoxia in WT PASMCs. One alternative mechanism could be that Cox4i2 decreases the proton slip of complex IV or increases the proton pumping stoichiometry (53), mechanisms that are, however, not generally accepted (17) and should be investigated further.
In contrast to the effect of Cox4i2 deficiency in acute hypoxia, we found only minor effects on sustained and chronic hypoxic responses of the pulmonary vasculature. This could be caused by the fact that for sustained HPV additional sensitizing effects of the endothelium have been proposed (3), and in chronic hypoxia-induced vascular alterations oxygen-dependent stabilization of HIF-1α might be a predominant signaling mechanism. This hypothesis is in line with the finding that HIF-1α, a major driver of chronic hypoxia-induced PH, was increased to a similar extent in WT and Cox4i2−/− mice, which is consistent with a recent study showing that mitochondrial ROS production does not stabilize HIF-1α (54). Along these lines it has been suggested for the pulmonary vasculature that even a decrease of H2O2 can stabilize HIF in PASMCs (55). This is, however, in conflict with substantial literature suggesting that specifically mitochondrial ROS can stabilize HIF-1α (27, 56, 57). One explanation is, that different ROS sources exist in the pulmonary vasculature exerting different effects. In this regard ROS derived from NADPH oxidases may contribute to HIF-1α stabilization in hypoxia in PASMCs (58). We can only speculate that our model may induced a counterregulation of different ROS sources in chronic hypoxia and thus maintain ROS and HIF-1α stabilization in the Cox4i2−/− PASMCs. However, further studies will be necessary to address the above discrepancies. Moreover, our study does not exclude that mitochondria regulate HIF-1α protein levels by ROS-independent mechanisms, e.g. increased availability of Krebs cycle substrates (59). The tendency of reduced proliferation in isolated PASMCs under PDGF-stimulation in hypoxia is in line with the only slightly lesser degree of muscularization in vivo. In vivo other cell types than PASMCs, particularly endothelial cells releasing vasoactive and proliferative factors, may influence vascular remodeling and result in the observed in vivo differences of vascular remodeling. Moreover, differences in shear stress due to different HPV may cause the observed alterations in WT and Cox4i2−/− mice in chronic hypoxia. Our study thus supports the concept that distinct mechanisms underlie acute and chronic oxygen sensing and are in accordance with previous studies showing that HPV and hypoxia-induced PH are mediated by different mechanisms (22, 60).
In summary, our study revealed that the mitochondrial COX subunit 4i2 in mouse and human PASMCs is essential for acute oxygen sensing in the pulmonary vasculature. Our data indicate that in response to hypoxia, Cox4i2 is required to hyperpolarize mitochondria of PASMCs with a subsequent superoxide increase, contributing to the cellular membrane depolarization that eventually results in intracellular calcium increase and HPV (Figure 7). Interestingly, chronic hypoxia-induced pulmonary vascular remodeling was only mildly affected. These findings substantially expand the understanding of hypoxia sensing and signaling in HPV, mechanisms with fundamental significance for lung biology and disease.
Figure 7. Proposed mechanism for oxygen sensing in HPV.
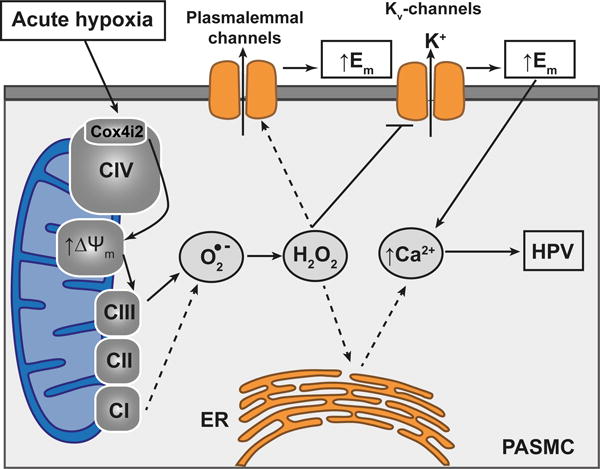
The hypoxia-induced effects require Cox4i2 resulting in ΔΨm hyperpolarization and superoxide production preferentially at complex III of the ETC, thereby linking the hitherto described mechanism of hypoxia-induced superoxide production at complex III (Waypa et al., 2013; Schumacker, 2011) and the novel oxygen sensing mechanism of Cox4i2. Superoxide can be converted to hydrogen peroxide and interact with intracellular and plasma membrane ion channels, e.g. TRPC6, directly or indirectly, and can inhibit Kv-channels, leading to cellular membrane depolarization and finally the intracellular calcium increase resulting in HPV. CI, CII, and CIII: mitochondrial complexes I, II and III; C109: cysteine residue at position 109, Cox4i2: cytochrome c oxidase subunit 4 isoform 2, O2−: superoxide anion; H2O2: hydrogen peroxide; ER: endoplasmic reticulum; PASMC: pulmonary arterial smooth muscle cell; HPV: hypoxia-induced pulmonary vasoconstriction; Ca2+: calcium, ΔΨm: mitochondrial membrane potential; K+: potassium, Kv: voltage gated potassium channels; Em: cellular membrane potential. Solid lines indicate pathways that have been shown by the current investigation.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
Vasoconstriction of precapillary vessels in response to alveolar hypoxia, known as hypoxic pulmonary vasoconstriction (HPV), is unique for the pulmonary circulation and essential to maintain arterial oxygenation.
HPV is mediated by cellular membrane depolarization, which is caused by inhibition of voltage gated potassium (Kv) channels, leading to an intracellular calcium increase in pulmonary arterial smooth muscle cells (PASMCs).
Mitochondria are essential for acute HPV.
What New Information Does This Article Contribute?
The hitherto incompletely clarified oxygen sensing and signaling mechanism underlying HPV essentially depends on the isoform 2 of subunit 4 of the cytochrome c oxidase (Cox4i2) in mitochondrial complex IV.
The main downstream mediator of HPV is superoxide which, after conversion to hydrogen peroxide (H2O2) inhibits Kv channels.
Chronic hypoxic pulmonary vascular responses are regulated differently than those of acute hypoxia.
HPV controls lung gas exchange by adaptation of blood perfusion to local alveolar ventilation. It was first described in 1946 by von-Euler and Liljestrand. A disturbance of HPV (e.g. caused by pneumonia) can result in life-threatening hypoxemia. The underlying oxygen sensing and signaling mechanisms are still not fully resolved. Mitochondria have been proposed to trigger HPV by reactive oxygen species (ROS) release. Previous studies provided conflicting data, suggesting either increased or decreased release of ROS from mitochondrial complex I or III during hypoxia. Our study now identified Cox4i2, a subunit of mitochondrial complex IV, as a possible new oxygen sensor for acute HPV. Cox4i2 is needed for induction of superoxide release during acute hypoxia in PASMCs, the site of HPV. We propose that superoxide, after conversion to H2O2, inhibits voltage-dependent potassium membrane channels, inducing PASMC membrane depolarization and an increase in intracellular calcium concentration, known to induce HPV. Thus, we now link previous evidence for an increase in ROS by mitochondria to Cox4i2 of mitochondrial complex IV. These findings help to understand the acute hypoxic signaling pathway and may facilitate the development of therapeutics to re-activate disturbed HPV.
Acknowledgments
The authors thank K. Quanz, K. Homberger, E. Kappes, N. Schupp and I. Breitenborn-Mueller for technical assistance.
SOURCES OF FUNDING
Research described in this article was supported by the Center for Molecular Medicine and Genetics, Wayne State University School of Medicine (M.H.), the Henry L. Brasza Endowment, a grant supplement from the NIH (GM48517; L.I.G.), and by the DFG (WE 1978/4-2 and SFB 1213 project A06; N.W.; N. S.).
Nonstandard Abbreviations and Acronyms
- ΔΨm
mitochondrial membrane potential
- C
cysteine
- Ca2+
calcium
- CMT
carcinoma cell line
- CO2
carbon dioxide
- Cox4i2
COX subunit 4
- ESR
electron spin resonance
- ETC
electron transport chain
- H2O2
hydrogen peroxide
- HIF-1α
Hypoxia–inducible factor 1-alpha
- HPV
hypoxic pulmonary vasoconstriction
- KCl
potassium–chloride
- Kv channel
potassium channel
- N2
nitrogen
- O2
oxygen
- PAP
pulmonary arterial pressure
- PASMCs
pulmonary arterial smooth muscle cells
- PDGF
platelet-derived growth factor
- PH
pulmonary hypertension
- ROS
reactive oxygen species
- S3QEL2
1-(3,4-Dimethylphenyl)-N,N-dipropyl-1H-pyrazolo[3,4-d]pyrimidin-4-amine
- SOD
superoxide dismutase
- TRPC
transient receptor potential channel
- WT
wild type
Footnotes
Portions of the doctoral thesis of Felix Jonas are incorporated into this report.
DISCLOSURE
None.
References
- 1.Ward JP. Oxygen sensors in context. Biochim Biophys Acta. 2008;1777:1–14. doi: 10.1016/j.bbabio.2007.10.010. [DOI] [PubMed] [Google Scholar]
- 2.Sylvester JT, Shimoda LA, Aaronson PI, Ward JP. Hypoxic pulmonary vasoconstriction. Physiol Rev. 2012;92:367–520. doi: 10.1152/physrev.00041.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sommer N, Strielkov I, Pak O, Weissmann N. Oxygen sensing and signal transduction in hypoxic pulmonary vasoconstriction. Eur Respir J. 2016;47:288–303. doi: 10.1183/13993003.00945-2015. [DOI] [PubMed] [Google Scholar]
- 4.Von Euler US, Liljestrand G. Observations on the pulmonary arterial blood pressure in the cat. Acta Physiol Scand. 1946;12:301–320. [Google Scholar]
- 5.Puthiyachirakkal M, Mhanna MJ. Pathophysiology, management, and outcome of persistent pulmonary hypertension of the newborn: a clinical review. Front Pediatr. 2013;1:23. doi: 10.3389/fped.2013.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Veit F, Pak O, Brandes RP, Weissmann N. Hypoxia-dependent reactive oxygen species signaling in the pulmonary circulation: focus on ion channels. Antioxid Redox Signal. 2015;22:537–552. doi: 10.1089/ars.2014.6234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Waypa GB, Marks JD, Guzy R, Mungai PT, Schriewer J, Dokic D, Schumacker PT. Hypoxia triggers subcellular compartmental redox signaling in vascular smooth muscle cells. Circ Res. 2010;106:526–535. doi: 10.1161/CIRCRESAHA.109.206334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Waypa GB, Marks JD, Guzy RD, Mungai PT, Schriewer JM, Dokic D, Ball MK, Schumacker PT. Superoxide generated at mitochondrial complex III triggers acute responses to hypoxia in the pulmonary circulation. Am J Respir Crit Care Med. 2013;187:424–432. doi: 10.1164/rccm.201207-1294OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu SS. Cooperation of a “reactive oxygen cycle” with the Q cycle and the proton cycle in the respiratory chain–superoxide generating and cycling mechanisms in mitochondria. J Bioenerg Biomembr. 1999;31:367–376. doi: 10.1023/a:1018650103259. [DOI] [PubMed] [Google Scholar]
- 10.Bleier L, Drose S. Superoxide generation by complex III: from mechanistic rationales to functional consequences. Biochim Biophys Acta. 2013;1827:1320–1331. doi: 10.1016/j.bbabio.2012.12.002. [DOI] [PubMed] [Google Scholar]
- 11.Sommer N, Pak O, Schorner S, Derfuss T, Krug A, Gnaiger E, Ghofrani HA, Schermuly RT, Huckstorf C, Seeger W, Grimminger F, Weissmann N. Mitochondrial cytochrome redox states and respiration in acute pulmonary oxygen sensing. Eur Respir J. 2010;36:1056–1066. doi: 10.1183/09031936.00013809. [DOI] [PubMed] [Google Scholar]
- 12.Michelakis ED, Hampl V, Nsair A, Wu X, Harry G, Haromy A, Gurtu R, Archer SL. Diversity in mitochondrial function explains differences in vascular oxygen sensing. Circ Res. 2002;90:1307–1315. doi: 10.1161/01.res.0000024689.07590.c2. [DOI] [PubMed] [Google Scholar]
- 13.Schumacker PT. Lung cell hypoxia: role of mitochondrial reactive oxygen species signaling in triggering responses. Proc Am Thorac Soc. 2011;8:477–484. doi: 10.1513/pats.201103-032MW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dromparis P, Sutendra G, Michelakis ED. The role of mitochondria in pulmonary vascular remodeling. J Mol Med (Berl) 2010;88:1003–1010. doi: 10.1007/s00109-010-0670-x. [DOI] [PubMed] [Google Scholar]
- 15.Ball MK, Waypa GB, Mungai PT, Nielsen JM, Czech L, Dudley VJ, Beussink L, Dettman RW, Berkelhamer SK, Steinhorn RH, Shah SJ, Schumacker PT. Regulation of hypoxia-induced pulmonary hypertension by vascular smooth muscle hypoxia-inducible factor-1alpha. Am J Respir Crit Care Med. 2014;189:314–324. doi: 10.1164/rccm.201302-0302OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Veith C, Schermuly RT, Brandes R, Weissmann N. Molecular mechanisms of HIF-induced pulmonary arterial smooth muscle cell alterations in pulmonary hypertension. J Physiol. 2015 doi: 10.1113/JP270689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kadenbach B. Intrinsic and extrinsic uncoupling of oxidative phosphorylation. Biochim Biophys Acta. 2003;1604:77–94. doi: 10.1016/s0005-2728(03)00027-6. [DOI] [PubMed] [Google Scholar]
- 18.Huttemann M, Lee I, Gao X, Pecina P, Pecinova A, Liu J, Aras S, Sommer N, Sanderson TH, Tost M, Neff F, Aguilar-Pimentel JA, Becker L, Naton B, Rathkolb B, Rozman J, Favor J, Hans W, Prehn C, Puk O, Schrewe A, Sun M, Hofler H, Adamski J, Bekeredjian R, Graw J, Adler T, Busch DH, Klingenspor M, Klopstock T, Ollert M, Wolf E, Fuchs H, Gailus-Durner V, Hrabe de Angelis M, Weissmann N, Doan JW, Bassett DJ, Grossman LI. Cytochrome c oxidase subunit 4 isoform 2-knockout mice show reduced enzyme activity, airway hyporeactivity, and lung pathology. FASEB J. 2012;26:3916–3930. doi: 10.1096/fj.11-203273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huttemann M, Lee I, Liu J, Grossman LI. Transcription of mammalian cytochrome c oxidase subunit IV-2 is controlled by a novel conserved oxygen responsive element. FEBS J. 2007;274:5737–5748. doi: 10.1111/j.1742-4658.2007.06093.x. [DOI] [PubMed] [Google Scholar]
- 20.Huttemann M, Kadenbach B, Grossman LI. Mammalian subunit IV isoforms of cytochrome c oxidase. Gene. 2001;267:111–123. doi: 10.1016/s0378-1119(01)00385-7. [DOI] [PubMed] [Google Scholar]
- 21.Aras S, Pak O, Sommer N, Finley R, Jr, Huttemann M, Weissmann N, Grossman LI. Oxygen-dependent expression of cytochrome c oxidase subunit 4-2 gene expression is mediated by transcription factors RBPJ, CXXC5 and CHCHD2. Nucleic Acids Res. 2013;41:2255–2266. doi: 10.1093/nar/gks1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weissmann N, Dietrich A, Fuchs B, Kalwa H, Ay M, Dumitrascu R, Olschewski A, Storch U, Mederos y Schnitzler M, Ghofrani HA, Schermuly RT, Pinkenburg O, Seeger W, Grimminger F, Gudermann T. Classical transient receptor potential channel 6 (TRPC6) is essential for hypoxic pulmonary vasoconstriction and alveolar gas exchange. Proc Natl Acad Sci U S A. 2006;103:19093–19098. doi: 10.1073/pnas.0606728103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weissmann N, Kuzkaya N, Fuchs B, Tiyerili V, Schafer RU, Schutte H, Ghofrani HA, Schermuly RT, Schudt C, Sydykov A, Egemnazarow B, Seeger W, Grimminger F. Detection of reactive oxygen species in isolated, perfused lungs by electron spin resonance spectroscopy. Respir Res. 2005;6:86. doi: 10.1186/1465-9921-6-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dikalov SI, Li W, Mehranpour P, Wang SS, Zafari AM. Production of extracellular superoxide by human lymphoblast cell lines: comparison of electron spin resonance techniques and cytochrome C reduction assay. Biochem Pharmacol. 2007;73:972–980. doi: 10.1016/j.bcp.2006.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kojonazarov B, Sydykov A, Pullamsetti SS, Luitel H, Dahal BK, Kosanovic D, Tian X, Majewski M, Baumann C, Evans S, Phillips P, Fairman D, Davie N, Wayman C, Kilty I, Weissmann N, Grimminger F, Seeger W, Ghofrani HA, Schermuly RT. Effects of multikinase inhibitors on pressure overload-induced right ventricular remodeling. Int J Cardiol. 2013;167:2630–2637. doi: 10.1016/j.ijcard.2012.06.129. [DOI] [PubMed] [Google Scholar]
- 26.Rocco-Machado N, Cosentino-Gomes D, Meyer-Fernandes JR. Modulation of Na+/K+ ATPase Activity by Hydrogen Peroxide Generated through Heme in L. amazonensis. PLoS One. 2015;10:e0129604. doi: 10.1371/journal.pone.0129604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Orr AL, Vargas L, Turk CN, Baaten JE, Matzen JT, Dardov VJ, Attle SJ, Li J, Quackenbush DC, Goncalves RL, Perevoshchikova IV, Petrassi HM, Meeusen SL, Ainscow EK, Brand MD. Suppressors of superoxide production from mitochondrial complex III. Nat Chem Biol. 2015;11:834–836. doi: 10.1038/nchembio.1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gao Y, Raj JU. Regulation of the pulmonary circulation in the fetus and newborn. Physiol Rev. 2010;90:1291–1335. doi: 10.1152/physrev.00032.2009. [DOI] [PubMed] [Google Scholar]
- 29.Sommer N, Dietrich A, Schermuly RT, Ghofrani HA, Gudermann T, Schulz R, Seeger W, Grimminger F, Weissmann N. Regulation of hypoxic pulmonary vasoconstriction: basic mechanisms. Eur Respir J. 2008;32:1639–1651. doi: 10.1183/09031936.00013908. [DOI] [PubMed] [Google Scholar]
- 30.Murray CI, Van Eyk JE. Chasing cysteine oxidative modifications: proteomic tools for characterizing cysteine redox status. Circ Cardiovasc Genet. 2012;5:591. doi: 10.1161/CIRCGENETICS.111.961425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koritzinsky M, Levitin F, van den Beucken T, Rumantir RA, Harding NJ, Chu KC, Boutros PC, Braakman I, Wouters BG. Two phases of disulfide bond formation have differing requirements for oxygen. J Cell Biol. 2013;203:615–627. doi: 10.1083/jcb.201307185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Trnka J, Blaikie FH, Logan A, Smith RA, Murphy MP. Antioxidant properties of MitoTEMPOL and its hydroxylamine. Free Radic Res. 2009;43:4–12. doi: 10.1080/10715760802582183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Firth AL, Remillard CV, Platoshyn O, Fantozzi I, Ko EA, Yuan JX. Functional ion channels in human pulmonary artery smooth muscle cells: Voltage-dependent cation channels. Pulm Circ. 2011;1:48–71. doi: 10.4103/2045-8932.78103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ko EA, Wan J, Yamamura A, Zimnicka AM, Yamamura H, Yoo HY, Tang H, Smith KA, Sundivakkam PC, Zeifman A, Ayon RJ, Makino A, Yuan JX. Functional characterization of voltage-dependent Ca(2+) channels in mouse pulmonary arterial smooth muscle cells: divergent effect of ROS. Am J Physiol Cell Physiol. 2013;304:C1042–1052. doi: 10.1152/ajpcell.00304.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cogolludo A, Frazziano G, Cobeno L, Moreno L, Lodi F, Villamor E, Tamargo J, Perez-Vizcaino F. Role of reactive oxygen species in Kv channel inhibition and vasoconstriction induced by TP receptor activation in rat pulmonary arteries. Ann N Y Acad Sci. 2006;1091:41–51. doi: 10.1196/annals.1378.053. [DOI] [PubMed] [Google Scholar]
- 36.Fernandez-Aguera MC, Gao L, Gonzalez-Rodriguez P, Pintado CO, Arias-Mayenco I, Garcia-Flores P, Garcia-Perganeda A, Pascual A, Ortega-Saenz P, Lopez-Barneo J. Oxygen Sensing by Arterial Chemoreceptors Depends on Mitochondrial Complex I Signaling. Cell Metab. 2015;22:825–837. doi: 10.1016/j.cmet.2015.09.004. [DOI] [PubMed] [Google Scholar]
- 37.Park MK, Lee SH, Ho WK, Earm YE. Redox agents as a link between hypoxia and the responses of ionic channels in rabbit pulmonary vascular smooth muscle. Exp Physiol. 1995;80:835–842. doi: 10.1113/expphysiol.1995.sp003891. [DOI] [PubMed] [Google Scholar]
- 38.Reeve HL, Weir EK, Nelson DP, Peterson DA, Archer SL. Opposing effects of oxidants and antioxidants on K+ channel activity and tone in rat vascular tissue. Exp Physiol. 1995;80:825–834. doi: 10.1113/expphysiol.1995.sp003890. [DOI] [PubMed] [Google Scholar]
- 39.Olschewski A, Hong Z, Peterson DA, Nelson DP, Porter VA, Weir EK. Opposite effects of redox status on membrane potential, cytosolic calcium, and tone in pulmonary arteries and ductus arteriosus. Am J Physiol Lung Cell Mol Physiol. 2004;286:L15–22. doi: 10.1152/ajplung.00372.2002. [DOI] [PubMed] [Google Scholar]
- 40.Schach C, Xu M, Platoshyn O, Keller SH, Yuan JX. Thiol oxidation causes pulmonary vasodilation by activating K+ channels and inhibiting store-operated Ca2+ channels. Am J Physiol Lung Cell Mol Physiol. 2007;292:L685–698. doi: 10.1152/ajplung.00276.2006. [DOI] [PubMed] [Google Scholar]
- 41.Osipenko ON, Tate RJ, Gurney AM. Potential role for kv3.1b channels as oxygen sensors. Circ Res. 2000;86:534–540. doi: 10.1161/01.res.86.5.534. [DOI] [PubMed] [Google Scholar]
- 42.Firth AL, Gordienko DV, Yuill KH, Smirnov SV. Cellular localization of mitochondria contributes to Kv channel-mediated regulation of cellular excitability in pulmonary but not mesenteric circulation. Am J Physiol Lung Cell Mol Physiol. 2009;296:L347–360. doi: 10.1152/ajplung.90341.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dawson TL, Gores GJ, Nieminen AL, Herman B, Lemasters JJ. Mitochondria as a source of reactive oxygen species during reductive stress in rat hepatocytes. Am J Physiol. 1993;264:C961–967. doi: 10.1152/ajpcell.1993.264.4.C961. [DOI] [PubMed] [Google Scholar]
- 44.Srinivasan S, Avadhani NG. Cytochrome c oxidase dysfunction in oxidative stress. Free Radic Biol Med. 2012;53:1252–1263. doi: 10.1016/j.freeradbiomed.2012.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen Q, Vazquez EJ, Moghaddas S, Hoppel CL, Lesnefsky EJ. Production of reactive oxygen species by mitochondria: central role of complex III. J Biol Chem. 2003;278:36027–36031. doi: 10.1074/jbc.M304854200. [DOI] [PubMed] [Google Scholar]
- 46.Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. 2003;552:335–344. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Misiak M, Singh S, Drewlo S, Beyer C, Arnold S. Brain region-specific vulnerability of astrocytes in response to 3-nitropropionic acid is mediated by cytochrome c oxidase isoform expression. Cell Tissue Res. 2010;341:83–93. doi: 10.1007/s00441-010-0995-3. [DOI] [PubMed] [Google Scholar]
- 49.Singh S, Misiak M, Beyer C, Arnold S. Cytochrome c oxidase isoform IV-2 is involved in 3-nitropropionic acid-induced toxicity in striatal astrocytes. Glia. 2009;57:1480–1491. doi: 10.1002/glia.20864. [DOI] [PubMed] [Google Scholar]
- 50.Selivanov VA, Zeak JA, Roca J, Cascante M, Trucco M, Votyakova TV. The role of external and matrix pH in mitochondrial reactive oxygen species generation. J Biol Chem. 2008;283:29292–29300. doi: 10.1074/jbc.M801019200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Arnold S. Cytochrome c oxidase and its role in neurodegeneration and neuroprotection. Adv Exp Med Biol. 2012;748:305–339. doi: 10.1007/978-1-4614-3573-0_13. [DOI] [PubMed] [Google Scholar]
- 52.Fukuda R, Zhang H, Kim JW, Shimoda L, Dang CV, Semenza GL. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell. 2007;129:111–122. doi: 10.1016/j.cell.2007.01.047. [DOI] [PubMed] [Google Scholar]
- 53.Hinkle PC, Kumar MA, Resetar A, Harris DL. Mechanistic stoichiometry of mitochondrial oxidative phosphorylation. Biochemistry. 1991;30:3576–3582. doi: 10.1021/bi00228a031. [DOI] [PubMed] [Google Scholar]
- 54.Chua YL, Dufour E, Dassa EP, Rustin P, Jacobs HT, Taylor CT, Hagen T. Stabilization of hypoxia-inducible factor-1alpha protein in hypoxia occurs independently of mitochondrial reactive oxygen species production. J Biol Chem. 2010;285:31277–31284. doi: 10.1074/jbc.M110.158485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Paulin R, Michelakis ED. The metabolic theory of pulmonary arterial hypertension. Circ Res. 2014;115:148–164. doi: 10.1161/CIRCRESAHA.115.301130. [DOI] [PubMed] [Google Scholar]
- 56.Martinez-Reyes I, Diebold LP, Kong H, Schieber M, Huang H, Hensley CT, Mehta MM, Wang T, Santos JH, Woychik R, Dufour E, Spelbrink JN, Weinberg SE, Zhao Y, DeBerardinis RJ, Chandel NS. TCA Cycle and Mitochondrial Membrane Potential Are Necessary for Diverse Biological Functions. Mol Cell. 2016;61:199–209. doi: 10.1016/j.molcel.2015.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Guzy RD, Hoyos B, Robin E, Chen H, Liu L, Mansfield KD, Simon MC, Hammerling U, Schumacker PT. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005;1:401–408. doi: 10.1016/j.cmet.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 58.Veith C, Schermuly RT, Brandes RP, Weissmann N. Molecular mechanisms of hypoxia-inducible factor-induced pulmonary arterial smooth muscle cell alterations in pulmonary hypertension. J Physiol. 2016;594:1167–1177. doi: 10.1113/JP270689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tormos KV, Chandel NS. Inter-connection between mitochondria and HIFs. J Cell Mol Med. 2010;14:795–804. doi: 10.1111/j.1582-4934.2010.01031.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Malczyk M, Veith C, Fuchs B, Hofmann K, Storch U, Schermuly RT, Witzenrath M, Ahlbrecht K, Fecher-Trost C, Flockerzi V, Ghofrani HA, Grimminger F, Seeger W, Gudermann T, Dietrich A, Weissmann N. Classical transient receptor potential channel 1 in hypoxia-induced pulmonary hypertension. Am J Respir Crit Care Med. 2013;188:1451–1459. doi: 10.1164/rccm.201307-1252OC. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.