

Abstract
Introduction
Daily dipeptidyl peptidase-4 (DPP-4) inhibitors are commonly used with other orally administered antihyperglycemic agents (AHA), as combination therapy, to treat Japanese patients with type 2 diabetes. When combination therapy is indicated, use of a once-weekly (q.w.) orally administered DPP-4 inhibitor might be an appropriate therapeutic option for some patients.
Methods
A 52-week trial was conducted to assess the safety and tolerability (primary objectives) and glycemic efficacy (secondary objectives) of the q.w. DPP-4 inhibitor omarigliptin as add-on therapy to five different classes of orally administered AHA [sulfonylurea (SU), glinide (GL), biguanide (BG), thiazolidinedione (TZD), or α-glucosidase inhibitor (AGI)] commonly used in Japan and having different mechanisms of drug action from DPP-4 inhibitors. The trial consisted of an initial 24-week double-blind, placebo-controlled period during which patients (stratified by background AHA) were randomized to omarigliptin 25 mg q.w. or placebo, followed by a 28-week open-label period during which patients on placebo were switched to omarigliptin.
Results
After 24 weeks, the percentages of patients with adverse events (AEs), serious AEs, drug-related AEs, AEs of symptomatic hypoglycemia, or who discontinued from trial medication because of an AE were generally similar in the omarigliptin and placebo groups, in all background AHA strata and in the overall population. From a mean baseline HbA1c of approximately 8.0%, the placebo-adjusted least-squares mean changes from baseline ranged from −0.80% (AGI stratum) to −1.16% (TZD stratum); p < 0.001 for all background AHA strata. During the open-label period, no safety signals emerged with longer-term treatment. At week 52, the change from baseline in HbA1c in the omarigliptin/omarigliptin group was similar to that of the placebo/omarigliptin group.
Conclusions
Addition of once-weekly omarigliptin to AHA therapy with an SU, GL, BG, TZD, or AGI for up to 52 weeks was generally safe and well tolerated, and provided persistent efficacy.
Clinical trial registration
ClinicalTrials.gov: NCT01697592.
Funding
MSD K.K., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA.
Electronic supplementary material
The online version of this article (doi:10.1007/s13300-017-0270-7) contains supplementary material, which is available to authorized users.
Keywords: α-Glucosidase inhibitor, Biguanide, Dipeptidyl peptidase-4, DPP-4, Glinide, Incretins, MK-3102, Oral antihyperglycemic agent, Sulfonylurea, Thiazolidinedione
Introduction
Type 2 diabetes (T2D) is a progressive disease, and with disease progression patients often require more than one antihyperglycemic agent (AHA) to maintain glycemic control. In this clinical scenario, addition of a second AHA with a complementary mechanism of action is a rational therapeutic approach. In Japan, daily DPP-4 inhibitors are commonly used as first-line therapy and often combined with other classes of orally administered AHA for the treatment of patients with T2D [1]. As a class, DPP-4 inhibitors improve glycemic control in patients with T2D by stabilizing glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic peptide, peptide hormones which enhance insulin secretion and (in the case of GLP-1) suppress glucagon release in a glucose-dependent manner [2].
Omarigliptin (MK-3102) is a highly specific, orally administered DPP-4 inhibitor with pharmacokinetic properties that allow once-weekly (q.w.) dosing [3, 4] that is presently approved in Japan. In clinical trials that compared omarigliptin to both placebo and active comparators, including the daily DPP-4 inhibitor sitagliptin, omarigliptin has been shown to have a safety, tolerability, and efficacy profile similar to that of daily DPP-4 inhibitors [5–10]. One of those trials, which supported approval in Japan, assessed omarigliptin as monotherapy in Japanese patients with T2D [6].
Herein we report the results of a clinical trial that supported approval in Japan that assessed the safety and efficacy of adding omarigliptin to the treatment of Japanese patients with T2D and inadequate glycemic control while on monotherapy with any of five different classes of orally administered AHA. Each of the five monotherapy background AHAs have a mechanism of action different from that of DPP-4 inhibitors, and all are frequently used in clinical practice in Japan [sulfonylurea (SU), glinide (GL), biguanide (BG), thiazolidinedione (TZD), and α-glucosidase inhibitor (AGI)].
Methods
Patients
Eligible patients were Japanese, male or female, treated with diet and exercise and a stable dose of an SU, GL, BG, TZD, or AGI (background AHA) as monotherapy and having an HbA1c (NGSP value) of ≥7.0% and ≤10.0%, or treated with dual combination therapy with one of the aforementioned background AHAs and another orally administered AHA and having an HbA1c of ≥6.5% and ≤9.0%. Patients treated with monotherapy were required to be on a stable dose of background AHA for at least 10 weeks if on an SU, GL, BG, or AGI and at least 14 weeks if on a TZD. Patients on dual combination therapy were required to be on a stable dose of background AHA for at least 4 weeks if on an SU, GL, BG, or AGI and at least 8 weeks if on a TZD. Patients were at least 20 years of age if not on a BG and restricted to at least 20 and less than 75 years of age if on a BG. Patients could be on one of the following background AHAs: in the SU stratum, gliclazide, glibenclamide (glyburide), or glimepiride; in the GL stratum, nateglinide, mitiglinide, or repaglinide; and in the AGI stratum, acarbose, voglibose, or miglitol. The BG and TZD strata consisted of patients on metformin and pioglitazone, respectively.
Patients were excluded from the study if they had type 1 diabetes, a history of ketoacidosis, active liver disease, significant cardiovascular disease, a history of malignancy, hematological disorders, or had been previously treated with insulin within 12 weeks prior to screening, or omarigliptin at any time.
Laboratory exclusion criteria included an estimated glomerular filtration rate (eGFR) <30 mL/min/1.73 m2 (or <60 mL/min/1.73 m2 if on a BG background AHA), alanine aminotransferase or aspartate aminotransferase more than two times the upper limit of normal, thyroid-stimulating hormone outside the central laboratory normal range, hemoglobin <11 g/dL (male) or <10 g/dL (female), triglycerides >600 mg/dL, or C-peptide <0.6 ng/mL.
Study Design
This multicenter trial consisted of two treatment periods. The initial period was a 24-week double-blind, placebo-controlled, parallel-group treatment period in which patients were randomized to omarigliptin 25 mg q.w. or matching placebo. The second period was a 28-week open-label period in which all patients were treated with omarigliptin (patients on placebo in the initial period were switched to omarigliptin). After a screening period of up to 2 weeks, patients on AHA monotherapy entered a 2-week single-blind placebo run-in period; patients on dual AHA therapy entered an 8-week period (which included a 2-week single-blind placebo run-in) during which they were discontinued from the second AHA. During the 2-week single-blind placebo run-in period, all patients took omarigliptin placebo once weekly to ensure proper understanding of the weekly dosing regimen. In addition, 2 weeks prior to randomization (week-2), all patients were required to have an HbA1c (NGSP value) ≥7.0% and ≤10.0%. At week-2, patients in all background AHA strata were required to have an fasting plasma glucose (FPG) ≤230 mg/dL and those on an SU were also required to have a FPG ≥126 mg/dL.
Patients were stratified according to their background AHA (SU, GL, BG, TZD, or AGI) and randomized in a 2:1 ratio to omarigliptin 25 mg q.w. or matching placebo. Randomization was performed using a sponsor-generated allocation schedule and an interactive voice response or integrated Web response system. Throughout the trial, patients continued on a stable dose of their background AHA, unless glycemic rescue was required or, in the case of the SU stratum, hypoglycemia occurred. Patients exceeding prespecified glycemic thresholds post-randomization (repeatedly confirmed central laboratory value of FPG >240 mg/dL for week 4–24 and FPG >200 mg/dL for week 24–52) were rescued, first with up-titration of their background AHA, then either with metformin (patients on SU or GL as background AHA) or glimepiride (patients on BG, TZD, or AGI as background AHA). If recurrent hypoglycemia occurred (defined as an FPG or fingerstick glucose ≤70 mg/dL with symptoms of hypoglycemia, or FPG or fingerstick glucose <50 mg/dL with or without symptoms of hypoglycemia) without a reasonable explanation (such as increased physical activity or skipped meal), patients whose background AHA was an SU could have the SU down-titrated; patients on other background AHAs were discontinued from trial.
All procedures followed in this trial (Omarigliptin Protocol 015; ClinicalTrials.gov: NCT01697592) were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1964, as revised in 2013. Informed consent was obtained from all patients in the trial.
Study Evaluations
The primary, secondary, and exploratory objectives of this trial were assessed for each class of background AHA. A pooled analysis of adverse events (AEs) in the overall population (i.e., across background AHA strata) was also performed. The primary objectives were to assess the safety and tolerability of the addition of omarigliptin through 24 weeks and for up to 52 weeks. Secondary objectives were to assess the effect of omarigliptin compared with placebo on change from baseline in HbA1c at week 24 and to assess the change from baseline in HbA1c for up to 52 weeks. The associated HbA1c hypothesis was that after 24 weeks, the addition of omarigliptin, compared with placebo, would provide a greater reduction from baseline in HbA1c. Exploratory objectives were to assess the effect of the addition of omarigliptin on change from baseline in FPG after 24 weeks and for up to 52 weeks.
Safety Endpoints
Safety endpoints included AEs, mean change from baseline at week 24 and week 52 in laboratory parameters, electrocardiogram (ECG), percentages of patients meeting predefined limits of change (PDLC) for laboratory and ECG parameters, vital signs, and body weight. A predefined AE of interest was symptomatic hypoglycemia.
In both periods of this trial, potential cases of pancreatitis and prespecified hypersensitivity AEs (anaphylactic reaction, angioedema, asthma-bronchospasm, erythema multiforme, Stevens–Johnson syndrome, toxic epidermal necrolysis, and drug rash with eosinophilia and systemic symptoms) were evaluated in a blinded manner by external clinical adjudication committees.
Efficacy Endpoints
Efficacy endpoints were changes from baseline in HbA1c and FPG after 24 and 52 weeks and percentages of patients at HbA1c goal of <7.0% and <6.5% for each background AHA at weeks 24 and 52.
Statistical Analyses
To avoid the confounding influence of rescue therapy, all analyses treated data collected after the initiation of rescue therapy as missing.
The population for analysis of safety data was all randomized patients who received at least one dose of trial treatment. Safety and tolerability were assessed by clinical review of all relevant parameters including AEs, laboratory tests, ECG, PDLCs, vital signs, and body weight. Events of symptomatic hypoglycemia were prespecified as AEs of interest and p values and 95% confidence intervals (CIs) for between-treatment group comparisons were calculated. For AEs with incidence of at least four patients in any treatment group, 95% CIs were calculated for between-group comparisons. In both cases the method of Miettinen and Nurminen [11] was used. For other safety endpoints, summary statistics were generated. Change from baseline in body weight at weeks 24 and 52 were analyzed using the longitudinal data analysis (LDA) method described below for HbA1c, substituting baseline body weight for HbA1c.
The primary population for efficacy analyses included all randomized patients who received at least one dose of study treatment and had a baseline or a post-randomization measurement. For analyses of the change from baseline in HbA1c at week 24, an LDA model [12] was used. The model had terms for treatment, background AHA (SU/GL/BG/TZD/AGI), prior AHA status except background AHA (yes/no), time, and the interaction of time × treatment, time × background AHA, time × prior AHA status except background AHA, background AHA × time × treatment, background AHA × time × prior AHA status except background AHA, background AHA × time × treatment × prior AHA status except background AHA, with a constraint that true mean at baseline is common to all treatment groups (which is valid owing to randomization). An unstructured covariance matrix was used to model the correlation among repeated measurements.
Changes from baseline in FPG were analyzed using the LDA method described for HbA1c, substituting the FPG values for HbA1c.
To determine the change from baseline in HbA1c at week 52, the LDA model was separately applied to each background AHA stratum as a result of non-convergence of the model when all the AHA strata were included simultaneously.
For analysis of percentages of individuals at the HbA1c goals of <7.0% and <6.5% at weeks 24 and 52, the LDA model used for the analysis of HbA1c change from baseline at weeks 24 and 52 was used to impute missing data for weeks 24 and 52 HbA1c. Each patient was categorized as a responder (satisfying the HbA1c (NGSP) specific goal of <7.0% or <6.5%) or non-responder at week 24 or week 52.
To ensure that approximately 100 patients from the SU stratum and approximately 50 patients from other background AHA strata were exposed to omarigliptin for 52 weeks and assuming an approximate 20% discontinuation rate, it was calculated that 568 patients would need to be randomized with a stratification ratio of 2:1:1:1:1 for SU, GL, BG, TZD, and AGI, respectively.
Results
Patient Disposition and Characteristics
Of 772 patients screened, 585 were randomized at 67 sites in Japan (Fig. 1). The most common reasons for patients not being randomized were meeting laboratory exclusion criteria for parameters including eGFR, AST, ALT, TSH, hemoglobin, TG, or C-peptide (50.6%) and/or not meeting inclusion criteria related to AHA regimen and HbA1c levels. The first patient was screened on October 26, 2012 and the last visit of the last patient was on May 8, 2014. Five hundred and seventy-one (97.6%) of the randomized patients completed the placebo-controlled period and 555 (94.9%) completed the open-label period on study medication. At week 24, in the overall population (all strata), 0.5% (2/389 patients) in the omarigliptin group and 2.0% (4/196 patients) in the placebo group required glycemic rescue. Through week 52, 2.8% (11/389 patients) and 2.6% (5/191 patients) required glycemic rescue.
Fig. 1.
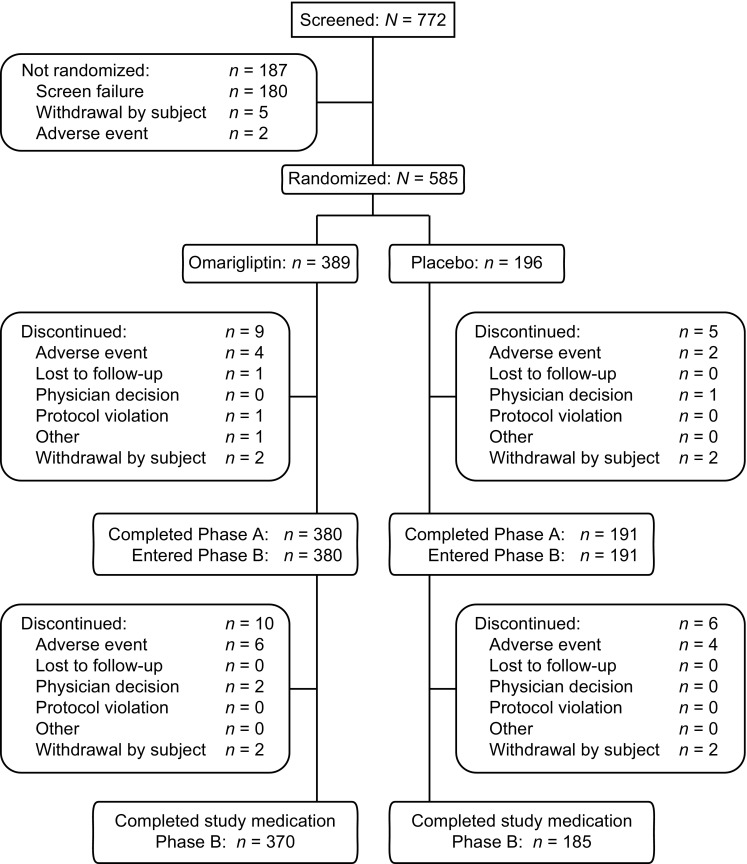
Patient disposition weeks 0–52
Baseline anthropometric and disease characteristics were generally balanced between omarigliptin and placebo treatment groups for each background AHA (Table 1). The mean age of all study patients was approximately 60 years and approximately 70% of patients were male; the mean body mass index was 25 kg/m2, the mean HbA1c level was 8.0%, the mean FPG was approximately 160 mg/dL, and the mean duration of diabetes was approximately 9.5 years. Among patients randomized, 25.6% were discontinued from dual AHA therapy (washed-off of a second AHA). The mean baseline dose and dose range of each background AHA can be found in Supplementary Table S1.
Table 1.
Baseline demographic and anthropometric characteristics of the study population
| Characteristic | Sulfonylurea | Glinide | Biguanide | Thiazolidinedione | α-Glucosidase inhibitor | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Omarigliptin n = 126 | Placebo n = 63 | Omarigliptin n = 65 | Placebo n = 34 | Omarigliptin n = 66 | Placebo n = 33 | Omarigliptin n = 65 | Placebo n = 34 | Omarigliptin n = 67 | Placebo n = 32 | |
| Age, years | 63 ± 9 | 63 ± 11 | 59 ± 11 | 61 ± 10 | 59 ± 9 | 57 ± 9 | 61 ± 10 | 61 ± 9 | 61 ± 11 | 61 ± 11 |
| Male, n (%) | 91 (72.2) | 45 (71.4) | 46 (70.8) | 27 (79.4) | 46 (69.7) | 23 (69.7) | 42 (64.6) | 27 (79.4) | 46 (68.7) | 23 (71.9) |
| Body weight, kg | 65 ± 11 | 67 ± 14 | 68 ± 13 | 69 ± 14 | 69 ± 14 | 70 ± 12 | 72 ± 14 | 71 ± 13 | 68 ± 13 | 65 ± 11 |
| BMI, kg/m2 | 24.5 ± 3.4 | 24.8 ± 3.5 | 25.1 ± 4.3 | 25.3 ± 4.1 | 25.6 ± 4.4 | 25.9 ± 3.6 | 27.0 ± 4.2 | 27.0 ± 4.9 | 25.3 ± 4.0 | 24.4 ± 3.7 |
| HbA1c, % | 8.1 ± 0.7 | 8.1 ± 0.6 | 8.0 ± 0.6 | 8.0 ± 0.8 | 7.8 ± 0.6 | 8.0 ± 0.6 | 8.2 ± 0.8 | 7.9 ± 0.8 | 7.9 ± 0.6 | 7.9 ± 0.8 |
| Range | 6.9–10.0 | 7.1–9.6 | 6.9–10.3 | 6.7–9.8 | 6.8–9.9 | 6.9–10.2 | 7.0–10.0 | 6.8–10.5 | 6.9–9.6 | 7.0–10.1 |
| FPG, mg/dL | 165.4 ± 32.0 | 170.5 ± 34.0 | 163.5 ± 29.4 | 164.4 ± 34.7 | 155.8 ± 26.1 | 157.6 ± 20.6 | 163.4 ± 33.6 | 152.4 ± 24.5 | 157.7 ± 24.3 | 159.8 ± 30.7 |
| Duration of type 2 diabetes, years | 10.7 ± 5.6 | 11.5 ± 6.3 | 8.5 ± 6.2 | 8.3 ± 5.2 | 8.4 ± 4.8 | 6.9 ± 3.5 | 9.1 ± 6.4 | 10.3 ± 5.4 | 8.4 ± 5.5 | 9.7 ± 5.8 |
| Prior AHA use, yes | 43 (34.1) | 21 (33.3) | 9 (13.8) | 4 (11.8) | 16 (24.2) | 3 (9.1) | 19 (29.2) | 6 (17.6) | 19 (28.4) | 10 (31.3) |
Data are expressed as mean ± standard deviation or frequency [n (%)]
BMI body mass index, FPG fasting plasma glucose, AHA antihyperglycemic agent
Safety
Double-Blind Placebo-Controlled Period (Day 1–Week 24)
During the double-blind, placebo-controlled period, the percentage of patients with one or more AEs, drug-related AEs, serious AEs, or who were discontinued from trial medication because of an AE was generally similar between the omarigliptin and placebo groups in all of the background AHA strata and in the overall population (Table 2).
Table 2.
Adverse events summary, specific AEs by system organ class (SOC) with incidence ≥5% in ≥1 treatment group, and incidences of symptomatic hypoglycemia during the 24-week double-blind period in the overall population and for each background AHA stratum
| Patients, n (%) | Sulfonylurea | Glinide | Biguanide | Thiazolidinedione | α-Glucosidase inhibitor | Overall | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Omarigliptin n = 126 | Placebo n = 63 | Omarigliptin n = 65 | Placebo n = 34 | Omarigliptin n = 66 | Placebo n = 33 | Omarigliptin n = 65 | Placebo n = 34 | Omarigliptin n = 67 | Placebo n = 32 | Omarigliptin n = 389 | Placebo n = 196 | |
| With one or more | ||||||||||||
| AEs | 75 (59.5) | 38 (60.3) | 35 (53.8) | 18 (52.9) | 36 (54.5) | 21 (63.6) | 39 (60.0) | 18 (52.9) | 34 (50.7) | 17 (53.1) | 219 (56.3) | 112 (57.1) |
| Drug-relateda AEs | 9 (7.1) | 5 (7.9) | 5 (7.7) | 2 (5.9) | 3 (4.5) | 0 (0.0) | 4 (6.2) | 0 (0.0) | 0 (0.0) | 1 (3.1) | 21 (5.4) | 8 (4.1) |
| Serious AEsb | 2 (1.6) | 0 (0.0) | 1 (1.5) | 1 (2.9) | 1 (1.5) | 1 (3.0) | 4 (6.2) | 0 (0.0) | 0 (0.0) | 2 (6.3) | 8 (2.1) | 4 (2.0) |
| Serious drug-relateda AEs | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Who died | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Who discontinued due to | ||||||||||||
| An AE | 0 (0.0) | 0 (0.0) | 2 (3.1) | 1 (2.9) | 0 (0.0) | 0 (0.0) | 2 (3.1) | 0 (0.0) | 0 (0.0) | 1 (3.1) | 4 (1.0) | 2 (1.0) |
| A drug-relateda AE | 0 (0.0) | 0 (0.0) | 1 (1.5) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (0.3) | 0 (0.0) |
| A serious AE | 0 (0.0) | 0 (0.0) | 1 (1.5) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (1.5) | 0 (0.0) | 0 (0.0) | 1 (3.1) | 2 (0.5) | 1 (0.5) |
| A serious drug-relateda AE | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| With specific AEs with incidence ≥5% in ≥1 treatment group, by SOCc | ||||||||||||
| Gastrointestinal disorders | ||||||||||||
| Dental caries | 4 (3.2) | 1 (1.6) | 1 (1.5) | 0 (0.0) | 0 (0.0) | 2 (6.1) | 2 (3.1) | 0 (0.0) | 1 (1.5) | 0 (0.0) | 8 (2.1) | 3 (1.5) |
| Infections and infestations | ||||||||||||
| Bronchitis | 3 (2.4) | 1 (1.6) | 0 (0.0) | 3 (8.8) | 0 (0.0) | 0 (0.0) | 1 (1.5) | 2 (5.9) | 2 (3.0) | 0 (0.0) | 6 (1.5) | 6 (3.1) |
| Gastroenteritis | 2 (1.6) | 0 (0.0) | 0 (0.0) | 2 (5.9) | 4 (6.1) | 0 (0.0) | 1 (1.5) | 1 (2.9) | 0 (0.0) | 0 (0.0) | 7 (1.8) | 3 (1.5) |
| Gingivitis | 2 (1.6) | 1 (1.6) | 0 (0.0) | 0 (0.0) | 2 (3.0) | 0 (0.0) | 0 (0.0) | 2 (5.9) | 0 (0.0) | 0 (0.0) | 4 (1.0) | 3 (1.5) |
| Influenza | 2 (1.6) | 1 (1.6) | 0 (0.0) | 1 (2.9) | 0 (0.0) | 0 (0.0) | 3 (4.6) | 2 (5.9) | 1 (1.5) | 0 (0.0) | 6 (1.5) | 4 (2.0) |
| Nasopharyngitis | 15 (11.9) | 15 (23.8) | 14 (21.5) | 7 (20.6) | 11 (16.7) | 9 (27.3) | 11 (16.9) | 5 (14.7) | 13 (19.4) | 8 (25.0) | 64 (16.5) | 44 (22.4) |
| Pharyngitis | 2 (1.6) | 1 (1.6) | 0 (0.0) | 0 (0.0) | 1 (1.5) | 2 (6.1) | 0 (0.0) | 1 (2.9) | 1 (1.5) | 0 (0.0) | 4 (1.0) | 4 (2.0) |
| Investigations | ||||||||||||
| Alanine aminotransferase increased | 0 (0.0) | 0 (0.0) | 2 (3.1) | 0 (0.0) | 2 (3.0) | 2 (6.1) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 4 (1.0) | 2 (1.0) |
| Metabolism and nutrition disorders | ||||||||||||
| Hyperglycemia | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (2.9) | 0 (0.0) | 2 (6.3) | 0 (0.0) | 3 (1.5) |
| Hypoglycemia | 7 (5.6) | 3 (4.8) | 1 (1.5) | 0 (0.0) | 3 (4.5) | 0 (0.0) | 1 (1.5) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 12 (3.1) | 3 (1.5) |
| Musculoskeletal and connective tissue disorders | ||||||||||||
| Myalgia | 0 (0.0) | 1 (1.6) | 1 (1.5) | 1 (2.9) | 0 (0.0) | 3 (9.1) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (0.3) | 5 (2.6) |
| Respiratory, thoracic, and mediastinal disorders | ||||||||||||
| Upper respiratory tract inflammation | 0 (0.0) | 0 (0.0) | 1 (1.5) | 2 (5.9) | 0 (0.0) | 1 (3.0) | 1 (1.5) | 0 (0.0) | 1 (1.5) | 1 (3.1) | 3 (0.8) | 4 (2.0) |
| With one or more AE of symptomatic hypoglycemiad | 6 (4.8) | 2 (3.2) | 1 (1.5) | 0 (0.0) | 2 (3.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 9 (2.3) | 2 (1.0) |
| Between-group difference in percentage (omarigliptin − placebo) | 1.6 (−6.5, 7.5) | – | 1.5 (−8.8, 8.3) | – | 3.0 (−7.6, 10.4) | – | 0.0 (−10.2, 5.6) | – | 0.0 (−10.8, 5.5) | – | 1.3 (−1.5, 3.5) | – |
aAssessed by the investigator to be related to the drug
bAfter database lock for the 24-week double-blind period, additional information for the non-serious AE of “cataract” in the omarigliptin group was reported. The new information included that this patient was hospitalized for “cataract” which changed this AE from non-serious to serious. Therefore this patient was not counted as “with serious adverse event” in this table. The change is reflected in the long-term analysis table
cSystem organ class defined by the Medical Dictionary for Regulatory Activities (MedDRA) classification system
dPrespecified AE of interest; symptomatic hypoglycemia: episode with clinical symptoms consistent with hypoglycemia, without regard to glucose level
The percentage of patients with AEs in any Medical Dictionary for Regulatory Activities (MedDRA) system organ class (SOC) (not shown) or in the percentage of patients with specific AEs with incidence of at least 5% in at least one treatment group in any SOC (Table 2) was also similar between the omarigliptin groups and the placebo groups in all background AHA strata.
When assessed in the overall population, the percentage of patients with AEs in any SOC was generally similar between the omarigliptin group and the placebo group, with the exception of the eye disorders SOC [3.1% (12/389 patients) versus 0.5% (1/196 patients)] and skin and subcutaneous tissue disorders SOC [5.4% (21/389 patients) versus 1.0% (2/196 patients)] where the percentage of patients with AEs was higher in the omarigliptin group compared with the placebo group (i.e., the lower limit of 95% CIs around the between-group difference versus placebo exceeded 0).
In the eye disorders SOC, the only specific AE that occurred in at least four patients in at least one treatment group was diabetic retinopathy which occurred in 1.5% (6/389) of patients in the omarigliptin group compared with 0.0% (0/196) of patients in the placebo group. The six events of diabetic retinopathy in the omarigliptin group (three patients in SU stratum, one patient in GL stratum, one patient in BG stratum, and one patient in TZD stratum) were all mild in intensity and none were assessed by the investigator to be related to trial medication (omarigliptin) or led to the discontinuation of trial medication. No other AE terms related to diabetic retinopathy (e.g., retinopathy) were reported in either treatment period.
In the skin and subcutaneous tissue disorders SOC, specific AEs that occurred in at least four patients in one or more treatment groups (omarigliptin group versus placebo group) included contact dermatitis [1.0% (4/389 patients) versus 0.5% (1/196 patients)] and eczema [1.3% (5/389 patients) versus 0.5% (1/196 patient)]. All of the AEs of contact dermatitis and eczema were nonserious, mild in intensity, and did not lead to discontinuation from trial medication. All of the AEs of contact dermatitis and three of the five cases of eczema resolved on trial medication (two cases of eczema recovered after the last dose); none of the AEs of contact dermatitis and only one AE of eczema were assessed by the investigator to be related to trial medication. In addition, the incidence of the AE of allergic dermatitis (a term similar to contact dermatitis) was 0.5% (2/389 patients) in the omarigliptin group and 0.0% (0/196 patients) in the placebo group. One of two patients experienced two events of allergic dermatitis; all of the AEs were nonserious, mild in intensity, and did not lead to discontinuation from trial medication (two of the three AEs resolved on trial medication) and none of the AEs were assessed by the investigator to be related to trial medication.
The percentage of patients with AEs of symptomatic hypoglycemia in the omarigliptin and placebo groups was generally similar in all of the background AHA strata and in the overall population (Table 2). The incidences of hypoglycemia were generally low in all background AHA strata, with the highest incidences being observed in the SU stratum. No severe hypoglycemia was reported in any background AHA stratum in either treatment group.
Open-Label Period (Week 24–Week 52)
Because of the differences in length of exposure to omarigliptin (52 weeks versus 28 weeks), no between-group comparisons can be made between the omarigliptin/omarigliptin groups and the placebo/omarigliptin groups. In the open-label period, AE incidences reported for the placebo/omarigliptin group reflect only those AEs that occurred during the 28 weeks in which patients were exposed to omarigliptin. Of the 196 patients in the placebo group, 191 were switched to omarigliptin at week 24 (see Fig. 1 for reasons for discontinuation of the five subjects).
After 52 weeks, the percentage of patients with AEs in the omarigliptin/omarigliptin groups increased in all background AHA strata compared to 24 weeks, as would be expected from the accumulation of AEs over time (Table 3). No specific AEs emerged in any of the background AHA strata that represented a safety signal associated with longer-term treatment.
Table 3.
Adverse events summary, specific AEs by SOC with incidence ≥5% and incidences of symptomatic hypoglycemia in the omarigliptin/omarigliptin treatment group through week 52 in the overall population and for each background AHA stratum
| Patients, n (%) | Sulfonylurea + omarigliptin (through week 52) n = 126 |
Glinide + omarigliptin (through week 52) n = 65 |
Biguanide + omarigliptin (through week 52) n = 66 |
Thiazolidinedione+ omarigliptin (through week 52) n = 65 |
α-Glucosidase inhibitor + omarigliptin (through week 52) n = 67 |
Overall + omarigliptin (through week 52) n = 389 |
|---|---|---|---|---|---|---|
| With one or more | ||||||
| AEs | 96 (76.2) | 49 (75.4) | 53 (80.3) | 55 (84.6) | 46 (68.7) | 299 (76.9) |
| Drug-relateda AEs | 11 (8.7) | 7 (10.8) | 2 (3.0) | 4 (6.2) | 0 (0.0) | 24 (6.2) |
| Serious AEs | 6 (4.8) | 3 (4.6) | 2 (3.0) | 4 (6.2) | 1 (1.5) | 16 (4.1) |
| Serious drug-relateda AEs | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Who died | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Who discontinued due to | ||||||
| An AE | 1 (0.8) | 3 (4.6) | 1 (1.5) | 4 (6.2) | 0 (0.0) | 9 (2.3) |
| A drug-relateda AE | 0 (0.0) | 1 (1.5) | 0 (0.0) | 1 (1.5) | 0 (0.0) | 2 (0.5) |
| A serious AE | 1 (0.8) | 2 (3.1) | 0 (0.0) | 1 (1.5) | 0 (0.0) | 4 (1.0) |
| A serious drug-relateda AE | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| With specific AEs with incidence ≥5% in ≥1 treatment group, by SOCb | ||||||
| Gastrointestinal disorders | ||||||
| Constipation | 7 (5.6) | 5 (7.7) | 1 (1.5) | 1 (1.5) | 2 (3.0) | 16 (4.1) |
| Infections and infestations | ||||||
| Bronchitis | 10 (7.9) | 1 (1.5) | 0 (0.0) | 2 (3.1) | 3 (4.5) | 16 (4.1) |
| Gastroenteritis | 2 (1.6) | 0 (0.0) | 4 (6.1) | 1 (1.5) | 1 (1.5) | 8 (2.1) |
| Influenza | 4 (3.2) | 0 (0.0) | 0 (0.0) | 4 (6.2) | 1 (1.5) | 9 (2.3) |
| Nasopharyngitis | 34 (27.0) | 23 (35.4) | 24 (36.4) | 19 (29.2) | 19 (28.4) | 119 (30.6) |
| Metabolism and nutrition disorders | ||||||
| Hypoglycemia | 11 (8.7) | 2 (3.1) | 3 (4.5) | 2 (3.1) | 0 (0.0) | 18 (4.6) |
| Musculoskeletal and connective tissue disorders | ||||||
| Arthralgia | 1 (0.8) | 5 (7.7) | 1 (1.5) | 3 (4.6) | 1 (1.5) | 11 (2.8) |
| Back pain | 6 (4.8) | 4 (6.2) | 4 (6.1) | 5 (7.7) | 4 (6.0) | 23 (5.9) |
| Nervous system disorders | ||||||
| Headache | 2 (1.6) | 0 (0.0) | 2 (3.0) | 4 (6.2) | 3 (4.5) | 11 (2.8) |
| With one or more AE of symptomatic hypoglycemiac | 10 (7.9) | 2 (3.1) | 2 (3.0) | 1 (1.5) | 0 (0.0) | 15 (3.9) |
aAssessed by the investigator to be related to the drug
bSystem organ class defined by the Medical Dictionary for Regulatory Activities (MedDRA) classification system
cPrespecified AE of interest; symptomatic hypoglycemia: episode with clinical symptoms consistent with hypoglycemia, without regard to glucose level
In the omarigliptin/omarigliptin group (all strata) the incidences of the AEs of diabetic retinopathy, contact dermatitis, and eczema over the 52-week treatment period (placebo-controlled and open-label periods) were 3.1% (12/389 patients), 1.8% (7/389 patients), and 3.3% (13/389 patients), respectively. In the omarigliptin/omarigliptin group the incidence of allergic dermatitis was 0.8% (3/389 patients) and the incidence of dermatitis was 0.3% (1/389 patients).
During the open-label period (28 weeks), the incidences of the AEs of diabetic retinopathy, contact dermatitis, and eczema in the placebo/omarigliptin group were 0.5% (1/191 patients), 0.5% (1/191 patient), and 2.6% (5/191 patients), respectively. In the placebo/omarigliptin group the incidence of dermatitis was 0.5% (1/191 patients) and there were no AEs of allergic dermatitis.
Over 52 weeks, the incidence of hypoglycemia in the omarigliptin/omarigliptin groups remained generally low, with the highest incidence observed in the SU stratum, compared to other background AHA strata (Table 3). No severe hypoglycemia was reported in any of the background AHA strata.
The types of AEs observed in the placebo/omarigliptin groups in the open-label period (28 weeks) after their switch to omarigliptin 25 mg q.w. were consistent with those observed in the omarigliptin group during the double-blind period (Supplementary Table S2).
Other Safety Evaluations
In both treatment periods, in each background AHA stratum and in the overall population, there were no clinically meaningful changes from baseline in laboratory safety measures, including lipids, liver function, renal function, muscle enzymes, or hematological parameter, and there were no clinically meaningful between-group differences in the percentages of patients meeting the predefined limits of change (PDLC) for safety measures. Omarigliptin had a neutral effect on body weight (Supplementary Table S3). There were no clinically meaningful changes from baseline in heart rate, blood pressure, or ECG intervals (including QTc). There were no adjudication-confirmed cases of pancreatitis or predefined hypersensitivity reaction AEs.
Efficacy
Double-Blind Placebo-Controlled Period (Day 1–Week 24)
Changes from baseline in HbA1c are summarized in Table 4. In all strata, the difference from placebo in change from baseline in HbA1c with omarigliptin treatment was significant (p < 0.001). The profile of change from baseline in HbA1c over time showed that in all strata the majority of the treatment effect was observed by weeks 12–16 (Fig. 2). The percentages of patients achieving an HbA1c of <7.0% and HbA1c of <6.5% at 24 weeks are shown in Supplementary Table S4.
Table 4.
Efficacy endpoints at weeks 24 and 52
| Parameter week 24 | SU + omarigliptin n = 126 |
SU + placebo n = 63 |
GL + omarigliptin n = 65 |
GL + placebo n = 34 |
BG + omarigliptin n = 66 |
BG + placebo n = 33 |
TZD + omarigliptin n = 65 |
TZD + placebo n = 34 |
AGI + omarigliptin n = 67 |
AGI + placebo n = 32 |
|---|---|---|---|---|---|---|---|---|---|---|
| HbA1c, % | ||||||||||
| Baseline | 8.1 ± 0.7 | 8.1 ± 0.6 | 8.0 ± 0.6 | 8.0 ± 0.8 | 7.8 ± 0.6 | 8.0 ± 0.6 | 8.2 ± 0.8 | 7.9 ± 0.8 | 7.9 ± 0.6 | 7.9 ± 0.8 |
| Change from baselinea | −0.84 (−0.94, −0.73) | 0.09 (−0.06, 0.24) | −0.68 (−0.89, −0.48) | 0.30 (−0.04, 0.64) | −0.94 (−1.10, −0.78) | −0.02 (−0.35, 0.31) | −0.88 (−1.04, −0.73) | 0.28 (0.03, 0.53) | −0.74 (−0.89, −0.59) | 0.06 (−0.16, 0.28) |
| Change vs. placebob | −0.93c (−1.10, −0.75) | – | −0.98c (−1.37, −0.60) | – | −0.92c (−1.29, −0.56) | – | −1.16c (−1.45, −0.88) | – | −0.80c (−1.06, −0.54) | – |
| FPG, mg/dL | ||||||||||
| Baseline | 165.4 ± 32.0 | 170.5 ± 34.0 | 163.5 ± 29.4 | 164.4 ± 34.7 | 155.8 ± 26.1 | 157.6 ± 20.6 | 163.4 ± 33.6 | 152.4 ± 24.5 | 157.7 ± 24.3 | 159.8 ± 30.7 |
| Change from baselinea | −24.4 (−28.9, −19.9) | −6.8 (−12.7, −0.9) | −19.3 (−27.9, −10.7) | 2.0 (−11.9, 16.0) | −29.0 (−36.0, −22.0) | −14.4 (−27.4, −1.4) | −28.4 (−35.0, −21.8) | −4.5 (−14.4, 5.3) | −20.4 (−26.8, −14.0) | −8.3 (−17.0, 0.4) |
| Change vs. placebob | −17.6c (−24.3, −10.8) | – | −21.3d (−36.5, −6.1) | – | −14.6d (−28.4, −0.8) | – | −23.9c (−34.7, −13.0) | – | −12.1d (−21.9, −2.3) | – |
| Parameter week 52 | SU + omarigliptin/omarigliptin | SU + placebo/omarigliptin | GL + omarigliptin/omarigliptin | GL + placebo/omarigliptin | BG + omarigliptin/omarigliptin | BG + placebo/omarigliptin | TZD + omarigliptin/omarigliptin | TZD + placebo/omarigliptin | AGI + omarigliptin/omarigliptin | AGI + placebo/omarigliptin |
|---|---|---|---|---|---|---|---|---|---|---|
| HbA1c, % | ||||||||||
| Change from baselinea | −0.45 (−0.58, −0.32) | −0.63 (−0.82, −0.45) | −0.59 (−0.84, −0.34) | −0.43 (−0.88, 0.02) | −0.63 (−0.82, −0.45) | −0.62 (−0.99, −0.24) | −0.63 (−0.82, −0.44) | −0.65 (−0.93, −0.36) | −0.60 (−0.77, −0.43) | −0.79 (−1.03, −0.55) |
| FPG, mg/dL | ||||||||||
| Change from baselinea | −8.3 (−14.1, −2.4) | −19.7 (−27.5, −11.9) | −14.7 (−24.1, −5.4) | −4.8 (−21.1, 11.6) | −16.6 (−23.6, −9.7) | −23.4 (−36.7, −10.1) | −17.2 (−23.6, −10.8) | −9.7 (−19.7, 0.4) | −9.3 (−15.1, −3.4) | −22.3 (−30.5, −14.0) |
aLeast-squares (LS) mean [95% confidence interval (CI)] based on longitudinal data analysis models described in the “Statistical Analyses” section
bDifference in LS means (95% CI)
c p < 0.001
d p < 0.05
SU sulfonylurea, GL glinide, BG biguanide, TZD thiazolidinedione, AGI α-glucosidase inhibitor
Fig. 2.
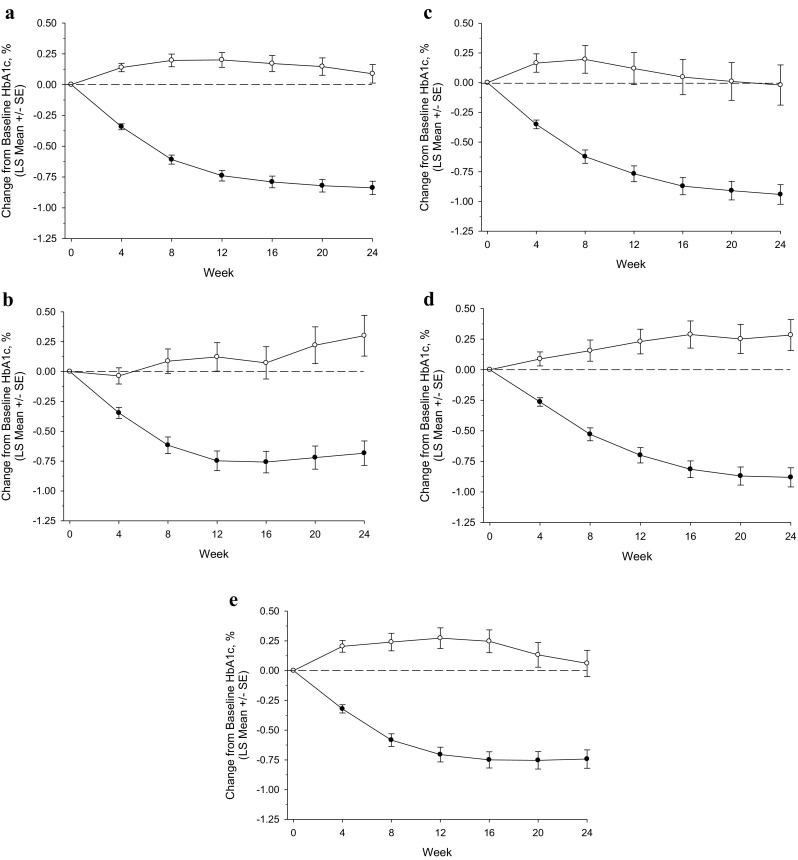
HbA1c change from baseline (%) through week 24 by background AHA; a sulfonylurea; b glinide; c biguanide; d thiazolidinedione; e α-glucosidase inhibitor; filled circle omarigliptin, open circle placebo; based on the longitudinal data analysis model described in the “Statistical Analyses” section
Changes from baseline in FPG are summarized in Table 4. The profile of change from baseline in FPG over time showed that in all strata the majority of the treatment effect was observed by week 4 and significant reductions were maintained throughout the remainder of the 24-week treatment period (data not shown).
Open-Label Period (Week 24–Week 52)
Changes from baseline in HbA1c in the omarigliptin/omarigliptin groups are summarized in Table 4 and shown in Fig. 3. At week 52, persistent reductions in HbA1c levels were observed in the omarigliptin/omarigliptin groups regardless of background AHA. After the switch from placebo to omarigliptin, reductions in HbA1c were observed in all placebo/omarigliptin groups (Table 4, Supplementary Fig. S1). At week 52, reductions in FPG levels were observed in the omarigliptin/omarigliptin groups regardless of background AHA. In the placebo/omarigliptin groups, FPG levels at week 52 were similar to the omarigliptin/omarigliptin groups (Table 4). The percentages of patients achieving an HbA1c of <7.0% and HbA1c of <6.5% at 52 weeks are shown in Supplementary Table S4.
Fig. 3.

HbA1c change from baseline (%) through week 52 in the omarigliptin/omarigliptin treatment groups by background AHA; filled circle sulfonylurea; open circle glinide; filled inverted triangle biguanide; filled triangle thiazolidinedione; filled square α-glucosidase inhibitor; based on the longitudinal data analysis model described in the “Statistical Analyses” section
Discussion
In the present trial in Japanese patients with T2D and inadequate glycemic control on monotherapy with one of five orally administered AHA classes (SU, GL, BG, TZD, or AGI), the addition of treatment with once-weekly omarigliptin for up to 52 weeks was generally safe and well tolerated and provided persistent glycemic efficacy. The orally administered background AHAs included in this study were those commonly used in Japan at the time of the planning and initiation of the trial.
The initial 24-week double-blind period provided an assessment of safety, tolerability, and efficacy compared with placebo, which is a comparator that is considered to provide the most rigorous assessment of the intrinsic safety and efficacy of a test compound. The second period of the trial, a 28-week open-label period, allowed the assessment of safety, tolerability, and efficacy over a longer time period in the omarigliptin/omarigliptin groups.
During the 24-week double-blind period, no notable between-group differences were observed in the percentage of patients with AEs in any background AHA stratum or in the overall population. However, in the overall population, a higher incidence of AEs in the eye disorder SOC and skin and subcutaneous disorder SOC in the omarigliptin group compared with the placebo group led to a review of AEs with the highest incidences in those SOCs (which included diabetic retinopathy, contact dermatitis, and eczema) across the omarigliptin phase 2/3 program.
In a pooled analysis of omarigliptin phase 2/3 trials (conducted both in Japan and outside of Japan) that were placebo- and active-controlled and that included 4467 omarigliptin-exposed patients and 3900 non-exposed, with a median duration of exposure of approximately 398 days, the incidences of AEs of diabetic retinopathy, contact dermatitis, and eczema were reviewed and found to be similar in the omarigliptin-exposed and non-exposed groups (unpublished data; for a description of the trials included in the pooled analysis see Supplemental Appendix).
In that pooled analysis of phase 2/3 omarigliptin trials, there were no notable between-group differences in the incidences of AEs of diabetic retinopathy (0.5% in both treatment groups) or the related AE term retinopathy (0.0% in the omarigliptin-exposed versus 0.2% in the non-exposed group; Supplementary Table S5).
In the pooled analysis there were no imbalances in the incidences of the AE of contact dermatitis or the related AE terms dermatitis, atopic dermatitis, and allergic dermatitis (Supplementary Table S5). The incidences of the AE of eczema in the omarigliptin-exposed and non-exposed groups in the pooled analysis were 0.6% versus 0.4% (Supplementary Table S5). The pooled analysis suggests that the imbalances in diabetic retinopathy and contact dermatitis observed in this trial are not clinically meaningful. The AE eczema is noted in the labels of one or more of the presently marketed daily DPP-4 inhibitors in Japan.
During the treatment period (24-week placebo-controlled period + 28-week open-label period), no specific AEs emerged that represented a safety signal associated with longer-term drug administration. In the open-label period, the types of AEs observed in the placebo/omarigliptin group were qualitatively similar to those observed in the omarigliptin group during the double-blind period.
A low incidence of hypoglycemia was observed in both treatment periods (double-blind and open-label) across the background AHA strata, with the exception of the SU stratum. This observation is consistent with the glucose-dependent mechanism of action of DPP-4 inhibitors and the observed higher incidence of hypoglycemia when DPP-4 inhibitors are administered with agents that cause hypoglycemia, such as sulfonylureas [13–15].
The magnitude of reductions of HbA1c and FPG were consistent with that observed for daily DPP-4 inhibitors in previous trials in Japanese patients with T2D [16–25]. The reductions from baseline in HbA1c and FPG at week 52 were clinically meaningful, but slightly less than that observed at week 24. However, the reductions at week 52 in the omarigliptin/omarigliptin groups were comparable to those observed in the placebo/omarigliptin groups despite the difference in duration of treatment with omarigliptin (52 weeks versus 28 weeks). This latter fact suggests that the observed increase in HbA1c at week 52 relative to week 24 was possibly due to a loss of trial effect and/or disease progression rather than a loss of omarigliptin efficacy. Similar changes in HbA1c over 52 weeks have been reported in several trials that assessed daily DPP-4 inhibitors as monotherapy and add-on therapy in Japanese patients with T2D [22–25].
The main limitation of this trial is that the conclusions that can be drawn about the safety and tolerability of the addition of omarigliptin to the five classes of AHA in Japanese patients with T2D are based on the relatively limited numbers of randomized patients. In addition, longer-term safety as defined by 52 weeks of exposure was only assessed in the omarigliptin/omarigliptin group. A wider clinical experience with omarigliptin will further define the safety, tolerability, and efficacy of this agent.
Conclusions
The safety and tolerability profile of once-weekly omarigliptin observed in this trial appears to be consistent with the known safety and tolerability profile of daily DPP-4 inhibitors [26, 27]. The results of this trial suggest that omarigliptin has the potential to be a useful therapeutic option when combination therapy is indicated for the treatment of T2D. In recent years, a patient-centered approach has been recommended for the treatment of T2D [28, 29]. A once-weekly, orally administered AHA has the potential to be a therapeutic option for the individualized care of patients with T2D (e.g., when the patient prefers a weekly regimen; in cases where poor adherence to daily medication has been identified as a barrier to achieving therapeutic goals; or when home healthcare is required and provided only intermittently).
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
Financial and editorial support for this study was provided by MSD K.K., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA. Merck & Co, Inc., Kenilworth, NJ, USA provided funding for this manuscript along with all related article processing fees. Editorial assistance in preparation of this manuscript was provided by Jennifer Rotonda and Michele McColgan of Merck & Co., Inc., Kenilworth, NJ, USA. All authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this manuscript, take responsibility for the integrity of the work as a whole, and have given final approval for the version to be published. Ira Gantz, Taro Okamoto, Yuka Ito, Asako Sato, Kotoba Okuyama, Edward A O’Neill, Samuel S. Engel, and Eseng Lai are responsible for the work described in this paper. Ira Gantz, Taro Okamoto, Yuka Ito, Asako Sato, Kotoba Okuyama, Samuel S. Engel, and Eseng Lai conceived, designed, and/or planned the study. Kotoba Okuyama analyzed the data. Ira Gantz, Taro Okamoto, Yuka Ito, Kotoba Okuyama, Edward A O’Neill, Samuel S. Engel, and Eseng Lai interpreted the results. Ira Gantz, Taro Okamoto, and Edward A O’Neill drafted the manuscript. Ira Gantz, Taro Okamoto, Yuka Ito, Asako Sato, Kotoba Okuyama, Samuel S. Engel, and Eseng Lai critically reviewed and/or revised the manuscript for important intellectual content. All authors provided final approval of the version to be published and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Disclosures
Ira Gantz is an employee of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA, and own stock and/or hold stock options in the company. Edward A. O’Neill is an employee of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA, and own stock and/or hold stock options in the company. Samuel S. Engel is an employee of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA, and own stock and/or hold stock options in the company. Eseng Lai is an employee of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA, and own stock and/or hold stock options in the company. Taro Okamoto is an employee of MSD K.K., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA, and own stock and/or hold stock options in the Company. Yuka Ito is an employee of MSD K.K., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA, and own stock and/or hold stock options in the Company. Asako Sato is an employee of MSD K.K., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA, and own stock and/or hold stock options in the Company. Kotoba Okuyama is an employee of MSD K.K., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA, and own stock and/or hold stock options in the Company. Omarigliptin Study 015 Group investigators are listed in Appendix 1.
Compliance with Ethics Guidelines
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1964, as revised in 2013. Informed consent was obtained from all patients included in the study.
Data Availability
Merck & Co., Inc.’s data sharing policy, including restrictions, is available at http://engagezone.merck.com/ds_documentation.php. Requests for access to the study data can be submitted through the EngageZone site or via email to dataaccess@merck.com.
Open Access
This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Footnotes
Omarigliptin Study 015 Group investigators are listed in Electronic Supplementary Material Appendix 1.
Enhanced content To view enhanced content for this article go to http://www.medengine.com/Redeem/A498F060150613D2.
References
- 1.Seino Y, Kuwata H, Yabe D. Incretin-based drugs for type 2 diabetes: focus on East Asian perspectives. J Diabetes Investig. 2016;7(Suppl 1):102–109. doi: 10.1111/jdi.12490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pratley RE, Salsali A. Inhibition of DPP-4: a new therapeutic approach for the treatment of type 2 diabetes. Curr Med Res Opin. 2007;23:919–931. doi: 10.1185/030079906X162746. [DOI] [PubMed] [Google Scholar]
- 3.Krishna R, Addy C, Tatosian D, et al. Pharmacokinetics and pharmacodynamics of omarigliptin, a once-weekly dipeptidyl peptidase-4 (DPP-4) inhibitor, after single and multiple doses in healthy subjects. J Clin Pharmacol. 2016;56:1528–1537. doi: 10.1002/jcph.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Biftu T, Sinha-Roy R, Chen P, et al. Omarigliptin (MK-3102): a novel long-acting DPP-4 inhibitor for once-weekly treatment of type 2 diabetes. J Med Chem. 2014;57:3205–3212. doi: 10.1021/jm401992e. [DOI] [PubMed] [Google Scholar]
- 5.Sheu WH, Gantz I, Chen M, et al. Safety and efficacy of omarigliptin (MK-3102), a novel once-weekly DPP-4 inhibitor for the treatment of patients with type 2 diabetes. Diabetes Care. 2015;38:2106–2114. doi: 10.2337/dc15-0109. [DOI] [PubMed] [Google Scholar]
- 6.Gantz I, Okamoto T, Ito Y, Okuyama K, Engel SS. Effect of omarigliptin, a novel once-weekly DPP-4 inhibitor, in Japanese patients with type 2 diabetes: a placebo- and sitagliptin-controlled trial. Diabetologia. 2014;57:S55. doi: 10.1111/dom.12988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goldenberg R, Gantz I, Andryuk PJ, et al. Randomized clinical trial comparing the efficacy and safety of treatment with the once-weekly dipeptidyl peptidase-4 (DPP-4) inhibitor omarigliptin or the once-daily DPP-4 inhibitor sitagliptin in patients with type 2 diabetes inadequately controlled on metformin monotherapy. Diabetes Obes Metab. 2017;19:394–400. doi: 10.1111/dom.12832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Handelsman Y, Lauring B, Gantz I, et al. A randomized, double-blind, non-inferiority trial evaluating the efficacy and safety of omarigliptin, a once-weekly DPP-4 inhibitor, or glimepiride in patients with type 2 diabetes inadequately controlled on metformin monotherapy. Curr Med Res Opin. 2017:1–19. doi:10.1080/03007995.2017.1335638. [DOI] [PubMed]
- 9.Addition of omarigliptin (MK-3102) to participants with type 2 diabetes mellitus who have inadequate glycemic control on combination therapy with glimepiride and metformin (MK-3102-022). Results posted at ClinicalTrials.gov: NCT01704261. https://clinicaltrials.gov/ct2/show/NCT01704261. Accessed 30 May 2017.
- 10.Shankar RR, Inzucchi SE, Scarabello V, et al. A randomized clinical trial evaluating the efficacy and safety of the once-weekly dipeptidyl peptidase-4 inhibitor omarigliptin in patients with type 2 diabetes inadequately controlled on metformin monotherapy. Curr Med Res Opin. 2017:1–20. doi:10.1080/03007995.2017.1335637. [DOI] [PubMed]
- 11.Miettinen O, Nurminen M. Comparative analysis of two rates. Stat Med. 1985;4:213–226. doi: 10.1002/sim.4780040211. [DOI] [PubMed] [Google Scholar]
- 12.Liang KY, Zeger SL. Longitudinal data analysis of continuous and discrete responses for pre-post designs. Indian J Stat. 2000;62:134–148. [Google Scholar]
- 13.Owens DR, Swallow R, Dugi KA, Woerle HJ. Efficacy and safety of linagliptin in persons with type 2 diabetes inadequately controlled by a combination of metformin and sulphonylurea: a 24-week randomized study. Diabet Med. 2011;28:1352–1361. doi: 10.1111/j.1464-5491.2011.03387.x. [DOI] [PubMed] [Google Scholar]
- 14.Moses RG, Round E, Shentu Y, et al. A randomized clinical trial evaluating the safety and efficacy of sitagliptin added to the combination of sulfonylurea and metformin in patients with type 2 diabetes mellitus and inadequate glycemic control. J Diabetes. 2016;8:701–711. doi: 10.1111/1753-0407.12351. [DOI] [PubMed] [Google Scholar]
- 15.Hermansen K, Kipnes M, Luo E, Fanurik D, Khatami H, Stein P. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor, sitagliptin, in patients with type 2 diabetes mellitus inadequately controlled on glimepiride alone or on glimepiride and metformin. Diabetes Obes Metab. 2007;9:733–745. doi: 10.1111/j.1463-1326.2007.00744.x. [DOI] [PubMed] [Google Scholar]
- 16.Iwamoto Y, Taniguchi T, Nonaka K, et al. Dose-ranging efficacy of sitagliptin, a dipeptidyl peptidase-4 inhibitor, in Japanese patients with type 2 diabetes mellitus. Endocr J. 2010;57:383–394. doi: 10.1507/endocrj.K09E-272. [DOI] [PubMed] [Google Scholar]
- 17.Tajima N, Kadowaki T, Odawara M, Nishii M, Taniguchi T, Arjona Ferreira JC. Addition of sitagliptin to ongoing glimepiride therapy in Japanese patients with type 2 diabetes over 52 weeks leads to improved glycemic control. Diabetol Int. 2011;2:32–44. doi: 10.1007/s13340-011-0022-2. [DOI] [Google Scholar]
- 18.Tajima N, Kadowaki T, Odawara M, et al. Safety and efficacy of addition of sitagliptin to rapid-acting insulin secretagogues for glycemic control, including post-prandial hyperglycemia, among Japanese with type 2 diabetes mellitus. Diabetol Int. 2016;7:155–166. doi: 10.1007/s13340-015-0230-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kadowaki T, Tajima N, Odawara M, Nishii M, Taniguchi T, Ferreira JC. Addition of sitagliptin to ongoing metformin monotherapy improves glycemic control in Japanese patients with type 2 diabetes over 52 weeks. J Diabetes Investig. 2013;4:174–181. doi: 10.1111/jdi.12001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kashiwagi A, Kadowaki T, Tajima N, et al. Sitagliptin added to treatment with ongoing pioglitazone for up to 52 weeks improves glycemic control in Japanese patients with type 2 diabetes. J Diabetes Investig. 2011;2:381–390. doi: 10.1111/j.2040-1124.2011.00120.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tajima N, Kadowaki T, Okamoto T, et al. Sitagliptin added to voglibose monotherapy improves glycemic control in patients with type 2 diabetes. J Diabetes Investig. 2013;4:595–604. doi: 10.1111/jdi.12116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Araki E, Kawamori R, Inagaki N, et al. Long-term safety of linagliptin monotherapy in Japanese patients with type 2 diabetes. Diabetes Obes Metab. 2013;15:364–371. doi: 10.1111/dom.12039. [DOI] [PubMed] [Google Scholar]
- 23.Konya H, Yano Y, Matsutani S, et al. Profile of saxagliptin in the treatment of type 2 diabetes: focus on Japanese patients. Ther Clin Risk Manag. 2014;10:547–558. doi: 10.2147/TCRM.S46076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Seino Y, Miyata Y, Hiroi S, Hirayama M, Kaku K. Efficacy and safety of alogliptin added to metformin in Japanese patients with type 2 diabetes: a randomized, double-blind, placebo-controlled trial with an open-label, long-term extension study. Diabetes Obes Metab. 2012;14:927–936. doi: 10.1111/j.1463-1326.2012.01620.x. [DOI] [PubMed] [Google Scholar]
- 25.Seino Y, Hiroi S, Hirayama M, Kaku K. Efficacy and safety of alogliptin added to sulfonylurea in Japanese patients with type 2 diabetes: a randomized, double-blind, placebo-controlled trial with an open-label, long-term extension study. J Diabetes Investig. 2012;3:517–525. doi: 10.1111/j.2040-1124.2012.00226.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Engel SS, Round E, Golm GT, Kaufman KD, Goldstein BJ. Safety and tolerability of sitagliptin in type 2 diabetes: pooled analysis of 25 clinical studies. Diabetes Ther. 2013;4:119–145. doi: 10.1007/s13300-013-0024-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.US prescribing information for JANUVIA (sitagliptin) Tablets, 2006. http://www.merck.com/product/usa/pi_circulars/j/januvia/januvia_pi.pdf. Updated 01/2017.
- 28.Inzucchi SE, Bergenstal RM, Buse JB, et al. Management of hyperglycemia in type 2 diabetes: a patient-centered approach: position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD) Diabetes Care. 2012;35:1364–1379. doi: 10.2337/dc12-0413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.American Diabetes Association. ADA standards of medical care in diabetes—2017. Diabetes Care. 2017;40(Suppl 1):S1–135.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Merck & Co., Inc.’s data sharing policy, including restrictions, is available at http://engagezone.merck.com/ds_documentation.php. Requests for access to the study data can be submitted through the EngageZone site or via email to dataaccess@merck.com.