

Summary
Heart failure is highly prevalent and is associated with substantial mortality and morbidity, despite the many treatment options currently available. We propose a potential new approach to treating heart disease: promoting cardiogenesis from existing cardiac myocytes by inhibiting the Hippo pathway, a “stop growth” pathway in the heart.
Keywords: congestive heart failure, cardiogenesis, new paradigm for treating heart disease
Heart failure (HF) is the third leading cause of cardiovascular death in the United States. It is a syndrome associated with high mortality, considerable morbidity, and an economic burden that is often relentlessly progressive. The American Heart Association and American College of Cardiology classify HF into 4 stages: Stage A, presence of risk factors, but with normal cardiac function and structure; Stage B, subclinical changes in left ventricular (LV) structure and/or function; Stage C, clinical HF; and Stage D, advanced HF.
Coronary artery disease is present in approximately half of patients with new-onset clinical HF and is especially prevalent in patients with a reduced LV ejection fraction. Data from Framingham show a rise in post–myocardial infarction (MI) clinical HF rates between 1971 and 2000, which is primarily due to an increase in HF incidence in the early post-MI period. It has been speculated that because intervention and thrombolytic therapy have improved survival in patients with MI, the population of patients at risk of post-MI HF has grown.
Major clinical risk factors for HF include age, male sex, hypertension, electrocardiographic evidence of LV hypertrophy, MI, diabetes mellitus, valvular heart disease, and obesity. Cardiac toxins that sometimes cause HF include chemotherapeutic drugs (doxorubicin, daunorubicin, cyclophosphamide, and 5-fluorouracil). Structural risk factors that contribute to HF include LV dilation, increased LV mass, LV systolic dysfunction, and LV diastolic dysfunction. Viral infection of the heart can lead to HF, and genetic risk factors, including family history of cardiomyopathy and genetic mutations and polymorphisms, can cause HF.
More than half a million new cases of HF are recognized in the United States each year, and their incidence is expected to increase further to 772,000 new cases each year by 2040. This increase is expected because of the aging of the population and the fact that both men's and women's risk of HF doubles with each successive decade after the age of 45 years. The lifetime risk of developing HF is estimated to be 20% for men and women. Medical therapy for heart failure resulting from LV systolic dysfunction includes diuretics, angiotensin-converting enzyme (ACE) inhibitors, angiotensin receptor blockers (ARBs), beta blockers (metoprolol or carvedilol), and salt-restricted diets.
In part because cardiomyocytes have long been considered terminally differentiated, cell therapy has been investigated as an alternative approach to treating HF. Intracardiac injections of allogenic mesenchymal stem cells (from the bone marrows of youthful donors),1 resident cardiac C-kit expressing cells,2 or both are being actively investigated. The notion that cardiomyocytes are unable to renew has recently been reevaluated. Rigorous C-14 dating approaches reveal that human cardiomyocytes have a measurable, although inefficient, capacity to renew during normal homeostasis.3 Interestingly, manipulation of important genetic pathways in mice indicates that renewal capacity can be significantly improved.4-6
We wish to propose a potential new approach to the future treatment of the damaged human heart. This strategy is to promote cardiogenesis from existing cardiac myocytes by inhibiting a “stop growth” pathway in the heart: the Hippo pathway, which is activated postnatally in mammals, to prevent continuous increase in heart size.
The mammalian core Hippo signaling components include the STE20-like kinases MST1 and MST2. These MST kinases, complexed with the Salvador scaffold protein, phosphorylate the large tumor suppressor (LATS) homolog kinases. YAP and TAZ, two related transcriptional co-activators that are downstream Hippo signaling components, are phosphorylated by LATS kinases, resulting in their removal from the nucleus and inhibition of transcriptional activity. YAP and TAZ partner with transcription factors, including TEADs, to transcriptionally activate genes that promote cardiomyocyte proliferation (Figure 1A).
Figure 1. Hippo activity in adult cardiomyocytes.
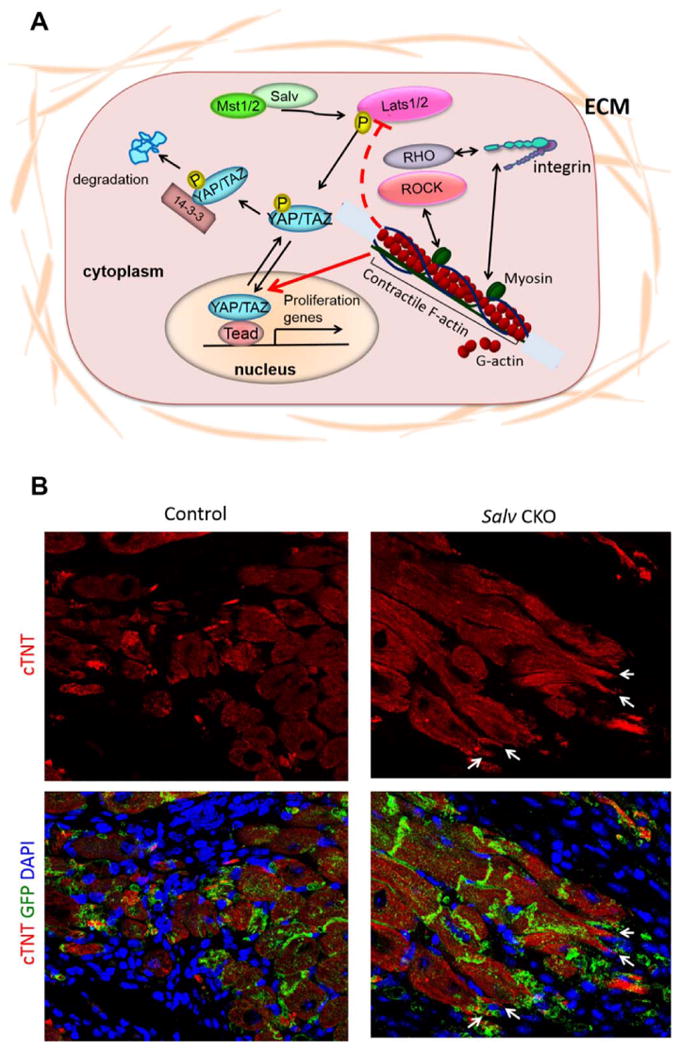
A) Diagram of the Hippo pathway. The Hippo pathway is a kinase cascade composed of the core Mst and Lats kinases that regulate the phosphorylation status of downstream effectors Yap and Taz. Physiologic inputs that regulate Hippo pathway activity include signals from the extracellular matrix. The actin cytoskeleton also regulates Yap activity. Phospho-Yap is excluded from the nucleus and transcriptionally inactive. B) Phenotypes of Hippo deficient border zone cardiomyocytes. After experimental myocardial infarction in adult mouse hearts, Hippo-deficient (Salv CKO) cardiomyocytes extend sarcomere-filled protrusions. Control cardiomyocytes never make protrusions, suggesting that protrusion formation is required for efficient cardiomyocyte renewal. GFP marks the Myh6-expressing cardiomyocyte lineage.
Studies in mice have shown that Hippo signaling inhibits cardiomyocyte proliferation during development to control heart size.7-9 Salvador-deficient hearts develop severe cardiomegaly from cardiomyocyte hyperplasia, increasing 2.5-fold in size.5,7 When Hippo signaling pathway components are “knocked out” genetically, or YAP is overexpressed, before an MI is experimentally created in the murine heart, cardiac myocytes regenerate through cardiogenesis, resulting in near-complete regeneration of the injured heart muscle (Figure 1B).7,10 In Hippo pathway loss-of-function hearts, cardiomyocytes increase progression through mitosis, and the proportion of mononucleated cardiomyocytes increases.5 The cytoskeletal phenotype of Hippo deficient border zone cardiomyocytes is very different from control cardiomyocytes (Figure 2). In Hippo mutants, cardiomyocytes extend sarcomere filled protrusions into the scar suggesting a potential beneficial interaction between cardiomyocytes and fibroblasts. Similar pre-clinical studies will also be necessary in a larger animal model, such as the porcine model with experimentally created MI and HF.
Although safety needs to be demonstrated, we believe that stimulating cardiogenesis directly by inhibiting the Hippo signaling pathway will prove to be an important addition to the options for treating HF. In a broader context, further studies should be undertaken to identify new intracardiac pathways that inhibit cardiac myogenesis and those capable of promoting cardiac myogenesis directly. Other strategies that are being used in preclinical studies include the identification of micro-RNAs that stimulate cardiogenesis11 and preclinical efforts to convert fibroblasts and other noncardiac cells into contracting myocytes in vitro and in vivo.12 Others are attempting to develop cardiac patches and scaffolds with cells that contribute to cardiomyogenesis.13,14 The cell-based and gene therapies currently being evaluated for treating heart disease may be complementary to the effort we encourage here to identify strategies that stimulate cardiogenesis directly.
Acknowledgments
Stephen N. Palmer, PhD, ELS, of the section of Scientific Publications at the Texas Heart Institute, contributed to the editing of the manuscript.
Sources of Funding: Supported by grants from the National Institutes of Health (DE 023177, HL 127717, HL 130804, and HL 118761 to J.F.M.) and the Vivian L. Smith Foundation (to J.F.M.). J.F.M. was supported by the Transatlantic Network of Excellence Award LeDucq Foundation Transatlantic Networks of Excellence in Cardiovascular Research 14CVD01: “Defining the genomic topology of atrial fibrillation.” The Cardiovascular Cell Therapy Research Network (CCTRN), sponsored by the National Heart, Lung, and Blood Institute, supports the Stem Cell Center at the Texas Heart Institute (E.C.P., J.T.W.).
Abbreviations and Nonstandard Acronyms
- ACE
Angiotensin-converting enzyme
- ARB
Angiotensin receptor blocker
- CAD
Coronary artery disease
- HF
Heart failure
- LATS
Large tumor suppressor
- LV
Left ventricular
- MI
Myocardial infarction
Footnotes
Disclosures: None.
Contributor Information
James F. Martin, Cardiomyocyte Renewal Laboratory, Texas Heart Institute, Houston, Texas, Department of Molecular Physiology and Biophysics, Baylor College of Medicine, Houston, Texas.
Emerson C. Perin, Division of Cardiology Research, Texas Heart Institute, Houston, Texas.
James T. Willerson, Division of Cardiology Research, Texas Heart Institute, Houston, Texas.
References
- 1.Perin EC, Borow KM, Silva GV, DeMaria AN, Marroquin OC, Huang PP, Traverse JH, Krum H, Skerrett D, Zheng Y, Willerson JT, Itescu S, Henry TD. A phase II dose-escalation study of allogeneic mesenchymal precursor cells in patients with ischemic or nonischemic heart failure. Circ Res. 2015;117:576–584. doi: 10.1161/CIRCRESAHA.115.306332. [DOI] [PubMed] [Google Scholar]
- 2.Bolli R, Chugh AR, D'Amario D, et al. Cardiac stem cells in patients with ischaemic cardiomyopathy (SCIPIO): initial results of a randomised phase 1 trial. Lancet. 2011;378:1847–1857. doi: 10.1016/S0140-6736(11)61590-0. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 3.Bergmann O, Zdunek S, Felker A, et al. Dynamics of cell generation and turnover in the human heart. Cell. 2015;161:1566–1575. doi: 10.1016/j.cell.2015.05.026. [DOI] [PubMed] [Google Scholar]
- 4.D'Uva G, Aharonov A, Lauriola M, et al. ERBB2 triggers mammalian heart regeneration by promoting cardiomyocyte dedifferentiation and proliferation. Nat Cell Biol. 2015;17:627–638. doi: 10.1038/ncb3149. [DOI] [PubMed] [Google Scholar]
- 5.Heallen T, Morikawa Y, Leach J, Tao G, Willerson JT, Johnson RL, Martin JF. Hippo signaling impedes adult heart regeneration. Development. 2013;140:4683–4690. doi: 10.1242/dev.102798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tao G, Kahr PC, Morikawa Y, Zhang M, Rahmani M, Heallen TR, Li L, Sun Z, Olson EN, Amendt BA, Martin JF. Pitx2 promotes heart repair by activating the antioxidant response after cardiac injury. Nature. 2016;534:119–123. doi: 10.1038/nature17959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heallen T, Zhang M, Wang J, Bonilla-Claudio M, Klysik E, Johnson RL, Martin JF. Hippo pathway inhibits Wnt signaling to restrain cardiomyocyte proliferation and heart size. Science. 2011;332:458–461. doi: 10.1126/science.1199010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.von Gise A, Lin Z, Schlegelmilch K, Honor LB, Pan GM, Buck JN, Ma Q, Ishiwata T, Zhou B, Camargo FD, Pu WT. YAP1, the nuclear target of Hippo signaling, stimulates heart growth through cardiomyocyte proliferation but not hypertrophy. Proc Natl Acad Sci U S A. 2012;109:2394–2399. doi: 10.1073/pnas.1116136109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xin M, Kim Y, Sutherland LB, Qi X, McAnally J, Schwartz RJ, Richardson JA, Bassel-Duby R, Olson EN. Regulation of insulin-like growth factor signaling by Yap governs cardiomyocyte proliferation and embryonic heart size. Sci Signal. 2011;4:ra70. doi: 10.1126/scisignal.2002278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin Z, von Gise A, Zhou P, et al. Cardiac-specific YAP activation improves cardiac function and survival in an experimental murine MI model. Circ Res. 2014;115:354–363. doi: 10.1161/CIRCRESAHA.115.303632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang J, Martin JF. Macro advances in microRNAs and myocardial regeneration. Curr Opin Cardiol. 2014;29:207–213. doi: 10.1097/HCO.0000000000000050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Srivastava D, DeWitt N. In vivo cellular reprogramming: the next generation. Cell. 2016;166:1386–1396. doi: 10.1016/j.cell.2016.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gao L, Kupfer ME, Jung JP, Yang L, Zhang P, Da Sie Y, Tran Q, Ajeti V, Freeman BT, Fast VG, Campagnola PJ, Ogle BM, Zhang J. Myocardial tissue engineering with cells derived from human-induced pluripotent stem cells and a native-like, high-resolution, 3-dimensionally printed scaffold. Circ Res. 2017;120:1318–1325. doi: 10.1161/CIRCRESAHA.116.310277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Taylor DA, Chandler AM, Gobin AS, Sampaio LC. Maximizing cardiac repair: Should we focus on the cells or on the matrix? Circ Res. 2017;120:30–32. doi: 10.1161/CIRCRESAHA.116.309959. [DOI] [PubMed] [Google Scholar]