

Abstract
The spliceosome machinery is composed of several proteins and multiple small RNA molecules that are involved in gene regulation through the removal of introns from pre-mRNAs in order to assemble exon-based mRNA containing protein-coding sequences. Splice-switching oligo-nucleotides (SSOs) are genetic control elements that can be used to specifically control the expression of genes through correction of aberrant splicing pathways. A current limitation with SSO methodologies is the inability to achieve conditional control of their function paired with high spatial and temporal resolution. We addressed this limitation through site-specific installation of light-removable nucleobase-caging groups as well as photocleavable backbone linkers into synthetic SSOs. This enables optochemical OFF → ON and ON → OFF switching of their activity and thus precise control of alternative splicing. The use of light as a regulatory element allows for tight spatial and temporal control of splice switching in mammalian cells and animals.
Graphical abstract
INTRODUCTION
Alternative splicing (AS) plays a large role in the regulation of gene expression,1 as the spliceosome is responsible for processing of pre-mRNA into coding sequence by eliminating introns and assembling the correct exons.2 The splicing of pre-mRNA in humans was documented in the mid 1980s,3 and the field has dramatically expanded since. It has been estimated that 95% of all human genes show AS pathways, highlighting the importance of spliceosome activity in RNA regulation.4–6 Additionally, up to 50% of mutations associated with genetic disorders result in altered pre-mRNA splicing pathways,7 linking aberrant mRNA splicing to a wide variety of disease states, including cystic fibrosis and Duchenne muscular dystrophy.8 Splice-switching oligonucleotides (SSOs) act by hybridizing to the pre-mRNA and blocking splice sites in a sequence-specific manner, which prevents interaction with components of the spliceosome such that splicing pathways are altered. SSOs are commonly used tools to control gene expression via alternative mRNA splicing pathways through the removal of aberrant introns or exons as well as exon retention or skipping.9 For example, the coding region of a protein can be interrupted via a mutant intron gene (Supporting Information Figure 1A). Through AS pathways, the aberrant sequences can be blocked with SSOs, allowing for corrected exon splicing and resulting in the expression of a functional gene product (Supporting Information Figure 1B). Besides applications as research tools, SSOs are proposed for use as therapeutic agents to correct splice mutations10–12 and have been applied to genetic disorders for the initiation of several human clinical trials13–17 because these oligonucleotides are able to restore proper gene function in disease states.
However, no conditional control of SSO activity has been reported, and neither temporal nor spatial regulation of SSO function and AS has been achieved. The specific timing and location of AS play very important roles in many regulatory pathways in cells across the animal kingdom, for example, during neuronal cell differentiation in the human brain.18,19 Similar spatial and temporal activity of AS has been observed in fruit flies,20 mice,21 zebrafish,22,23 and nematodes,24 demonstrating that distinct AS patterns are responsible for many essential biological functions. Methods to accurately perturb the spatial and temporal patterns of AS are critical for understanding the complex mechanisms that underlie gene regulatory control by the spliceosome.25 However, traditional SSOs are constitutively in an ON state, i.e., the splicing pathways are immediately altered and cannot be controlled with any spatial and temporal resolution, precluding precise investigation of AS in cells and multicellular organisms. In order to achieve conditional control of alternative splicing, we developed optochemical tools based on the introduction of caged nucleobases and photocleavable linkers into SSOs.26,27 This approach enabled both optochemical activation and deactivation of splicing pathways, leading to efficient two-directional genetic control through OFF → ON as well as ON → OFF light switches. These optochemical regulation tools provide conditional control of SSO activity with high resolution in cellular and embryonic environments, providing spatial and temporal capabilities for dissection of AS pathways as well as the general control of gene function.
RESULTS AND DISCUSSION
Before applying our light-controlled SSO approach to a complex biological system, such as a developing zebrafish embryo, we first selected the β-globin intron 1 as a proof-of-principle target, since it contains an aberrant splice site that contributes to the genetic blood disorder β-thalassaemia and that can be corrected with SSOs.28–32 HeLa cells stably expressing an enhanced green fluorescent protein (EGFP) gene interrupted with the mutant β-globin intron (HeLa:EGFP654)12 were used to analyze the optochemical regulation of SSOs and the ability to control AS pathways with light. The EGFP reporter gene contains an altered coding region with an aberrant splice site to impair functional fluorescent protein expression until it is blocked by a sequence-specific SSO. The activity of control SSOs was analyzed using TAMRA-labeled oligonucleotides to track transfection, for both a noncaged SSO and a five-base-mismatched oligo (Supporting Information Table 1). EGFP expression was observed only in the presence of the positive control SSO (Supporting Information Figure 2), and no detrimental effect on the SSO-controlled expression of EGFP in the presence of UV light was detected, ensuring that the reporter construct itself was not UV responsive (Supporting Information Figure 3). In order to optochemically regulate SSO activity, oligonucleotides with light-responsive modifications were synthesized containing 2′OMe nucleotides and phosphor-othioate linkages, which are highly effective oligonucleotide chemistries for cellular applications of SSOs.9,33 First, a nucleobase-caged SSO was engineered that contained light-removable protecting groups that inhibit hybridization to the complementary pre-mRNA target. A 2′OMe-NPOM-caged uridine phosphoramidite was synthesized34 (Supporting Information Figure 4A) and incorporated into the SSO at four positions throughout the sequence, as indicated in Supporting Information Table 1, to achieve full inhibition of duplex formation. This light-activated SSO (LASSO) was designed to be inactive when transfected into cells until briefly exposed to UV light, thus enabling efficient OFF → ON photoswitching for aberrant splice correction (Figures 1A,B). The LASSO was synthesized using phosphoramidite chemistry, gel extracted to obtain a high-purity full-length oligomer (Supporting Information Figures 4B and 5), and subsequently transfected into the HeLa reporter cell line. In the absence of UV irradiation, no EGFP expression was observed, but after a short UV exposure the AS pathway was activated through SSO decaging, leading to removal of the mutant intron and recovery of EGFP reporter gene expression (Figure 1D). Only cells that were UV-irradiated exhibit splice-switching activity and EGFP expression. In order to maximize light activation, the parameters for UV irradiation and LASSO concentration were further analyzed, revealing a 200 nM concentration and a 2 min irradiation time as being optimal for light-activated splice correction (Supporting Information Figure 6). Diminished LASSO activation is observed with shorter irradiation times. Importantly, no background leakiness of the LASSO before irradiation was observed, confirming excellent OFF to ON switching behavior.
Figure 1.

Optochemical regulation of splice switching with the LASSO for an OFF → ON gene expression light switch. Schematics for the two RNA splicing pathways (black) and the mutant intron (red) are shown. (A) In the absence of irradiation, the LASSO does not bind to the target site due to inhibition of base hybridization from the nucleobase caging groups. (B) UV decaging of the LASSO enables conditional splice correction of the aberrant mutant intron and activation of gene expression. (C) Dark blue boxes represent 2′OMe-NPOM-caged uridine residues, with the light-removable group indicated in red. (D) HeLa:EGFP654 cells were transfected with the LASSO, irradiated for 2 min or kept in the dark, and imaged for EGFP expression after 24 h. Images of the EGFP channel (left) and the EGFP channel merged with a brightfield image (right) are shown. Scale bars indicate 0.2 mm.
As a second approach, a light-cleavable oligonucleotide was synthesized to enable ON → OFF photoswitching of gene expression using an ortho-nitrobenzyl (ONB) linker35 (Supporting Information Figure 4A). The ONB group was incorporated at two locations internally within the SSO sequence, as indicated in Supporting Information Table 1, both to maintain binding with the target site and to ensure complete cleavage of the full-length oligonucleotide through a brief UV exposure. This light-deactivated SSO (LDSSO) was designed to be active in the absence of UV irradiation, but it can be deactivated with UV light, inducing oligonucleotide cleavage (Figures 2A,B). The successful synthesis and UV-dependent cleavage of the LDSSO was confirmed by gel analysis prior to cellular applications (Supporting Information Figure 4B and 5). Transfection of the LDSSO confirmed that the installation of ONB linkers did not interfere with the SSO activity and that splice-switching correction for the expression of EGFP occurred in the absence of UV exposure (Figure 2D). In contrast, cells that were UV-irradiated showed no EGFP expression due to deactivation of splice-switching oligonucleotide as a result of light-induced oligonucleotide fragmentation. Importantly, splice correction with the LDSSO was completely deactivated through UV irradiation, demonstrating an excellent ON → OFF switching behavior of the SSO pathway. Thus, in conjunction with the LASSO described above, both optical activation and deactivation of splice-switching pathways are possible.
Figure 2.

Optochemical regulation of splice switching with the LDSSO for an ON → OFF gene expression light switch. Schematics for the two RNA splicing pathways (black) and the mutant intron (red) are shown. (A) In the absence of irradiation, the LDSSO binds to the target site for aberrant splice correction and activation of gene expression. (B) UV cleavage of the LDSSO deactivates splice correction, and gene expression is interrupted by the mutant intron. (C) Red circles represent ONB linker residues, with the photo-cleavable group indicated in red. (D) HeLa:EGFP654 cells were transfected with the LDSSO, irradiated for 2 min or kept in the dark, and imaged for EGFP expression after 24 h. Images of the EGFP channel (left) and the EGFP channel merged with a brightfield image (right) are shown. Scale bars indicate 0.2 mm.
The effect of optochemical regulation of SSO activity and aberrant intron splicing on EGFP expression was then quantified through flow cytometry. Conditions for cell transfection and fluorescent cell counting were optimized both for the LASSO and LDSSO (Supporting Information Figure 7), followed by analysis of the optical OFF → ON and ON → OFF switching with the light-regulated SSOs and comparison to a noncaged positive control (Figure 3). The EGFP reporter expression for the LASSO was fully restored, showing nearly identical expression levels as that of the control after a 2 min UV irradiation for light-activation of SSO function. The light-deactivated SSO showed similarly high levels of EGFP expression as that of the noncaged SSO in the absence of UV irradiation but greatly reduced expression levels after UV exposure. Both SSO light switches exhibited >10-fold changes in EGFP expression between the OFF and ON states. These findings are in agreement with the observations from cellular micrographs presented in Figures 1 and 2, validating the application of photocaged bases and photocleavable linkers for optochemical activation and deactivation of aberrant splicing pathways.
Figure 3.
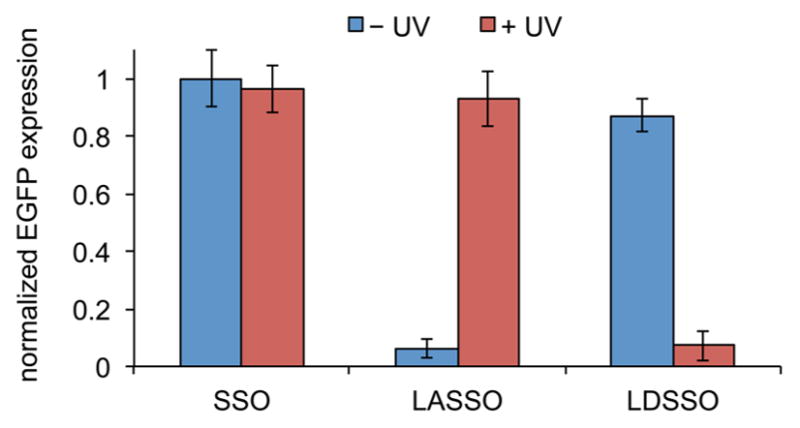
Flow cytometry quantification of optically regulated splice-switching with LASSO and LDSSO. HeLa:EGFP654 cells were transfected with the SSOs, irradiated for 2 min or kept in the dark, and analyzed for EGFP expression after 24 h. The gated EGFP positive cells were normalized to the noncaged control. Error bars represent standard deviations from three independent experiments through counts of 20 000 cells each.
The ability to specifically control the location of SSO optochemical regulation through localized UV illumination was then investigated. Irradiations for the LASSO were performed using irradiation masks as well as microscope optics. When UV irradiation was applied in a spatially defined area in conjunction with the light-activated SSO, only the cells that were exposed to 365 nm light exhibited EGFP expression through an OFF → ON light switch of SSO activity (Figure 4). The time of irradiation was decreased from 2 min to 30 s for localized irradiations because these were performed through focusing microscope optics using a Xe/Hg lamp. The tight spatial control conferred with this method can potentially be used to analyze splice switching and aberrant intron correction in small subsets of cellular populations as well as within defined locations in multicellular organisms.
Figure 4.
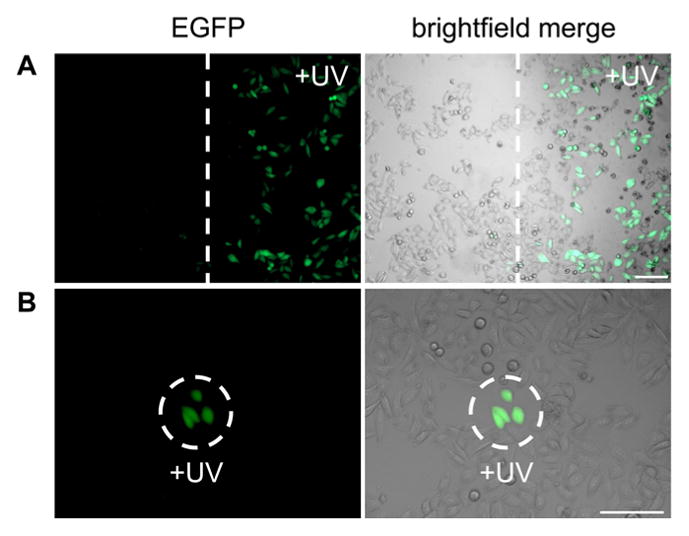
Spatial control of EGFP expression through optical regulation of splice-switching using the LASSO. Localized irradiations were performed using a vertical mask (A) or a partially closed microscope shutter (B). Cells were imaged after 24 h. Images of the EGFP channel (left) and the EGFP channel merged with a brightfield image (right) are shown. Scale bars indicate 0.2 mm.
In order to translate our methodology for the optical regulation of splice-switching from cell culture experiments to multicellular organisms, we targeted an endogenous gene in zebrafish to demonstrate conditional control of RNA splicing. The zebrafish embryo was selected as a model system because it has been shown that UV-A exposure does not affect zebrafish embryonic development, hatch rate, mortality, or global gene expression,36 making this translucent and ex utero developing animal highly suited for photoactivation studies with micro-injected caged oligonucleotides.37,38 We designed a light-activated splice-switching oligonucleotide targeting sox31 (also known as sox19b), a member of the B1 Sox gene family that is responsible for many critical processes during zebrafish development, both before and after the midblastula transition (MBT).39–41 During the blastulation stage, sox31 is expressed throughout the blastoderm, where it then assists in epiboly formation during the gastrulation stage.42 Splice variants of sox31 have been previously generated with morpholino oligonucleotides (MO) targeting cryptic splice sites in sox31.43 Alternative splicing-based inhibition of sox31 resulted in normal development during the first 3 hpf, followed by arrested embryonic development and loss of epiboly formation for zebrafish that failed to undergo gastrulation. Although these findings were made with MOs, there is precedence for the use of 2′OMe-modified oligonucleotides as antisense agents in zebrafish.44 As such, a 2′OMe sox31 LASSO was synthesized with NPOM-caged uridine residues incorporated at four positions throughout the sequence, as indicated in Supporting Information Table 1, and purified for injection into zebrafish embryos (Supporting Information Figure 4B). The sox31 LASSO was designed to be inactive in the absence of UV exposure, allowing for correct RNA splicing and functional protein production (Figure 5A). However, after a brief UV exposure, the sox31 LASSO will be activated through photolysis of the nucleobase caging groups and will bind the RNA target site, initiating AS and production of a nonfunctional interrupted RNA (Figure 5B). To this end, embryos were injected with the oligonucleotides (5 ng) and either irradiated (365 nm, 2 min) or kept in the dark. At 8 h postfertilization (hpf), the embryos were imaged during the gastrulation stage when ~75% epiboly formation is expected (Figure 5C), and the frequency of embryos exhibiting epiboly defects was determined (Figure 5D). The noninjected embryos were not affected by UV exposure, as no significant changes in development were observed. Injection with the noncaged positive control sox31 SSO showed developmental arrest and high frequency of the no epiboly phenotype. A negative control scrambled 2′OMe oligonucleotide shows low levels of developmental arrest, slightly above those observed for noninjected embryos. The sox31 LASSO showed normal epiboly formation and development through gastrulation for the injected embryos in the absence of UV irradiation, similar to the negative control. However, activation of the LASSO with UV irradiation inhibited sox31 expression and induced the no epiboly phenotype to the same high levels as that observed for injection of the noncaged control. Thus, the conditional control of splice switching of an endogenous gene was successfully demonstrated in a living animal. Additionally, UV exposures at different time points from 1 to 7 hpf were performed to determine temporal requirements for the activity of sox31 during early embryonic development. Only embryos irradiated after 4 hpf (after the MBT) showed a distinct decrease in epiboly defects (Supporting Information Figure 8), supporting functional sox31 as a requirement for epiboly formation.43 Thus, the sox31 LASSO enabled OFF → ON photoswitching of a dominant negative splice variant, thereby inhibiting sox31 and other SoxB1 genes in the developing zebrafish embryo.
Figure 5.

Optical regulation of splice switching in living zebrafish embryos. Schematic for the RNA splicing (black) and its deactivation by the LASSO. Dark blue boxes represent 2′OMe-NPOM-caged uridine residues. (A) In the absence of irradiation, the sox31 LASSO does not bind to the target site and the sox31 mRNA is correctly spliced, resulting in gene expression. (B) UV activation of the sox31 LASSO enables conditional splice blocking of the sox31 RNA, resulting in inhibition of gene expression. (C) Zebrafish embryos were imaged at 8 hpf, showing the major phenotype observed from treatment with the light-activated and control splice-switching oligonucleotides, both without (top) and with (bottom) UV irradiation. Arrows indicate the absence of epiboly formation. (D) Zebrafish embryo scores were determined for the frequency of no epiboly formation with and without UV exposure (365 nm, 2 min). Error bars represent standard deviations from three independent experiments. N = 18–44.
SUMMARY
Alternative splicing (AS) is an important factor in the regulation of gene expression, responsible for the processing of pre-mRNA in many organisms. Misregulation of the spliceosome resulting in aberrant splicing is associated with a wide range of human genetic disorders, and distinct changes in endogenous alternative splicing can lead to gene expression patterns that are tightly regulated in many biological pathways. Splice-switching oligos (SSOs) provide a method to investigate AS and control splicing pathways through hybridization with the pre-mRNA, which blocks splice sites and prevents interaction with components of the spliceosome, thus modulating gene expression and protein function. In order to address the lack of tools to conditionally regulate SSO function with spatial and temporal resolution, we developed a method to regulate AS pathways with light as an external control element. The installation of caged nucleobases in synthetic SSOs enabled optochemical activation of splice switching and alternative expression pathways in live cells and animals. Light-triggered deactivation of splice switching was demonstrated through the installation of photocleavable linkers into the oligonucleotide backbone. The NPOM-caged nucleobases and ONB-linker groups site-specifically introduced into SSOs were used to develop both OFF → ON and ON → OFF light switches for gene splicing, with excellent switching behavior and no detectable background activity before light exposure. These findings are supported by cellular micrographs as well as florescent cell quantification using an EGFP reporter system in human cells. In addition, spatial control of SSO function in a cellular monolayer was achieved through localized UV exposure, showing distinct splice-switch regulation and activation of the caged SSO in a subset of cells. The conditional control of splice switching was demonstrated with the correction of a mutant intron, but it could be readily applied to other mechanisms of AS, such as mutant exon removal or processing of genes that contain multiple splice variants. To this end, we applied the developed methodology to the regulation of mRNA processing for an endogenous gene in a living organism, by targeting sox31 splicing in zebrafish embryos. The induction of developmental arrest through the inhibition of epiboly formation during zebrafish gastrulation was conditionally controlled with injection of a sox31 light-activated SSO, successfully demonstrating temporal regulation of AS pathways in a complex animal model. By conveying optochemical regulation to SSOs, we have added a new level of precision for conditional control over these gene regulatory tools and potential therapeutic reagents. The developed methodology will aid in the investigation of spatial and temporal mechanisms underlying spliceosome correction, aberrant splice switching, and their function in cells as well as multicellular organisms.
METHODS
Custom DNA Synthesis
The noncaged and negative control SSOs were gifted from Rudolph Juliano. The negative control scramble 2′OMe oligonucleotide was purchased from Ambion. DNA synthesis of modified oligonucleotides was performed using standard β-cyanoethyl phosphoramidite chemistry, with 2′OMe bases and phosphorothioate linkages on an Applied Biosystems model 394 automated DNA/RNA synthesizer at a 0.2 mM scale with solid-phase supports. All commercial reagents were obtained from Glen Research. The 2′OMe 6-nitropiperonyloxymethyl (NPOM)-caged uridine34 and the ortho-nitrobenzyl (ONB) linker35 phosphoramidites were synthesized as previously described and dissolved in anhydrous acetonitrile to a final concentration of 0.05 M. Standard synthesis cycles provided by Applied Biosystems were used with 10 min coupling times for all 2′OMe bases. The sulfurization step was performed using the Beaucage sulfurizing reagent (3H-1,2-benzodithiole-3-one-1,1-dioxide) at 0.05 M in acetonitrile for 15 min. Oligonucleotides were eluted from the solid-phase supports with 1 mL of ammonium hydroxide methylamine (AMA, 1:1) and deprotected at 65 °C for 2 h. The full-length caged oligonucleotides were purified with Nap-10 columns (GE Healthcare) followed by PAGE gel band excision and elution into PBS buffer, pH 7.4. Electrospray ionization mass spectrometry (ESI-MS) was performed by Novatia (Newtown, PA).
Mammalian Cell Culture
HeLa:EGFP654 cells were grown in 10 cm dishes with DMEM growth media supplemented with 10% FBS and 2% penicillin/streptomycin at 37 °C in 5% CO2. The cell line was passaged when confluence reached >90% at a 1:10 dilution of approximately 1 × 106 cells into a new 10 cm plate containing 10 mL of growth media.
Cellular Transfection of SSOs
HeLa:EGFP654 cells were seeded at ~10 000 cells per well into black clear bottom 96-well plates (BD Falcon) and incubated overnight in DMEM growth media supplemented with 10% FBS and 2% penicillin/streptomycin at 37 °C in 5% CO2. Transfections were performed with 50–200 nM of each SSO using 1 μL of X-tremeGENE siRNA reagent (Roche) in 200 μL of Opti-Mem (Invitrogen) at 37 °C for 4 h. After 4 h, the Opti-Mem transfection mixtures were removed from the cells and replaced with DMEM growth media.
Optochemical Regulation of SSOs
Cellular irradiations were performed with a 365 nm UV transilluminator (6.3 mW/cm2) for 30 s to 2 min. Spatially distinct UV irradiations were performed through precut vertical slits in tinfoil 96-well plate covers and irradiated for 2 min. Additionally, localized irradiations were performed with a Zeiss Observer Z1 microscope (40× objective, NA 0.75 plan-apochromat; Zeiss) and a DAPI (68 HE) filter (ex: BP377/28) with a partially closed shutter to irradiate a specific subset of cells for 30 s.
Fluorescence Imaging of EGFP
Fluorescent imaging was performed after a 24 h incubation. Media was replaced with clear DMEM-high modified growth media (Thermo Scientific) for microscopy imaging. Expression of EGFP was observed using a Zeiss Observer Z1 microscope (20× objective, NA 0.8 plan-apochromat; Zeiss) and a GFP (38 HE) filter (ex: BP470/40; em: BP525/50). The EGFP signal was normalized to a standard setting for fluorescent intensity (black = 300; white = 3000; gamma = 0.8) in Zen Pro 2011 imaging software. Fluorescent and brightfield merged images are shown with scale bars.
Fluorescent Cell Counting
HeLa:EGFP64 cells12 were seeded at ~100 000 cells per well into 6-well plates (BD Falcon) and incubated overnight in DMEM growth media supplemented with 10% FBS and 2% penicillin/streptomycin at 37 °C in 5% CO2. Transfections were performed with 50–200 nM of each SSO using 5 μL of X-tremeGENE siRNA reagent (Roche) in 1 mL of Opti-Mem (Invitrogen) at 37 °C for 4 h, at which point the transfection mixtures were removed from the cells and replaced with DMEM growth media. After trans-fection, UV irradiations were performed as described above followed by 24 h incubation. Cells were then trypsinized with 500 μL of TrypLE (Invitrogen), washed, and resuspended in 300 μL of PBS, pH 7.4, for fluorescent analysis. Flow cytometry was performed on a FACSCalibur (Becton-Dickinson) instrument (488 nm argon laser, 530/50 nm BPF) and analyzed using Cellquest Pro software. Cells were gated for EGFP fluorescence (above 102.5 RFUs) and analyzed until 20 000 cells had been counted for each condition tested. The frequency of EGFP positive cells (gated/total) was normalized to the noncaged control SSO. Error bars represent the standard deviation of experimental triplicates.
Zebrafish Maintenance and Injections
All zebrafish experiments were performed with the University of Pittsburgh Institutional Animal Care and Use Committee approval. The Oregon AB* strain was maintained under standard conditions at the University of Pittsburgh School of Medicine in accordance with Institutional and Federal guidelines. Embryos from natural matings were obtained and microinjected at the 1 to 2 cell stage with 5 ng of the oligonucleotides using a World Precision Instruments Pneumatic PicoPump injector. Embryos were then irradiated immediately following injection for 2 min with a 365 nm UV transilluminator and incubated in the dark at 28 °C for 8 h. Imaging was performed on a Leica MZ16FA stereo fluorescence microscope with a 1× objective (NA 0.14) at 60× zoom and collected with a QImaging Retiga-EXi Fast 1394 digital camera. Phenotype scores were calculated with embryo counts of [(no epiboly/alive) × 100]. For each of the replicates, the data were averaged, and standard deviations were calculated.
Supplementary Material
Acknowledgments
This work was supported in part by the National Institutes of Health (GM079114, GM108952, and CA151964), North Carolina State University, and the University of Pittsburgh.
Footnotes
Notes
The authors declare no competing financial interest.
Oligonucleotide sequences, splice-switching schematic, SSO controls, UV irradiations in cells, specialized phosphoramidites, oligonucleotide synthesis product gels and analyses, UV irradiation and SSO concentration optimizations, flow cytometry optimization, and zebrafish irradiation time points. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Scherrer K, Imaizumi-Scherrer MT, Reynaud CA, Therwath A. Mol Biol Rep. 1979;5:5. doi: 10.1007/BF00777484. [DOI] [PubMed] [Google Scholar]
- 2.Lamond AI. BioEssays. 1993;15:595. doi: 10.1002/bies.950150905. [DOI] [PubMed] [Google Scholar]
- 3.Leff SE, Rosenfeld MG, Evans RM. Annu Rev Biochem. 1986;55:1091. doi: 10.1146/annurev.bi.55.070186.005303. [DOI] [PubMed] [Google Scholar]
- 4.Ozsolak F, Milos PM. Nat Rev Genet. 2011;12:87. doi: 10.1038/nrg2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Nat Genet. 2008;40:1413. doi: 10.1038/ng.259. [DOI] [PubMed] [Google Scholar]
- 6.Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB. Nature. 2008;456:470. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cartegni L, Chew SL, Krainer AR. Nat Rev Genet. 2002;3:285. doi: 10.1038/nrg775. [DOI] [PubMed] [Google Scholar]
- 8.Tazi J, Durand S, Jeanteur P. Trends Biochem Sci. 2005;30:469. doi: 10.1016/j.tibs.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 9.Kole R, Krainer AR, Altman S. Nat Rev Drug Discovery. 2012;11:125. doi: 10.1038/nrd3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sazani P, Kole R. Prog Mol Subcell Biol. 2003;31:217. doi: 10.1007/978-3-662-09728-1_8. [DOI] [PubMed] [Google Scholar]
- 11.Sazani P, Kole R. J Clin Invest. 2003;112:481. doi: 10.1172/JCI19547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sazani P, Kang SH, Maier MA, Wei C, Dillman J, Summerton J, Manoharan M, Kole R. Nucleic Acids Res. 2001;29:3965. doi: 10.1093/nar/29.19.3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van Deutekom JC, Janson AA, Ginjaar IB, Frankhuizen WS, Aartsma-Rus A, Bremmer-Bout M, den Dunnen JT, Koop K, van der Kooi AJ, Goemans NM, de Kimpe SJ, Ekhart PF, Venneker EH, Platenburg GJ, Verschuuren JJ, van Ommen GJ. N Engl J Med. 2007;357:2677. doi: 10.1056/NEJMoa073108. [DOI] [PubMed] [Google Scholar]
- 14.Kinali M, Arechavala-Gomeza V, Feng L, Cirak S, Hunt D, Adkin C, Guglieri M, Ashton E, Abbs S, Nihoyannopoulos P, Garralda ME, Rutherford M, McCulley C, Popplewell L, Graham IR, Dickson G, Wood MJ, Wells DJ, Wilton SD, Kole R, Straub V, Bushby K, Sewry C, Morgan JE, Muntoni F. Lancet Neurol. 2009;8:918. doi: 10.1016/S1474-4422(09)70211-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goemans NM, Tulinius M, van den Akker JT, Burm BE, Ekhart PF, Heuvelmans N, Holling T, Janson AA, Platenburg GJ, Sipkens JA, Sitsen JM, Aartsma-Rus A, van Ommen GJ, Buyse G, Darin N, Verschuuren JJ, Campion GV, de Kimpe SJ, van Deutekom JC. N Engl J Med. 2011;364:1513. doi: 10.1056/NEJMoa1011367. [DOI] [PubMed] [Google Scholar]
- 16.Cirak S, Arechavala-Gomeza V, Guglieri M, Feng L, Torelli S, Anthony K, Abbs S, Garralda ME, Bourke J, Wells DJ, Dickson G, Wood MJ, Wilton SD, Straub V, Kole R, Shrewsbury SB, Sewry C, Morgan JE, Bushby K, Muntoni F. Lancet. 2011;378:595. doi: 10.1016/S0140-6736(11)60756-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mendell JR, Rodino-Klapac LR, Sahenk Z, Roush K, Bird L, Lowes LP, Alfano L, Gomez AM, Lewis S, Kota J, Malik V, Shontz K, Walker CM, Flanigan KM, Corridore M, Kean JR, Allen HD, Shilling C, Melia KR, Sazani P, Saoud JB, Kaye EM, Group ES. Ann Neurol. 2013;74:637. doi: 10.1002/ana.23982. [DOI] [PubMed] [Google Scholar]
- 18.Grabowski P. Curr Opin Genet Dev. 2011;21:388. doi: 10.1016/j.gde.2011.03.005. [DOI] [PubMed] [Google Scholar]
- 19.Grabowski PJ, Black DL. Prog Neurobiol. 2001;65:289. doi: 10.1016/s0301-0082(01)00007-7. [DOI] [PubMed] [Google Scholar]
- 20.Goodwin SF, Taylor BJ, Villella A, Foss M, Ryner LC, Baker BS, Hall JC. Genetics. 2000;154:725. doi: 10.1093/genetics/154.2.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Strehler EE, Zacharias DA. Physiol Rev. 2001;81:21. doi: 10.1152/physrev.2001.81.1.21. [DOI] [PubMed] [Google Scholar]
- 22.Ott EB, Te Velthuis AJ, Bagowski CP. Gene Expression Patterns. 2007;7:620. doi: 10.1016/j.modgep.2006.12.005. [DOI] [PubMed] [Google Scholar]
- 23.Rissone A, Sangiorgio L, Monopoli M, Beltrame M, Zucchi I, Bussolino F, Arese M, Cotelli F. Dev Dyn. 2010;239:688. doi: 10.1002/dvdy.22196. [DOI] [PubMed] [Google Scholar]
- 24.Mullen GP, Rogalski TM, Bush JA, Gorji PR, Moerman DG. Mol Biol Cell. 1999;10:3205. doi: 10.1091/mbc.10.10.3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nilsen TW, Graveley BR. Nature. 2010;463:457. doi: 10.1038/nature08909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu Q, Deiters A. Acc Chem Res. 2014;47:45. doi: 10.1021/ar400036a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brieke C, Rohrbach F, Gottschalk A, Mayer G, Heckel A. Angew Chem, Int Ed. 2012;51:8446. doi: 10.1002/anie.201202134. [DOI] [PubMed] [Google Scholar]
- 28.Dominski Z, Kole R. Proc Natl Acad Sci USA. 1993;90:8673. doi: 10.1073/pnas.90.18.8673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kotula JW, Pratico ED, Ming X, Nakagawa O, Juliano RL, Sullenger BA. Nucleic Acid Ther. 2012;22:187. doi: 10.1089/nat.2012.0347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ming X, Alam MR, Fisher M, Yan Y, Chen X, Juliano RL. Nucleic Acids Res. 2010;38:6567. doi: 10.1093/nar/gkq534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nakagawa O, Ming X, Huang L, Juliano RL. J Am Chem Soc. 2010;132:8848. doi: 10.1021/ja102635c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ming X, Carver K, Fisher M, Noel R, Cintrat JC, Gillet D, Barbier J, Cao C, Bauman J, Juliano RL. Nucleic Acids Res. 2013;41:3673. doi: 10.1093/nar/gkt066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Resina S, Kole R, Travo A, Lebleu B, Thierry AR. J Gene Med. 2007;9:498. doi: 10.1002/jgm.1044. [DOI] [PubMed] [Google Scholar]
- 34.Connelly CM, Uprety R, Hemphill J, Deiters A. Mol BioSyst. 2012;8:2987. doi: 10.1039/c2mb25175b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ordoukhanian P, Taylor JS. Bioconjugate Chem. 2000;11:94. doi: 10.1021/bc9900993. [DOI] [PubMed] [Google Scholar]
- 36.Dong Q, Svoboda K, Tiersch T, Monroe W. J Photochem Photobiol B. 2007;88:137. doi: 10.1016/j.jphotobiol.2007.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Holder N, Xu Q. Methods Mol Biol. 1999;97:487. doi: 10.1385/1-59259-270-8:487. [DOI] [PubMed] [Google Scholar]
- 38.Xu Q. Methods Mol Biol. 1999;127:125. doi: 10.1385/1-59259-678-9:125. [DOI] [PubMed] [Google Scholar]
- 39.Hu S, Wu Z, Yan Y, Li Y. Acta Biochim Biophys Sin. 2011;43:387. doi: 10.1093/abbs/gmr025. [DOI] [PubMed] [Google Scholar]
- 40.Okuda Y, Ogura E, Kondoh H, Kamachi Y. PLoS Genet. 2010;6:e1000936. doi: 10.1371/journal.pgen.1000936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Okuda Y, Yoda H, Uchikawa M, Furutani-Seiki M, Takeda H, Kondoh H, Kamachi Y. Dev Dyn. 2006;235:811. doi: 10.1002/dvdy.20678. [DOI] [PubMed] [Google Scholar]
- 42.Girard F, Crémazy F, Berta P, Renucci A. Mech Dev. 2001;100:71. doi: 10.1016/s0925-4773(00)00491-3. [DOI] [PubMed] [Google Scholar]
- 43.Hu SN, Yu H, Zhang YB, Wu ZL, Yan YC, Li YX, Li YY, Li YP. FEBS Lett. 2012;586:222. doi: 10.1016/j.febslet.2011.12.008. [DOI] [PubMed] [Google Scholar]
- 44.Schneider PN, Olthoff JT, Matthews AJ, Houston DW. Genesis. 2011;49:117. doi: 10.1002/dvg.20689. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.