

Abstract
[FeFe]-hydrogenases catalyze the reversible reduction of protons to molecular hydrogen with extremely high efficiency. The active site (“H-cluster”) consists of a [4Fe–4S]H cluster linked through a bridging cysteine to a [2Fe]H subsite coordinated by CN− and CO ligands featuring a dithiol-amine moiety that serves as proton shuttle between the protein proton channel and the catalytic distal iron site (Fed). Although there is broad consensus that an iron-bound terminal hydride species must occur in the catalytic mechanism, such a species has never been directly observed experimentally. Here, we present FTIR and nuclear resonance vibrational spectroscopy (NRVS) experiments in conjunction with density functional theory (DFT) calculations on an [FeFe]-hydrogenase variant lacking the amine proton shuttle which is stabilizing a putative hydride state. The NRVS spectra unequivocally show the bending modes of the terminal Fe–H species fully consistent with widely accepted models of the catalytic cycle.
The unique architecture of the H-cluster in all [FeFe]-hydrogenases1,2 is depicted in Figure 1a. It features a classical [4Fe–4S] cluster linked through a bridging cysteine ligand to a binuclear [2Fe]H subsite that is coordinated by two CN− and three CO ligands as well as an azadithiolate (ADT) ligand that bridges the two Fe atoms. The amine group of ADT shuttles protons to and from the distal iron site (Fed). In contrast to the situation in [NiFe]-hydrogenase, where hydride substrates are stabilized in the bridging position between the two metals,2,3 [FeFe]-hydrogenase is postulated to operate via terminal hydride intermediates.4-7 Studies on synthetic molecular catalysts indeed point to the intermediacy of terminal hydrides, which interact with the pendant amine of the ADT ligand.8
Figure 1.
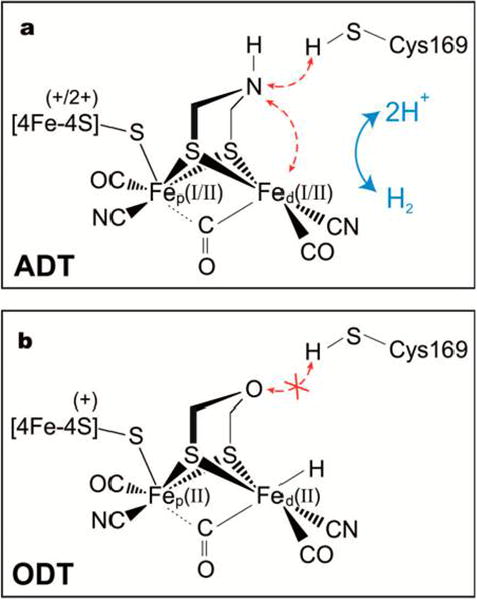
Schematic structure of the H-cluster containing (a) the native azadithiolate (ADT) bridging ligand and (b) the oxadithiolate (ODT) bridging ligand for which the proton transfer is blocked.
Several redox and protonation states assumed to be relevant for the catalysis have been identified in the enzyme from Chlamydomonas reinhardtii (CrHydA1) by FTIR.2,9-11 The active oxidized state Hox is represented as [4Fe–4S]H2+-Fe(I)Fe(II). Two singly reduced states have recently been identified: Hred=[4Fe–4S]H+-Fe(I)Fe(II) and HredH+=[4Fe–4S]H2+Fe(I)Fe(I)(NH2+) where protonation of the ADT amine is coupled to electronic rearrangement within the H-cluster.9 The doubly reduced state is characterized as HsredH+=[4Fe–4S]H+-Fe(I)Fe(I)(NH2+). According to widely accepted models of the catalytic cycle,4-7 Hox interacts with molecular hydrogen which is deprotonated by the pendant ADT amine in concert with electron transfer from the [2Fe]H subsite to the [4Fe–4S]H2+ cluster yielding an Fe(II)Fe(II)-bound hydride.
Recent EPR/FTIR/Mössbauer studies by King et al. identified an H-cluster species stabilized in CrHydA1 under strongly reducing conditions and in a C169S mutant with restricted proton transport properties.10,11 The spectral properties of this “Hhyd” species could best be modeled by density functional theory (DFT) calculations assuming a [4Fe–4S]H+-Fe(II)Fe(II) (NH)[H−] configuration, i.e., a reduced [4Fe-4S]H cluster and a homovalent diferrous [2Fe]H subsite with a terminal hydride bound to Fed. Although the analysis of this species is convincing, the presence of the hydride is only indirectly inferred from its influence on the electronic structure of the H-cluster. Spectroscopic observation of the hydride itself is, however, still lacking. To address this problem, we developed a strategy to accumulate the hydride species in high concentration and identify it directly through nuclear resonance vibrational spectroscopy (NRVS). This technique has been recently used to identify the bridging hydride in [NiFe]-hydrogenases.3
An effective strategy to accumulate the putative hydride intermediate is to obstruct the flow of protons to and from the active site by blocking the proton relay pathway.10,12 Our approach is based on the modification of the [2Fe]H subcluster itself. We exploit the recently established technique to directly reconstitute the apoprotein with a synthetic binuclear subcluster,13–15 thus bypassing the native maturation system.16,17 These studies had shown that incorporation of [Fe2(ADT)(CN)2(CO)4]2− produced fully functional CrHydA1 enzyme.14 This artificial maturation approach allows selective 57Fe-labeling of the diiron subsite. The resulting enzyme can be exquisitely characterized using the synchrotron-based NRVS technique that probes vibrational sidebands of the 57Fe Mössbauer nuclear resonance. NRVS is well suited for selectively investigating the [257Fe]H site of CrHydA1 because only the 57Fe nuclei contribution to vibrational motion is observed.18
Of the many analogues of [2Fe]H that have been introduced into the enzyme,13,15 the ODT variant in which the bridging amine group is replaced by an oxygen, is distinct. The ether group in ODT is much less basic than the amine of ADT, and thus is a poor proton relay.19 The effect of ODT/ADT exchange is shown by the observation that CrHydA1([2Fe]H-ODT) almost exclusively forms an “Htrans-like” state upon artificial maturation.13 The Htrans state is an oxidized inactivated state characterized by a reduced [4Fe–4S]H cluster and an Fe(II)Fe(II) subsite.20,21 Because the ODT-variant, similarly to the C169S mutant,12 features a disrupted proton transfer apparatus as shown in Figure 1b, it is reasonable to assume that the active site of CrHydA1([2Fe]H-ODT) is trapped in an intermediate state that is transient when the proton coupled electron transfer (PCET) apparatus is intact. The oxidized iron core may indicate the presence of a substrate, i.e. a terminal hydride, bound to Fed. To characterize this state in detail, we have prepared CrHydA1 containing the [257Fe]H-ODT subsite and analyzed these samples in H2O/H2 and D2O/D2 buffers using FTIR and NRVS. The spectroscopic results have been rationalized by DFT calculations (using BP86 and B3LYP functionals) on a model of the [2Fe]H subsite (see Supporting Information 1.6).
As shown in Figure 2a,b, the FTIR spectra of the two samples (H2O/D2O) suggest that, according to the relative band intensities, more than 70% of the H-clusters adopt the Htrans-like state. Its FTIR signature is virtually identical to that of the putative Hhyd species reported by King et al.10,11 Of particular interest is the band at 1869 cm−1 assigned to the semibridging ν(C≡O) stretching mode. This vibration is expected to effectively couple to the apical ligand on Fed in trans position to CO, if present. Upon H/D exchange, the semibridging CO band is red-shifted by 8 cm−1. This isotope shift clearly indicates the involvement of a hydrogen species in the Htrans-like (“Hhyd”) state. Indeed, as shown in Figure 2c, DFT modeling of the binuclear [257Fe]H-ODT subsite (see Supporting Information 1.6 and Figure 3a) predicts a shift of −5 cm−1 for the IR-active semibridging ν(C≡O) normal mode upon H/D exchange. Although the ν(C≡O) and v(57Fed—H) stretching vibrations are coupled, this coupling is predicted to be effectively eliminated via isotope labeling in the D-variant (see Supporting Information for the vibrational mode animations).
Figure 2.
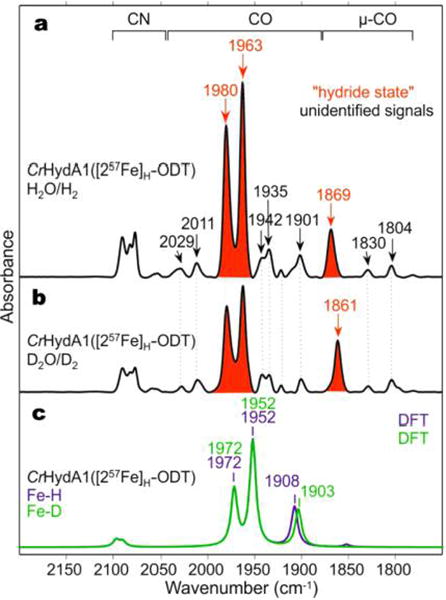
FTIR spectra of CrHydA1([257Fe]H-ODT) in the CO/CN region (a) recorded in H2O/H2, (b) recorded in D2O/D2, and (c) calculated using DFT for the H/D variants. Experimental CO bands assigned to the hydride state are marked in red.
Figure 3.
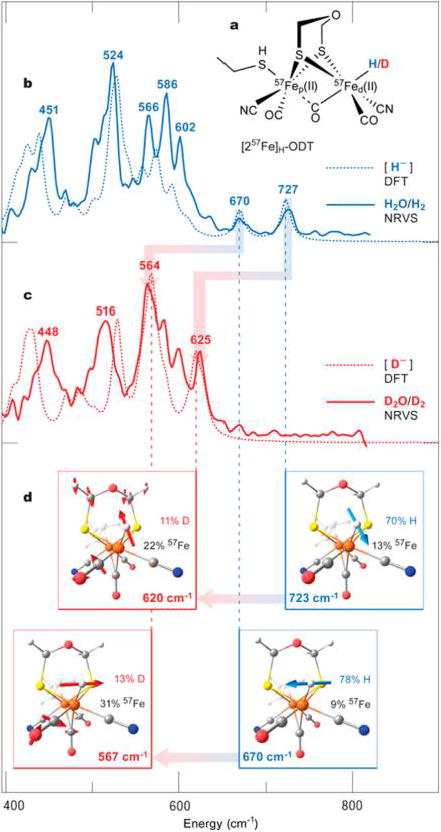
Experimental and DFT calculated NRVS spectra of CrHydA1 maturated with [257Fe]H-ODT. (a) Schematic representation of the [257Fe]H-ODT DFT model. (b,c) NRVS spectra (solid lines) of CrHydA1([257Fe]H-ODT) in the 400–800 cm−1 range recorded on samples in (b) H2O/H2 (blue) and (c) D2O/D2 (red), shown overlaid with the corresponding DFT spectra (broken lines) of [257Fe]H-ODT; the NRVS bands are labeled with their positions (cm−1). (d) Representations of the DFT δ(57Fed–H/D) normal modes assigned to the individual NRVS bands; mode frequencies and contributions (%) to the vibrational energy from the 57Fe/H/D nuclei are given. The calculated NRVS band positions were scaled with factor of 0.96. For the full-range (0–800 cm−1) spectra, see Figures S2 and S3.
To identify unequivocally the presence of the Fed-H/D species, the very same CrHydA1([257Fe]H-ODT) samples were examined by NRVS experiments. In the region below 400 cm−1, the spectra for both samples (H/D) are nearly identical, with features between 160 and 400 cm−1 assigned to 57Fe—S bending (δ) and stretching (ν) modes of the [2Fe]H subsite. Above 400 cm−1, the major bands are representing ν/δ(57Fe–CO/CN) modes (see Supporting Information 4.2, and Figures S2 and S3, as well as vibrational mode animations).
As illustrated in Figure 3b,c, the H/D-variant NRVS spectra are also quite similar in the region from 400 to 600 cm−1. From previous work,18,22 we assign the H/D sample features at 451/ 448 cm−1 primarily to ν(57Fe–CN) motion, whereas the bands observed from 470 to 600 cm−1 correspond to ν/δ(57Fe–CO) motions. Most remarkably, the H/D spectra are completely different above 600 cm−1. The H2O/H2CrHydA1([257Fe]H-ODT) sample exhibits a well-resolved band at 727 cm−1 and a smaller feature at 670 cm−1, whereas the D2O/D2 sample instead shows a sharp peak at 625 cm−1 and enhanced intensity of the peak at 564 cm−1.
The nature of these higher energy features is quantitatively explained by DFT calculations. The 727 and 670 cm−1 NRVS bands for the H2O/H2 sample are assigned to bending modes of the 57Fed-H unit. The DFT calculations predict corresponding modes of ≈80–90%δ(Fed–H) contribution at 723 and 670 cm−1, with the hydride motion respectively in or perpendicular to the Fep-Fed-H plane as indicated in Figure 3d. The relative intensities of these δ(Fed–H) bands can be explained by the amount of 57Fe motion in each mode. Conversely, CrHydA1-([257Fe]H-ODT) in D2O/D2 is predicted to have related modes of ≈30–40%δ(Fed–D) contribution respectively at 620 and 567 cm−1, both strongly mixed with δ(Fed–CO) motion. The 620 cm−1 mode also involves bending of the ODT unit. The isotope-dependent spectral changes in the 400–800 cm−1 region from the NRVS experiment coupled with 57Fed-H/D DFT calculations clearly confirm our mode assignments (see Supporting Information 4.3 and 4.4 for a detailed discussion).
NRVS measurements combined with DFT analysis (along with previous IR and EPR studies) therefore unambiguously validate that the Htrans-like state is associated with an Fed-H species that can be denoted as “Hhyd” in CrHydA1([2Fe]H-ODT), characterized by a reduced [4Fe–4S]H cluster coupled to the oxidized binuclear subsite, Fe(II)Fe(II). The presence of the reduced [4Fe–4S]H cluster is confirmed by the characteristic EPR spectrum of CrHydA1([257Fe]H-ODT) (Figure S4). The “Hhyd” state identified in the ODT variant is virtually identical to the species reported by King et al.10,11 Moreover, our DFT calculations (Supporting Information 4.6 and 4.7, Tables S2 and S3) indicate that the electronic structure of the [2Fe]H-ODT subsite is identical to that of the native[2Fe]H(-ADT) variant. In addition, a recent study23 indicated that the Hhyd FTIR signature is found in [FeFe]-hydrogenases from multiple organisms. Thus, the hydride state that we have directly characterized in this work appears to be universal in [FeFe]-hydrogenases.
The identification of the hydride-containing state now provides a basis for analyzing and integrating various mechanistic studies. Previous work9,24 on CrHydA1 identified the doubly reduced state of the H-cluster “HsredH+” assumed to contain both electrons needed to reduce a proton to a hydride. If the ADT in Hhyd is in the amine form, as the recent work of King et al. suggests,11 HsredH+ is logically related to Hhyd by proton transfer from the ADT ammonium to the distal iron Fed. According to the DFT analysis by Finkelmann et al. on a model of the [2Fe]H subsite, this step is strongly downhill.25 Protonation of the amine in Hhyd should then trigger H2 production.
DFT calculations on the native enzyme and inorganic model complexes suggest that H2 can only be liberated from the H-cluster in the mixed valence Fe(I)Fe(II) state of the [2Fe]H subsite.25,26 Therefore, one could speculate that protonation of Hhyd to HhydH+ triggers electron transfer from the reduced [4Fe–4S]H+ cluster to the [2Fe]H subsite producing the shortlived [4Fe–4S]2+-Fe(I)Fe(II)(NH2+)[H−] state immediately forming Hox(H2), from which H2 is finally dissociating. This PCET mechanism is supported by our DFT calculations on the native CrHydA1(ADT) H-cluster including the [4Fe–4S]H cluster (see Supporting Information 4.6). In the native enzyme, we cannot exclude the occurrence of a homovalent Fe(II)Fe(II) ammonium hydride state H*hydH+. Recent FTIR studies indicate that Hhyd can be found in [FeFe]-hydrogenase of multiple native organisms not only under strongly reducing conditions but also at low pH (<6) and H2 exposure.23 Furthermore, an inorganic mimic for the [2Fe]H subsite has been crystallized in the Fe(II)Fe(II)(NH2+)[H−] configuration matching the H*hydH+species.8
Taking into account the relevant states of the H-cluster that were experimentally observed in all known [FeFe]-hydrogenases, i.e., Hox, Hred, HredH+, HsredH+, as well as the key hydride species Hhyd (and possibly HhydH+), we can formulate a new working model for the catalytic mechanism as displayed in Figure 4. One of the key features of this mechanism is the role of PCET,27 which avoids the accumulation of hydride intermediates in the native enzyme. Clearly, some open questions still remain, such as the factors that affect the isomerization equilibrium between HsredH+ and Hhyd. These could be answered by kinetic (time-resolved) experiments. The general mechanistic insights gained here will be of great importance for the design of synthetic hydrogen conversion catalysts.28–30
Figure 4.
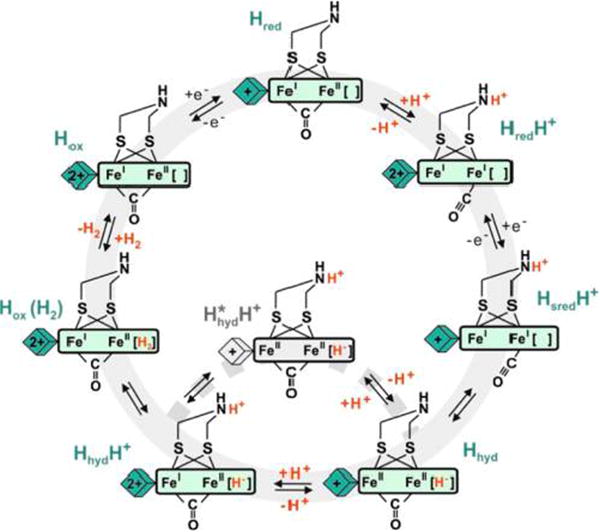
Proposed catalytic cycle for [FeFe]-hydrogenase including the putative hydride states.
Supplementary Material
Acknowledgments
This work was supported by the Max Planck Society (E.J.R., A.A.V., J.F.S., W.L.), the Cluster of Excellence “Unifying Concepts in Catalysis” initiative of DFG (VP), and grants NIH GM-65440 (SPC), NIH GM-0611 (TBR). C.C.P. acknowledges UC Davis and the U.S. Department of Education for a GAANN fellowship (P200A120187). We acknowledge Jiyong Zhao, Ercan Alps, and Michael Hu at 3-ID-D, Advance Photon Source (APS) at Argonne National Laboratory for their help collecting preliminary data for CrHydA1([257Fe]H-ODT) in D2O.
Footnotes
Supporting Information: The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b00686.
Notes: The authors declare no competing financial interest.
References
- 1.Frey M. Chem Bio Chem. 2002;3:153. [Google Scholar]
- 2.Lubitz W, Ogata H, Ruediger O, Reijerse E. Chem Rev. 2014;114:4081. doi: 10.1021/cr4005814. [DOI] [PubMed] [Google Scholar]
- 3.Ogata H, Kraemer T, Wang H, Schilter D, Pelmenschikov V, van Gastel M, Neese F, Rauchfuss TB, Gee LB, Scott AD, Yoda Y, Tanaka Y, Lubitz W, Cramer SP. Nat Commun. 2015;6:7890. doi: 10.1038/ncomms8890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fan HJ, Hall MB. J AM Chem Soc. 2001;123:3828. doi: 10.1021/ja004120i. [DOI] [PubMed] [Google Scholar]
- 5.Siegbahn PEM, Tye JW, Hall MB. Chem Rev. 2007;107:4414. doi: 10.1021/cr050185y. [DOI] [PubMed] [Google Scholar]
- 6.Bruschi M, Greco C, Kaukonen M, Fantucci P, Ryde U, De Gioia L. Angew Chem, Int Ed. 2009;48:3503. doi: 10.1002/anie.200900494. [DOI] [PubMed] [Google Scholar]
- 7.Greco C, De Gioia L. Inorg, Chem. 2011;50:6987. doi: 10.1021/ic200297d. [DOI] [PubMed] [Google Scholar]
- 8.Carroll ME, Barton BE, Rauchfuss TB, Carroll PJ. J Am Chem Soc. 2012;134:18843. doi: 10.1021/ja309216v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sommer C, Adamska-Venkatesh A, Pawlak K, Birrell J, Rüdiger O, Reijerse E, Lubitz W. J Am Chem Soc. 2017;139:1440. doi: 10.1021/jacs.6b12636. [DOI] [PubMed] [Google Scholar]
- 10.Mulder DW, Ratzloff MW, Bruschi M, Greco C, Koonce E, Peters JW, King PW. J Am Chem Soc. 2014;136:15394. doi: 10.1021/ja508629m. [DOI] [PubMed] [Google Scholar]
- 11.Mulder DW, Guo Y, Ratzloff MW, King PW. J Am Chem Soc. 2017;139:83. doi: 10.1021/jacs.6b11409. [DOI] [PubMed] [Google Scholar]
- 12.Cornish AJ, Gaertner K, Yang H, Peters JW, Hegg EL. J Biol Chm. 2011;286:38341. doi: 10.1074/jbc.M111.254664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berggren G, Adamska A, Lambertz C, Simmons T, Esselborn J, Atta M, Gambarelli S, Mouesca J, Reijerse E, Lubitz W, Happe T, Artero V, Fontecave M. Nature. 2013;499:66. doi: 10.1038/nature12239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Esselborn J, Lambertz C, Adamska-Venkatesh A, Simmons T, Berggren G, Noth J, Siebel J, Hemschemeier A, Artero V, Reijerse E, Fontecave M, Lubitz W, Happe T. Nat Chem Biol. 2013;9:607. doi: 10.1038/nchembio.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Siebel JF, Adamska-Venkatesh A, Weber K, Rumpel S, Reijerse E, Lubitz W. Biochemistry. 2015;54:1474. doi: 10.1021/bi501391d. [DOI] [PubMed] [Google Scholar]
- 16.Kuchenreuther JM, Myers WK, Suess DLM, Stich TA, Pelmenschikov V, Shiigi SA, Cramer SP, Swartz JR, Britt RD, George S. J Science. 2014;343:424. doi: 10.1126/science.1246572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Suess DLM, Bürstel I, De La Paz L, Kuchenreuther JM, Pham CC, Cramer SP, Swartz JR, Britt RD. Proc Natl Acad Sci, U S A. 2015;112:11455. doi: 10.1073/pnas.1508440112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gilbert-Wilson R, Siebel JF, Adamska-Venkatesh A, Pham CC, Reijerse E, Wang H, Cramer SP, Lubitz W, Rauchfuss TBI. Am Chem Soc. 2015;137:8998. doi: 10.1021/jacs.5b03270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barton BE, Olsen MT, Rauchfuss TB. J Am Chem Soc. 2008;130:16834. doi: 10.1021/ja8057666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roseboom W, De Lacey AL, Fernandez VM, Hatchikian EC, Albracht SPJ. J Biol Inorg Chem. 2006;11:102. doi: 10.1007/s00775-005-0040-2. [DOI] [PubMed] [Google Scholar]
- 21.Pereira AS, Tavares P, Moura I, Moura JJG, Huynh BH. J Am Chem Soc. 2001;123:2771. doi: 10.1021/ja003176+. [DOI] [PubMed] [Google Scholar]
- 22.Kamali S, Wang H, Mitra D, Ogata H, Lubitz W, Manor BC, Rauchfuss TB, Byrne D, Bonnefoy V, Jenney FE, Jr, Adams MWW, Yoda Y, Alp E, Zhao J, Cramer SP. Angew Chem, Int Ed. 2013;52:724. doi: 10.1002/anie.201204616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Winkler M, Happe T. Personal Communication [Google Scholar]
- 24.Adamska A, Silakov A, Lambertz C, Ruediger O, Happe T, Reijerse E, Lubitz W. Angew Chem, Int Ed. 2012;51:11458. doi: 10.1002/anie.201204800. [DOI] [PubMed] [Google Scholar]
- 25.Finkelmann AR, Stiebritz MT, Reiher M. Chem Sci. 2014;5:215. doi: 10.1021/ic501049z. [DOI] [PubMed] [Google Scholar]
- 26.Huynh MT, Wang W, Rauchfuss TB, Hammes-Schiffer S. Inorg Chem. 2014;53:10301. doi: 10.1021/ic5013523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Warren JJ, Mayer JM. Biochemistry. 2015;54:1863. doi: 10.1021/acs.biochem.5b00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wright JA, Pickett CJ. In: Bioinspired Catalysis. Weigand W, Schollhammer P, editors. Wiley-VCH Verlag GmbH & Co; KGaA, Weinheim, Germany: 2015. p. 161. [DOI] [Google Scholar]
- 29.Esswein AJ, Nocera DG. Chem Rev. 2007;107:4022. doi: 10.1021/cr050193e. [DOI] [PubMed] [Google Scholar]
- 30.McKone JR, Marinescu SC, Brunschwig BS, Winkler JR, Gray HB. Chem Sci. 2014;5:865. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.