

Abstract
Determinants of multidrug resistance (MDR) are often encoded on mobile elements, such as plasmids, transposons, and integrons, which have the potential to transfer among foodborne pathogens, as well as to other virulent pathogens, increasing the threats these traits pose to human and veterinary health. Our understanding of MDR among Salmonella has been limited by the lack of closed plasmid genomes for comparisons across resistance phenotypes, due to difficulties in effectively separating the DNA of these high-molecular weight, low-copy-number plasmids from chromosomal DNA. To resolve this problem, we demonstrate an efficient protocol for isolating, sequencing and closing IncA/C plasmids from Salmonella sp. using single molecule real-time sequencing on a Pacific Biosciences (Pacbio) RS II Sequencer. We obtained six Salmonella enterica isolates from poultry, representing six different serovars, each exhibiting the MDR-Ampc resistance profile. Salmonella plasmids were obtained using a modified mini preparation and transformed with Escherichia coli DH10Br. A Qiagen Large-Construct kit™ was used to recover highly concentrated and purified plasmid DNA that was sequenced using PacBio technology. These six closed IncA/C plasmids ranged in size from 104 to 191 kb and shared a stable, conserved backbone containing 98 core genes, with only six differences among those core genes. The plasmids encoded a number of antimicrobial resistance genes, including those for quaternary ammonium compounds and mercury. We then compared our six IncA/C plasmid sequences: first with 14 IncA/C plasmids derived from S. enterica available at the National Center for Biotechnology Information (NCBI), and then with an additional 38 IncA/C plasmids derived from different taxa. These comparisons allowed us to build an evolutionary picture of how antimicrobial resistance may be mediated by this common plasmid backbone. Our project provides detailed genetic information about resistance genes in plasmids, advances in plasmid sequencing, and phylogenetic analyses, and important insights about how MDR evolution occurs across diverse serotypes from different animal sources, particularly in agricultural settings where antimicrobial drug use practices vary.
Keywords: antimicrobial resistance, IncA/C plasmid, Salmonella enterica, next generation sequencing
Introduction
Salmonella enterica is one of the most common bacterial causes of foodborne illness, resulting in over 2,100 hospitalizations, 30 deaths, and over 3 billion dollars (US) in direct medical costs each year (Scallan et al., 2011; USDA, 2014). Increasingly, Salmonella strains, which can affect both humans, livestock and other animals, are exhibiting multidrug resistance (MDR) to many of the currently available antibiotic therapies, with consequences for consumers, farmers, and public health (Lu et al., 2014). Strains displaying MDR are associated with more severe infections (Varma et al., 2005). In particular, the emergence of MDR strains resistant to both fluoroquinolone and third-generation cephalosporins greatly limits options for treating human salmonellosis (Fair and Tor, 2014), which usually manifests as self-limiting diarrhea, but can progress to life-threatening infections requiring medical intervention (White et al., 2002). The National Antimicrobial Resistance Monitoring System (NARMS) monitors antimicrobial resistance in Salmonella isolated from humans, retail meats, and food animals in the United States (FDA, 2016). Some of the MDR Salmonella strains identified display resistance to more than 8 antimicrobials. Clinically-relevant antimicrobial resistances among Salmonella include the MDR-AmpC phenotype, which is resistant to ampicillin, chloramphenicol, streptomycin, sulfonamide, tetracycline (ACSSuT), as well as to amoxicillin/clavulanic acid, cefoxitin, ceftiofur, and ceftriaxone (Gilbert et al., 2007). While actions have been taken to reduce the use of antibiotics in animal husbandry, variations in use patterns are likely to result in variations in the persistence of MDR phenotypes in different geographic regions.
Although, the composition of bacterial genomes can change rapidly and dramatically in response to antibiotic exposure, the most important changes in the genomes Salmonella and many other genera occur via horizontal gene transfer. Determinants of MDR are often encoded on mobile elements, such as plasmids, transposons, and integrons, which can be transferred from foodborne pathogens to more virulent human pathogens (Winokur et al., 2000; Fricke et al., 2011). This process has greatly contributed to the rapid dissemination of antimicrobial resistance among multiple bacterial genera of human and veterinary importance (Fernandez-Alarcon et al., 2011).
Ideally, we could use comparative analyses of MDR plasmids to understand how resistance evolves among diverse serotypes from different animal sources, particularly in agricultural settings where variations in antimicrobial drug use could exert different selective pressures on Salmonella strains. For this reason, complete plasmid sequence information is critical for understanding the nature of antibiotic resistance. The presence of single genes (such as can be detected by PCR) or transferable phenotypes (identified by conjugation experiments) does not provide the detailed genetic context needed to understand the molecular mechanisms involved in bacterial evolution at the allelic level. The same allele might be present in a plasmid as part of a transposon or as an independent coding sequence flanked by degenerate recombination machinery. Interpreting the evolution of antibiotic resistance will differ according to the molecular pathways deduced from the DNA sequence.
However, our understanding about the evolution of MDR is hampered by having only a few closed plasmids available for analysis. Plasmid DNA is often difficult to separate from chromosomal DNA contigs in next generation sequencing (NGS) analyses, particularly if a plasmid is only present in low copy numbers. Furthermore, plasmid sequences frequently contain multiple repetitive regions, which can be difficult to assemble properly from short reads.
One means of resolving that challenge is to use third-generation, single molecule, real-time DNA sequencing on the Pacific Biosciences (PacBio) RS II Sequencer, which can provide high-quality reads that are longer than those from any other sequencing platform available as of the beginning of 2017. The use of very long reads greatly facilitates distinguishing plasmid sequences from chromosomal DNA. However, the PacBio technology requires sufficiently high concentrations of high-molecular-weight, low copy number, naturally-occurring plasmids, which has been difficult to achieve using most commercially available isolation kits. Therefore, our project was to develop an efficient plasmid isolation protocol for Salmonella enterica serovars that could yield the necessary amounts of high quality plasmid DNA, specifically of large plasmids with a low copy number, which are known to carry genes conferring resistance to a myriad of antibiotics and several toxic heavy metals. To evaluate this method, we prepared plasmid DNA from six different Salmonella serovars and analyzed the resulting sequences including (1) 14 additional IncA/C plasmids derived from S. enterica and (2) 38 IncA/C additional plasmids derived from different taxa to provide an evolutionary picture of antimicrobial resistance development in IncA/C plasmids.
By analyzing the sequences of these highly similar IncA/C plasmids, we may better understand how antimicrobial resistance is shaped by regional selection pressures, host species, and patterns of antibiotic use, which may provide important insights that could improve future traceback investigations.
Materials and methods
Bacterial strains, growth conditions, and antimicrobial susceptibility testing
Six different S. enterica isolates, collected from poultry and belonging to serovars Newport, Typhimurium, Infantis, Agona, Kentucky, and Heidelberg, were used in this study (Table 1). These isolates were cultured on trypticase soy agar (TSA; Becton, Dickinson, NJ, USA) and in trypticase soy broth (TSB; Becton, Dickinson, NJ, USA) overnight at 37°C, after which the isolates were stored in TSB containing 20% glycerol at −80°C. All isolates were serotyped by conventional methods, including the standardized NARMS method of in vitro antimicrobial susceptibility-testing which uses a panel of 15 antimicrobial agents (amikacin, ampicillin, amoxicillin-clavulanic acid, cefoxitin, ceftiofur, ceftriaxone, chloramphenicol, ciprofloxacin, gentamicin, kanamycin, nalidixic acid, streptomycin, sulfisoxazole, tetracycline, and trimethoprim-sulfamethoxazole), and the TREK Sensititre® automated antimicrobial susceptibility system (Thermo Fisher Scientific, Waltham, MA). Results were interpreted using clinical breakpoints previously established by the Clinical and Laboratory Standards Institute (CLSI).
Table 1.
List of the metadata for the strains used in this study.
| Accession # | Plasmid name | Species | Location | Source | Year |
|---|---|---|---|---|---|
| IncA/C PLASMIDS SEQUENCED IN THIS STUDY | |||||
| CP009409 | pCFSAN000405 | Salmonella Heidelberg | USA_NM | ground turkey | 2004 |
| CP009410 | pCFSAN007405 | Salmonella Typhimurium var. O 5 | USA_CA | ground turkey | 2003 |
| CP009411 | pCFSAN007425 | Salmonella Newport | USA_MD | ground turkey | 2002 |
| CP009412 | pCFSAN007426 | Salmonella Agona | USA_CO | ground turkey | 2008 |
| CP009413 | pCFSAN007427 | Salmonella Infantis | USA_NM | ground turkey | 2009 |
| CP009414 | pCFSAN007428 | Salmonella Kentucky | USA_NM | chicken breast | 2006 |
| IncA/C PLASMID SEQUENCES OBTAINED FROM NCBI | |||||
| CP009560 | pCVM22425 | Salmonella Newport | USA_AZ | cattle | 2003 |
| CP009562 | pCVM22513 | Salmonella Newport | USA_NC | cattle | 2003 |
| CP009563 | pCVM21538 | Salmonella Newport | USA_GA | chicken breast | n/a |
| CP009564 | pCVM21550 | Salmonella Newport | USA_TX | swine | n/a |
| CP009567 | pCFSAN000934 | Salmonella Newport | USA_AZ | canine | 2003 |
| CP009570 | pCFSAN000941 | Salmonella Newport | USA_GA | ground beef | 2003 |
| FJ621587 | pAM04528 | Salmonella Newport | USA_KS | human | 1998 |
| CP000604 | pSN254 | Salmonella Newport | USA_MN | human | 2000 |
| JN983043 | pSH111_166 | Salmonella Heidelberg | USA_OH | cattle | 2001 |
| JN983045 | psH163_135 | Salmonella Heidelberg | USA_OH | swine | 2002 |
| JN983048 | psH696_135 | Salmonella Heidelberg | USA | turkey | 2000 |
| JF267651 | pSD_174 | Salmonella Dublin | USA_WI | cattle | n/a |
| KF056330 | p1643_10 | Salmonella Kentucky | Polen | n/a | n/a |
| KM670336 | pSRC119-A/C | Salmonella Senftenberg | Australia | n/a | 2000 |
| AP012208 | pNDM-1_Dok01 | Escherichia coli | Japan | human | 2009 |
| HQ023861 | pPG010208 | Escherichia coli | Chile | cattle | 2004 |
| HQ023862 | pUMNK88 | Escherichia coli | USA_MN | swine | 2007 |
| HQ023863 | pAPEC1990_61 | Escherichia coli | USA | turkey | 1995 |
| HQ023864 | pAR060302 | Escherichia coli | USA_IL | cattle | 2002 |
| JF503991 | pNDM10505 | Escherichia coli | Canada | human | 2010 |
| JF714412 | pNDM102337 | Escherichia coli | Canada | human | 2010 |
| FJ621586 | peH4H | Escherichia coli | USA_WA | cattle | 2002 |
| KF152885 | pSCEC2 | Escherichia coli | China | swine | 2010 |
| CP003225 | pKPHS3 | Klebsiella pneumoniae | China | human | 2011 |
| JN157804 | pNDM-KN | Klebsiella pneumoniae | Kenya | human | 2009 |
| JN861072 | pNDM10469 | Klebsiella pneumoniae | Canada | human | 2010 |
| JQ010984 | pR55 | Klebsiella pneumoniae | France | human | 1969 |
| JX442976 | pIncA/C-LS6 | Klebsiella pneumoniae | Italy | human | 2011 |
| KF976462 | pRMH760 | Klebsiella pneumoniae | Australia | human | 1997 |
| FJ705807 | pRA1 | Aeromonas hydrophila | Japan | fish | 1971 |
| JX141473 | pR148 | Aeromonas hydrophila | Thailand | fish | 2008 |
| AB277723 | pP99-018 | Photobacterium damselae | Japan | fish | 1999 |
| AB277724 | pP91278 | Photobacterium damselae | Japan | fish | 1991 |
| CP000602 | pYR1 | Yersinia ruckeri | USA | fish | 1996 |
| CP000603 | pIP1202 | Yersinia pestis | Madagascar | human | 1995 |
| CP007636 | 2012EL-2176 | Vibrio cholerae | Haiti | human | 2012 |
| JN687470 | pMR0211 | Providencia stuartii | Afghanistan | human | 2011 |
| FN667743 | pXNC1 | Xenorhabdus nematophila | USA | nematode | 1965 |
For comparative analysis, in addition to the six S. enterica plasmids sequenced in this study, we included sequence data from 14 IncA/C plasmids derived from S. enterica and 24 IncA/C plasmids derived from different taxa (Escherichia coli, Klebsiella pneumonia, Aeromonas hydrophila, Photobacterium damselae, Yersinia ruckeri, Yersinia pestis, Vibrio cholera, Providencia stuartii, and Xenorhabdus nematophila) available at the National Center for Biotechnology Information (NCBI); these are described in Table 1.
Plasmid isolation
Plasmids were isolated from 5 mL overnight cultures of each strain, grown at 37°C in Luria Bertani (LB) broth containing the appropriate antibiotics at a concentration that assured plasmid maintenance. Bacterial cells were pelleted by centrifugation at 6,000 g/10:00 min/4°C. The resulting bacterial pellet was resuspended in 350 μL resuspension buffer (1X PBS, 40 mM Tris (pH8), 1 mM EDTA) and 200 μg RNaseA was added. This mixture was transferred to 2 mL microfuge tubes, 350 μL lysis buffer (0.2 M NaOH, 1% SDS) was added, and each tube was mixed by inversion several times. After 2 min of incubation at room temperature, 500 μL of neutralization buffer (1.32 M potassium acetate) were added; the mixtures were incubated at room temperature for another 5 min, then centrifuged (20,800 g/10:00 min/4°C). Each supernatant was transferred to a 2 mL microfuge tube and extracted one time using an equal volume of a phenol:chloroform:isoamyl alcohol solution (25:24:1) and tube inversion until the preparation was a homogenous milky color in appearance. Each preparation was centrifuged (20,800 g/8:00 min/4°C) and the aqueous phase transferred to a new 2 mL microfuge tube, avoiding precipitate band present at the interface. The aqueous phase was then centrifuged (20,800 g/10:00 min/4°C) to pellet any residual white precipitate present, then the supernatant (~750 μL) transferred to a 1.5 mL microfuge tube. Next, 0.1 volume (80 μL) of 3M NaOAc (pH5.5) was added and mixed prior to adding 500 μL isopropanol (room temperature), and mixing by inversion (~40X). The solution was centrifuged, as above, to collect the DNA precipitate and washed once with 70% ethanol (room temperature). This last supernatant was discarded and the dried DNA resuspended in 12 μL dIH2O. The quantity and quality of this DNA was determined by electrophoresis of a 2 μL aliquot on a 0.6% TBE agarose gel at 120 V (constant).
Plasmid transformation
We used 40 μL phage-resistant E. coli (E. coli) DH10B-T1 [Invitrogen catalog number 12033-015. F-mcrA Δ(mrr-hsdRMS-mcrBC) f80lacZ ΔM15 ΔlacX74 recA1 endA1 araD139 Δ(ara, leu)7697 galU galK λ—rpsL nupG tonA] as host cells for transformation. Transformation was done by electroporation with 1.5–2.0 μL plasmid preparation using large construct parameters, 1.3 kV, 100 Ω, and 25 μFd in a 0.1 cm electroporation cuvette. Typically, time constants ranged from 2.3 to 2.5 ms. Cells were rescued in 750 μL SOC broth at 37°C for 1 h prior to plating on LB agar containing 20 μg/mL chloramphenicol (MP Biomedicals catalog number 02190321, OH, USA) or 100 μg/mL ampicillin (Sigma-Aldrich catalog number A9518, MO, USA). DH10Br-T1 cells containing the desired Salmonella antibiotic-resistance plasmids were processed using the Qiagen® Large Plasmid Construct kit (Qiagen catalog number 12462, CA, USA), according to manufacturer's protocol, to obtain suitable concentrations of sequence-quality plasmids. At the completion of each plasmid isolation protocol, DNA purity, and quantity were determined by electrophoresis of (2 μL) each sample on a 0.6% TBE agarose gel at 120 V constant.
Genome sequencing, assembly, and annotation
We then sequenced these six plasmids using third-generation, single molecule, real-time DNA sequencing on the PacBio RS II Sequencer, as previously described (Hoffmann et al., 2013, 2014). Six different 10-kb libraries were prepared following the PacBio sample preparation methods. Each library was sequenced using the C2/P4 chemistry on one single molecule real-time (SMRT) cell with a 120-min collection protocol. The continuous-long-read (CLR) data was assembled de novo using the PacBio hierarchical genome assembly process (HGAP2.0). The assemblies outputs from HGAP contain overlapping regions at the end, which can be identified using dot plots in Gepard (Krumsiek et al., 2007). Afterwards, the improved consensus sequences were uploaded in SMRT Analysis, and final consensus and accuracy scores were determined using the Quiver consensus algorithm (Chin et al., 2013). All assembled plasmids were annotated using the NCBI's Prokaryotic Genomes Automatic Annotation Pipeline (PGAAP; Klimke et al., 2009).
Ideogram of six plasmids
The ideogram (Figure 1) was drawn based on the presence and absence of genes among our six IncA/C plasmids of S. enterica. The genes for the composite outer ring and each individual plasmid were put into BED format as separate files using a custom Python 2.7 script. We used CP009409 as a template for the composite outer ring, preferentially placing the genes found in other plasmids at the same location as the homolog present in CP009409. Genes were only added to the outer ring if their homolog was absent from CP009409. When it was necessary to add genes to the composite, they were added bordering a gene matching the one they resided next to in their native context. Data from this composite and each plasmid were then loaded into R in BED format as separate files. The ideogram was drawn using Circlize17 with some additional scripting in R to refine the positions of the labels.
Figure 1.

Ideogram showing positions and presence-absence of genes between the six IncA/C plasmids of S. enterica. The order of the genes in the composite outer ring was modeled after CP009409. Genes not present in that plasmid were added to the composite next to genes they would border in their natural context.
Heat map representation of antimicrobial resistance genes
We identified the antimicrobial resistance genes by mapping the closed plasmids to a custom database consisting of 1,379 known resistance genes (Hoffmann et al., 2014). The abundance profile of these genes was then used to construct heatmaps (Figure 2) and clustering using the Heatmap.2 function of the gplots (Warnes et al., 2015) package within R (R Development Core Team, 2015). This enabled us to visualize both the raw counts within each sample and the phenetic relationships among samples, based on Manhattan distances.
Figure 2.
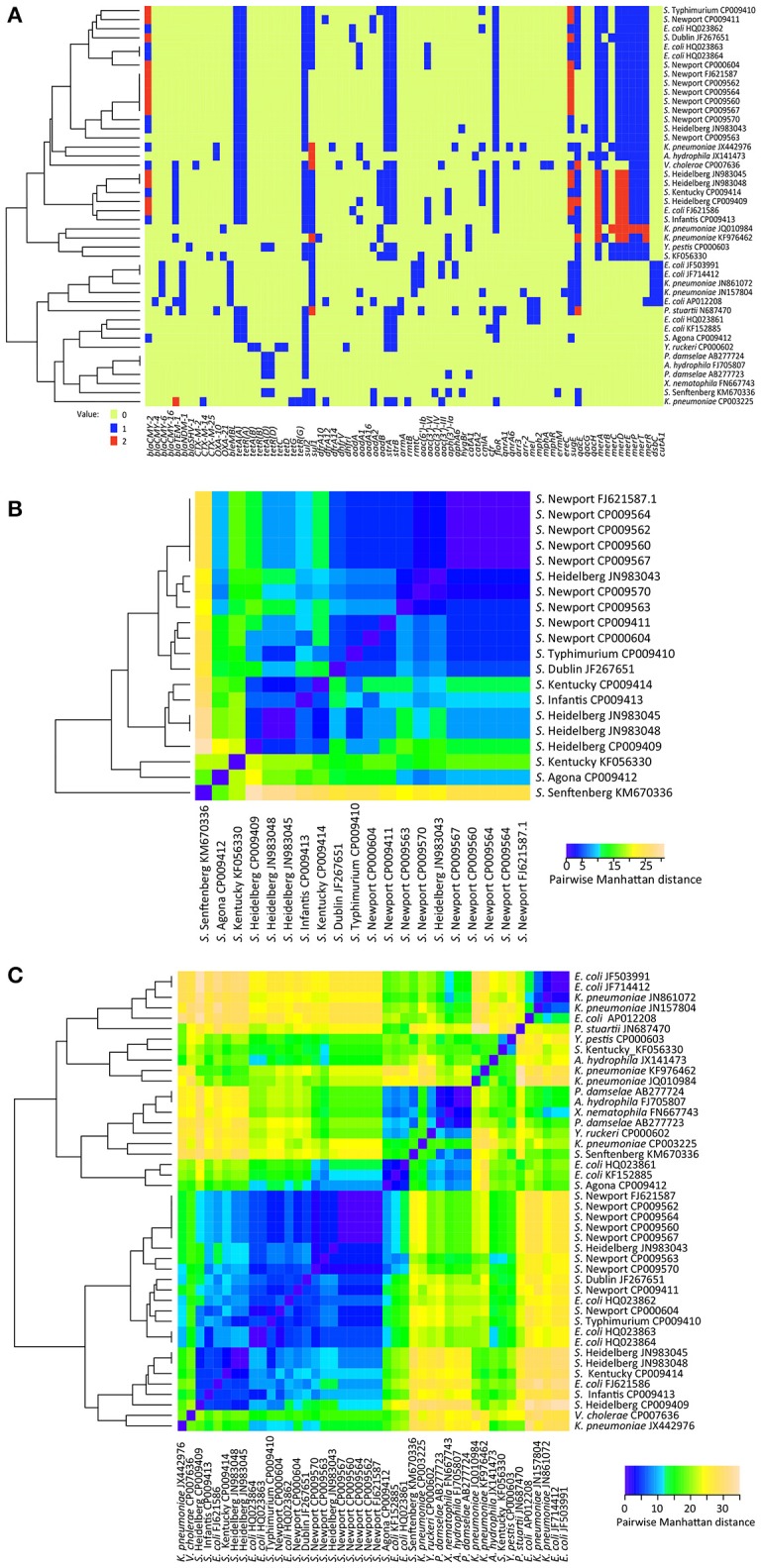
(A) Heatmap of presence, absence, and abundance of antimicrobial, metal, and quaternary ammonium compounds resistance genes between 44 IncA/C plasmids. (B) Heatmap based on calculating the distance between 20 Salmonella IncA/C plasmids in the presence, absence, and abundance of antimicrobial, metal, and quaternary ammonium compounds resistance genes in them. (C) Heatmap based on calculating the distance between 44 IncA/C plasmids in the presence, absence, and abundance of antimicrobial, metal, and quaternary ammonium compounds resistance genes in them. The heatmaps were generated using Heatmap.2 in R.
Comparative plasmid sequences analyses
We then performed comparative analyses of the six plasmids with sequence data from 38 complete IncA/C plasmids available at NCBI: 14 extracted from S. enterica and 24 isolated from other taxa (Table 1), using the Mauve (Darling et al., 2010) genome alignment v.2 and Geneious R7 v.7.1.3 software packages. The core genes were identified using Ridom SeqSphere+ software v3.0.0 (Ridom GmbH, Münster, Germany). The maximum-likelihood (ML) method of the Genetic Algorithm for Rapid Likelihood Inference (GARLI) software (Zwickl, 2006) was used to construct evolutionary relationships among the closed Salmonella plasmid genomes and the wider taxonomic dataset.
We used a reference-free k-mer based approach, embedded in the kSNP v2.1 software(Gardner and Hall, 2013), to identify phylogenetically informative SNP sites (i.e., SNPs shared by two or more strains in the alignment). Briefly, kSNP indexes plasmids into k-mers and then identifies (1) those k-mers that are unique within each plasmid and (2) those k-mers that have a SNP difference in the middle of the k-mer.
Genbank accession numbers
The plasmid sequences determined in this study have been deposited in the GenBank database under following range of accession numbers: CP009409-CP009414 (Table 1).
Results and discussion
Antimicrobial resistance
As shown in Table 2, in vitro antimicrobial susceptibility testing results confirmed that our initial set of six Salmonella isolates all exhibited the MDR-AmpC resistance profile: resistance to amoxicillin, ampicillin, cefoxitin, ceftriaxone, chloramphenicol, streptomycin, sulfisoxazole, tetracycline, and ceftiofur. Our serovars of S. Heidelberg (CFSAN000405), S. Infantis (CFSAN007427) and S. Kentucky (CFSAN007428) demonstrated additional resistances to kanamycin and gentamicin, while S. Typhimurium (CFSAN007405) and S. Newport (CFSAN007425) showed additional resistances to kanamycin, nalidixic acid, and cephalexin, and cephalexin and trimethoprim/sulfamethoxazole, respectively. In the next section we will confirm that the genes enabling these resistance patterns are contained on each respective plasmid.
Table 2.
List of the resistance genotype-phenotype identified for the six IncA/C plasmids.
| S. Kentucky CFSAN007428 | S. Newport CFSAN007425 | S. Typhimurium CFSAN007405 | S. Agona CFSAN007426 | S. Heidelberg CFSAN000405 | S. Infantis CFSAN007427 | |
|---|---|---|---|---|---|---|
| Resistance Phenotype | AMC, AMP, FOX, AXO, CHL, GEN, KAN, STR, FIS, TET, TIO | AMC, AMP, FOX, AXO, CEP, CHL, STR, FIS, TET, COT, TIO | AMC, AMP, FOX, AXO, CEP, CHL, KAN, NAL, STR, FIS, TET, TIO | AMC, AMP, FOX, AXO, CHL, STR, FIS, TET, TIO | AMC, AMP, FOX, AXO, CHL, GEN, KAN, STR, FIS, TET, TIO | AMC, AMP, FOX, AXO, CHL, GEN, KAN, STR, FIS, TET, TIO |
| Beta-Lactams | blacmy−2, blatem−1 | blacmy−2* | blacmy−2* | blacmy−2 | blacmy−2*, blatem−1 | blacmy−2, blatem−1 |
| Aminoglycosides | strA, strB, aadB aph(3)-Ia | strA, strB, aadA2 | strA, strB, aadB | strA, strB | strA, strB, aadA1, aadB, aac(3')-VI, aph(3')-Ia | strA, strB, aadA1, aac(3)-VI |
| Tetracyclines | tetR(A), tetA | tetR(A), tetA | tetR(A), tetA | tetR(A), tetA | tetR(A), tetA | tetR(A), tetA |
| Chloramphenicols | floR, cmlA | floR | floR, cmlA | floR | floR, cmlA | floR |
| Folate synthesis inhibitors | sul1, sul2 | sul1, sul2, dfra12 | sul1, sul2 | sul2 | sul1, sul2 | sul1, sul2 |
| Quaternary ammonium compounds | quacE, sugE | quacE, sugE* | quacE, sugE* | sugE | quacE*, sugE* | quacE, sugE |
| Mercury ions | merA*, merB, merC, merD*, merE*, merP, merT, merR | merA, merB, merC, merD, merE, merP, merT, merR | merA, merB, merC, merD, merE, merP, merT, merR | merA*, merB, merC, merD*, merE*, merP, merT, merR | merA*, merB, merC, merD*, merE*, merP, merT, merR |
Gene is present twice.
Plasmid purification and sequencing
All six of the MDR S. enterica isolates contained a large IncA/C plasmid, which we identified using replicon typing, as described by Carattoli et al. (2006). We devised an efficient two step protocol for plasmid isolation and extraction: (1) using an osmotically stable resuspension buffer in the initial step to avoid lysing the cells prematurely, a problem observed when using commercially-available buffers, and (2) using E. coli and commercially-available high molecular weight plasmid isolation kits and their accompanying protocols. The average yield was ~20 μg of high quality DNA.
Analysis and characterization of the six IncA/C plasmids
All six plasmids were successfully sequenced on the Pacbio RS II sequencer, achieving an average coverage between 970X and 3,783X (Table 3). The G+C contents varied from 51.9 to 53.2%. Although, these plasmids differed in size, ranging from 104 in pCFSAN007426 (S. Agona) to 191 kb in pCFSAN000405 (S. Heidelberg) with between 128 and 226 genes, the genetic organization of each plasmid was very similar (Figure 1). The BLAST analysis of the repA gene from these complete sequences confirmed that all six plasmids belonged to the IncA/C incompatibility group. However, in pCFSAN007427 (S. Infantis) we identified a large inversion of ~110 kb between the transposase insB2 (locus_tag JV44_00185), which occurs upstream of the floR region, and the transposase insB3 (locus_tag JV44_00790), which occurs downstream of the aadA region.
Table 3.
General characteristics of the IncA/C plasmids sequenced in this study.
| Serovar | Plasmid name | Location | Source | Year | Size | #Genes | Coverage | Accession # |
|---|---|---|---|---|---|---|---|---|
| Heidelberg | pCFSAN000405 | USA NM | ground turkey | 2004 | 190,923 | 226 | 970X | CP009409 |
| Typhimurium | pCFSAN007405 | USA CA | ground turkey | 2003 | 132,146 | 163 | 1322X | CP009410 |
| Newport | pCFSAN007425 | USA MD | ground turkey | 2002 | 166,496 | 203 | 1137X | CP009411 |
| Agona | pCFSAN007426 | USA CO | ground turkey | 2008 | 103,586 | 128 | 1484X | CP009412 |
| Infantis | pCFSAN007427 | USA NM | ground turkey | 2009 | 175,517 | 217 | 3783X | CP009413 |
| Kentucky | pCFSAN007428 | USA NM | chicken breast | 2006 | 164,924 | 205 | 995X | CP009414 |
We identified 98 genes that were shared across all six IncA/C plasmids; this core plasmid shared 99.9% nucleotide identity and was virtually identical to the plasmid backbone of S. Newport SL254, which is very common among MDR isolates from food sources (Welch et al., 2007; Fricke et al., 2009). This backbone includes the replication initiation protein gene, repA, and a variety of maintenance genes, such as DNA helicase, DNA binding genes, plasmid stabilization genes, methyltransferase, restriction endonuclease, and the type IV secretion-like conjugative plasmid transfer system (16 genes). Only six of those 98 core genes showed intragenic variability—specifically in the type VI secretion gene, florfenicol/chloramphenicol resistance gene (floR), DNA topoisomerase III (topB), DNA binding gene (ner), DNA helicase, and the plasmid stability gene (stbA); each variation consisted of a single SNP. Five of those six SNPs were non-synonymous SNPs that would result in changes to the amino acid sequences of the proteins produced (Supplementary Table S1).
Five out of the six IncA/C plasmids contained three type IV conjugative transfer genes regions: (I) traIDLEKBVA, (II) traCWUN, and (III) traFHG. Only pCFSAN007405 (S. Typhimurium) did not carry (II) traCWUN. Instead, plasmid pCFSAN007405 had an additional region containing a second copy of conjugative transfer genes traA, traV, and traB that were identical to the type IV conjugative transfer genes traA, traV, and traB from region (I) traIDLEKBVA, although in reverse order (Figure 1). The main differences among these plasmids appeared to be their accessory regions, most likely a consequence of horizontal gene transfer (Fernandez-Alarcon et al., 2011). These accessory regions tend to be comprised of mobile elements, such as integrons and insertion sequences, carrying transposases, and a variety of resistance genes, which here included genes confering resistance to antimicrobials, quaternary ammonium compounds, and mercury-based antibiotics (Figure 1).
All six plasmids contained the modules floR-tetA-strAB-sul2 and blaCMY−2-blc-sugE, and all but pCFSAN007426 (S. Agona) also carried the sulfonamide resistance gene, sul1 (Table 2). However, certain plasmids contained additional copies of modules or additional genes of interest. Notably, three plasmids, pCFSAN000405 (S. Heidelberg), pCFSAN007405 (S. Typhimurium), and pCFSAN007425 (S. Newport), carried two copies of the blaCMY−2-blc-sugE region, confirming previous reports (Welch et al., 2007). Plasmids pCFSAN000405 (S. Heidelberg) and pCFSAN007427 (S. Infantis) also contained genes for streptomycin resistance, aadA1 and gentamycin, aac(3′)-VI. The plasmids pCFSAN000405 (S. Heidelberg), pCFSAN007405 (S. Typhimurium) and pCFSAN007428 (S. Kentucky) all also contained an additional streptomycin resistance gene, aadB, along with the chloramphenicol resistance gene, cmlA. The plasmids from pCFSAN000405 S. Heidelberg and pCFSAN007428 S. Kentucky also carried the kanamycin resistance gene, aph(3′)-Ia. Furthermore, pCFSAN007425 (S. Newport) contains the streptomycin resistance gene, aadA2, as well as the trimethoprim resistance gene, dfra12. Collectively, the total number of antibiotic resistance determinants in each of these isolates varied between 7 and 14 [S. Newport pCFSAN007425 (10), S. Typhimurium pCFSAN007405 (10), S. Heidelberg pCFSAN000405 (14), S. Infantis pCFSAN007427 (11), S. Agona pCFSAN007426 (7), and S. Kentucky pCFSAN007428 (12)] with all resistance phenotypes correlating positively with the genotypes identified in all six Salmonella isolates (Table 2).
In addition to the antibiotic resistance determinants, these plasmids also contained resistance genes protecting against quaternary ammonium and mercury compounds (Figure 1). With the exception of pCFSAN007426, all plasmids contained the quaternary ammonium compound-resistance gene, qacE; pCFSAN000405 (S. Heidelberg) carried two copies of qacE. The same five plasmids carried the merEDBAPTR mercury resistance operon with pCFSAN000405 (S. Heidelberg), pCFSAN007428 (S. Kentucky), and pCFSAN007427 (S. Infantis) carrying two copies of the mercury resistance genes, merA, merD, and merE.
Every resistance gene was surrounded by transposases and/or integrases, suggesting that resistance genes could be transferred easily from one genetic locus to another. Each of our six plasmids contained between 6 and 20 transposition and recombination genes, encoding transposases and integrases; the largest number of these genes were in plasmid pCFSAN000405, which also carried the greatest number of resistance genes.
To complete our comparative analysis of the presence or absence of resistance genes in these plasmids, we constructed heat maps that included data from 38 additional IncA/C plasmids whose sequences were available at NCBI. These heat maps allowed us to generate a color visualization of the data (1) indicating the presence or absence of antimicrobial, metal, and quaternary ammonium compounds resistance genes (Figure 2A), and (2) calculating the distance between IncA/C plasmids in the presence, absence, and abundance of the resistance genes within them (Figures 2B,C). In this analysis, 35 different resistance genes were detected among the 20 Salmonella IncA/C plasmids used in this analysis (Figure 2A). Only one resistance gene, sul2, was found across all 20 plasmids. Interestingly, two geographically-separated isolates, S. Senftenberg KM670336 (Australia) and S. Kentucky KF056330 (Poland), showed a distinct resistance gene absence/presence profile when compared to each other, and were also well-separated from the 18 Salmonella isolates collected in the USA (Figures 2A,B). The nine S. Newport strains isolated in the USA shared similar resistance gene absence/presence profiles (Figures 2A,B) despite coming from different sources (human, swine, cattle, canine, chicken breast, ground turkey, and ground beef). This is especially important, since it demonstrates the stability of the resistance genes within IncA/C plasmids collected from specific geographical locations.
A third heatmap was constructed to establish or determine trends, incorporating the above 20 IncA/C plasmids from Salmonella isolates, and adding 24 different IncA/C plasmids from other taxa, available from NCBI at the time of our study. This multi-taxa analysis revealed 76 different resistance genes could be detected among the expanded set of 44 IncA/C plasmids (Figure 2A). This heat map showed that no unique resistance gene profile could be detected for a particular species of bacteria, i.e., there was no cluster within the heat map that included only individual plasmids from the same species (Figures 2A,C). Nonetheless, plasmids isolated from the USA tended to have a similar resistance gene absence/presence profile that could be distinguished from the profiles of plasmids that had been isolated elsewhere in the world. Additionally, the pairwise distance dendrogram demonstrated that plasmids isolated from fish had a similar resistance gene absence/presence profile different from those of plasmids isolated from terrestrial animals that carry more antibiotic resistance genes (Figure 2C). This suggests that each isolate has accumulated resistance genes, which favor survival in its specific environment. Looking at the 14 IncA/C plasmids isolated from humans, 10 of these exhibited a resistance profile similar to the plasmids isolated from fish while four plasmids exhibited a resistance profile more similar to plasmids isolated from land animals.
Phylogenetic analyses
By comparing the genomes of the six plasmids sequenced in this study with those from publically-available (NCBI) IncA/C plasmids derived from Salmonella (Table 1), we identified 67 core genes (Supplementary Table S2). Of these core genes, 44 were 100% identical and 23 had variations in their sequences. We concatenated (total length 14,076 bp), and aligned those 23 variable core genes in order to construct a Maximum Likelihood (ML) tree. The gene matrix consisted of 149 SNP positions 123 of which were informative. The resulting ML tree partitioned the 20 Salmonella IncA/C plasmids into two distinct lineages, separated by a mean distance of 132 SNPS, with 100% bootstrap support (Figure 3A).
Figure 3.

(A) ML tree based for 20 IncA/C plasmids isolated from Salmonella. A total of 23 genes with 149 variable SNPs were found using Ridom (SeqSphere+). The ML tree was generated in GARLI v.2.0 under the GTR + Γ model of nucleotide evolution and visualized using Figtree v1.3.1. To the right of the tree, a Distruct plots were reconstructed with the gene matrix. The Distruct plot was generated using a model-based Bayesian clustering method implemented in STRUCTURE v2.3.2 and visualized with DISTRUCT v1.1. Different colors represent the different clusters and each bar represents an individual isolate. The fraction of the bar that is a given color represents the coefficient of membership to that cluster (e.g., multicolored bars indicate membership to multiple groups indicative of admixture). The taxa of source for each isolate, geographic location, and date were mapped onto the tree. (B) Minimum spanning tree based for 20 IncA/C plasmids isolated from Salmonella. The spanning tree was generated in Beast. The numbers of SNPs are shown on each branch.
Lineage I included 18 plasmids having an intra-SNP diversity of 2 SNPs, while Lineage II included only two plasmids, one derived from S. Kentucky and the other from S. Senftenberg, having a much broader intra-SNP diversity of 19 SNPs. The IncA/C plasmids from Lineage I all belonged to sequence type (ST) 1 A/C2 plasmids (Harmer and Hall 2015) and had all been obtained in the USA. Isolates from Lineage II all belonged to ST2 A/C2 plasmids (Harmer and Hall, 2015). Based on earlier observations that all of the isolates from a shared geographical area tend to cluster, it seems likely that the plasmids would most likely tend to cluster by geographical location rather than by source or bacterial taxa. The majority of the plasmids from Lineage I could be further divided into two sublineages (65% bootstrap support). Sublineage (SL IA) consisted of S. Typhimurium, S. Kentucky, S. Dublin, S. Agona, S. Dublin and the isolates of S. Heidelberg that had been isolated from chicken breast (3), ground turkey, cattle, and pigs with an intra- diversity of only 1.3 SNPs. Sublinage (SL IIA) consisted of all nine S. Newport IncA/C plasmids that had been isolated from different sources, with an intra-SNP diversity of only 0.2.
Although the 23 variable core genes identified within the 20 plasmids had 123 informative SNPs, the haplotype diversity was fairly low, consisting of only 9 haplotypes (Figure 3B). Consequently, there were only a few unique sequences among the core genes from IncA/C Salmonella plasmids that were congruent with our tree topology. To provide another way of identifying the number of lineages into which our set of plasmids could be segregated, we used the program STRUCTURE (Falush et al., 2003) to construct groups and then visualized the results using DISTRUCT (Rosenberg, 2004; Figure 3A). Those results (Figure 3A) were consistent with the lineages established by our phylogenetic analyses: two distinct groups, each represented by a different color.
To provide more precise picture of the evolution and diversity among IncA/C plasmids, we repeated the above analysis, this time including 38 additional IncA/C plasmids from 10 different taxa. Among these 44 IncA/C plasmids a total of 22 core genes could be identified, all containing SNPs (Supplementary Table S3). Those 22 core genes were concatenated (total length 13,299 bp), aligned, and used to create another ML tree (Figure 4A). This gene matrix consisted of 2,366 variable SNP positions, 480 of which were informative. The resulting ML tree partitioned the 44 IncA/C plasmids into three distinct lineages. For this set, Lineage I contained 7 type 2 A/C plasmids, representing three different species (K. pneumonia, A. hydrophila, and E. coli) isolated and sequenced between 1997 and 2010. Lineage I was divided into 2 sublineages. Sublineage (SL IB) represented plasmids isolated from human and tilapia sources obtained in Australia, Thailand, and Japan; Sublineage (SL IIB) represented four plasmids isolated from humans, obtained in Canada. Lineage II included 34 plasmids, representing eight different species (Klebsiella pneumoniae, A. hydrophila, Salmonella, P. damselae, Y. pestis, P. stuartii, Vibrio cholerae, and E. coli), isolated between 1991 and 2011. Lineage II could be further divided into three sublineages. Sublineage (SL IC) included nine isolates sequenced type 2 A/C2 plasmids from different taxa, isolated from fish and humans, obtained in China, Japan, Madagascar, France and Australia between 1969 and 2011. Sublineage (SL IIC) was comprised of 15 sequenced type 1 A/C2 plasmids isolated from human, meat, and animals, obtained mainly from the USA. Sublineage (SL IIIC) was comprised of 10 plasmids from different sources in the USA between 1995 and 2003 and one E. coli plasmid isolated from a cow in the USA in 2002. The most diverse lineage, Lineage III, was the most distant from the others; it contained three plasmids obtained from Y. ruckeri, A. hydrophila, and X. nematophila, isolated from fish (Y. ruckeri, A. hydrophila) and nematode (X. nematophila) sources. These three lineages were separated by two thousand SNPs; surprisingly, Lineages I and II had an intra-mean diversity of only two SNPs, even though those plasmids had been isolated from very different species at different locations and different times.
Figure 4.

(A) ML tree based for 44 IncA/C plasmids. A total of 22 genes with 2,373 variable SNPs were found using Ridom (SeqSphere+). The ML tree was generated as described in Figure 3A. (B) Minimum spanning tree based for 44 IncA/C plasmids. The spanning tree was generated in Beast. The numbers of SNPs are shown on each branch.
Among the complete set of 44 IncA/C plasmids, the 22 variable core genes contained 2,366 informative SNPs. Despite this, the haplotype diversity is fairly low, revealing only 12 haplotypes (Figure 4B), which demonstrated that were fewer unique core gene sequences than the number of IncA/C plasmids obtained across all taxa in our analyses. Using kSNP, we constructed a SNP matrix for each plasmid to further investigate the evolutionary relationships among the closed Salmonella plasmid genomes and the wider taxonomic dataset. In contrast to the core-genome approach, we used the full SNP matrix produced by kSNP, which allows for missing data in the matrix. The ML trees produced in this way provided even finer resolution within the Lineages described above (Supplementary Figure S1).
Conclusions
Although newer sequencing technologies have made it more affordable to perform WGS on bacteria, separating large plasmids from chromosomal contigs is still challenging, particularly if the plasmid is only present low copy numbers. Previous methods of high-molecular-weight plasmid isolation, such as cesium chloride density gradient centrifugation and Solid-Phase Reversible Immobilization (SPRI), involve special equipment and are often very time consuming, especially for a high-throughput and a high-yield approach. This becomes particularly important when one considers that a minimum of 5 μg DNA is required for sequence reads that will extend beyond the size of the repeat domains commonly observed on these plasmids. The problem becomes further exaggerated when the plasmids are to be isolated from Salmonella serovars, which have been found to be unusually recalcitrant to those procedures typically used to facilitate such isolations, often yielding little, if any, suitable plasmid DNA. This report presents a plasmid isolation protocol for use with Salmonella that is very efficient (yield > 20 μg high quality DNA), fast, and inexpensive.
This method allowed us to isolate and sequence six IncA/C plasmids from six different poultry-derived MDR Salmonella enterica serovars. In all six plasmids, we identified genes encoding proteins that correspond to antimicrobial resistance. Each of these antimicrobial-resistance phenotypes could be accounted for by the presence of corresponding resistance genes in these plasmids. There were no instances of resistance that occurred in the absence of a corresponding gene, which demonstrates the importance of plasmid-mediated resistance in Salmonella. Our results confirm earlier studies indicating that genetic prediction of phenotypic resistance is a reliable method for resistance monitoring (Hoffmann et al., 2014; Tyson et al., 2015; Zhao et al., 2015). This is especially important as clinical diagnostics move toward culture-independent technologies.
Our study has also provided results that allow a better understanding of the Inc A/C plasmid diversity and evolution in not only Salmonella but also across other taxa. The core genome of all the plasmids across the taxa analyzed in this study has a highly conserved backbone of 22 genes. Even though these plasmids were isolated from different bacterial species and from different sources, plasmids isolated from organisms of similar geographic origin tended to have low mutation rates in their core genes. Our phylogenetic tree could distinguish among plasmids obtained from the USA and those plasmids obtained from other parts of the world but did not differentiate among taxa, isolation source, or year. Interestingly, only S. Newport genomes clustered tightly together regardless of their source and year of isolation.
Further, differences in SNPs as well as in resistance gene absence/presence profiles between fish-derived plasmids and terrestrial animal-derived plasmids were noticed. Given these patterns, it is likely that local environmental selective pressures are the driving force that gives rise to the diversity among IncA/C plasmids. Although, the majority of the sequence data in our study was acquired from NCBI, and we do not have information regarding the origins of those strains, our analyses suggest that some patients had been infected by strains bearing plasmids with characteristics similar to those from water-borne sources, while other individuals were made ill by strains associated with terrestrial sources. In the future, perhaps resistance gene absence/presence profiles could be used to help guide outbreak investigations of multidrug resistant strains.
Data availability
GenBank accession numbers for all new sequences are listed in Table 1.
Author contributions
All authors played an integral part of project conception. Each author has read and approved the final version of the manuscript. Specifically, conceived and designed the experiments: MH, SZ, MA, PM, EB, SM. Performed the experiments: MH, SA, SM. Analyzed the data: MH, JP, NG, JM. Wrote the manuscript: MH, SZ, MA, PM, EB, SM.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Lili Fox Vélez, Ph.D. for scientific writing support.
The views expressed in this presentation are those of the author(s) and do not necessarily reflect the official policy of the Department of Health and Human Services, the U.S. Food andDrug Administration, or the U.S. Government. Reference to any commercial materials, equipment, or process does not in any way constitute approval, endorsement, or recommendation by the Food and Drug Administration.
Footnotes
Funding. This project was supported by internal FDA/CFSAN and FDA/CVM research funding.
Supplementary material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.01459/full#supplementary-material
List of the six variable core genes identified between the six IncA/C plasmids sequenced in this study.
List of the 67 core genes identified between all 20 Salmonella IncA/C plasmids Ridom (SeqSphere+).
List of the 22 core genes identified between all 44 taxa IncA/C plasmids Ridom (SeqSphere+).
(A) ML tree based for 20 IncA/C plasmids isolated from Salmonella. (B) ML tree based for 44 IncA/C plasmids isolated from different taxa. The SNPs were found based on k-mer analysis using kSNP. ML trees were generated as described in Figure 3.
References
- Carattoli A., Miriagou V., Bertini A., Loli A., Colinon C., Villa L., et al. (2006). Replicon typing of plasmids encoding resistance to newer beta-lactams. Emerging Infect. Dis. 12, 1145–1148. 10.3201/eid1207.051555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin C. S., Alexander D. H., Marks P., Klammer A. A., Drake J., Heiner C., et al. (2013). Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 10:563–569. 10.1038/nmeth.2474 [DOI] [PubMed] [Google Scholar]
- Darling A. E., Mau B., Perna N. T. (2010). progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 5:e11147. 10.1371/journal.pone.0011147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fair R. J., Tor Y. (2014). Antibiotics and bacterial resistance in the 21st century. Perspect. Medicin. Chem. 6, 25–64. 10.4137/PMC.S14459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falush D., Stephens M., Pritchard J. K. (2003). Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164, 1567–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FDA (2016). The National Antimicrobial Resistance Monitoring System. Available online at: https://wwwfdagov/AnimalVeterinary/SafetyHealth/AntimicrobialResistance/NationalAntimicrobialResistanceMonitoringSystem/defaulthtm (Accessed May 26, 2017).
- Fernandez-Alarcon C., Singer R. S., Johnson T. J. (2011). Comparative genomics of multidrug resistance-encoding IncA/C plasmids from commensal and pathogenic Escherichia coli from multiple animal sources. PLoS ONE 6:e23415. 10.1371/journal.pone.0023415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fricke W. F., Mammel M. K., McDermott P. F., Tartera C., White D. G., Leclerc J. E., et al. (2011). Comparative genomics of 28 Salmonella enterica isolates: evidence for CRISPR-mediated adaptive sublineage evolution. J. Bacteriol. 193, 3556–3568. 10.1128/JB.00297-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fricke W. F., Welch T. J., McDermott P. F., Mammel M. K., LeClerc J. E., White D. G., et al. (2009). Comparative genomics of the IncA/C multidrug resistance plasmid family. J. Bacteriol. 191, 4750–4757. 10.1128/JB.00189-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner S. N., Hall B. G. (2013). When whole-genome alignments just won't work: kSNP v2 software for alignment-free SNP discovery and phylogenetics of hundreds of microbial genomes. PLoS ONE 8:e81760. 10.1371/journal.pone.0081760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert J. M., White D. G., McDermott P. F. (2007). The US national antimicrobial resistance monitoring system. Future Microbiol. 2, 493–500. 10.2217/17460913.2.5.493 [DOI] [PubMed] [Google Scholar]
- Harmer C. J., Hall R. M. (2015). The A to Z of A/C plasmids. Plasmid 80, 63–82. 10.1016/j.plasmid.2015.04.003 [DOI] [PubMed] [Google Scholar]
- Hoffmann M., Muruvanda T., Allard M. W., Korlach J., Roberts R. J., Timme R., et al. (2013). Complete genome sequence of a multidrug-resistant Salmonella enterica Serovar Typhimurium var. 5- strain isolated from chicken breast. Genome Announc. 1:e01068–13. 10.1128/genomeA.01068-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M., Zhao S., Pettengill J., Luo Y., Monday S. R., Abbott J., et al. (2014). Comparative genomic analysis and virulence differences in closely related Salmonella enterica serotype Heidelberg isolates from humans, retail meats, and animals. Genome Biol. Evol. 6, 1046–1068. 10.1093/gbe/evu079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klimke W., Agarwala R., Badretdin A., Chetvernin S., Ciufo S., Fedorov B., et al. (2009). The National Center for Biotechnology Information's Protein Clusters Database. Nucleic Acids Res. 37, D216–D223. 10.1093/nar/gkn734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krumsiek J., Arnold R., Rattei T. (2007). Gepard: a rapid and sensitive tool for creating dotplots on genome scale. Bioinformatics 23, 1026–1028. 10.1093/bioinformatics/btm039 [DOI] [PubMed] [Google Scholar]
- Lu Y., Zhao H., Sun J., Liu Y., Zhou X., Beier R. C., et al. (2014). Characterization of multidrug-resistant Salmonella enterica serovars Indiana and Enteritidis from chickens in Eastern China. PLoS ONE 9:e96050. 10.1371/journal.pone.0096050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team R. C. (2015). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing. [Google Scholar]
- Rosenberg N. A. (2004). Distruct: a program for the graphical display of population structure. Mol. Ecol. 4, 137–138. 10.1046/j.1471-8286.2003.00566.x [DOI] [Google Scholar]
- Scallan E., Hoekstra R. M., Angulo F. J., Tauxe R. V., Widdowson M. A., Roy S. L., et al. (2011). Foodborne illness acquired in the United States–major pathogens. Emerging Infect. Dis. 17, 7–15. 10.3201/eid1701.P11101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyson G. H., McDermott P. F., Li C., Chen Y., Tadesse D. A., Mukherjee S., et al. (2015). WGS accurately predicts antimicrobial resistance in Escherichia coli. J. Antimicrob. Chemother. 70, 2763–2769. 10.1093/jac/dkv186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- USDA (2014). Cost Estimates of Foodborne Illnesses. United States Department of Agriculture, Economic Research Service (ERS). Available online at: http://ersusdagov/data-products/cost-estimates-of-foodborne-illnessesaspx (Accessed May 26th, 2017).
- Varma J. K., Greene K. D., Ovitt J., Barrett T. J., Medalla F., Angulo F. J. (2005). Hospitalization and antimicrobial resistance in Salmonella outbreaks, 1984-2002. Emerging Infect. Dis. 11, 943–946. 10.3201/eid1106.041231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnes G. R., Bolker B., Bonebakker L., Gentleman R., Huber W., Liaw A., et al. (2015). gplots: Various R Programming Tools for Plotting Data. R package version 2.17.0. Available online at: http://CRANR-projectorg/package=gplots
- Welch T. J., Fricke W. F., McDermott P. F., White D. G., Rosso M. L., Rasko D. A., et al. (2007). Multiple antimicrobial resistance in plague: an emerging public health risk. PLoS ONE 2:e309. 10.1371/journal.pone.0000309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White D. G., Zhao S., Simjee S., Wagner D. D., McDermott P. F. (2002). Antimicrobial resistance of foodborne pathogens. Microbes Infect. 4, 405–412. 10.1016/S1286-4579(02)01554-X [DOI] [PubMed] [Google Scholar]
- Winokur P. L., Brueggemann A., DeSalvo D. L., Hoffmann L., Apley M. D., Uhlenhopp E. K., et al. (2000). Animal and human multidrug-resistant, cephalosporin-resistant Salmonella isolates expressing a plasmid-mediated CMY-2 AmpC beta-lactamase. Antimicrob. Agents Chemother. 44, 2777–2783. 10.1128/AAC.44.10.2777-2783.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S., Tyson G. H., Chen Y., Li C., Mukherjee S., Young S., et al. (2015). Whole-genome sequencing analysis accurately predicts antimicrobial resistance phenotypes in Campylobacter spp. Appl. Environ. Microbiol. 82, 459–466. 10.1128/AEM.02873-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwickl D. J. (2006). Genetic Algorithm Approaches for the Phylogenetic Analysis of Large Biological Sequence Datasets under the Maximum Likelihood Criterion. Ph.D. dissertation, The University of Texas at Austin. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
List of the six variable core genes identified between the six IncA/C plasmids sequenced in this study.
List of the 67 core genes identified between all 20 Salmonella IncA/C plasmids Ridom (SeqSphere+).
List of the 22 core genes identified between all 44 taxa IncA/C plasmids Ridom (SeqSphere+).
(A) ML tree based for 20 IncA/C plasmids isolated from Salmonella. (B) ML tree based for 44 IncA/C plasmids isolated from different taxa. The SNPs were found based on k-mer analysis using kSNP. ML trees were generated as described in Figure 3.
Data Availability Statement
GenBank accession numbers for all new sequences are listed in Table 1.