


Abstract
Long range oxidative damage as a result of charge transport is shown to occur through single crossover junctions assembled from four semi-complementary strands of DNA. When a rhodium complex is tethered to one of the arms of the four-way junction assembly, thereby restricting its intercalation into the π-stack, photo-induced oxidative damage occurs to varying degrees at all guanine doublets in the assembly, though direct strand scission only occurs at the predicted site of intercalation. In studies where the Mg2+ concentration was varied, so as to perturb base stacking at the junction, charge transport was found to be enhanced but not to be strongly localized to the arms that preferentially stack on each other. These data suggest that the conformations of four-way junctions can be relatively mobile. Certainly, in four-way junctions charge transport is less discriminate than in the more rigidly stacked DNA double crossover assemblies.
INTRODUCTION
DNA can both mediate and participate in long range electron transport. As a result, oxidative damage can occur in DNA at a distance from the site of radical introduction (1,2). Because oxidative damage to DNA appears to play significant roles in cancer and aging, understanding the molecular basis of genetic degradation is an important research goal (3–5). In addition, recent nanotechnological research has focused on the possible application of DNA duplexes as one-dimensional quantum wires (6–8) and as templates for directed material deposition (9). In the same arena, larger nucleotide assemblies, like crossover junctions, have been used as building blocks for nanoscale structures and signaling devices (10–14). Understanding the molecular mechanisms of charge transport for these nanoscale building blocks is of fundamental importance to designing DNA microarray electronics. Finally, because DNA charge transport is sensitive to base pair stacking, measurements of charge transport also provide a means to explore the local variations in nucleic acid structure that can occur both statically and dynamically.
To investigate long range charge transport in DNA, we prepared assemblies containing the covalently tethered intercalator Rh(phi)2bpy′3+, a potent photooxidant (phi, phenanthrenequinone diimine; bpy′, 4-butyric acid, 4′-methylbipyridine). Photolysis at 365 nm of the tethered photooxidant leads to injection of an electron hole directly into the DNA base stack (15). Subsequent hole migration through the base stack results in localization and irreversible damage to the 5′-G of a 5′-GG-3′ doublet, the most oxidatively sensitive site in DNA (16,17). This lesion can be revealed by treatment with hot piperidine or base excision repair proteins (18). In contrast, rhodium complexes of phi promote direct strand scission at sites of intercalation without chemical treatment when irradiated at 313 nm (19). Thus, covalently tethering Rh(phi)2bpy′3+ to the end of a base stack should also restrict the damage that occurs by photo-irradiation at 313 nm to the end of the duplex to which the metal complex is tethered (15–20).
Using this strategy, long range charge transport has been shown to occur with a variety of photooxidants (15,21–25) and at distances up to 200 Å from a site of hole injection in duplex DNA (26–27). Similar charge transport reactions have been observed in RNA–DNA hybrid duplexes (D.T.Odom, and J.K.Barton, unpublished results; 28) and DNA triple helices (29,30). In addition, the ability of charge transport to migrate through regions of unusual structure, such as A tracts, has also been demonstrated (31). Interruptions in the π-stack, such as base bulges or insertion of aliphatic protein side chains in place of nucleobases, attenuates radical migration (32,33).
Four-way DNA junctions have long been known in biology as flexible intermediates during homologous recombination of sister chromatids. Originally proposed by Holliday in 1964 (and thus often called Holliday junctions) (34), they are composed of four strands of DNA that are partially complementary to form parallel stacks of bases that can interchange between different stacking isomers. In nature these junctions are unstable to migration and, when formed during recombination events, can rapidly migrate via a zippering mechanism. Joining two four-way junctions together into a double crossover (DX) assembly greatly rigidifies the base stacks of DNA (35). When in isolation, however, single four-way junctions have dramatically different stacking and stability than do DX assemblies (36–38). In the absence of magnesium the four-way junctions adopt an extended conformation with an open core to minimize negative charge localization from the backbone strands. In the presence of >100 µM magnesium or other highly charged cations the four helix arms fold into two coaxial π-stacks in an anti-parallel fashion.
In order to study the structural architecture of these molecules, synthetic four-way junctions can be made immobile to base pairing migration by constructing each arm from non-complementary sequences (36–39). The location of the crossover junction is dictated by the complementarity of the arms. These immobilized four-way junctions, however, retain high conformer exchange rates. Solution studies have shown that the rate of interconversion between the two junction isomers and their relative abundance are critically dependent on the sequence of the 3 bp region in each arm that flanks the crossover junction itself (40–43). By judicious choice of core, the four-way junction can be made to partition evenly between crossover isomers (43) or strongly prefer one or the other of these isomers (40).
At present, two crystal structures have been reported in the literature for all-DNA four-way junctions (44,45). Importantly, the base pairs are well-stacked through the junction. Though the sugar–phosphate backbone is severely distorted at the crossover, the bases across the junction are parallel and a base pair rise of 3.4 Å per step is evident. Furthermore, in both crystal structures the alignment of the two base stacks is anti-parallel and the angle between the two stack axes is ∼40°. These angle measurements are slightly smaller than those predicted in previous studies. Atomic force microscopy of arrays of non-covalently associated four-way junctions (46) and other biophysical studies (47–50) had suggested angles between the helical axes of ∼60°. However, studies using fluorescence resonance energy transport (FRET) (30) and restriction nuclease analysis (38) have shown that in solution these four-way junctions are fluxional and are stacked in both possible anti-parallel conformations, though in differing proportions of each.
Interest in using various DNA constructs to assemble architectural features in nanoconstruction has prompted us to investigate the electronic structure of crossover junctions. Charge transport has been seen in DNA DX assemblies and, remarkably, hole migration in DX assemblies only occurs through the base stack into which an electron hole is injected (51). Charge migration does not occur across the junction between the stacks. In addition, introduction of a double mismatch into an otherwise properly paired DX assembly does not disrupt radical migration; a similar disruption in the proper base pairing of identically sequenced duplex DNA causes complete disruption of charge migration. The different reactivity of the two DNAs is likely due to the stabilization of base dynamics that occurs in DX assemblies from the packing of the sugar–phosphate backbones into the major groove of the adjacent base stack.
Here we explore charge migration in single four-way junctions. We have designed four-way junctions using core residues that were shown previously to preferentially maintain one isomeric stacking form (40). Charge transport experiments employing this four-way junction were conducted with both non-covalently bound metallointercalator and tethered metal complex. Studies here contrast with previous results found for much more rigid DX assemblies and highlight the critical requirements for stable, well-defined architecture to control pathways of long range charge transport.
MATERIALS AND METHODS
Oligodeoxyribonucleotide preparation
Oligodeoxyribonucleotides were prepared in an ABI392 DNA synthesizer using standard phosphoramidite chemistry. DNA was synthesized with a 5′-dimethoxytrityl (DMT) protective group and purified on a Dynamax 300 Å reverse phase C4 column (10 mm i.d. × 25 cm length) from Rainin. The DMT group was removed using an aqueous 80% acetic acid solution to suspend the dried, protected DNA for 20 min. After evaporation in vacuo of the acetic acid the DNA was repurifed using the same Rainin C4 column. The purified DNA was dissolved into 10 mM Tris, pH 7.4, with 1 mM EDTA (TE) to a standardized concentration of 100 µM using extinction coefficients of: (ɛ260, M–1cm–1) adenine (A) = 15 000; guanine (G) = 12 300; cytosine (C) = 7400; thymine (T) = 6700. Synthesis and tethering of Rh(phi)2bpy′3+ to DNA has been described elsewhere (52).
DNA techniques and photocleavage
In separate experiments, each strand except the one tethered to the rhodium complex was 5′-labeled with [γ-32P]ATP using polynucleotide kinase. BioRad P6 Microspin columns were used to remove excess, unreacted [γ-32P]ATP. This desalted solution was treated with 10% piperidine at 90°C for 30 min to cleave damaged oligonucleotides. After drying in vacuo each full-length oligonucleotide was purified on a 20% denaturing polyacrylamide gel and the parent band extracted from the gel by elution at 37°C into TE. All rinses were pooled, dried and resuspended in 50 µl of TE and then passed through a Bio-Rad P6 column to remove urea and salts. Four-way junctions and duplexes were assembled by annealing equimolar amounts of each strand using a linear gradient of 90–4°C over 90 min on a Perkin-Elmer Cetus Gene Amplifier. These four-way junctions were characterized by their mobilities on 10% non-denaturing gels in 10 mM Mg(OAc)2 and 45 mM Tris–acetate, 1 mM EDTA, pH 7.6, (1× TAEMg) buffer. In general it was found that the four-way junction comprised >90% of the species visualized by their mobilities, regardless of the labeled strand; the remaining species were of smaller size. Non-covalent metallointercalators were added after annealing.
Irradiations were performed at 15°C on 10–30 µl samples in 1.7 ml presilanized Eppendorf tubes using a 1000 W Hg/Xe arc lamp equiped with a monochromator. Unless otherwise noted all reactions were performed in 1× TAE. When magnesium was present it was 10 mM as Mg(OAc)2. After irradiation samples were dried in vacuo and samples to be treated with piperidine (i.e. all 365 nm irradiations and dark controls) were resuspended in 100 µl of 10% piperidine and heated to 90°C for 30 min. These samples were then analyzed on a 20% denaturing gel followed by phosphorimagery. For quantitation, lanes were first normalized for loading, and cleavage in the dark controls was subtracted. Data from at least three trials were used for all analyses. Sequencing reactions followed standard protocols (53).
RESULTS
Design of four-way junctions to investigate long range charge transport
We constructed a four-way junction that would preferentially assume one isomeric conformation of an anti-parallel stacked bundle of helices (40). Figure 1 shows the sequences employed. The backbone strands are solvent exposed at the crossover point when the junction is properly folded in the presence of Mg2+. Rhodium complexes are covalently attached to the 5′-end of a backbone strand, thus allowing spatial control of radical injection into the base stack. The crossover strands, which are tightly constrained and thus largely solvent excluded in the presence of Mg2+ (44,45,48), were designed to incorporate oxidatively sensitive guanine doublets at well-spaced locations along the helix. Because these strands extend across the junction and into both stacks, oxidative damage on different arms can be easily visualized in a single experiment.
Figure 1.

Schematic illustrations of four-way junctions. (A) A folded four-way junction in the presence of magnesium ion, adapted from the crystal structure (45). (B) The non-covalent four-way junction assembly with its strands color coded and labeled with lower case letters. The stacked arms are labeled in Roman numerals. (C) The four-way junction with covalently attached metal complex. The structure of the appended metallointercalator, which is shown schematically in this rendering, is given at the bottom of this figure. (D) Duplex of the same base-stacked sequence as the combined arm I/II from (C) with covalently attached metallointercalator. In all sequences of DNA assemblies used, guanine doublets to be oxidized are shown in red.
Photo-induced cleavage of DNA four-way junctions by non-covalently bound Rh(phi)2DMB3+
Many organic intercalators, like MPE·Fe(II), Cu(I)-(o-phenanthroline)2 and Stains-All, preferentially bind to the core of four-way junctions (54–57). To distinguish preferred binding sites for non-covalently intercalated Rh(phi)2DMB3+, where DMB is 4,4′-dimethylbipyridine, this metallointercalator (58–59) was incubated with an equimolar amount of the four-way junction shown in Figure 1A. By sequentially 5′-32P-labeling each strand and irradiating at 313 nm in the presence and absence of Mg2+ the binding location(s) of the metal complex within the DNA assembly could be easily visualized. It was found that in all cases, with and without magnesium ion, the metal complex bound preferentially to the core of the crossover junctions.
The pattern and amount of reactivity depends on the concentration of Mg2+ and, thus, the preferred stacking geometry. In the absence of magnesium ion the metal complex tightly binds to the core of the junction and all of the strands show equal damage at the crossover location (Fig. 2, –Mg2+ 313 nm lanes). Upon introduction of Mg2+, however, the direct strand scission seen in crossover strands b and c when irradiated at 313 nm in the presence of Rh(phi)2DMB3+ dramatically decreased, whereas the damage in backbone strand d barely decreased. This variation in reactivity among the strands is consistent with crossover strands b and c being less solvent accessible in the presence of Mg2+ and, hence, less accessible to Rh(phi)2DMB3+, while backbone strand d, which remains on the outside face of the crossover junction, is accessible to photocleavage. Tight packing of the core can occur when Mg2+ is present to neutralize the core junction phosphate charges. This allows the arms that are extended and separate in the absence of magnesium ion to condense into two coaxial, anti-parallel base stacks (38).
Figure 2.

Direct photocleavage and piperidine-induced cleavage of four-way junction DNA with non-covalently bound Rh(phi)2bpy′3+ as a function of magnesium concentration. (A) Phosphorimagery after electrophoresis in denaturing 20% polyacrylamide gel with 5′-32P-end-labeling of strand b of the four-way junction in Figure 1 after photo-irradiation. Lane 1, Maxam–Gilbert A+G; lane 2, C>T sequencing reactions (42); lane 3, photolysis at 313 nm for 10 min; lane 4 photolysis at 365 nm for 2 h followed by piperidine treatment; lane 5, the same assembly without photolysis (dark control, DC) followed by piperidine treatment. Oxidatively sensitive sites are bracketed to the left of the sequencing lanes and the site of the crossover junction is shown by a hollow triangle. The presence or absence of 5 mM Mg(OAc)2 is indicated above the respective lanes and the arm locations are indicated to the left of each gel. (B) Phosphorimagery of strand c. Lanes as in (A). (C) Phosphorimagery of strand d. Lanes as in (A).
The oxidative damage seen when these assemblies are irradiated at 365 nm, with the notable exception of crossover strand b in Figure 2A, generally remains localized to the 5′-G of 5′-GG-3′ doublets. Furthermore, this oxidative damage is largely independent of Mg2+ concentration.
Long range charge transport through duplex DNA with covalently bound Rh(phi)2bpy′3+
We then constructed a duplex assembly (Fig. 1C) as a positive control for photo-induced long range oxidative damage in the sequence used to assemble four-way junctions. This duplex was designed to be identical in sequence to the arm I/arm II base stack in the four-way junction shown in Figure 1. As expected, the damage found with photolysis at 313 nm is localized to the end of the helix that bears covalently tethered Rh(phi)2bpy′3+. Guanine oxidation is apparent at all of the 5′-G of 5′-GG-3′ doublets in the labeled strand complementary to the rhodium-bearing strand (Fig. 3) upon irradiation at 365 nm and piperidine treatment, consistent with long range charge transport through the duplex.
Figure 3.
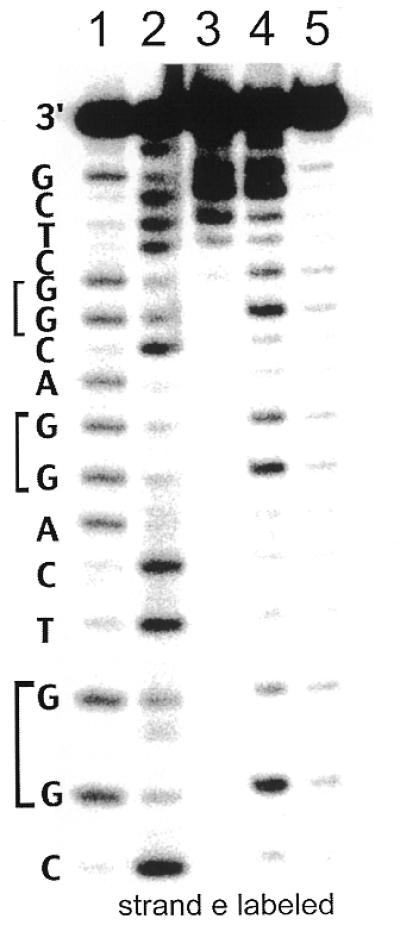
Oxidation of a metallointercalator-bearing duplex representing stacked arms I and II from the four-way junction shown in Figure 1. Lanes are as described in Figure 2.
Long range charge transport through the crossover junction with covalently bound Rh(phi)2bpy′3+
Having verified that duplex DNA of the same sequence can mediate long range charge transport, we assembled the four-way junction shown in Figure 1B and performed photo-irradiation on this assembly. By covalently tethering the metallointercalator to the 5′-end of backbone strand a, the location of radical injection into the assembly was restricted to within 3 bp of the terminus of arm I.
This restricted binding of Rh(phi)2bpy′3+ was evident in that the only site of photocleavage in these covalent assemblies, with or without Mg2+, is the end of arm I (Fig. 4). Importantly, the level of photocleavage damage observed was independent of the amount of magnesium ion in solution. This Mg2+ independence demonstrates that the photocleavage decrease in the core seen previously with non-covalent Rh(phi)2DMB3+ was a function of the architectural changes upon Mg2+ addition that sterically preclude metal complex access to the crossover and not the result of non-specific magnesium ion binding to the phosphate backbone.
Figure 4.
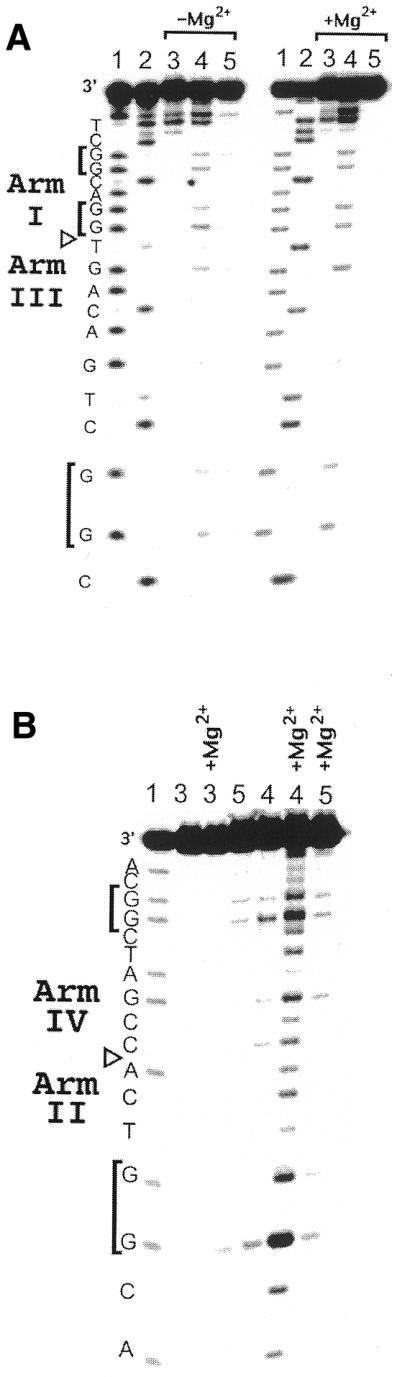
(A) Long range oxidation of DNA by covalently tethered Rh(phi)2bpy′3+ in four-way junctions as a function of magnesium concentration. The gels are labeled as in Figure 2. Strand b of the covalently tethered four-way junction is shown to be cleaved at the 5′-end on arm I by the covalently attached metal complex upon 313 nm irradiation and long range charge transport is evident upon 365 nm photo-irradiation in the absence and presence of magnesium. (B) Long range oxidation of DNA by covalently tethered Rh(phi)2bpy′3+ in four-way junctions as a function of magnesium concentration. Strand c of the covalently tethered four-way junction is shown to be unaffected by 313 nm irradiation, but long range charge transport is evident upon 365 nm photo-irradiation in the absence and, particularly, in the presence of magnesium.
In contrast, irradiating the covalent assemblies at 365 nm causes oxidative damage to all the guanines on crossover strands b and c. In the absence of magnesium ion, oxidative damage occurs at all three 5′-GG-3′ sites in crossover strand b. When Mg2+ was present, oxidative damage increases at all of the guanines and guanine doublets. Strand c shows even more interesting behavior (Fig. 4). In the absence of magnesium ion, the oxidative damage is rather weak and evenly distributed between the two guanine doublets located on this strand. With Mg2+ there is an increase in the level of damage at the guanine doublet on the 3′-side of this strand, located in arm IV. Most dramatically, however, the oxidative damage at the guanine doublet on the 5′-end of strand c in arm II is greatly increased. This increase may reflect the stacking of this arm onto the rhodium-bearing arm I, allowing electronic coupling to occur between the site of hole injection on arm I and the affected guanine on arm II.
We sought to verify whether guanine oxidation occurred as a result of intermolecular interactions. A radioactively labeled four-way junction with no tethered rhodium complex and an unlabeled four-way junction bearing a covalently bound metallointercalator were annealed separately, mixed in equimolar amounts and then photo-irradiated. No direct strand scission or guanine oxidation was observed in the labeled assembly by Rh(phi)2bpy′3+ tethered to a separately annealed four-way junction. Thus, long range charge transport proceeds only within an individual crossover assembly.
DISCUSSION
Intercalator exclusion from the crossover core in the presence of Mg2+
Direct photocleavage of the four-way junction by non-covalently bound Rh(phi)2DMB3+ shows that under all circumstances and salt concentrations the preferred binding site is the core of the four-way junction. However, the affinity of the metal complex for the crossover assembly depends on the concentration of magnesium ion. In the absence of Mg2+ the metal complex cleaves at the crossover junction on all of the inspected strands. By way of comparison, the addition of Mg2+, which induces crossover strands to assume distorted conformations to bridge between the two separate base stacks, also leads to greatly diminished photocleavage of these strands. Notably, however, backbone strand d shows photocleavage that appears to be independent of the magnesium ion concentration.
The differing susceptibility of each of the strands to metal complex binding in the presence of magnesium ion is consistent with the previously established stacking preferences of this junction. FRET experiments have demonstrated that the sequences used here favor the conformation shown in Figure 1, where arm I and arm II are stacked and arm III and arm IV are stacked, by 20:1 over the alternative anti-parallel conformation where arm I is stacked over arm III and arm II over arm IV (40). Clearly, this preferred stacking pattern is reflected in the photocleavage experiments described above; when Mg2+ is present and the two extended stacks are folded across one another, strands b and c are inaccessible to the metal complex but strand d remains solvent exposed.
In contrast to Rh(phi)2DMB3+, organic intercalators bind to the core of the crossover junction in the presence of magnesium ion (43–46). The metal complex tris(4,7-diphenylphenanthroline)rhodium(III) [Rh(DIP)33+] was also shown to target cruciforms (60). When bound to a plasmid in low salt conditions and irradiated at 315 nm, this bulky, sterically demanding metal complex induced single- and double-strand breaks at a cruciform extrusion. This reactivity was found to be entirely independent of the sodium chloride concentration. In a result that is remarkably similar to what was found in this study, cleavage of the cruciform site occured at Mg2+ concentrations of 1 mM but cleavage completely disappeared when magnesium ion was present in 10 mM concentration.
Long range charge transport in single crossover DNA assemblies
Next, we tethered the metallointercalator onto a duplex of the same sequence as the Rh(phi)2bpy3+-bearing stack of the preferred conformer of the four-way junction. In contrast to four-way junction DNA, this duplex has a continous sugar–phosphate backbone on the strand complementary to the metal complex (Fig. 1C). As expected, direct photocleavage mapped the rhodium-binding site to the end of the helix. When photo-irradiated at 365 nm, followed by piperidine treatment to reveal oxidative lesions, the duplex showed damage at all 5′-G in 5′-GG-3′ doublets. This experiment is in effect a positive control for the types of long range charge transport that would occur in single crossovers if four-way junction stacking afforded constructs as stable as duplex DNA.
In the absence of Mg2+ four-way junctions assume an extended conformation where the arms are not stacked beyond their canonical base pairing regions (Figs 5 and 6). Thus, long range charge transport experiments in the absence of magnesium would be expected only to yield damage at the guanine doublet in arm I, as radical migration should not occur across the empty, open junction. Instead, guanine oxidation is found to occur at 5′-G of guanine doublets in all arms. As damage is found in arm III, the four-way junction must be transiently sampling conformations that permit even the highly disfavored parallel alignment of arm I over arm III to occur. Clearly, radical migration is occuring across the junction. The binding location of the tethered metallointercalator, as revealed by direct strand scission at 313 nm, is exclusively at the terminus of arm I, hence, any damage seen in other locations is caused at long range.
Figure 5.

Schematic illustration of the damage pattern, location and intensity of direct photocleavage and photolysis-induced oxidative damage in four-way junction assemblies by the non-covalent metal complex derived from Figure 4A. Direct photocleavage damage is shown by black arrows, whereas the long range charge transport at 5′-GG-3′ doublets is shown with red arrows. The assemblies are shown with their presumed structural forms, open in the absence of magnesium, and with stacked arms in the presence of magnesium.
Figure 6.

Schematic illustration of the damage pattern, location and intensity of direct photocleavage and photolysis-induced oxidative damage in four-way junction assemblies by the covalently tethered metal complex derived from Figure 4B. The relative intensity of damage here is represented qualitatively by the size of arrows.
In the presence of Mg2+, hole transport across four-way junctions with a covalently tethered metallointercalator should cause oxidative lesions in arms I and II and, to a lesser extent, in arm IV, which in general make up the favored, anti-parallel stacking partners. The arm I/arm III stacking arrangement should be strongly disfavored regardless of magnesium ion concentration and thus the guanine doublet located in arm III would not be expected to show oxidative damage.
The results summarized in Figure 6 show that addition of magnesium to this system, which presumably causes folding of the otherwise separated arms into coaxial base stacks, increases guanine oxidation overall, but especially in the guanine doublets that are located in the stacked arms I and II. Most dramatically, the guanine doublet in arm II is much more damaged by radical migration when Mg2+ is present. As predicted above, this increase is probably a result of the favored stacking of arm I with arm II, which would electronically couple these two base stacks much more efficiently and thus allow radical migration across the junction. Damage also increases, though more modestly, to the guanine doublet in arm IV, again consistent with the above analysis of the arm I/arm IV stacking conformer as the minor species. Furthermore, the differential increase, where arm II is more intensely damaged than arm IV, reflects the trend, if not the absolute numbers, of the previously determined 20:1 partition ratio of the stacking arrangements of these arms.
Notably, however, guanines are also oxidized on arm III, the highly disfavored stacking partner for arm I. This result may reflect transient sampling of this strongly disfavored stacking arrangement by the four-way junction. This suggestion would explain the guanine oxidation seen in arm III in the absence of Mg2+, as in the absence of highly charged cations the four-way junction is extremely fluxional. It is more surprising, however, when considering the conformation of the four-way junction in the presence of Mg2+, where the separate arms are generally presumed to be in a more tightly packed arrangement and the overall assembly to be much more rigid.
In some cases, especially crossover strand b, damage also occurs in guanines at the 3′ position of doublets and single guanines as well, even ones that would be expected to be entirely orthogonal to the oxidation caused by the tethered metallointercalator. As suggested previously, the flexible junctions likely sample conformations that permit long range charge transport to proceed, but may also sample conformations that weaken the stacking geometry that concentrates HOMO localization onto the 5′-G of 5′-GG-3′ doublets (17). The damage seen on single guanines and 3′-G in 5′-GG-3′ doublets on strand b, otherwise unexpected, could be the result.
Magnesium ion effects on long range transport
These results suggest that, in general, the addition of Mg2+ strongly stabilizes tertiary structures with well-stacked arms and enhances charge transport to guanines by rigidifying the base stack. This effect is especially notable when metal complexes are covalently tethered to the end of one of the base stacks. Probing by direct photoinduced cleavage shows that the tethered metallointercalator binds to the end of arm I independent of the Mg2+ concentration. In contrast, in the presence of Mg2+ the level of guanine damage observed (Fig. 6) is greatly increased at all positions, despite the difference in relative amount at each location. However, the largest increases are at the predicted sites of greatest oxidative sensitivity, the 5′-G of guanine doublets.
Similar increases in guanine oxidation are observed when a non-covalent rhodium complex is used, reflecting an increased rigidity of the duplex arms (Fig. 5). This increase occurs on strand b not only at the 5′-G of 5′-GG-3′ doublets, but at all guanines regardless of location. Remarkably, and in contrast to the results with tethered metallointercalator, direct strand scission in the presence of magnesium ion is much lower with non-covalently bound Rh(phi)2DMB3+. In other words, it appears that lower levels of binding are yielding greater amounts of oxidative damage.
In general, then, while Mg2+ must rigidify the individual duplex arms, there is still apparently significant flexibility between the arms with Mg2+. This may be concluded based upon our finding of significant long range charge transport to disfavored sites in the assemblies containing tethered rhodium. While a duplex stack may be more rigid with Mg2+, there are still significant transient changes in folding among the arms that must occur.
Comparison of charge transport in four-way junctions to DX assemblies
DNA single crossover assemblies are known to be more flexible than their DNA DX counterparts. This fact accounts for why a large body of work exists exploring the nanostructural architecture of DX assemblies (10–13), but to date few papers have been published regarding the attempted use of single crossovers as architectural elements (46,61). However, considerable effort has gone into delineating the different folding behaviors of single crossover junctions (36–38).
Previously investigated DX assemblies showed remarkable fidelity in radical migration through single stacks, even across crossover junctions, due to their rigid and inflexible structures (51). Notably, this rigid structure is only made possible by the presence of 10 mM Mg(OAc)2. Resistance to crosstalk in separate but adjoining stacks within DX molecules implies that they may be used successfully in nanotechnological systems that involve charge migration.
Similar considerations point to single crossover molecules as far ‘leakier’ systems towards charge migration. The results described here on single four-way junction assemblies suggest that there is a transient sampling of conformers that allows perhaps brief, but significant, base stacking to occur in normally disfavored alignments and, thus, radical migration throughout the assembly. These data suggest that single crossover assemblies, although interesting from a structural and architectural standpoint, would not make effective components in nanoconstruction, which demands controlled charge migration.
Acknowledgments
ACKNOWLEDGEMENTS
We are grateful to the NIH (GM49216) for financial support. We also thank the Parsons Foundation for pre-doctoral support of D.T.O. and the Caltech SURF office for a summer fellowship to E.A.D.
References
- 1.Núñez M.E. and Barton,J.K. (2000) Probing DNA charge transport with metallointercalators. Curr. Opin. Chem. Biol., 4, 199–206. [DOI] [PubMed] [Google Scholar]
- 2.Kelley S.O. and Barton,J.K. (1998) Radical migration through the DNA helix: chemistry at a distance. Metal Ions Biol. Syst., 26, 211–249. [PubMed] [Google Scholar]
- 3.Ames B. (1983) Dietary carcinogens and anticarcinogens—oxygen radicals and degenerative diseases. Science, 221, 1256–1264. [DOI] [PubMed] [Google Scholar]
- 4.Piette J. (1991) Mechanism of DNA cleavage mediated by photoexcited nonsteroidal antiinflammatory drugs. J. Photochem. Photobiol. B, 11, 241–260. [DOI] [PubMed] [Google Scholar]
- 5.Simon M.I. and Van Vunakis,H. (1962) The photodynamic reaction of methylene blue with DNA. J. Mol. Biol., 4, 488–499. [DOI] [PubMed] [Google Scholar]
- 6.Fink H.W. and Schonenberger,C. (1999) Electrical conduction through DNA molecules. Nature, 398, 407–410. [DOI] [PubMed] [Google Scholar]
- 7.Porath D., Bezryadin,A., de Vries,S. and Dekker,C. (2000) Direct measurement of electrical transport through DNA molecules. Nature, 403, 635–638. [DOI] [PubMed] [Google Scholar]
- 8.Okahata Y., Kobayashi,T., Tanaka,K. and Shimomura,M. (1998) Anisotropic electric conductivity in an aligned DNA cast film. J. Am. Chem. Soc., 120, 6165–6166. [Google Scholar]
- 9.Storhoff J.J. and Mirkin,C.A. (1999) Programmed materials synthesis with DNA. Chem. Rev., 99, 1849–1862. [DOI] [PubMed] [Google Scholar]
- 10.Li X., Yang,X., Qi,J. and Seeman,N.C. (1996) Antiparallel DNA double crossover molecules as components for nanoconstruction. J. Am. Chem. Soc., 118, 6131–6140. [Google Scholar]
- 11.Mao C., Sun,W., Shen,Z. and Seeman,N.C. (1999) A nanomolecular device based on the B–Z transition of DNA. Nature, 397, 144–146. [DOI] [PubMed] [Google Scholar]
- 12.Winfree E., Liu,F., Wenzler,L.A. and Seeman,N.C. (1998) Design and self-assembly of two-dimensional DNA crystals. Nature, 394, 539–544. [DOI] [PubMed] [Google Scholar]
- 13.Yang X., Wenzler,L.A., Qi,L., Li,X. and Seeman,N.C. (1998) Ligation of DNA triangles containing double crossover molecules. J. Am. Chem. Soc., 120, 9779–9786. [Google Scholar]
- 14.Yurke B., Turberfield,A.J., Mills,A.P., Simmel,F.C. and Neumann,J.L. (2000) A DNA-fuelled molecular machine made of DNA. Nature, 406, 605–608. [DOI] [PubMed] [Google Scholar]
- 15.Hall D.B., Holmlin,R.E. and Barton,J.K. (1996) Oxidative DNA damage through long range electron transfer. Nature, 382, 731–735. [DOI] [PubMed] [Google Scholar]
- 16.Saito I., Takayama,M., Sugiyama,H., Nakatani,K., Tsuchida,A. and Yamamoto,M. (1995) Photoinduced DNA cleavage via electron transfer—demonstration that guanine residues located 5′ to guanine are the most electron donating sites. J. Am. Chem. Soc., 117, 6406–6407. [Google Scholar]
- 17.Sugiyama H. and Saito,I. (1996) Theoretical studies of GG-specific photocleavage of DNA via electron transfer: significant lowering of ionization potential and 5′-localization of HOMO of stacked GG bases in B-form DNA. J. Am. Chem. Soc., 118, 7063–7068. [Google Scholar]
- 18.Burrows C.J. and Muller,J.G. (1998) Oxidative nucleobase modifications leading to strand scission. Chem. Rev., 98, 1109–1151. [DOI] [PubMed] [Google Scholar]
- 19.Sitlani A., Long,E.C., Pyle,A.M. and Barton,J.K. (1992) DNA photocleavage by phenanthenequinone diimine complexes of rhodium (III)—shape-selective recognition and reaction. J. Am. Chem. Soc., 114, 2303–2311. [Google Scholar]
- 20.Nunez M.E., Rajski,S.R. and Barton,J.K. (2000) Damage to DNA by long range charge transport. Methods Enzymol., 319, 165–188. [DOI] [PubMed] [Google Scholar]
- 21.Giese B. (2000) Long distance charge transport in DNA: the hopping mechanism. Acc. Chem. Res., 33, 631–636. [DOI] [PubMed] [Google Scholar]
- 22.Schuster G.B. (2000) Long range charge transfer in DNA: transient structural distortions control the distance dependence. Acc. Chem. Res., 33, 253–260. [DOI] [PubMed] [Google Scholar]
- 23.Nakatani K., Dohno,C. and Saito,I. (1999) Chemistry of sequence-dependent remote guanine oxidation: photoreaction of duplex DNA containing cyanobenzophenone-substituted uridine. J. Am. Chem. Soc., 121, 10854–10855. [Google Scholar]
- 24.Arkin M.R., Stemp,E.D.A., Coates-Pulver,S. and Barton,J.K. (1997) Long range oxidation of guanine by Ru(II) in duplex DNA. Chem. Biol., 4, 389–400. [DOI] [PubMed] [Google Scholar]
- 25.Hall D.B., Kelley,S.O. and Barton,J.K. (1998) Long-range and short-range oxidative damage to DNA: photoinduced damage to guanines in ethidium-DNA assemblies. Biochemistry, 37, 15933–15940. [DOI] [PubMed] [Google Scholar]
- 26.Núñez M.E., Hall,D.B. and Barton,J.K. (1999) Long-range oxidative damage to DNA: effects of distance and sequnce. Chem. Biol., 6, 85–97. [DOI] [PubMed] [Google Scholar]
- 27.Henderson P.T., Jones,D., Hampikian,G., Kan,Y. and Schuster,G.B. (1999) Long-distance charge transport in duplex DNA: the phonon-assisted polaron-like hopping mechansim. Proc. Natl Acad. Sci. USA, 96, 8353–8358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sartor V., Henderson,P.T. and Schuster,G.B. (1999) Radical cation transport and reaction in RNA/DNA hybrid duplexes: effect of global structure on reactivity. J. Am. Chem. Soc., 121, 11027–11033. [Google Scholar]
- 29.Núñez M.E., Noyes,K.T., Gianolio,D.A., McLaughlin,L.W. and Barton,J.K. (2000) Long-range guanine oxidation in DNA restriction fragments by a triplex-directed naphthalene diimide intercalator. Biochemistry, 39, 6190–6199. [DOI] [PubMed] [Google Scholar]
- 30.Kan Y. and Schuster,G.B. (1999) Radical cation transport and reaction in triplex DNA: long range guanine damage. J. Am. Chem. Soc., 121, 11607–11614. [Google Scholar]
- 31.Williams T.T., Odom,D.T. and Barton,J.K. (2000) Variations in DNA charge transport with nucleotide composition and sequence. J. Am. Chem. Soc., 122, 9048–9049. [Google Scholar]
- 32.Hall D.B. and Barton,J.K. (1997) Sensitivity of DNA-mediated electron transfer to the intervening pi-stack: a probe for the integrity of the DNA base stack. J. Am. Chem. Soc., 119, 5045–5046. [Google Scholar]
- 33.Rajski S.R., Kumar,S., Roberts,R.J. and Barton,J.K. (1999) Protein-modulated DNA electron transfer. J. Am. Chem. Soc., 121, 5615–5616. [Google Scholar]
- 34.Holliday R. (1964) A mechanism for gene conversion in fungi. Genet. Res., 5, 282–304. [DOI] [PubMed] [Google Scholar]
- 35.Fu T.-J. and Seeman,N.C. (1993) DNA double crossover molecules. Biochemistry, 32, 3211–3220. [DOI] [PubMed] [Google Scholar]
- 36.Lilley D.M.J. and Clegg,R.M. (1993) The structure of the 4-way junction in DNA. Annu. Rev. Biophys. Biomol. Struct., 22, 299–328. [DOI] [PubMed] [Google Scholar]
- 37.Lilley D.M.J. and Clegg,R.M.,(1993) The structure of DNA branched species. Q. Rev. Biophys., 26, 131–175. [DOI] [PubMed] [Google Scholar]
- 38.Seeman N.C. and Kallenbach,N.R. (1994) DNA branched junctions. Annu. Rev. Biophys. Biomol. Struct., 23, 53–86. [DOI] [PubMed] [Google Scholar]
- 39.Lilley, D.M.J. (1999) In Neidle,S. (ed.), Nucleic Acid Structure. Oxford University Press, Oxford, UK, pp. 471–498.
- 40.Miick S.M., Fee,R.S., Millar,D.P. and Chazin,W.J. (1997) Crossover isomer bias is the primary sequence-dependent property of immobilized Holliday junctions. Proc. Natl Acad. Sci. USA, 94, 9080–9084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Seeman N.C., Chen,J.H. and Kallenbach,N.R. (1989) Gel electrophoretic analysis of DNA branched junctions. Electrophoresis, 10, 345–354. [DOI] [PubMed] [Google Scholar]
- 42.Chen J.H., Churchill,M.E.A., Tullius,T.D., Kallenbach,N.R. and Seeman,N.C. (1988) Construction and analysis of monomobile DNA junctions. Biochemistry, 27, 6032–6038. [DOI] [PubMed] [Google Scholar]
- 43.Grainger R.J., Murchie,A.I.H. and Lilley,D.M.J. (1998) Exchange between stacking conformers in a four-way DNA junction. Biochemistry, 37, 23–32. [DOI] [PubMed] [Google Scholar]
- 44.Ortiz-Lombardía M., González,A., Eritja,R., Aymamí,J., Azorín F. and Coll,M. (1999) Crystal structure of a DNA Holliday junction. Nat. Struct. Biol., 6, 913–917. [DOI] [PubMed] [Google Scholar]
- 45.Eichman B.F., Vargason,J.M., Mooers,B.H.M. and Ho,P.S. (2000) The Holliday junction in an inverted repeat DNA sequence: sequence effects on the structure of four-way junctions. Proc. Natl Acad. Sci. USA, 97, 3971–3976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mao C.D., Sun,W.Q. and Seeman,N.C. (1999) Designed two-dimensional DNA Holliday junction arrays visualized by AFM. J. Am. Chem. Soc., 121, 5437–5443. [Google Scholar]
- 47.Duckett D.R., Murchie,A.I.H., Diekmann,S., von Kitzing,E., Kemper,B. and Lilley,D.M.J. (1988) The structure of the Holliday junction and its resolution. Cell, 55, 79–89. [DOI] [PubMed] [Google Scholar]
- 48.Churchill M.E.A., Tullius,T.D., Kallenbach,N.R. and Seeman,N.C. (1988) A Holliday recombination intermediate is two-fold symmetric. Proc. Natl Acad. Sci. USA, 85, 4653–4656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cooper J.P. and Hagerman,P.J. (1989) Geometry of a branched DNA-structure in solution. Proc. Natl Acad. Sci. USA, 86, 7336–7340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Murchie A.I.H., Clegg,R.M., von Kitzing,E., Duckett,D.R., Diekmann,S. and Lilley,D.M.J. (1989) Fluorescence energy transfer shows that the 4-way junction is a right-handed cross of antiparallel molecules. Nature, 341, 763–766. [DOI] [PubMed] [Google Scholar]
- 51.Odom D.T., Dill,E.A. and Barton,J.K. (2000) Robust charge transport in DNA double crossover assemblies. Chem. Biol., 7, 475–481. [DOI] [PubMed] [Google Scholar]
- 52.Holmlin R.E., Dandliker,P.J. and Barton,J.K. (1999) Synthesis of metallointercalator–DNA conjugates on a solid support. Bioconjugate Chem., 10, 1122–1130. [DOI] [PubMed] [Google Scholar]
- 53.Maxam A.M. and Gilbert,W. (1986) Sequencing DNA by labeling the end and breaking at the bases—DNA segments, end labels, cleavage reactions, polyacrylamide gels and strategies. Mol. Biol., 20, 461–509. [Google Scholar]
- 54.Guo Q., Seeman,N.C. and Kallenbach,N.R. (1989) Site-specific interaction of intercalating drugs with a branched DNA molecule. Biochemistry, 28, 2355–2359. [DOI] [PubMed] [Google Scholar]
- 55.Lu M., Guo,Q., Pasternack,R.F., Wink,D., Seeman,N.C. and Kallenbach,N.R. (1990) Drug-binding by branched DNA—selective intercalation of tetrapyridyl porphyrins with an immobile junction. Biochemistry, 29, 1614–1624. [DOI] [PubMed] [Google Scholar]
- 56.Guo Q., Lu,M., Seeman,N.C. and Kallenbach,N.R. (1990) Drug-binding by branched DNA-molecules—analysis by chemical footprinting of intercalation into an immobile junction. Biochemistry, 29, 570–578. [DOI] [PubMed] [Google Scholar]
- 57.Lu M., Guo,Q., Seeman,N.C. and Kallenbach,N.R. (1990) Drug-binding by branched DNA—selective interaction of the dye Stains-All with an immobile junciton. Biochemistry, 29, 3407–3412. [DOI] [PubMed] [Google Scholar]
- 58.Kielkopf C.L., Erkkila,K.E., Hudson,B.P., Barton,J.K. and Rees,D.C. (2000) Structure of a photoactive rhodium complex intercalated into DNA. Nat. Struct. Biol., 7, 117–121. [DOI] [PubMed] [Google Scholar]
- 59.Erkkila K.E., Odom,D.T. and Barton,J.K. (1999) Recognition and reaction of metallointercalators with DNA. Chem. Rev., 99, 2777–2795. [DOI] [PubMed] [Google Scholar]
- 60.Kirshenbaum M.R., Tribolet,R. and Barton,J.K. (1988) Rh(DIP)33+—a shape-selective metal-complex which targets cruciforms. Nucleic Acids Res., 16, 7943–7960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Petrillo M.L., Newton,C.J., Cunningham,R.P., Ma,R.I., Kallenbach,N.R. and Seeman,N.C. (1988) The ligation and flexibility of 4-arm junctions. Biopolymers, 27, 1337–1352. [DOI] [PubMed] [Google Scholar]