

Abstract
The Standard for Exchange of Nonclinical Data (SEND), introduced by the US Food and Drug Administration (FDA), is a scheme for the computerization, electronic application, and screening of preclinical data. Since its establishment, related organizations have been working together to implement SEND. However, it is difficult for individual pharmaceutical companies that often outsource to achieve complete compliance with SEND; hence, the cooperation of contract research organizations (CROs) and SEND Registered Solution Providers (RSPs) is indispensable. In SEND, most data, including those on pathology findings, are converted into controlled terminology (CT), but it is not a simple process to convert findings or levels of severity in the field of pathology, which is a descriptive science. The authors have successfully completed an FDA trial submission for a toxicology test conducted at a CRO and in doing so acquired important knowledge. This article presents a clear picture of such important knowledge from a pathologist’s viewpoint.
Keywords: SEND, INHAND, controlled terminology, CDISC, PhUSE
Introduction
Established by the Clinical Data Interchange Standards Consortium (CDISC)1, the Study Data Tabulation Model (SDTM) provides a basis for data models for not only clinical but also nonclinical studies. The rules for creating data for nonclinical studies are specified by the Standard for Exchange of Nonclinical Data (SEND)2. Effective as of December 2016, use of the SDTM is required by the US Food and Drug Administration (FDA) for the electronic application/review of nonclinical and clinical studies. The SEND and SDTM require controlled terminology (CT) mapping, however, and pathological terminology represents a major issue. Relevant organizations have been working together to develop CT for use in SEND, and the International Harmonization of Nomenclature and Diagnostic Criteria for Lesions in Rats/Mice and Non-rodent Species (INHAND)3 is leading the way in standardizing terminology in toxicological pathology. Complete compliance with SEND cannot be achieved by pharmaceutical companies alone, and it is necessary to establish a model for task-sharing among different organizations, including nonclinical contract research organizations (CROs) and SEND Registered Solutions Providers (RSPs)4, which is being undertaken by the Pharmaceutical Users Software Exchange (PhUSE). These organizations, however, do not offer practical guidelines for individual pharmaceutical companies and researchers who work for such companies, pathologists, or toxicologists as to what is necessary to prepare for individual studies or how to handle actual study data. Considering that pathology is a descriptive science, the appropriate response to SEND is required for pathologist with respect to such things as how to handle CT and how to establish compatible terminology and a severity grading system for use at individual institutions.
The authors have recently submitted several SEND datasets based on previously published procedures5 to the US FDA in a trial submission. The FDA reviewed one of these datasets, and it successfully passed the screening. By organizing the wealth of information acquired through this experience, not only with the study that was included in the review but also those that were not, we have developed a set of procedures. We have also identified common challenges in the handling of pathology findings that many pathologists should be aware of when creating SEND data, such as what terms to use.
Procedures for Trial Submission
Before the commencement of two 13-week repeated-dose studies on rats (Good laboratory practice [GLP] studies), we prepared for the creation of SEND data at Ina Research Inc. The major parameters included general conditions, body weight, food consumption, hematology, blood biochemistry, autopsy, organ weight, and histopathology.
Data conversion
In general, the following two methods were used to extract the original data without making changes to the systems used at the testing institution to convert the data into SEND-ready form:
1) Excel-based data were extracted directly from an in-house computer system and converted using TranSENDTM (PDS Life Sciences) with adaptors.
2) Pathological data (macroscopic and histological findings) were input directly into Ascentos® PathData® (PDS Life Sciences) Version 1.1 and converted into raw data using TranSENDTM.
Guide used
Data were converted in accordance with the SEND Implementation Guide (SENDIG) Version 3.06.
SEND conversion of pathological terms
To create a SEND dataset, the pathological data entered into Ascentos® PathData® Version 1.1 (PDS Life Science) were migrated directly into a SEND conversion tool called TranSENDTM and converted automatically in real time. The SEND datasets for pathological data were divided roughly into the following two domains (files):
∙MA domain: macroscopic (autopsy) findings
∙MI domain: microscopic (pathology) findings
Additional modifier information for the autopsy and pathology findings was then stored in the SUPP domains (SUPPMA and/or SUPPMI). The additional modifier information contained information on size, color, and other factors.
Creation of a Define.xml file
For the studies included in this submission, first a Define.xml file was created automatically using the relevant TranSEND™ component (PDS Life Sciences), and then the file was edited manually for passing through Pinnacle 21 ver.2.1.1 (Pinnacle 21 LLC).
Preparation of a Study Data Reviewer’s Guide (SDRG)
The SEND scheme requires that an SDRG be submitted as supporting material for each study for review by the FDA. SDRGs were created for each study in this submission.
FDA trial submission
The created SEND dataset was submitted to the FDA by PDS Life Sciences, acting on behalf of the authors, based on previously published procedures5. Although it was initially announced that a SEND trial submission could be made using the FDA’s dedicated gateway, the FDA required submission of compact discs, which we did accordingly.
Finalizing and storing SEND data
Once the FDA trial submission was completed successfully, the SEND datasets were finalized and stored within the specified GLP storage facilities at the testing institution.
Overview of the Trial Submission Results
Figure 1 and 2 show examples of pathology findings included in the SEND dataset created by TranSEND™. The raw data for the findings and associated CT-mapped data were stored in the SEND dataset. Data on each parameter were stored in the same manner and filed with the FDA, along with the manually adjusted Define.xml file, SDRG, and file and SDRG and other test data as part of the trial submission. According to the Pinnacle 21 validation report subsequently sent from the FDA, the result was “zero errors.” Over the course of these processes, from the creation of the SEND data to the successful completion of the FDA trial submission, we identified critical points of note before, during, and after the data conversion, as described below.
Fig. 1.

Examples of data conversion and storage of microscopic findings (this was used for an actual submission). Examples of raw data conversion and storage, where the data refer to cell infiltration, inflammatory cells, pelvis, unilateral, and Grade 1.
Fig. 2.

Third party-provided sample findings (this was not used for submission). Examples of raw data conversion and storage, where the data refer to inflammation, chronic, pelvis, bilateral, and slight.
SEND Compliance Model
Figure 3 “SEND compliance scheme” illustrates how Japanese or other non-US countries’ pharmaceutical companies can overcome challenges by using multiple CROs and RSPs. One of this scheme’s characteristics is that the phases from SEND dataset preparation to final confirmation are described based on the use of CROs with different nationalities and capacities to use SEND. The pathologist should be aware of the interorganizational complexity in SEND.
Fig.3.

SEND compliance scheme (Anzai et al.). Determine in advance the methods for finalization and storage of SEND data through thorough discussion at individual facilities.
Tasks to Perform before Conversion
Before creating SEND data, it is important not only to study the relevant SEND-related guidelines7891011 but also to make decisions on the following matters.
A. In-house pathology terminology control policy
Check the procedures for control of pathology terms at the testing institution and define the institution’s glossary and terminology where possible. The systems for grading pathology findings vary among different institutions; they may be 4- or 5-grade scales, for instance, as shown in Table 1. If there is any discrepancy between the institutional and SEND grading systems, refer to the CDISC definitions and National Cancer Institute (NCI) terms shown in Table 2 and review the system to ensure consistency so that grades may be converted in an evidence-based manner.
Table 1. Examples of Discrepancies between Different Grading Systems.
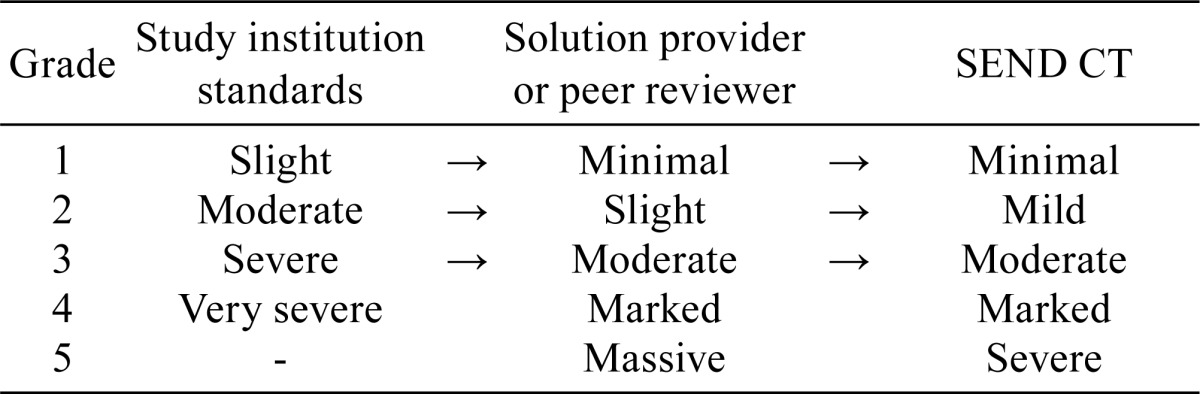
Table 2. CDISC Definitions and NCI Grades.

B. Ensuring the reliability of findings based on pathology peer review
Ensure consistency in use of terms and grading of findings within institutions by performing pathology peer review on study reports before they are finalized. Refer to the OECD guidance on pathology peer review12.
C. Understanding CT
Understand the CT before starting a study and make sure to use the latest set. The usefulness of the INHAND terminology is recognized by SEND, which adopted INHAND terminology as CT in SEND. However, it should be noted that INHAND terminology does not necessarily cover every finding.
D. Choosing appropriate tools
Determine what software to use for conversion, taking into consideration the track record, reliability assurance, and ability to automatically create a Define.xml file.
E. Liaising with the CRO
Determine in advance who is responsible for SEND pathology data conversion, either the study or the sponsor pathologist.
F. Coordination between the in-house study monitor and CRO study director
It is necessary to establish specifics concerning SEND data finalization and its conditions as well as storage procedures. As noted under 7. of Fig. 3, SEND data, once a dataset has been finalized, require handling with a level of care equal to that required to modify a final report.
G. SEND and GLP
While SEND data are not GLP data, the FDA base their review on SEND data, and therefore it requires a level of quality equal to that of GLP data. Seek the opinions of the quality assurance unit, and establish specifics concerning in-house handling of SEND data in advance.
H. Setting quality control (QC) procedures
Determine in advance who is responsible for data QC and how it will be performed. For instance, establish procedures for QC by the pathologist in charge and by a third party, as well as recording procedures.
Points to Note for SEND Conversion and Finalization
When creating SEND data, it is important for the pathologist to confirm the following matters.
A. Creating data consistent with the final report
The pathologist is responsible for checking the consistency between the pathology data contained in the SEND dataset and the final report. The pathologist must also check the descriptions of the pathology terms in the SDRG.
B. Data storage of appropriately converted variables according to domain
There are empirical approaches to data storage that are not specified in the relevant guidelines or information from CDISC or PhUSE; check with the RSP and make changes as necessary. As an example, in Fig. 2, it was necessary to ensure that the same grading system was used for MIORRES and MISEV; if it is not, sufficient explanations should be provided in the SDRG.
C. Adjusting the grading system
Although it is ideal to match the findings grading system to the 5-grade system required for SEND from the beginning, the number or names of grades used in the system in practice often vary among individual institutions. Convert the grades in an appropriate manner with the help of a third party such as the RSP and pathology peer reviewers. At Ina Research Inc., where the present study was conducted, the grading system specified in the Standard Operating Procedures (SOPs) was changed as shown in Fig. 4 after the completion of the study. An important point in adjusting the grading system was to divide the existing evaluation range covered by a grade, rather than adding a new range as a result of the increased number of grades, as illustrated in Fig. 4.
Fig. 4.

Changing the number of grades used at Ina Research. As a result of a change from a 4- to 5-grade rating system, the “slight” category in the 4-grade system was included in the “mild” category in the new 5-grade system.
D. Changes to the system
Make any necessary changes to the default grades and dictionary set in the toxicology study system used at an individual institution.
E. Creating appropriate additional modifier domains
The pathologist should understand what is required for an additional modifier for each of the pathology findings, as indicated in the additional modifier domains in Fig. 1 and 2.
F. Securing sufficient time
While the length of time required for mapping varies depending on the operator skill, it is ideal to allow an ample amount of time when scheduling.
G. Validation (QC) of CT mapping by the pathologist
The pathologist should perform validations and maintain a record in accordance with the predetermined QC procedures if the pathologist is to validate the mapping.
H. Ensuring mapping consistency at the institutional level
If a third person within an institution other than the pathologist is to perform QC, this third person should perform validation and maintain a record in accordance with the predetermined QC procedures.
I. Checking/liaising with the sponsor on mapping
If the sponsor is to perform QC of SEND data created by an RSP or CRO, the sponsor should understand the RSP’s/CRO’s QC procedures and endeavor to perform QC in an efficient manner.
J. Accurate explanations in the SDRG
Ensure that all SDRGs created at an institution follow the same format. As they are used to brief the reviewer, SDRGs should ideally be reviewed by an RSP or other experts.
K. Defining data finalization and storage procedures
As noted under 7 in Fig. 3, finalization of SEND data is an important procedure and should therefore be performed according to the predetermined procedures established by the sponsor, RSP, and CRO.
Conclusion
Pathologists play an important role in SEND data conversion. Unlike data such as body weight, in which the values remain unchanged after conversion, pathology findings are the product of a descriptive science; thus, there are always gray areas in expressing morphologic changes. In order to understand this particularity and create appropriate SEND data, it is essential not only to make use of pathology peer review but also to ensure that pathologists themselves are involved from the beginning in establishing the procedures for creating and validating SEND data, as has been demonstrated in the present article.
Footnotes
Disclosure of Potential Conflict of Interest: The authors do not have any conflict of interest to disclose in relation to this article.
References
- 1.CDISC SDTM TEAM. The Study Data Tabulation Model (SDTM) Implementation Guide version 3.2, November 26 2013, from CDISC website: https://www.cdisc.org/standards/foundational/sdtmig.
- 2.Standard for Exchange of Nonclinical Implementation Guide (IG) Version 3.1 for Public Review as of Dec. 4, 2014, CDISC SEND TEAM May 19 2011, from CDISC website: http://www.cdisc.org/send.
- 3.INHAND International Harmonization of Nomenclature and Diagnostic Criteria for Lesions in Rats and Mice, Collaboration with the FDA on SEND-Standard for the Exchange of Nonclinical Data, April 14, 2012, from STP website: https://www.toxpath.org/STP_inhand-84x42v2%20pdf.pdf.
- 4.CDISC. CDISC Solution Provider Chart, from CDISC website: http://www.cdisc.org/rsp-chart.
- 5.Anzai T, Kaminishi M, Sato K, Kaufman L, Iwata H, and Nakae D. Responses to the Standard for Exchange of Nonclinical Data (SEND) in non-US countries. J Toxicol Pathol. 28: 57–64. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.CDISC SEND TEAM, Clinical Data Interchange Standards Consortium. Standard for Exchange of Nonclinical Implementation Guide (IG) Version 3.0, from CDISC web site: http://www.fda.gov/downloads/ForIndustry/DataStandards/StudyDataStandards/UCM384744.pdf.
- 7.GDUFA Information Technology Plan (Draft) FY 2013. - FY 2017 from U.S. Food and Drug Administration web site: https://www.fda.gov/downloads/forindustry/userfees/genericdruguserfees/ucm379851.pdf.
- 8.Study Data Technical Conformance Guide v3.2, October 31,2016, from U.S. Food and Drug Administration website: https://www.fda.gov/downloads/forindustry/datastandards/studydatastandards/ucm384744.pdf.
- 9.Guidance for Industry Providing Regulatory Submissions in Electronic Format-Standard Study Data, Electronic Submissions, Revision 1, December 2014, from U.S. Food and Drug Administration website: https://www.fda.gov/downloads/drugs/guidances/ucm292334.pdf.
- 10.Guidance for Industry Providing Regulatory Submissions in Electronic Format- Submissions Under Section 745(a) of the Federal Food, Drug, and Cosmetic Act, Electronic Submission, from US Food and Drug Administration website: https://www.fda.gov/downloads/drugs/guidances/ucm 384686.pdf.
- 11.Guidance for Industry Providing Regulatory Submissions in Electronic Format- Receipt Date Electronic Submissions, February 2014, U.S. Food and Drug Administration website: https://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm072385.pdf.
- 12.Series on Principles of Good Laboratory Practice and Compliance Monitoring No 16, Advisory Document of the Working Group on Good Laboratory Practice Guidance on the GLP Requirements for Peer Review of Histopathology, ENV/JM/MONO(2014)30 26-Sep-2014, from OECD website: http://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?cote=env/jm/mono(2014)30&doclanguage=en.