

Abstract
Rifapentine is a highly active antituberculosis antibiotic with treatment‐shortening potential; however, exposure–response relations and the dose needed for maximal bactericidal activity have not been established. We used pharmacokinetic/pharmacodynamic data from 657 adults with pulmonary tuberculosis participating in treatment trials to compare rifapentine (n = 405) with rifampin (n = 252) as part of intensive‐phase therapy. Population pharmacokinetic/pharmacodynamic analyses were performed with nonlinear mixed‐effects modeling. Time to stable culture conversion of sputum to negative was determined in cultures obtained over 4 months of therapy. Rifapentine exposures were lower in participants who were coinfected with human immunodeficiency virus, black, male, or fasting when taking drug. Rifapentine exposure, large lung cavity size, and geographic region were independently associated with time to culture conversion in liquid media. Maximal treatment efficacy is likely achieved with rifapentine at 1,200 mg daily. Patients with large lung cavities appear less responsive to treatment, even at high rifapentine doses.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Rifapentine is a highly active antituberculosis antibiotic with possible treatment‐shortening potential. However, it is not clear what are the exposure–response relations, the dose with maximal bactericidal activity, and which patients are unlikely to respond to short‐term treatment.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ This pharmacokinetic/pharmacodynamic study investigated the dose of rifapentine with the greatest treatment‐shortening potential and the profile of patients unlikely to adequately respond to reduced therapy.
WHAT THIS STUDY ADDS TO OUR KNOWLEDGE
☑ Rifapentine exposures were lower in participants with HIV infection or who were black, male, or fasting. Optimal treatment efficacy with satisfactory safety in the study was achieved with 1,200‐mg daily rifapentine. Rifapentine exposure, large lung cavity, and geographic region were independently associated with time to culture conversion in liquid media. Patients with large lung cavities appeared less responsive to rifapentine treatment, even at high doses.
HOW THIS MIGHT CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE
☑ Pharmacokinetic/pharmacodynamic results supported level rifapentine dosing in adults and further evaluation of rifapentine at high daily doses as a TB treatment‐shortening strategy.
Development of better treatment for tuberculosis (TB) is an urgent global health need.1 Rifamycins have concentration‐dependent activity against Mycobacterium tuberculosis in vitro and in murine models.2 In a phase II trial comparing rifapentine to rifampin administered 5 days per week as part of a multidrug intensive‐phase therapy, the proportions of participants with stable sputum culture conversion to negative were similar after completion of 8 weeks of therapy.3 In a subsequent trial, antimicrobial activity and tolerability were evaluated with rifapentine doses of 10, 15, or 20 mg/kg administered 7 days per week with food.4 After 8 weeks of treatment, the proportions of stable culture conversions in liquid media were higher with rifapentine than rifampin; however, the study was not powered to compare efficacy across rifapentine groups, and the optimal dose for testing in phase III trials could not be established.
Sputum culture conversion to negative after completion of 2 months of intensive‐phase treatment is a widely used efficacy biomarker in phase II TB treatment trials.5, 6 However, this biomarker may not predict drug efficacy reliably in phase III trials.7, 8, 9 In addition, using a binary outcome measure of efficacy does not maximize use of the rich, longitudinal data, including serial microbiologic outcome measures and drug exposure data.10 Furthermore, clinical trials have not rigorously compared results obtained from solid medium cultures with those obtained from more sensitive liquid medium cultures.
Optimal drug dose and frequency can be efficiently estimated with population pharmacokinetic/pharmacodynamic (PK/PD) modeling methods that combine data on pharmacokinetic properties and efficacy outcomes.11, 12 The main objectives of this PK/PD study were to identify the rifapentine regimen that has the greatest potential to shorten the duration of TB treatment, and to describe a profile of patients unlikely to respond to shorter‐term therapy. To achieve the objectives, we characterized the population pharmacokinetics of rifapentine in participants with pulmonary TB treated with rifapentine as part of multidrug therapy and established the PK/PD relation between rifapentine exposure and time to stable sputum culture conversion.
RESULTS
Study population
Of the 668 adults who had smear‐positive pulmonary TB in the modified intention‐to‐treat group of TB Trial Consortium Studies 29 and 29X, 11 (1.6%) were excluded from the analysis due to missing liquid culture data. Of the remaining 657 participants, 405 participants who had been treated with rifapentine during intensive‐phase therapy were included in the PK/PD analyses and 252 participants who had been treated with rifampin during intensive‐phase therapy were included in pharmacodynamic analyses (Table 1).13 For 383 participants (95%), rifapentine pharmacokinetic parameters were computed using plasma concentrations. For the remaining 22 participants (5%) treated with rifapentine, pharmacokinetic values were estimated using model parameters and individual covariates (see Supplementary Table S1 and Supplementary Figure S1). The rifampin and rifapentine groups were similar in age, place of birth, race, and clinical features except that the rifapentine group had a higher frequency of sparse pharmacokinetic testing and lower Karnofsky score (Table 1).
Table 1.
Demographic, clinical, and sampling characteristics of study participants with culture results in liquid media
| Characteristic |
Rifampin n = 252 (38%) |
Rifapentine n = 405 (62%) |
Total | P ≤a |
|---|---|---|---|---|
| Demographic factors | ||||
| Age (y) | 33.0 (31.0, 36.0) | 31.0 (29.0, 33.0) | 32.0 (31.0, 33.0) | NS |
| Place of birth | NS | |||
| Africa | 140 (56%) | 221 (55%) | 361 (55%) | |
| South/Central America | 50 (20%) | 59 (15%) | 109 (17%) | |
| Asia/Pacific | 30 (12%) | 70 (17%) | 100 (15%) | |
| North America | 20 (8%) | 42 (10%) | 62 (9%) | |
| Europe | 12 (5%) | 13 (3%) | 25 (4%) | |
| Race | NS | |||
| Black | 149 (59%) | 243 (60%) | 392 (60%) | |
| White | 63 (25%) | 76 (19%) | 139 (21%) | |
| Asian | 30 (12%) | 66 (16%) | 96 (15%) | |
| Other | 1 (0.4%) | 3 (0.7%) | 4 (1%) | |
| Not reported | 9 (4%) | 17 (4%) | 26 (4%) | |
| Sex, male | 164 (65%) | 286 (71%) | 450 (68%) | NS |
| Clinical data | ||||
| Cavitation on chest radiograph | NS | |||
| Cavities ≥ 4 cm total | 92 (37%) | 141 (35%) | 233 (35%) | |
| Cavities < 4 cm total | 82 (33%) | 137 (34%) | 219 (33%) | |
| No cavity | 77 (31%) | 127 (31%) | 204 (31%) | |
| Dose, rifapentine | Not applicable | |||
| 10 mg/kg | 0 (0%) | 284 (70%) | 284 (43%) | |
| 15 mg/kg | 0 (0%) | 65 (16%) | 65 (10%) | |
| 20 mg/kg | 0 (0%) | 56 (14%) | 56 (9%) | |
| HIV positive | 34 (13%) | 35 (9%) | 69 (11%) | NS |
| Weight (kg) | 54.9 (53.5, 55.8) | 55.0 (53.7, 56.7) | 55.0 (54.0, 55.8) | NS |
| Body mass index (kg/m2) | 19.7 (19.2, 20.4) | 19.8 (19.4, 20.2) | 19.8 (19.5, 20.1) | NS |
| Pharmacokinetic testing | 0.0001 | |||
| Intensive (6‐7 samples) | 13 (100%)b | 79 (25%)b | 92 (14%) | |
| Sparse (1‐3 samples) | 0 (0%)b | 237(75%)b | 237 (36%) | |
| Karnofsky score | 0.02 | |||
| 100 | 37 (15%) | 36 (9%) | 73 (11%) | |
| ≤ 90 | 215 (85%) | 369 (91%) | 584 (89%) | |
| Cough before treatment | NS | |||
| Productive | 224 (89%) | 365 (90%) | 589 (90%) | |
| Nonproductive | 19 (8%) | 26 (6%) | 45 (7%) | |
| No cough | 9 (4%) | 14 (3%) | 23 (4%) | |
| Sputum AFB smear gradec | NS | |||
| 4+ (Highly bacillary) | 98 (39%) | 151 (38%) | 249 (38%) | |
| 3+ (Intermediate bacillary) | 56 (22%) | 96 (24%) | 152 (23%) | |
| 1+ (Paucibacillary) | 85 (34%) | 124 (31%) | 209 (32%) | |
| Negative | 12 (5%) | 31 (8%) | 43 (7%) |
Data reported as number (%) or median (interquartile range). AFB, acid‐fast bacilli; HIV, human immunodeficiency virus.
NS, not significant (P > 0.05).
Percentage in pharmacokinetic study participants.
At least one positive sputum AFB smear during screening was required for enrollment, but sputum smear status may have changed at the start of treatment (baseline). AFB sputum smear at baseline, quantified with light microscopy (Ziehl‐Neelsen stain, original magnification ×1,000) and shown as follows: negative (none); grade 1+ (1 per 100 fields to 9 per 10 fields); grade 3+ (1 to 9 per field); and grade 4+ (>9 per field).15
Rifapentine pharmacokinetic properties
Estimated model‐based pharmacokinetic parameters for rifapentine are shown in Table 2. Rifapentine area under the concentration–time curve from 0–24 h (AUC0‐24) and peak concentration increased with increasing daily dose (Table 3). Age and sex (Table 2 and Supplementary Table S2), but not body weight (Supplementary Figure S2), were significant covariates of apparent oral clearance. The AUC0‐24 increased 0.4% per year in age. Compared with a 450‐mg dose, bioavailability decreased 9%, 17%, and 26% with rifapentine doses of 600, 900, and combined 1,200 and 1,500 mg (comparison of four doses using log‐likelihood ratio test with 3 degrees of freedom; P = 0.004). Other significant covariates that affected bioavailability included race (Asian, 50% increase compared with black race; P ≤ 0.0001) and human immunodeficiency virus (HIV) infection (15% decrease; P = 0.001) (Table 2). For the same rifapentine dose, models suggested that the lowest rifapentine exposures would occur in younger (continuous variable), black, male participants taking rifapentine without food (Supplementary Figure S3). Interindividual variability in rifapentine AUC0‐24 was high (coefficient of variance (CV) of 21%), resulting in more than 4‐fold variation in rifapentine exposures for a given dose.
Table 2.
Estimated parameters for the integrated pharmacokinetic model for oral rifapentine in adults with tuberculosis
| Parameter |
Value (RSE, %) |
Between‐subject variability, CV% (RSE, %) |
|---|---|---|
| CL/F (L/h) | 1.86 (5) | 40 (15) |
| V/F (L) | 12.77 (5) | — |
| ka (h‐1) | 0.07 (3) | — |
| CLm/Fm (L/h) | 1.91 (6) | 44 (10) |
| Vm/Fm (L) | 8.83 (12) | |
| Bioavailability of 450‐mg dose with high fat food (reference) | 1 | 36 (11) |
| Bioavailability of 600‐mg dose (fraction) relative to reference dose | 0.91 (6) | |
| Bioavailability of 900‐mg dose (fraction) relative to reference dose | 0.83 (6) | — |
| Bioavailability of 1200‐mg dose (fraction) relative to reference dose | 0.74 (8) | — |
| Fasting effect (vs. high fat) on bioavailability (fraction) | 0.72 (7) | — |
| Effect of low‐fat food (vs. high‐fat) on bioavailability (fraction) | 0.83 (20) | — |
| White (vs. black) race effect on bioavailability (fraction) | 1.19 (36) | — |
| Asian (vs. black) race effect on bioavailability (fraction) | 1.51 (16) | — |
| HIV infection (vs. HIV‐uninfected) effect on bioavailability (fraction) | 0.85 (44) | — |
| Correlation CL‐F | 0.55 (21) | — |
| Correlation CLm‐F | 0.45 (19) | — |
| Correlation CL‐CLm | 0.69 (16) | — |
| Age effect on CL (yearly decrease from the median age 31 y)a | —0.00379 (50) | — |
| Sex effect on CL (fraction in female vs. male) | 0.81 (36) | — |
| Dose effect (> 600 mg) on fraction metabolized, Fm (fraction) | 1.34 (4) | — |
| Proportional residual error, rifapentine (CV%) | 19 (13) | — |
| Additive residual error, rifapentine (μg/mL) | 1.61 (28) | — |
| Proportional residual error, metabolite (CV%) | 14 (10) | — |
| Additive residual error, metabolite (μg/mL) | 1.33 (12) | — |
CL, clearance; CLm, clearance of desacetyl rifapentine; CV%, coefficient of variance; F, bioavailability; Fm, fraction metabolized; ka, absorption rate constant; m, metabolite (desacetyl rifapentine); RSE, relative standard error; V, rifapentine volume of distribution.
CL = CL/F × (1 + CLage × [age (y) –31]).
Table 3.
Relation between rifapentine daily dose, area under the concentration‐time curve from 0 to 24 h, and peak concentrationa
| Daily dose | No. of participants |
AUC0‐24
(μg × h/mL) |
Cmax
(μg/mL) |
||
|---|---|---|---|---|---|
| Dose (mg) | |||||
| 450 | 60 | 290 ± 123 | 255 (153, 510) | 14.7 ± 5.8 | 13.8 (8.2, 26.3) |
| 600 | 211 | 324 ± 143 | 295 (151, 579) | 17.0 ± 7.1 | 15.7 (7.7, 29.4) |
| 900 | 78 | 498 ± 149 | 503 (272, 721) | 25.1 ± 7.0 | 24.4 (13.8, 36.3) |
| 1200 | 30 | 587 ± 197 | 587 (317, 882) | 29.9 ± 9.1 | 31.4 (16.4, 43.1) |
| 1500 | 4 | 663 ± 84 | 666 (572, 748) | 33.2 ± 3.3 | 33.0 (29.8, 36.8) |
| Dose (mg/kg) | |||||
| 10 | 236 | 309 ± 132 | 285 (154, 570) | 16.1 ± 6.6 | 15.0 (7.86 27.8) |
| 15 | 74 | 434 ± 173 | 391 (203, 726) | 22.1 ± 8.2 | 20.5 (11.0, 35.7) |
| 20 | 73 | 546 ± 176 | 538 (290, 832) | 27.5 ± 8.2 | 28.0 (15.2, 40.4) |
AUC0‐24, area under the concentration‐time curve from 0 to 24 h; Cmax, peak concentration.
Data reported as mean ± SD or median (5th, 95th percentile).
Rifapentine PK/PD modeling
Time to stable conversion on either liquid or solid media was highly correlated with both rifapentine AUC0‐24 and peak concentration (Figure 1). Time to stable conversion was best described with a Weibull model with increasing probability (hazard) of culture conversion with time. The pharmacokinetics‐based predictors were related to the hazard parameter (P < 0.001), and participants with high AUC0‐24 or peak concentration showed significantly decreased treatment time to achieve stable culture conversion (Figure 1 and Supplementary Figure S4).
Figure 1.
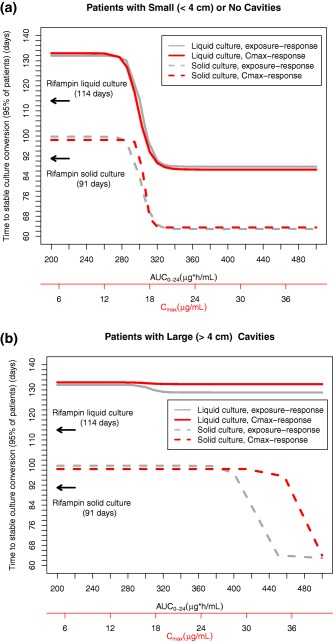
Relation between rifapentine area under the concentration–time curve from 0 to 24 h (AUC0‐24) and maximum concentration (Cmax) vs. estimated time required for 95% patients with no or small lung cavities (a) or large lung cavities (b) to achieve stable conversion to negative sputum culture. Rifapentine AUC0‐24 (gray) or Cmax (red) shown for liquid (continuous line) and solid (broken line) culture media. Estimated time to stable culture conversion for all control participants treated with rifampin during intensive‐phase therapy was 114 days in liquid media (top arrow) and 91 days on solid media (bottom arrow). Data for participants with large cavities on solid media were estimated for grade 4 acid‐fast bacilli smear from baseline sputum.
Because of the high interindividual variability in rifapentine AUC0‐24 for a given dose used in this study, no significant relation was found between stable culture conversion in liquid media and rifapentine study arm (doses: 10, 15, or 20 mg/kg; P = 0.62) or between stable culture conversion in liquid media and fixed rifapentine doses (range, 450–1,500 mg daily; P = 0.39) (Supplementary Table S3).
Liquid media rifapentine PK/PD modeling
Estimated rifapentine AUC0‐24 >350 μg × h/mL predicted maximum treatment efficacy in most patients (Table 4). Daily rifapentine AUC0‐24 >350 μg × h/mL was associated with stable conversion to negative sputum cultures in liquid media within 12.4 weeks (95% confidence interval (CI), 11.1–14.2 wk) of daily treatment in 95% of participants with no or small lung cavities; however, for the group with large lung cavities, the estimated time required for 95% of participants to achieve stable conversion was >16 weeks (mean, 18.6 wk; 95% CI, 15.7–22.7 wk) (Table 4). Significant independent covariates in models evaluating stable culture conversion in liquid media were rifapentine AUC0‐24, lung cavity aggregate diameter (≥ or <4 cm), geographic region of study site (Africa vs. non‐African region), and Karnofsky score (Table 4 and Supplementary Table S4). In participants who had cavities ≥4 cm, higher exposure did not reduce the estimated average time to stable culture conversion (Table 4, Figures 1 and 2).
Table 4.
Rifapentine and rifampin pharmacokinetic/pharmacodynamic outcomes in liquid media
| Rifapentine pharmacokinetic/pharmacodynamic outcomesa | ||||
|---|---|---|---|---|
| Rifapentine AUC0‐24 a (μg × h/mL) | Aggregate cavity size on chest radiograph (cm) | Study site in Africa | Percent participants with negative cultures in liquid media at completion of intensive‐phase therapy, mean [95% CI] |
Time (d) calculated for 50% participants to develop stable conversion to negative cultures in liquid media while receiving antituberculosis treatment [range: 5%, 95% participants] |
| > 350 | < 4 | Yes | 67 [53, 83] | 45 [14, 88] |
| ≥ 4 | Yes | 40 [20, 56] | 66 [20, > 120] b | |
| < 4 | No | 79 [70, 87] | 39 [12, 76] | |
| ≥ 4 | No | 48 [30, 70] | 57 [17, 111] | |
| 325 | < 4 | Yes | 61 [44, 78] | 48 [15, 94] |
| ≥ 4 | Yes | 36 [20, 56] | 66 [20, > 120] c | |
| < 4 | No | 73 [65, 83] | 42 [13, 81] | |
| ≥ 4 | No | 48 [30, 70] | 57 [17, 111] | |
| < 300 | < 4 | Yes | 37 [27, 48] | 68 [21, > 120] c |
| ≥ 4 | Yes | 37 [25, 49] | 68 [21, > 120] c | |
| < 4 | No | 47 [28, 63] | 58 [18, 114] | |
| ≥ 4 | No | 44 [22, 72] | 58 [18, 114] | |
| Rifampin pharmacodynamic outcomesd | |||
|---|---|---|---|
| Extent of lung infiltrate on chest radiographd | Productive cough at baseline | Percent of participants with negative cultures in liquid media at completion of intensive‐phase therapy, mean [95% CI] | Time (d) calculated for 50% participants to develop negative cultures in liquid media while receiving antituberculosis treatment [range: 5%, 95%] |
| < 25% | Yes | 67 [55, 79] | 46 [16, 85] |
| No | 93 [72, 100] | 32 [11, 58] | |
| ≥ 25% | Yes | 37 [30, 45] | 66 [22, 122] |
| No | 68 [42, 92] | 45 [15, 84] | |
AUC0‐24 computed as rifapentine dose/CL, and the AUC0‐24 targets refer to daily drug administration 7 days/week. Estimates of rifapentine AUC95 and inspection of Figure 1 and Supplementary Figure S4 were used to formulate target rifapentine AUC0‐24 cutoffs of >350 and <300 μg × h/mL. Participants with Karnofsky score ≤90 are grouped by the significant covariates of rifapentine exposure, aggregate cavity size on chest radiograph, and geographic origin of study site. To more simply display the most relevant pharmacokinetic/pharmacodynamic data from most of the study participants, data for 38 participants with Karnofsky score of 100 are separately presented in Supplementary Table S4. Proportion of participants with estimated treatment time >120 days were 7.7% (b) and 8.8% (c). AUC0‐24, area under the concentration‐time curve from 0 to 24 h; AUC95, area under the concentration‐time curve to achieve stable conversion in 95% of participants; CI, confidence interval; CL, clearance.
dParticipants grouped by the significant covariates of percentage area of extent of lung infiltrate on chest radiograph and baseline cough with or without sputum production.
Figure 2.
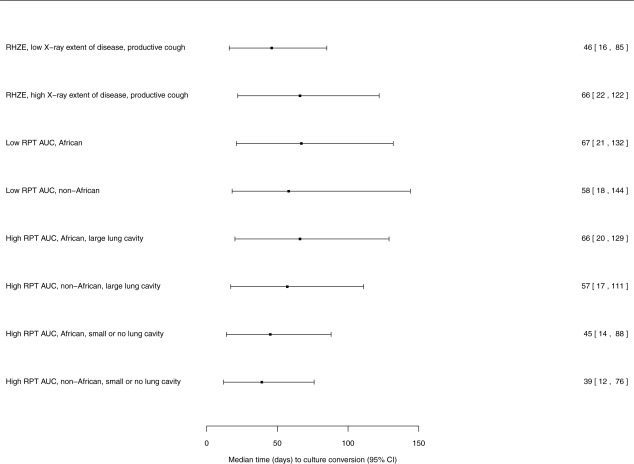
Forest plot of the relative effects of demographics, clinical covariates, and rifapentine area under the concentration–time curve from 0 to 24 h (AUC0‐24) on outcome of time (d) to culture conversion of sputum in liquid media culture. Median and 95% confidence interval are indicated by the square box and bars. Covariate effects are shown in patients taking rifampin‐based intensive‐phase therapy in the control group. Low (high) rifapentine (RPT) AUC, target rifapentine AUC0‐24 <300 (>350) μg × h/mL (AUC0‐24 from daily drug administration 7 days per week); low (high) radiographic extent of disease, <50% (≥50%) lung area by baseline chest radiograph; productive cough, productive cough at entry into the phase IIB treatment trials; large lung cavity (small or no lung cavity) on radiograph, aggregate size ≥4 cm (<4 cm); RHZE, Control regimen during intensive‐phase therapy of rifampin (R), isoniazid (H), pyrazinamide (Z), and ethambutol (E); RPT, rifapentine.
Solid media rifapentine PK/PD modeling
Significant independent covariates of time to stable culture conversion on solid media were rifapentine AUC0‐24, baseline aggregate lung cavity diameter (≥ or <4 cm) on chest radiographs, grade of acid‐fast bacilli smear of baseline sputum, and food intake with study drug. Covariates that affected treatment response with rifapentine differed somewhat with solid culture compared with liquid culture data (Supplementary Table S5). We found that 95% of participants with rifapentine AUC0‐24 >350 μg × h/mL and lung cavities <4 cm and those with AUC0‐24 >460 μg × h/mL and lung cavities ≥4 cm in aggregate size (e.g., AUC95) achieved stable conversion on solid media after completion of 2 months of daily treatment (Supplementary Table S6 and Supplementary Figure S4). In contrast with the exposure–response model using liquid media (Table 4, Supplementary Figure S4), higher rifapentine exposures conferred some benefit in patients with lung cavities ≥4 cm using solid culture data (see Supplementary Results, Supplementary Tables S6 and S7, and Supplementary Figure S4). Stable culture conversion on solid media required 52 to 62 days in 95% of participants with rifapentine exposure ≥AUC95 (Supplementary Table S6).
PK/PD modeling for safety endpoints
The event rates for evaluation of safety endpoints in participants were: 18.9% (n = 78) for rifapentine‐related adverse events of grade 3 and higher, and 15.6% (n = 40) for rifampin‐related adverse events. The probability by logistic regression of participants experiencing grade 3 and higher adverse events was not associated with rifapentine dose, AUC0‐24 or Cmax (P > 0.05). Similar results were obtained using a proportional odds model; no significant relationships between grade of adverse events and rifapentine dose or PK variables were observed (P > 0.05).
Achieving rifapentine target exposures
From clinical trial simulations of time‐to‐event maximal achievable effect models using data from liquid culture, target rifapentine AUC0‐24 ≥350 μg × h/mL (AUC95) was achieved with a daily dose of rifapentine 1,200 mg in ≥87% of participants who took drugs with high‐fat foods, ≥73% of participants who took study drugs with low‐fat (<27 g) foods, and 64% of participants who took study drugs when fasting. The rifapentine AUC0‐24 achieved was higher with higher rifapentine doses and foods higher in fat (Figure 3). To achieve target rifapentine exposures in most participants with drug taken with or without food, it was estimated that the rifapentine dose would need to be increased to 1,800 mg daily in participants of black race and remain at 1,200 mg in other participants. With these adjusted rifapentine doses, 96% of participants taking drug with high‐fat food would attain target AUC0‐24 vs. 85% if drug was taken without food (Supplementary Figure S5).
Figure 3.
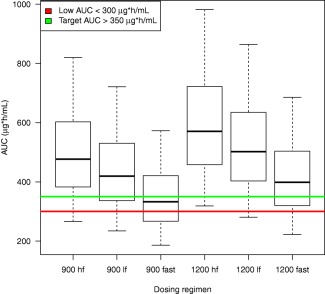
Relation between rifapentine area under the concentration–time curve (AUC) from 0 to 24 h (AUC0‐24) vs. dose (900 or 1,200 mg) and food type (high fat (hf), >27 g fat; lower fat (lf), 1 to 27 g fat; or fasting (fast)). Target rifapentine AUC0‐24 needed for 95% participants with no or small (<4 cm) lung cavities at baseline radiograph to achieve persistently negative cultures (AUC95) in liquid media indicated by the green horizontal line. Insufficient exposure indicated by the red line. Model estimates of rifapentine AUC95 and Figure 1 were used to formulate target cutoffs of rifapentine AUC0‐24 >350 μg × h/mL and low target rifapentine AUC0‐24 of <300 μg × h/mL using sputa cultures in liquid media.
Rifampin pharmacodynamics
Covariates independently associated with outcome in the time‐to‐event models for rifampin were extent of lung infiltrates and baseline cough but not lung cavities (Table 4). For rifampin models derived from data from liquid and solid cultures, the significant covariates were the same (Supplementary Table S8). The PK/PD outcomes were not directly comparable between rifampin and rifapentine because rifampin pharmacokinetic sampling was not performed, and lung cavitation was not a significant independent covariate of stable culture conversion in participants treated with rifampin. When compared with the participant group treated with rifapentine with AUC0‐24 ≥350 μg × h/mL and with no or small lung cavities, all control participants treated with rifampin (Figure 1 a, top arrow) were estimated to take an additional 3.7 weeks to develop stable culture conversion to negative in liquid media. However, participants with large cavities treated with rifapentine (irrespective of dose) took an additional 2 weeks in liquid media to develop stable culture conversion to negative compared with all control participants treated with rifampin (Figure 1 b, top arrow).
DISCUSSION
In this large PK/PD study, we identified a rifapentine dose that may potentially shorten TB treatment and described a profile of patients unlikely to respond to shorter‐term treatment. In support of these objectives, we characterized the population pharmacokinetics of rifapentine in participants who had TB. Interindividual variability of rifapentine was high. Although the bioavailability of rifapentine decreased with increasing dose as compared with the lowest dose administered (450 mg), high exposures could be achieved when rifapentine was given daily at high doses with food. Age and sex, but not body weight, affected clearance, supporting the use of a single rifapentine dose in adults with weights encountered in this study, rather than dosing based on weight for the treatment of adults who have TB. Significant predictors of lower exposures were fasting, black race, male sex, younger age, and HIV coinfection. Food increased rifapentine bioavailability by 40%, similar to that shown previously.14 Most participants (73%) achieved target rifapentine exposures by taking the drug while consuming food.
To improve the proportion of patients achieving target exposure, individualized rifapentine dosing regimens might be studied. Lower rifapentine bioavailability was observed in black vs. other participants, although race/region may have been confounded by the food type (bulk, protein, carbohydrate as well as fat) taken with study drug. Model simulations suggested that target rifapentine exposures in 96% of all participants would be attained when a very high daily dose of 1,800 mg rifapentine would be given with high‐fat food to black patients (or in 85% of all participants who were fasting). However, high doses of rifapentine are not well tolerated in healthy volunteers given rifapentine doses of 900 to 1,800 mg daily with food.15 A rifapentine dose of 1,800 mg was not evaluated in the analyzed trials; however, rifapentine 1,200 mg daily with food was well tolerated in trial participants, no significant relationships between grade of adverse events and rifapentine dose or PK variables were observed in this PK/PD study, and target drug exposures were attained in most participants. Only 34 participants received a rifapentine dose ≥1,200 mg in Study 29X; therefore, tolerability of higher doses of rifapentine warrants further evaluation. With a pharmacodynamic model of bactericidal activity, time to stable culture conversion in liquid media was highly associated with rifapentine exposure (P ≤ 0.001) but not with rifapentine dose used in this study (10, 15, and 20 mg/kg). This demonstrates the utility of PK/PD modeling to assess exposure–response relations and inform dose selection for future trials.
In our study we identified subgroups that took longer than others to convert their cultures to negative. Risk factors for slow treatment response included high mycobacterial disease burden at baseline, large lung cavities (≥4 cm), and enrollment from African trial sites. African participants in this study exhibited multiple features of more severe disease, including greater extent of disease and cavitation on chest radiography, higher‐grade acid‐fast bacilli smear in sputum, lower Karnofsky score, and lower body mass index. Prior studies showed that risk factors for treatment failure and relapse included more acid‐fast bacilli in sputum and greater radiographic extent of disease and cavitation.16, 17, 18 Furthermore, in two recent phase III trials to evaluate 4 months of TB treatment (using rifamycin plus fluoroquinolone‐containing regimens), cavitary lung disease was a significant risk factor for an unfavorable outcome.8, 9 HIV coinfection in our study was associated with a decrease in rifapentine bioavailability; however, this was not a risk factor (independent of exposure) for time to stable sputum culture conversion, similar to that shown in previous studies.8, 9
Rifapentine potency was markedly reduced in participants who had extensive cavitary disease in our study (Table 4); no rifapentine exposure–response relation was observed using liquid culture data in this group. Prior studies in animal models of TB that produced lung pathology similar to human disease, with granuloma, lung cavities, and caseation, similarly reported mixed results.19, 20, 21 Our results suggest that increased rifapentine exposure may not improve culture conversion over standardized rifampin doses in patients who have large lung cavities. Limited drug penetration into severely affected lung tissue and cavitary lesions could contribute to decreased efficacy.21 Although rifapentine demonstrates satisfactory total concentrations in patient plasma, high plasma protein binding (97–99%) reduces the free microbially active drug available for passive diffusion into the extravascular, necrotic extracellular space of a large cavity with caseation.22
The recovery of M. tuberculosis from sputum in the present study was greater using liquid than using solid media, similar to previous studies.23, 24 This may account for differences between PK/PD models using the different media. As expected, model‐predicted time to stable culture conversion was shorter in solid than in liquid media. Future phase III trials may evaluate whether factors associated with treatment response or target exposures are better predicted by PK/PD models that use liquid vs. solid culture results.
The present study has several limitations. Rifapentine was administered for only 8 weeks during intensive‐phase therapy and followed by rifampin in continuation‐phase therapy in both treatment trials. Further investigations are required to assess the effects of rifapentine treatment beyond 8 weeks. Another limitation of the PK/PD analyses was that only 34 participants received a rifapentine dose ≥1,200 mg in the dose‐ranging trial; however, the robust pharmacokinetic analyses of 405 participants receiving rifapentine treatment support our PK/PD findings. Also, HIV‐infected participants who were on antiretroviral therapy were underrepresented in this study. In addition, the pharmacodynamic endpoints were assessed using time to stable culture conversion during anti‐TB treatment; participants were not followed after treatment completion to assess long‐term cure. A final limitation was that concentrations of antitubercular drugs other than rifapentine were not examined. However, study participants received the same standard doses of nonrifamycin drugs per protocol. The effect of all antitubercular drugs are now being examined in a phase III randomized clinical trial of higher‐dose rifapentine administered for 4 months for tuberculosis treatment.
Strengths of the present study included the data collection as a component of two rigorously conducted clinical trials done at sites in Asia, Africa, North America, and Europe that compared rifapentine‐ to rifampin‐based intensive‐phase treatment administered by directly observed therapy. The pharmacokinetic samples were collected using standardized procedures and assayed by one laboratory. Furthermore, a novel PK/PD analytic model demonstrated proof of principle to establish rifapentine exposure‐efficacy response relations.
In summary, the present pharmacokinetic study supports level dosing of rifapentine in adults (dosing in mg instead of mg/kg) because rifapentine clearance was not affected within a range of common adult weights (aged 18 years and older). The PK/PD modeling demonstrated significant exposure–response relations. Risk factors for low rifapentine concentrations and reduced response to treatment were identified.
The potential utility of treatment shortening with rifapentine is unknown. The PK/PD model simulations suggested that stable sputum culture conversion can be achieved after completion of 4 months of therapy in most participants who have no or small lung cavities and who are treated with rifapentine 1,200 mg daily with food as part of multidrug therapy. However, the PK/PD analyses suggested poorer outcomes in patients with large lung cavities and highly positive sputum smears. These results support further evaluation of rifapentine at high daily doses as a TB treatment‐shortening strategy, with special attention to subgroups at higher risk of suboptimal rifapentine exposures and reduced response to therapy.
METHODS
Study design
We evaluated adults who had smear‐positive pulmonary TB enrolled in two randomized phase II clinical trials that compared rifapentine with rifampin during the first 8 weeks of anti‐TB therapy (TB Trial Consortium Studies 29 and 29X) conducted in Brazil, Hong Kong, Kenya, Peru, South Africa, Spain, Vietnam, Uganda, and the United States.3, 4 In both studies, rifapentine was given as 150‐mg tablets (Priftin, Sanofi‐Aventis, Anagni, Italy). Participants also received isoniazid, pyrazinamide, and ethambutol in weight‐based doses during the initial 8 weeks of treatment, in accordance with published guidelines.25 In both studies, participants were treated during the continuation‐phase with rifampin and isoniazid according to published guidelines.25 Methods and results from both trials have been published.3, 4 Both trials were registered at ClinicalTrials.gov (NCT00694629 and NCT01043575).
In Study 29, participants were randomized to receive rifapentine (10 mg/kg/dose) or rifampin (10 mg/kg/dose) 5 days per week for 8 weeks (intensive‐phase), and doses were taken on an empty stomach.3 In Study 29X, participants received rifampin (10 mg/kg/dose) or rifapentine (10, 15, or 20 mg/kg/dose) once daily with food, 7 days per week, for 8 weeks, and food consumption before PK sampling was documented with food histories (see Supplementary Methods).
The PK/PD analysis included participants from the modified intention‐to‐treat group (individuals with culture‐confirmed M. tuberculosis) from both treatment studies. This study was approved by the Institutional Review Boards of the United States Centers for Disease Control and Prevention and participating sites. Informed consent was obtained from all participants.
Rifapentine and desacetyl rifapentine assays and pharmacokinetic data
Blood samples were collected in either a sparse (1–3 samples per participant) or an intensive (7 samples per participant) pharmacokinetic sampling visit, conducted after ≥2 but ≤8 weeks of anti‐TB treatment (Supplementary Methods and Results). Plasma concentrations of rifapentine and its desacetyl rifapentine metabolite were determined using a validated high‐performance liquid chromatography assay (Pharmacokinetics Laboratory, University of Florida, Gainesville, FL).26
Microbiologic data
Sputum specimens were collected before the start of study therapy (baseline), after completion of 2, 4, 6, 8, and 12 weeks of treatment, and then monthly during continuation‐phase treatment unless any two consecutive prior sputum samples were documented as culture‐negative.3, 4
Population pharmacokinetic/pharmacodynamic modeling
Data were analyzed using a nonlinear mixed‐effects approach with software (NONMEM, v. 7, ICON, Dublin, Ireland) (Supplementary Methods and Supplementary Figure S1). For PK/PD modeling, individual AUC values from participants in Study 29 were adjusted (decreased by 28.6%) to account for drug administration on 5 of 7 days per week, compared with participants in Study 29X, where drugs were administered 7 days per week. Efficacy endpoints were characterized using serial sputum culture results on both solid and liquid media. Stable culture conversion was defined as conversion of sputum cultures from positive to negative during anti‐TB therapy in two consecutive sputum cultures. Efficacy endpoints used in exposure–response models included 1) percentage of participants who had negative sputum cultures at completion of intensive‐phase therapy and 2) days of anti‐TB treatment required for stable culture conversion. The latter was used to develop maximum effect time‐to‐event models. Covariates tested in exposure–response models included age, sex, weight, race, HIV status, body mass index, the summed diameter of all cavitary lesions on pretreatment chest radiographs, extent of lung infiltrate, baseline sputum smear grade, cough, and Karnofsky score (Supplementary Table S1). Covariates were tested on the following model parameters: maximal achievable effect, rifapentine area under the concentration–time curve to achieve 50% (AUC50) maximal achievable effect, and hazard function defined by scale and shape parameters (Supplementary Methods). Most culture conversion data in the PK/PD model were collected within the first 4 months of treatment; therefore, model predictions and simulations were restricted to ≤120 days to increase reliability.
Development of the pharmacokinetic/pharmacodynamic models
To describe the time to stable culture conversion, a parametric survival function was used, according to the equation:
The hazard was ht, and the survival St was a function of the cumulative hazard from time 0 to time t describing the probability of not converting the culture to negative within this time interval. The base model was developed by exploring different functions for the hazard ht, starting from a simple time‐independent constant hazard and gradually progressing to more complex functions, including Weibull function according to the equation:
where h0 was baseline hazard at time 0 and γ was a shape parameter.
Model building was guided by the likelihood ratio test, diagnostic plots, and internal model validation techniques, including visual and numeric predictive checks. Additional details for development of the PK/PD models and rifapentine and rifampin PK/PD model parameters are described in the Supplementary Methods (Supplementary Table S3).
Data from participants receiving rifampin were used for comparison in clinical trial simulations (Supplementary Methods).
CONFLICT OF INTEREST/DISCLOSURE
R.M.S. received research funding from Sanofi‐Aventis. The other authors declare no competing interests. Sanofi‐Aventis did not participate in the study design; collection, analysis, or interpretation of data; writing the article; or the decision to submit this article for publication.
AUTHOR CONTRIBUTIONS
M.W., R.S., W.R.M.K., J.L.J., K.E.D., and S.E.D. wrote the article; M.W., R.S., and W.R.M.K. designed the research; M.W., R.S., M.E., W.C.W., J.L.J., P.Ns., P.Na., N.V.N., and C.P. performed the research; M.W. and R.S. analyzed the data.
Supporting information
Supporting Information
ACKNOWLEDGMENTS
This work was supported by the United States Division of Tuberculosis Elimination, National Center for HIV/AIDS, Viral Hepatitis, STD, and TB Prevention, Centers for Disease Control and Prevention; the Veterans Administration; and the National Center for Advancing Translational Sciences, National Institutes of Health, through the Clinical and Translational Science Award (UL1 TR001120). Sanofi‐Aventis donated the rifapentine and rifampin used in the study, and, since 2007, has donated >$2.3 million to the Centers for Disease Control and Prevention Foundation to supplement available United States federal funding for rifapentine research of tuberculosis. References in this article to any specific commercial products, process, service, manufacturer, or company does not constitute endorsement or recommendation by the United States government, Centers for Disease Control and Prevention, Veterans Administration, or National Institutes of Health. The findings and conclusions are solely those of the authors and do not necessarily represent the official views of the Centers for Disease Control and Prevention, Veterans Administration, or National Institutes of Health.
The authors thank the study participants who contributed to this study. The authors are grateful to Philip LoBue and Kenneth Castro for their support of the Tuberculosis Trials Consortium within the Centers for Disease Control and Prevention, Erin Bliven‐Sizemore, Deron Burton, and Andrew Vernon (Tuberculosis Trials Consortium) for review of the article, and Elly Trepman for editorial assistance. The authors thank the local TB program staff who assisted in the clinical management of some study participants.
Participating clinical sites (principal investigators, study coordinators, and microbiologists, with numbers of participants in this study in parentheses) were as follows:
Uganda–Case Western Reserve University Research Collaboration, Kampala (245): John L. Johnson, M.D., Roy D. Mugerwa, M.B. Ch.B., M.Med., Harriet Mayanja‐Kizza, M.B. Ch.B., M.Med., Grace Muzanyi, M.B. Ch.B., Phineas Gitta, M.B. Ch.B., Alphonse Okwera, M.B. Ch.B., M.Sc., Dorcas Lamunu, B.S.N., Pheona Nsubuga, B.Pharm., Moses Joloba, M.B. Ch.B., Ph.D., Karen Morgan, M.S., M.P.H., Sam Ogwang, M.S., Yusuf Mulumba, M.S., Paul Mubiri, B.Stat., Joseph G. Nakibali, James Kimera, and Elias Ssaku.
South Texas‐Audie Murphy VA Hospital Research Collaboration, Harlingen (47): Marc H. Weiner, M.D., Richard Wing, M.D., Diane Wing, R.N., Juan Uribe, R.N., Josefina Gonzalez, L.V.N., Melissa Engle, C.R.T., C.C.R.C., Lee C. Sadkowski, M.T. (A.S.C.P.), Suzanne Salinas, M.T. (A.S.C.P.), S.M., and Mildred Manzano, A.M.T.
Vietnam National TB Program–University of California, San Francisco Research Collaboration, Hanoi (44): Nguyen Viet Nhung, M.D., Ph.D., Payam Nahid, M.D., M.P.H., Dinh Ngoc Sy, M.D., Ph.D., Luu Thi Lien, M.D., Pham Huu Thuong, M.D., Nguyen Van Hung, M.D., Ph.D., Vu Cao Cuong, M.D., M.Sc., Nguyen Phuong Hoang, M.D., Ha Phan, M.D., Dr.PH., Nguyen Thi Thuy Hanh, Nguyen Luu Van Trang, and Cindy Merrifield, R.N.
Wits Health Consortium, Soweto (42): Richard E. Chaisson, M.D., Neil Martinson, M.B. B.Ch., M.P.H., Bernardine Friese, B.Sc. Hons., Jennifer Hoffmann, M.S.N., M.P.H., Anne Efron, M.S.N., M.P.H., and Jessica Trussler, M.B. Ch.B., M.Med.
Spain TB Investigation Unit of Barcelona‐University of North Texas Research Collaboration, Barcelona (36): Joan A. Cayla, M.D., Ph.D., Jose M. Miró, M.D., Ph.D., Antonio Moreno, M.D., Joan P. Millet, M.D., Ph.D., Lucía del Baño, R.N., Laia Fina, M.Sc., Llanos Roldán, R.N., Àngels Orcau, M.D., José A. Martinez M.D., Ph.D., M. Antònia Sambeat M.D., Ph.D., Virginia Pomar, M.D., Hernando Knobel, M.D., Ph.D., M. Luiza de Souza, M.D., M. Angeles Jiménez, M.D., Celia Milà, M.D., Xavier Martínez Lacasa, M.D., Adrià Curran, M.D., Ph.D., Israel Molina, M.D., Miguel Santín, M.D., Ph.D., Laura Muñoz, M.D., Fernando Alcaide, M.D., Ph.D., Raquel Moure, Ph.D., Julian Gonzalez, M.D., Ph.D., Griselda Tudo Ph.D., M. Teresa Tórtola, M.D., Lucía González, R.N., Jessica Muñoz, R.N., Àngels Fontanet, R.N., Núria Saborit, R.N., Elisa Lara, R.N., Carmen Ligero, R.N., Neus Jové, R.N., Roser Font, R.N., Marisa Manzanares, and Adela Cantos, R.N.
Baylor College of Medicine, Houston (33): Elizabeth Guy, M.D., Richard Hamill, M.D., Ruby Nickson, R.N., and Kathleen Goodrich, M.T., M.S., S.M. (A.S.C.P.).
University of KwaZulu Natal‐Harlem Hospital Research Collaboration, Durban (29): Wafaa El‐Sadr, M.D., Nesri Padayatchi, M.D., M.S., Sheila Bamber, M.D., Surie T. Christop Chinappa, M.D., Yael Hirsch‐Moverman, M.P.H., Vikesh Naidoo, Nelisiwe Mnguni, Thokozani Mthethwa, Zevile Gumede, and Kaloshnee Ganas.
University of North Texas Health Science Center, Fort Worth (28): Michel Fernandez, M.D., Stephen Weis, D.O., Barbara King, R.N., Le Turk, R.N., Gloria Stevenson, Joseph Helal, R.Ph., Norma Shafer, L.V.N., Denise Dunbar, M. (A.S.C.P.), and Ken Jost, M.T. (A.S.C.P.).
Stellenbosch University, Cape Town (22): Anneke Hesseling, M.D., Ph.D., Mark Cotton, M.Med., Ph.D., Andreas Diacon, M.D., Ph.D., Norma Groenewald, Charlotte Lawn, Vera Simmonds, Zoja Noveljic, MB. Ch.B., M.Med., and Amour Venter.
KEMRI‐CDC, Kisumu, Kenya (21): Kevin Cain, M.D., Kayla Laserson, Sc.D., Lena Matata, M.D., Elisha Okeyo, M.D., Victor Mudhune, B.Pharm, Albert Okumu, Jeremiah Khayumbi, and Janet Agaya.
Audie L. Murphy VA Hospital, San Antonio (21): Marc H. Weiner, M.D., Melissa Engle, C.R.T., C.C.R.C., Hipolito Pavon, M.P.H., Jose Jimenez, B.S., Lee C. Sadkowski, M.T. (A.S.C.P.), Suzanne Salinas, M.T. (A.S.C.P.), S.M., and Mildred Manzano, A.M.T..
Columbia University College of Physicians and Surgeons and the New York City Department of Health and Mental Hygiene Tuberculosis Control Program, New York (18): Neil W. Schluger, M.D., Joseph Burzynski, M.D., M.P.H., Vilma Lozano, R.N., Magda Wolk, R.N., and Adeleh Ebrahimzadeh, Ph.D.
TB and Chest Service of Hong Kong, China (18): Chi‐Chiu Leung, M.B. B.S., Kwok‐Chiu Chang, M.B. B.S., M.Sc., Sik‐Wai Tam, Cheuk‐Ming Tam, M.B. B.S., M.Sc., Sau‐Yin Tam, Ka‐Yun Mak, Ka‐Lin Fong, Nai‐Chung Lee, Chi‐Wai Yip, Ph.D., Judy Yee‐Man Lam, M.B. B.S., Chi‐Wai Ng, M.B. B.S., Oi‐Wah Fong, Edman Tin‐Keung Lam, M.B. Ch.B., Chung‐Ying Wong, and Ka‐Ling Leung.
University of Medicine and Dentistry of New Jersey, Newark (9): Bonita T. Mangura, M.D., Lee B. Reichman, M.D., M.P.H., Alfred A. Lardizabal, M.D., Amee Patrawalla, M.D., Maria Corazon Leus, R.N., Veronica Anokute, R.N., Michelle Burday, Ph.D., and Debra Sickles, B.S.
University of California, San Francisco (9): Payam Nahid, M.D., M.P.H., Philip Hopewell, M.D., Cindy Merrifield, R.N., Irina Rudoy, M.D., Jill Israel, R.N., and Anna Babst.
Denver Health and Hospitals, Denver (8): Randall Reves, M.D., William Burman, M.D., Laurie Luna, R.N, Robert Belknap, M.D., Jacquie Moore, and Ginger Hildred, M.T. (A.S.C.P.) S.M.
Brazil‐Johns Hopkins University Research Collaboration, Rio de Janeiro (7): Susan Dorman, M.D., Marcus B. Conde, M.D., Anne Efron, M.S.N., M.P.H., Carla Loredo, Fernanda Mello, M.D., Rafael Duarte, Ph.D., M.D., Gisele Betzler de Oliveira Vieira, Milene Barty, and Derek Armstrong.
Public Health – Seattle & King County (6): Masahiro Narita, M.D., Monica Pecha, M.P.H., Margaret Ragland, M.S., Linh Le, and Jean Pass.
Universidad Peruana Cayetano Heredia, Lima (6): Eduardo Jose Gotuzzo, M.D., F.A.C.P., Carlos Zamudio, M.D., Carlos Seas, M.D., Cinthia Hurtado, M.D., Leslie Levano, M.D., and Celer Pantoja.
University of California, San Diego Medical Center and collaborating site San Diego County Department of Health and Human Services, TB Control (5): Antonino Catanzaro, M.D., Kathleen S. Moser, M.D., Mark J. Tracy, M.D., Vivien Peach Francisco, R.N., Judy Davis, Sharon Reed, M.D., and Christopher R. Peter, Ph.D.
Vanderbilt University Medical Center, Nashville (5): Timothy R. Sterling, M.D., Amy Kerrigan, M.S.N., R.N., C.C.R.P., Teresa Smith, and Alicia Wright, M.S.
Washington, DC VA Medical Center, Prince George's County and Montgomery County, MD TB Control Programs, District of Columbia TB Control Program, and the George Washington University Medical Center (3): Debra Benator, M.D., Donna Sepulveda Conwell, R.N., and Fred M. Gordin, M.D.
Montreal Chest Institute, Montreal (2): Dick Menzies, M.D., Kevin Schwartzman, M.D., M.P.H., Christina Greenaway, M.D., M.Sc., Marthe Pelletier, Chantal Valiquette, Paul Plaisir, and Louise Thibert, M.Sc.
FHI360/Duke University/VA Durham (2): Carol Dukes Hamilton, M.D., M.H.S., Jason Stout, M.D., M.H.S., David Holland, M.D., M.H.S., Ann Mosher, R.N., M.P.H., F.N.P.‐B.C., and Emily Hecker, R.N., M.S.N.
Johns Hopkins University, Baltimore (1): Susan Dorman, M.D., Richard E. Chaisson, M.D., Gina Maltas, R.N., James Fisher, L.P.N., Nancy Hooper, M.S.
University of Southern California (1): Brenda E. Jones, M.D., Ermelinda Rayos, C.W., B.S.N., R.N., Peregrina Molina, R.N., Celia Luken, and Samuel Sum.
References
- 1. Centers for Disease Control and Prevention Web site . Tuberculosis: data and statistics. <http://www.cdc.gov/tb/statistics/default.htm>. Accessed 18 September 2013.
- 2. Rosenthal, I.M. et al Daily dosing of rifapentine cures tuberculosis in three months or less in the murine model. PLoS Med. 4, e344 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dorman, S.E. et al Substitution of rifapentine for rifampin during intensive phase treatment of pulmonary tuberculosis: study 29 of the tuberculosis trials consortium. J. Infect. Dis. 206, 1030–1040 (2012). [DOI] [PubMed] [Google Scholar]
- 4. Dorman, S.E. et al Daily rifapentine for treatment of pulmonary tuberculosis. A randomized, dose‐ranging trial. Am. J. Respir. Crit. Care Med. 191, 333–343 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mitchison, D.A. Assessment of new sterilizing drugs for treating pulmonary tuberculosis by culture at 2 months. Am. Rev. Respir. Dis. 147, 1062–1063 (1993). [DOI] [PubMed] [Google Scholar]
- 6. Gler, M.T. et al Delamanid for multidrug‐resistant pulmonary tuberculosis. N. Engl. J. Med. 366, 2151–2160 (2012). [DOI] [PubMed] [Google Scholar]
- 7. Benator, D. et al Rifapentine and isoniazid once a week versus rifampicin and isoniazid twice a week for treatment of drug‐susceptible pulmonary tuberculosis in HIV‐negative patients: a randomised clinical trial. Lancet 360, 528–534 (2002). [DOI] [PubMed] [Google Scholar]
- 8. Merle, C.S. et al A four‐month gatifloxacin‐containing regimen for treating tuberculosis. N. Engl. J. Med. 371, 1588–1598 (2014). [DOI] [PubMed] [Google Scholar]
- 9. Gillespie, S.H. et al Four‐month moxifloxacin‐based regimens for drug‐sensitive tuberculosis. N. Engl. J. Med. 371, 1577–1587 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Horne, D.J. et al Sputum monitoring during tuberculosis treatment for predicting outcome: systematic review and meta‐analysis. Lancet Infect. Dis. 10, 387–394 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Morse, G.D. , Catanzaro, L.M. & Acosta, E.P. Clinical pharmacodynamics of HIV‐1 protease inhibitors: use of inhibitory quotients to optimise pharmacotherapy. Lancet Infect. Dis. 6, 215–225 (2006). [DOI] [PubMed] [Google Scholar]
- 12. de Kanter, C.T. et al Viral hepatitis C therapy: pharmacokinetic and pharmacodynamic considerations. Clin. Pharmacokinet. 53, 409–427 (2014). [DOI] [PubMed] [Google Scholar]
- 13. Kent, P.T. & Kubica, G.P. Public Health Mycobacteriology: A Guide for the Level III Laboratory. (U.S. Department of Health and Human Services, Public Health Service, Centers for Disease Control, Atlanta, GA, 1985). [Google Scholar]
- 14. Zvada, S.P. et al Effects of four different meals types on the population pharmacokinetics of single‐dose rifapentine in healthy male volunteers. Antimicrob. Agents Chemother. 54, 3390–3394 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dooley, K.E. et al Novel dosing strategies increase exposures of the potent antituberculosis drug rifapentine but are poorly tolerated in healthy volunteers. Antimicrob. Agents Chemother. 59, 3399–3405 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tam, C.M. , Chan, S.L. , Kam, K.M. , Goodall, R.L. & Mitchison, D.A. Rifapentine and isoniazid in the continuation‐phase of a 6‐month regimen. Final report at 5 years: prognostic value of various measures. Int. J. Tuberc. Lung Dis. 6, 3–10 (2002). [PubMed] [Google Scholar]
- 17. Weiner, M. et al Low isoniazid concentrations and outcome of tuberculosis treatment with once‐weekly isoniazid and rifapentine. Am. J. Respir. Crit. Care Med. 167, 1341–1347 (2003). [DOI] [PubMed] [Google Scholar]
- 18. Hesseling, A.C. et al Baseline sputum time to detection predicts month two culture conversion and relapse in non‐HIV‐infected patients. Int. J. Tuberc. Lung Dis. 14, 560–570 (2010). [PubMed] [Google Scholar]
- 19. Hoff, D.R. et al Location of intra‐ and extracellular M. tuberculosis populations in lungs of mice and guinea pigs during disease progression and after drug treatment. PLoS One 6, e17550 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rosenthal, I.M. et al Dose‐ranging comparison of rifampin and rifapentine in two pathologically distinct murine models of tuberculosis. Antimicrob. Agents Chemother. 56, 4331–4340 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dartois, V. The path of anti‐tuberculosis drugs: from blood to lesions to mycobacterial cells. Nat. Rev. Microbiol. 12, 159–167 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Egelund, E.F. et al Protein binding of rifapentine and its 25‐desacetyl metabolite in patients with pulmonary tuberculosis. Antimicrob. Agents Chemother. 58, 4904–4910 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hasegawa, N. , Miura, T. , Ishizaka, A. , Yamaguchi, K. & Ishii, K. Detection of mycobacteria in patients with pulmonary tuberculosis undergoing chemotherapy using MGIT and egg‐based solid medium culture systems. Int. J. Tuberc. Lung Dis. 6, 447–453 (2002). [PubMed] [Google Scholar]
- 24. Chihota, V.N. et al Liquid vs solid culture for tuberculosis: performance and cost in a resource‐constrained setting. Int. J. Tuberc. Lung Dis. 14, 1024–1031 (2010). [PubMed] [Google Scholar]
- 25. Blumberg, H.M. et al American Thoracic Society/Centers for Disease Control and Prevention/Infectious Diseases Society of America: treatment of tuberculosis. Am. J. Respir. Crit. Care Med. 167, 603–662 (2003). [DOI] [PubMed] [Google Scholar]
- 26. Savic, R.M. et al Population pharmacokinetics of rifapentine and desacetyl rifapentine in healthy volunteers: nonlinearities in clearance and bioavailability. Antimicrob. Agents Chemother. 58, 3035–3042 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information