

Abstract
Organs-on-a-chip platforms seek to recapitulate the complex microenvironment of human organs using miniaturized microfluidic devices. Besides modeling healthy organs, these devices have been used to model diseases, yielding new insights into pathophysiology. Hutchinson-Gilford progeria syndrome (HGPS) is a premature aging disease showing accelerated vascular aging, leading to the death of patients due to cardiovascular diseases. HGPS targets primarily vascular cells, which reside in mechanically active tissues. Here, we developed a progeria-on-a-chip model and examined the effects of biomechanical strain in the context of vascular aging and disease. Physiological strain induced a contractile phenotype in primary smooth muscle cells (SMCs), while a pathological strain induced a hypertensive phenotype similar to that of angiotensin II treatment. Interestingly, SMCs derived from human induced pluripotent stem cells of HGPS donors (HGPS iPS-SMCs), but not from healthy donors, showed an exacerbated inflammatory response to strain. In particular, we observed increased levels of inflammation markers as well as DNA damage. Pharmacological intervention reversed the strain-induced damage by shifting gene expression profile away from inflammation. The progeria-on-a-chip is a relevant platform to study biomechanics in vascular biology, particularly in the setting of vascular disease and aging, while simultaneously facilitating the discovery of new drugs and/or therapeutic targets.
Keywords: organ-on-a-chip, mechanotransduction, aging, vascular disease, progeria
Graphical Abstract
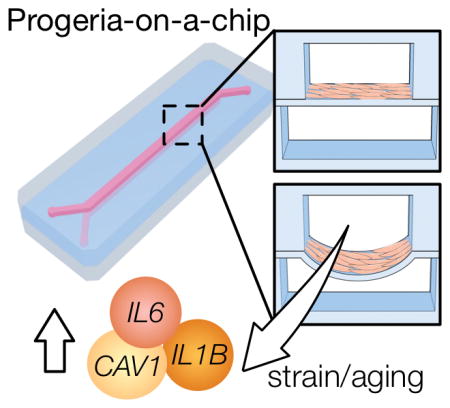
1. Introduction
The naturally occurring biomechanical strains in blood vessels translate via mechanotransduction into behavioral changes of vascular smooth muscles (SMCs) and endothelial cells (ECs). Whereas ECs are primarily exposed to fluid shear stress, SMCs are mainly exposed to cyclic biomechanical strain, which plays a key role in controlling the tone of the vessel and concomitant blood pressure [1]. In a healthy arterial wall, SMCs experience cyclic biomechanical strain of 9% [2], while SMCs under pathological conditions experience strains of up to approximately 15% [3]. In vitro, several studies conducted have utilized uniaxial strain values in the order of 5–25% [4]. These studies revealed that cyclical biomechanical strain in SMCs is transduced by integrins [5] and results in the acquisition of a contractile phenotype reminiscent of the in vivo phenotype [4, 6, 7]. Pathologic levels of biomechanical strain can increase reactive oxygen species (ROS) levels as well as induce expression of vascular injury and inflammation markers [4, 8]. Accumulated levels of such markers are hallmarks of vascular disease and progressively increase during aging [9, 10], leading to further worsening of pathology.
Currently there is no dedicated in vitro microfluidic system with SMCs to study the impact of biomechanics in aging and vascular diseases such as hypertension. In the past few years, the lack of appropriate in vitro models has motivated the need for the development of microfluidic organ-on-a-chip models [11–13] that are able to recapitulate the complex in vivo biological parameters. Integration with microfluidic devices makes these platforms uniquely suited to apply physiologically relevant biomechanical strain, shear stress, transmural pressure, and/or provide three-dimensional (3D) environments. To date, some microfluidic models have been developed to apply biomechanical strain mimicking the lung [14–16], gut [17–19], and blood vessels [20–24], but have not been applied in the context of human vascular aging.
Recently, stem cell technologies have facilitated the generation of aged cells [25]. Hutchinson-Gilford progeria syndrome (HGPS) is a rare genetic disorder caused by a mutant form of the nuclear protein lamin A – progerin [26, 27]. HGPS patients suffer from premature and accelerated aging [25, 28–31], while accumulation of progerin also occurs during physiological aging. Notably, HGPS targets primarily vascular cells [32], which are mechanically active tissues. Induced pluripotent stem cells (iPSCs) generated from HGPS fibroblasts have been used to recapitulate normal aging in an accelerated fashion [29, 30, 33–35], proving to be a valuable tool to study vascular aging and thus facilitate the discovery of novel treatments [36, 37]. However, accurate in vitro models must take into account the interplay between biomechanical strain and the behavior of aged cells.
Here, we set out to develop a novel progeria-on-a-chip model that would capture blood vessel biomechanical dynamics on-chip. Within this device, we exposed healthy iPS-SMCs and HGPS iPS-SMCs to normal and pathological strains to study the interplay between biomechanical strain and vascular aging. Models that combine biomechanics and vascular aging are crucial tools towards understanding vascular disease/aging and developing new therapies.
2. Results
2.1 Recapitulating blood vessel dynamics on-chip
Blood vessels are constantly exposed to cyclic mechanical stretch (Fig. 1A) varying from the normal 9% relative strain in healthy individuals [2] to the pathological 15% relative strain [3]. Studies conducted in vitro used values in the order of 5–25% [4], with the range of 5–10% considering physiological and >15% pathological strain. To recapitulate the cyclic mechanical deformation experienced by SMCs in the arterial wall of blood vessels we have developed a novel microfluidic polydimethylsiloxane (PDMS) device (Fig. 1B–E). The device consists of a top fluidic channel with an underlying vacuum channel that is separated by a 123.5 ± 3.0 μm PDMS membrane (Fig. 1B; Fig. S1). The fabrication process is simple (Fig. 1C–D) and relies on the creation of a three-layer device, the top fluidic channel, a middle thin PDMS membrane and a bottom vacuum channel. Cells are cultured on top of the membrane (Fig. 1F), which is deformed by applying different pressure drops on the bottom channel (Movie S1). Previous approaches used pressure drops on side channels to stretch a membrane over a central post [22, 38, 39], or positive pressure to bulge a membrane [40, 41]. The device measures 40 mm by 18 mm (Fig. 1E), is optically transparent, and fits a standard glass slide for easy microscopic visualization. The fluidic channel has a straight region measuring 25 mm by 1 mm. To facilitate channel alignment during assembly of the PDMS layers, the bottom layer was made 0.2 mm wider. The device was designed to be usable in any laboratory setting: pressure drops used are obtained from a laboratory vacuum line, and cell seeding onto the device can be achieved by manually pipetting a cell suspension inside the fluidic channel. We have used this novel device to characterize the vascular response of healthy and HGPS iPS-SMCs to different levels of biomechanical strain (Fig. 1G).
Figure 1. Recapitulation of blood vessel dynamics on chip.

(A) Blood vessels experience cyclic strain due to the pulsatile nature of blood flow. (B) Biomimetic microfluidic vascular model containing two overlapping channels. A cross-sectional view of the microfluidic device shows the cell layer cultured on top of the PDMS membrane and a view during vacuum stimulation shows the downward membrane deformation. (C) The first step of the chip fabrication containing the casting of PDMS (10:1 ratio) on an acetal resin mold and the spincoating of PDMS (20:1 ratio) to generate a thin membrane on top of a silicon wafer. (D) The top slab and membrane portions of the PDMS device are bonded using oxygen plasma and then peeled off the silicon wafer. The top part is then bonded to the bottom PDMS slab molded previously, and treated with fibronectin solution to allow cell culture. (E) Photograph of the microfluidic channel showing the media inlet, outlet and the vacuum port. (F) Micrographs of SMCs cultured in the microfluidic chip (scale bar represents 250μm). (G) Proposed methodology to unveil strain-related vascular changes in iPS-derived SMCs from healthy and HGPS donors.
To characterize the device, we determined the membrane deformation in situ and in silico. Cross-sectional views of the microfluidic device under different amounts of vacuum pressure ranging from 0 kPa to 50 kPa demonstrated a pressure-dependent increase in membrane deformation (Fig. 2A). The approach used to show in situ membrane deformation has not been demonstrated before and allows immediate and direct visualization of membrane deformation. The thickness and mechanical properties of the device’s membrane were tailored to function under low pressures while achieving the required strains. A pressure drop of 10 kPa was found to average to 9 ± 2% strain, 20 kPa to 16 ± 1%, and 30 kPa to 24 ± 2 % (Fig. 2B). In silico modeling of the membrane deformation demonstrated similar results in the range of 0 kPa to 30 kPa (Fig. S2). Indeed, a strong correlation was found between cross-sectional measurements and simulation data (Fig. 2B; Pearson’s correlation of r = 0.9825). Moreover, we mapped the variability in mechanical strain by characterizing the surface strain (Fig. S2). Additionally, we have simulated the effect of this spatial strain distribution on cells attached to the membrane using micrographs of nuclei within our devices (Fig. 2C). In line with our modeling, we observed a gradient strain similar to other reports for microfluidic strain devices [14]. Importantly, we have also verified that the strain applied was mostly uniaxial (Fig. 2D). We used the average strain values and analyzed the entire cell population inside the microfluidic device.
Figure 2. Characterization of stretch capabilities of microfluidic device.

(A) Cross-sectional view of microfluidic device and membrane deformation under different amounts of pressure drop (scale bar represents 500 μm). (B) Comparison of the overall strain on the y-axis between cross-sectional measurements and theoretical computational simulation (results represent mean ± SD of n=5). (C) Computational simulation of the strain on a 1 mm x 0.1mm membrane section overlaid with a representative nuclei image under different levels of pressure drop. (D) Two representative nuclear outlines of cells under 0 kPa and 30 kPa and respective vector displacement maps (thick red arrows in the top panels indicate strain direction).
2.2 Biomechanical strain induced cell re-orientation and a contractile phenotype in SMCs
Mechanical stimulation regulates morphology and function of SMCs [3]. The lack of mechanical strain in static cultures induces SMCs to shift towards a dedifferentiated phenotype, which is characterized by a higher proliferative state, higher protein synthesis, and relatively circular cell shape [3, 4, 42]. Upon mechanical stimulation, cells acquire a contractile phenotype that is reminiscent of the in vivo state. SMCs under cyclic mechanical strain reorient perpendicular to the strain direction [43, 44]. We examined the morphology, orientation, and expression of contractile markers by SMCs under normal and hypertensive mechanical strains. Cells were cultured for 24 hours under cyclic mechanical strain in a microfluidic channel coated with fibronectin. This coating was chosen because SMCs are more responsive to the cyclic strain on fibronectin-coated substrates rather than other ECM proteins [42]. We cultured cells for 24 hours to observe early effects of strain, whereas other studies cultured cells under 48 or 72 hours [42, 45]. SMCs under no mechanical strain exhibited a random angle orientation distribution (Fig. 3A). Strained cells reoriented perpendicular to the direction of strain and increased their aspect ratio (Fig. 3B–C), with a magnitude-dependent effect. Indeed, the angle orientation distribution of SMCs became narrower and closer to 0° as the stretch was increased from 9% to 16% strain (Fig. 3D–F). Cell shape analysis revealed a decrease in cell width (Fig. 3G, Fig. S3), while no change was observed in cell length (Fig. S3), resulting in an overall increase in the aspect ratio of the cells (Fig. 3H). SM22α and β1-integrin are enriched in contractile SMCs [6]. Besides, β1- and α5-integrins mediate intracellular signal mechanotransduction via adhesion to fibronectin [3–5, 42]. Therefore, to evaluate the impact of cyclic stretching in the phenotype of SMCs and their mechanotransduction responsiveness, we measured the expression levels of TAGLN (encoding SM22α), ITGB1 (encoding β1-integrin) and ITGA5 (encoding α5-integrin) gene transcripts by real-time quantitative polymerase chain reaction (qRT-PCR). We found TAGLN, ITGB1 and ITGA5 mRNA upregulated under 9% and 16% strain (Fig. 3I). Overall, we confirmed that using our on-chip system SMCs responded to mechanical strain by acquiring a more differentiated contractile phenotype.
Figure 3. Strain induces cytoskeletal reorientation and contractile phenotype markers on chip.

(A–C) Micrographs of f-actin stained cells under different amounts of strain for 24 hours. Cells exposed to different amounts of strain were quantified on (D–E) angle orientation distribution, (G) cell width and (H) aspect ratio (#, P<0.01; ns, not significant; scale bars represent 100 μm and red arrow indicates direction of strain). Relative mRNA expression levels of TAGLN (SM22α gene), ITGB1 (β1-integrin gene) and ITGA5 (α5-integrin gene) (I) [bars represent mean ± SD of n=5; ##, P<0.01].
2.3 Hypertensive strain recapitulates angiotensin II induced phenotype
To evaluate whether a threshold pathological strain induced vascular damage, we analyzed gene expression in cells cultured under strain as compared to static conditions treated with angiotensin II treatment. The renin-angiotensin-aldosterone system is implicated in the development of hypertension and regulates blood pressure in vivo by controlling the vascular tone of SMCs [46]. In this context, angiotensin II has been demonstrated to generate a hypertensive phenotype in SMCs [46]. Caveolin-1 (CAV1), a component of caveolae, is an important mediator of signal transduction [47] and plays a role in mechanotransduction in endothelial cells [48]. Microarray profiles of vascular tissues have identified caveolin-1 as a potential target marker of hypertension, showing overexpression in both spontaneous and adrenocorticotropic hormone–induced hypertensive rats [49]. Similarly, increased expression of caveolin-1 and caveolae has been reported in human pulmonary artery hypertension [50, 51]. IL-6 is a major pro-inflammatory cytokine that has been associated with essential hypertension [52], which is overexpressed in human serum of pulmonary hypertension patients [53] and induces hypertension in mice [54].
Our results showed that culturing SMCs under 9% strain led to minor changes of mRNA expression levels (Fig. 4A; Fig. S4), with an increase in the cytosolic superoxide dismutase (SOD1) and decrease in mitochondrial one (SOD2). Importantly, ROS has been implicated in vascular diseases, and angiotensin II is known to increase mitochondrial ROS [55–58]. Both pathological strain (Fig. 4B) and 100 nM of angiotensin II treatment (Fig. 4C; Fig. S5) showed a marked increase in mitochondrial SOD2, with a smaller increase in SOD1. Interestingly, this observation suggested that under physiological strain ROS was primarily produced in the cytosol. Using the NADPH oxidase inhibitor VAS2870 (20μM) [59] we showed downregulation of the NAPDH oxidase subunit p22phox and restoration SOD1 levels, while no change was observed for SOD2 (Fig. S6).
Figure 4. Strain-dependent activation of SMCs recapitulates angiotensin II vascular gene fingerprint.

Rose plots of the mRNA expression profiles of SMCs treated for 24 hours under (A) physiological strain (9%), (B) pathological strain (16%), or (C) 100 nM of angiotensin II [AngII] (mean values of n=5 for 9% and 16% strain, mean values of n=3 for AngII treatment; see Figures S4 and S5 for the bar plots and statistical significance).
Our results further showed that CAV1 was upregulated in a strain magnitude-dependent way (Fig. S4C), and a similar increase was shown with angiotensin II treatment (Fig. 4C; Fig. S5C). Moreover, we showed increase in ITGB1 with strain and angiotensin II (Fig. 4) similar to previously reported results [60]. The pro-inflammatory markers IL6 and IL1B were significantly increased with hypertensive strain (Fig. 4B) and angiotensin II (Fig. 4C), but not with physiological strain. Such difference suggested the existence of a strain-dependent threshold that gates the biomechanically induced upregulation of IL-6 expression in SMCs.
Overall, SMCs under pathological strain conditions exhibited a gene expression pattern similar to angiotensin II treatment, with similar responses of ROS, inflammation, and vascular injury genes, although strain alone may not fully recapitulate the effects of angiotensin II. Further studies are required to understand the regulatory pathways of strain and angiotensin II in SMCs, both alone and in combination.
2.4 Healthy and HGPS iPS-SMCs undergo cytoskeletal remodeling upon biomechanical strain
Vascular diseases and aging are intimately linked [61] yet rarely studied in an integrated approach. Due to the relation between vascular disease and aging, we examined the influence of biomechanics in an iPS-derived progeria-on-a-chip model. Stem cell-based models represent ideal candidates to study human diseases due to the practical capability of large scale expansion and differentiation [62]. HGPS patients exhibit premature aging, with increased arterial stiffening, expression of pro-inflammatory markers, the risk of atherosclerosis, calcification, and changes in systolic and pulse pressure [36]. HGPS and other accelerated aging syndromes have been established as aging models that recapitulate several aspects of cellular aging [29, 34, 36, 63]. In HGPS, both progerin and lamin A accumulate in the nucleus [36]. We generated SMCs through differentiation [64] of iPS cells derived from HGPS donors (and healthy controls) [35], and evaluated the interplay between biomechanical strain and aging (Fig. 5A) [36]. The generation of iPS cells from HGPS fibroblasts was previously shown to be similar to healthy fibroblast [35]. Importantly, we have characterized the differentiated SMCs derived from HGPS iPS cells, and showed that iPS-SMCs from HGPS donors express significantly higher levels of progerin mRNA (Fig. 5B). Considering the focus of the current work, the complete dataset on the characterization of HGPS iPS-SMCs has not been included here and will be reported in another publication in details. Concordant with the alignment results from SMCs (Fig. 3), iPS-derived SMCs from healthy and HGPS donors showed cytoskeletal reorientation upon mechanical stimulation, while non-stimulated cells showed a random distribution (Fig. 5C–F).
Figure 5. Biomechanical strain induces cytoskeletal reorientation of iPS-SMCs from healthy and HGPS donors.

(A) Schematic of the methodology used to explore biomechanical changes in a context of vascular aging. (B) mRNA expression levels of progerin confirmed its overexpression HGPS cells (bars represent mean ± SD of n=5). F-actin was stained for (C–D) healthy or (E–F) HGPS iPS-SMCs under 0% and 16% strain for 24 hours and the corresponding angle orientation distribution was determined (#, P<0.0001; scale bars represent 50 μm; red arrow indicates direction of strain). (G) HGPS iPS-SMCs showed increase levels of lamin A/C/progerin (antibody against epitope corresponding to aminoacids 231–340). (H) Quantification of lamin A/C/progerin levels.
The accumulation of nuclear lamins and progerin occurs naturally during aging, leading to stiffer and less compliant nuclei [29, 65, 66]. Besides promoting nuclear architecture changes [67], this accumulation can further result in alterations in transcription, changes in chromatin structure, and epigenetic changes [65]. However, the mechanism and effects of biomechanical strain in a context of lamin/progerin accumulation are still poorly understood. Using the same approach as Cao et al. [68], we distinctly evaluated the mRNA levels of LMNA and progerin with specific primers by qRT-PCR (Fig. S7). Interestingly, in HGPS iPS-SMCs, physiological strain slightly increased level of progerin while pathological strain decreased progerin and increased LMNA. Healthy iPS-SMCs showed an opposite trend with reduced levels of LMNA with physiological or pathological strain. We then used an antibody recognizing both lamin A/C and progerin (epitope corresponding to aminoacids 231–340) to immunostain (Fig. 5G) and quantify (Fig. 5H) the combined protein levels of lamin A/C and progerin. Similar to mRNA levels, we verified also lamin A/C/progerin accumulation in HGPS iPS-SMCs as compared to healthy iPS-SMCs (Fig. 5G–H). The specific accumulation of lamin A/C/progerin in the nucleus can lead to changes in the mechanical properties [67, 69], having implications for the cell response to strain.
2.5 Lovastatin and lonafarnib mitigated the inflammation response of HGPS iPS-SMCs
Several cellular pathways are shared between aging and hypertension, resulting in vascular alterations such as remodeling, stiffness, inflammation, and oxidative stress [70]. In particular, HGPS fibroblasts are mechanically sensitive, and under conditions of mechanical stimulation show decreased viability and increased apoptosis [66]. We used iPS-SMCs derived from healthy and HGPS donors, and compared the biomechanical response to strain. The markers CAV1, IL6, IL1B, and JUN drastically increased in HGPS iPS-SMCs under 16% strain (Fig. 6A–D). This observation demonstrated that pathological strain elicited an exacerbated inflammatory response in HGPS iPS-SMCs that did not occur in healthy iPS-SMCs. However, we showed increased IL6 mRNA expression in primary SMCs, but have not in healthy iPS-SMCs. CAV1 has been identified as a marker of hypertension across different animal models [49, 71] and is elevated in human serum of patients with vascular hypertension [51, 53]. The increased expression of the transcriptional factor JUN (Jun Proto-Oncogene) suggested further activation of cytokines, beyond the increases in IL6 and IL1B, and has been highlighted as a potential target for anti-inflammatory therapies [72].
Figure 6. Exacerbated response to biomechanical strain in HGPS iPS-SMCs is rescued by lovastatin and lonafarnib.

Relative mRNA expression levels of healthy and HGPS iPS-SMCs cultured under 0%, 9%, and 16% biomechanical strains for 24 hours. Injury marker (A) CAV1 and inflammation markers (B) IL6, (C) IL1B, and (D) JUN (* P<0.01 against 0% healthy iPS-SMCs, and # indicates P<0.01 against 0% HGPS iPS-SMCs; bars represent mean ± SD of n=5). DNA damage was evaluated in HGPS iPS-SMCs with H2A.X immunostaining (E) and quantified (F) (mean ± SD of n=3; **, P=0.0039). Senescence was measured in HGPS iPS-SMCs via β-galactosidase activity staining (G) and quantified (H) (mean ± SD of n=3; ns, not significant [P=0.0532]). Treatment of HGPS iPS-SMCs under 16% mechanical strain with lovastatin or lonafarnib prevented increased injury and inflammatory markers (I–L), while static control on a Petri dish failed to trigger a response [(§§, P<0.001; §, P=0.04; compared to no drug treatment; bars for lonafarnib and lovastatin represent mean ± SD of n=4; bars for static control represent mean ± SD of n=3].
Together, evidence suggested that progerin accumulation in HGPS might result in increased mechanosensitivity to pathological strain and led to an exacerbated inflammatory response. The combined action of transcription factor upregulation (JUN) and increased pro-inflammatory cytokines (IL6 and IL1B) suggested a cycle of propagation of inflammation that might occur in the vascular wall in HGPS patients and, at a slower pace, in physiological vascular aging. Furthermore, the observation of increased IL6 expression, together with similar observations in two mouse models of progeria suggested the potential role of the cytokine as a biomarker of disease. We then investigated whether exposure to pathological strain would induce further cellular damage in HGPS iPS-SMCs. DNA damage and cellular senescence have been established as hallmarks of aging [9, 73], and are upregulated in HGPS [33, 63, 74, 75]. In our experiments, HGPS iPS-SMCs showed increased DNA damage after 24 hours of biomechanical stimulation (Fig. 6E–F; P=0.0039). We additionally stimulated cells for 5 days under strain and observed a small increase in senescence (Fig. 6G–H; P=0.0532). This suggested that HGPS under dynamic pathological strain conditions altered their mRNA expression levels in favor of inflammation and vascular injury. In addition, cells presented endpoint markers of aging such as DNA damage and senescence. Importantly, chronic inflammation, which is observed in HGPS [29], can lead to increased senescence and DNA damage [76, 77] and ultimately accelerate aging [78].
Drug treatments for HGPS primarily aim to reduce levels of progerin. These include inhibitors of the lamin-processing pathway such as 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA; statins) [50, 79, 80], farnesyl transferase (FTI) [80–83], and mechanistic target of rapamycin (mTOR) [84]. In particular, lovastatin has been shown to improve the nuclear shape abnormalities in progeroid fibroblasts and to disrupt caveolae and decrease caveolin in SMCs [50, 79]. Additionally, statins have been shown to have anti-inflammatory effects [85–87]. We then hypothesized that lovastatin treatment would mitigate changes in mRNA expression levels that were associated with strain in HGPS iPS-SMCs. Indeed, we revealed that administration of 10μM of lovastatin was able to rescue HGPS iPS-SMCs exacerbated injury response on the transcriptional level by preventing increases in CAV1, IL6, IL1B, and JUN (Fig. 6I–L). Interestingly, 10μM treatment of lovastatin during 24 hours under 16% strain did not reduce the levels of progerin (Fig. S7). Lonafarnib is a classic FTI shown to reduce levels of progerin, restore nuclear abnormalities and improve vascular stiffness [81, 82, 88]. Treatment of HGPS iPS-SMCs for 24 hours under 16% strain with 2 μM of lonafarnib resulted in a decrease of progerin (Fig. S7), while it reduced the levels of CAV1, IL1B, and JUN (Fig. 6I–L). However, lonafarnib failed to decrease the levels of IL6. This might be due to the low exposure time to lonafarnib (24 hours) in comparison to other studies that have treated cells for 72 hours [81]. Overall, both lovastatin and lonafarnib were able to ameliorate the exacerbated inflammatory response to strain in HGPS iPS-SMCs, with lonafarnib being more efficient in downregulating the progerin mRNA levels. Importantly, culturing HGPS iPS-SMCs under conventional cell culture systems (Petri dish) failed to trigger inflammatory and vascular injury markers (Fig. 6I–L).
3. Discussion
Here, we have developed a microfluidic device that is easy to manufacture and enables the characterization of cellular responses across a range of physiological and pathological strain levels. Besides a standalone progeria-on-a-chip model, the developed device could potentially be integrated in a multi-organ-on-a-chip system to provide a biomimetic vascular platform and detect system-wide effects on the vasculature. We have improved important aspects of the device design in comparison to other previously reported designs [20, 22, 39, 40]. The fabrication methodology is simple, less expensive, and does not require highly trained operators. Our device can be conveniently scaled up to a large amount of parallel channels to serve as a high-throughput platform for vascular drug development. Additionally, the strain levels are fully characterized in situ and in silico, allowing the application of strain in a wide range. Within the device, the entire cell population can be analyzed and visualized under any conventional fluorescence microscope, and the cells can be removed from the device via trypsinization for further assays.
Using our device, we showed that primary SMCs acquire a more contractile phenotype in vitro following exposure to strain, thus better recapitulating the in vivo phenotype. Morphological changes and higher expression of TAGLN, ITGB1 and ITGA5 suggested a more contractile phenotype and pointed towards higher mechanotransduction sensitivity via increased integrin expression [5, 42]. These results are in agreement with previous reports [42, 44, 45] showing the alignment of SMCs under strain conditions. We next hypothesized whether a pathological strain level would induce cellular changes similar to hypertension. According to several studies, the hypertensive phenotype is characterized by an increase of mitochondrial ROS [55–58], caveolin-1 [47, 48], IL-6 and IL1-β [52, 53] expression. The exploration of strain-magnitude unveiled a gene expression profile similar to treatment with angiotensin II, a compound known to be implicated in the development of hypertension. We indirectly assessed the levels of ROS through mRNA expression of cytosolic and mitochondrial superoxide dismutase (SOD1 and SOD2 respectively). We observed a distinct regulation of SOD1 and SOD2 depending on the strain amount. Whereas normal strain levels elicited increased expression of cytosolic superoxide dismutase, pathological strain levels elicited increased expression of mitochondrial one. By inhibiting cytosolic ROS increase with a NADPH oxidase inhibitor, we were able to specifically decrease SOD1 under normal strain without affecting the levels of SOD2. This observation suggested that different strains might have distinct effects on ROS production, but further studies are required to elucidate the influence in vascular disease. Pathological levels of strain induced higher expression of vascular injury marker CAV1, which has been reported in patients with pulmonary hypertension [50, 51], identified in microarray screenings and proposed as a potential target marker of hypertension [49]. Also, we observed an increase in the pro-inflammatory cytokine mRNA levels of IL6 and IL1B under pathological strain levels. These have been implicated additionally in hypertension and aging [52–54]. Evidence suggests that pathological strain levels can indeed recapitulate some of the hallmarks observed in vascular disease and elicited by angiotensin II treatment. These alterations point to the existence of a threshold pathological strain that elicits an injury response. However, still more research is needed to understand the specific underlying pathways and potential identification of novel therapeutic targets, such as the Ras/MAPK/NF-κB pathways [89].
There is a strong association between aging and cardiovascular diseases, and aging alone is the single most important risk factor for the development of cardiovascular diseases [29, 61]. Additionally, inflammation is a potential mediator in the pathogenesis of several vascular diseases, including hypertension and atherosclerosis [8, 10, 90]. Mouse models that phenotypically recapitulate HGPS show increased activation of NF-κB with a concomitant increase in IL-6 at the transcriptional and protein levels [91]. Conversely, a mouse model of low-level chronic inflammation showed accelerated aging with increased expression of IL-6, IL1-β, and a decrease in SOD2 [78]. Together, these findings highlight a potential role for inflammation and oxidative stress in the vascular wall during aging and vascular disease [55, 56]. Our progeria-on-a-chip system is a crucial step towards the understanding of biomechanics of aging. HGPS targets primarily vascular cells, which are under constant mechanical stimulation. Due to lack of data specific for progeria blood vessel strain levels, we hypothesized an increase in pathological strain associated with premature aging. Here we explored a platform that combines both biomechanical stimulation and iPS-SMCs derived from HGPS patients. The HGPS iPS-SMCs expressed progerin in 15% of the cells, and were not further enriched. However, the pooled qPCR results indicated a ~30-fold increase in progerin mRNA levels in iPS-SMCs from HGPS donors. Additionally, artificially overexpressing progerin could result in a level higher than disease, resulting in higher cell death or an over response to strain, which could reduce the relevance of the work. We showed that iPS-SMCs from both healthy and HGPS go under cytoskeletal reorientation under mechanical strain. Furthermore, we showed that HGPS-derived vascular cells demonstrated an exacerbated effect following biomechanical strain, which was unobserved using conventional planar cell culture methods (Petri dish). With this platform, we showed a unique exacerbated increase in inflammatory mRNA levels of markers IL6, IL1B, and JUN, as well as the vascular injury marker CAV1. Importantly, there is a strong link between inflammation and vascular diseases and aging, which is highlighted in the current work. We showed additionally that a statin (lovastatin) was able to prevent biomechanically induced inflammatory response, likely through anti-inflammatory effects and not directly by reducing progerin levels. Lonafarnib treatment was able to reduce levels of progerin and rescue the inflammatory and injury gene expression profiles of IL1B, JUN, and CAV1. However, the results suggested differences between healthy iPS-SMCs and primary SMCs, and a direct comparison might be hindered by several factors. One possible explanation for such is the different cell culture media used. The cell culture medium that maintained the iPS-SMC phenotype was smooth muscle growth medium (SmGM), being also used to expand primary SMCs. However, primary SMCs tend to dedifferentiate in culture with SmGM medium, thus requiring a starvation medium [92]. The starvation medium used for primary SMCs is less rich, while iPS-SMCs experiments were performed in a richer media (SmGM). In addition, the primary SMCs used here were from aortic origin, and might have phenotypic differences compared to a generic SMC phenotype beyond the traditional SMC markers SM-MHC and calponin. Together, these factor could explain some of the differences observed, and more research is needed to shed light onto tissue-specific SMCs differences in mechanosensitivity.
The progeria-on-a-chip system allowed the unveiling of new strain-derived in vitro mechanism that leads to increased IL6 mRNA levels. Although the increase in IL6 mRNA has been reported for progeria models in vivo, we showed here similar responses in vitro. We highlight the potential role of the pro-inflammatory cytokine IL-6 as a marker of vascular disease and potentially useful in assessing progression of HGPS. Further studies are required for the validation of IL-6 as a potential marker of disease. We hypothesize that further utilization of this platform can lead to an improved understanding of biomechanics in vascular biology. In particular, the exploration of combined effects of strain frequency, periodicity, and shear stress is expected to yield novel biological insights. Gaining deeper understanding of the molecular pathways regulating inflammation during vascular aging might pave the way for new strategies to minimizing cardiovascular risk with age. Finally, we expect the newly developed tool to serve as a standardized platform technology to study the effects of biomechanics in vascular biology, disease, and aging, while facilitating the discovery of new drugs.
4. Experimental Section
Device fabrication
The microfluidic device was made with PDMS (Sylgard, Dow Corning) at a ratio of 10:1 (w/w) monomer to curing agent. Hard molds of the device were custom-made by laser cutting (VersaLaser) 800-μm polyoxymethylene (DuPont) sheets and glued to the bottom of petri dishes. PDMS was cast onto the molds and cured for 24 hours at 80 °C. PDMS membranes were produced by spin-coating PDMS 20:1 (w/w) on silanized silicon wafers at 950 rpm for 20 seconds, and cured at 80 °C for 24 hours. The bottom layer PDMS slab was bonded to the PDMS membrane with oxygen plasma (Plasma Etch PE-25), and the resulting set was peeled from the wafer. The top PDMS slab was then bonded to the set of bottom-membrane with oxygen plasma and aligned manually under a microscope. The surface of the fluidic channel was treated with fibronectin (Sigma-Aldrich) at a concentration of 50 μg mL−1 to allow for cell attachment.
Computational simulation
Computational finite-element models were developed using COMSOL® to simulate the experimental results and to represent the mechanical deformation and allow for stress analysis of the PDMS membrane. The device structure was modeled as two PDMS flexible bodies sandwiching a flexible PDMS membrane with a thickness of 100μm. The Young’s Moduli used were 2.5 MPa for the flexible bodies and 500 kPa for the membrane respectively, as determined from mechanical characterization by Instron® tensile mechanical measurements. The Poisson’s ratio used for both PDMS compositions was 0.49 [93]. The interfaces between the different PDMS layers were modeled as a bonded contact. The base of the model was constrained as fixed and a linearly increasing pressure increase, to a value of 0, 10, 20, 30, or 50 kPa, was applied to the top surface of the PDMS membrane. The simulation took into account the presence of SMCs, which were uniformly distributed along the top surface of the PDMS membrane. The interfaces between the cells and the PDMS membrane were modeled as bonded contact. The cells were shaped according to a morphological evaluation of in vitro studies through confocal imaging [94]. For the cells, literature values for the Elastic modulus (100 kPa) and the Poisson’s ratio (0.49) were used [95]. The strains generated on the top surface of the membrane, as well as on the cells attached to the membrane surface, were analyzed.
Mechanical stimulation
Cells were stimulated for 24 hours with different percentages of cyclic strain. To ensure media exchange, the fluidic channel was perfused with cell culture media at a flow rate of 100 μL hour−1. To stimulate the cells, the vacuum inlet of the microfluidic pump was connected to a computer-controlled solenoid system and stimulated at a frequency of 0.5Hz. The vacuum pressure was adjusted with a pressure regulator and used in the range of 0 kPa (=0% strain), 10 kPa (=9% strain), and 20 kPa (=16% strain).
Cell culture
Aortic SMCs (Lonza) were grown in Smooth Muscle Growth Media-2 BulletKit (SmGM-2; Lonza) at 37 °C and 5% CO2 in a humidified incubator. Cells were trypsinized from cell culture flasks and seeded in the microfluidic channels at a density of 1.6 million cells mL−1. Prior to the start of strain experiments, cells were maintained in media containing DMEM/F12 1:1 mixture (Thermo Fisher Scientific) supplemented with Insulin-Transferrin-Selenium (ITS; Thermo Fisher Scientific) [92]. iPS generated from healthy and HGPS donors were kindly provided by Xavier Nissan and previously characterized [35]. Differentiation was performed according to a previously described protocol [64]. At the end, 95% of both differentiated cells express a-SMA, SMMHC and calponin proteins. Moreover, HGPS-iPSC SMCs express progerin protein (15% of the cells). Healthy and HGPS iPS-derived SMCs were grown in SmGM-2 media at 37 °C and 5% CO2 in a humidified incubator. Due to the smaller size, iPS-SMCs were seeded in the microfluidic device at a density of 3.2 million cells mL−1, which yielded a cell confluence that was identical to those used for the SMCs. Treatment with 10μM lovastatin (Sigma-Aldrich) was administered 2 hours before the start of mechanical stimulation and continued through the 24 hours of cyclic strain. Treatment with 2μM lonafarnib (Sigma-Aldrich) was administered together with mechanical stimulation and continued through the 24 hours of cyclic strain.
Gene expression
Cells were trypsinized from the microfluidic devices 24 hours after mechanical stimulation. RNA was extracted using an RNeasy Micro kit (Qiagen). cDNA was synthesized from a total of 500 ng of RNA using the QuantiTec Reverse Transcription kit (Qiagen) following the manufacturer’s protocol. qRT-PCR was performed in an iQ5 thermocylcer using SYBR green probe (Biorad). Gene expressions were normalized using housekeeping GAPDH. All used primer sequences are listed in Table 1 (Supplementary Information). Results follow the 2-ΔΔCt method and are reported as fold change as compared with the no strain (0%) control, unless otherwise indicated.
Immunocytochemistry
Cells were immediately fixed in 4% paraformaldehyde (Sigma-Aldrich) for 15 minutes at room temperature. Cells were then permeabilized with 1% (v/v) Triton X-100 (Sigma-Aldrich) for 10 minutes, followed by blocking with 1% bovine albumin serum (Sigma Aldrich) for 45 minutes at room temperature. Primary antibody against lamin A/C (Santa Cruz, sc-20681; 1:50; reacts against both lamin A/C and progerin) was incubated at room temperature for 1 hour. Primary antibody against the phosphor-histone H2A.X (Cell Signaling Technology, 9718, 1:400) was incubated overnight at 4°C. The channels were washed with phosphate buffer saline (PBS; Thermo Fisher Scientific) five times and solutions of fluorescently labeled secondary antibodies were introduced (Alexa 546 anti-mouse, Thermo Fisher Scientific; Alexa 594 anti-rabbit, Thermo Fisher Scientific). Secondary antibodies were incubated for 1 hour and nuclei counterstained with DAPI (Thermo Fisher Scientific) for 5 minutes. F-actin staining was achieved by incubating cells with Alexa 488 phalloidin solution according to manufacturer’s protocol. Images were acquired with a Zeiss Observer D1 microscope.
Cell senescence
Senescent cells (n = 3 experiments) were detected through histochemical staining of β-galactosidase (Senescence Cells Histochemical Staining Kit, Sigma-Aldrich) according to the manufacturer’s protocol. Briefly, after 5 days of cyclic mechanical stimulation, cells were washed with PBS and fixed with 1X fixation buffer for 7 min at room temperature. The staining 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside (X-Gal) solution was prepared accordingly and the cells were incubated for 24 hours at 37 °C. Stained cells were washed in PBS and imaged on an inverted brightfield microscope.
Image quantification
Cell orientation, length and width were determined from n = 3 experiments of f-actin stained cells. Three microscopic images for each n were analyzed in ImageJ to determine angle of orientation, length, and width. Nuclei images obtained after computer simulations were used to determine the vector displacement maps upon strain, using the Particle Image Velocimetry (PIV) plugin of ImageJ.
Statistical analysis
Results are presented as mean ± standard deviation (SD) unless otherwise indicated. Group data analysis was performed with one-way ANOVA and a Tukey’s post hoc test against control group. Comparison between two groups was performed using a student t test.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge funding by the Defense Threat Reduction Agency (DTRA) under Space and Naval Warfare Systems Center Pacific (SSC PACIFIC) Contract No. N66001-13-C-2027. The authors also acknowledge funding from the Office of Naval Research Young National Investigator Award, the National Institutes of Health (EB012597, AR057837, DE021468, HL099073, R56AI105024), and the Presidential Early Career Award for Scientists and Engineers (PECASE). The publication of this material does not constitute approval by the government of the findings or conclusions herein. Ribas acknowledges the support from the Portuguese Foundation for Science and Technology (SFRH/BD/51679/2011). Dr. Leijten acknowledges financial support from Innovative Research Incentives Scheme Veni #14328 of the Netherlands Organization for Scientific Research (NWO). Dr. Rouwkema acknowledges financial support by the People Programme (Marie Curie Actions) under REA Grant Agreement no. 622294 (PreVascIn).
Footnotes
Competing Financial Interest
The authors declare no conflict of interest.
Supporting Information is available from the Wiley Online Library.
Contributor Information
João Ribas, Biomaterials Innovation Research Center, Division of Engineering in Medicine, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Cambridge, MA 02139, USA, Harvard-MIT Division of Health Sciences and Technology, Massachusetts Institute of Technology, Cambridge, MA 02139, USA. Doctoral Program in Experimental Biology and Biomedicine, Center for Neuroscience and Cell Biology, Institute for Interdisciplinary Research, University of Coimbra, 3030-789 Coimbra, Portugal.
Dr. Yu Shrike Zhang, Biomaterials Innovation Research Center, Division of Engineering in Medicine, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Cambridge, MA 02139, USA, Harvard-MIT Division of Health Sciences and Technology, Massachusetts Institute of Technology, Cambridge, MA 02139, USA. Wyss Institute for Biologically Inspired Engineering, Harvard University, Boston, MA 02115, USA
Dr. Patrícia R. Pitrez, CNC-Centre for Neuroscience and Cell Biology, University of Coimbra, Coimbra, Portugal, Institute for Interdisciplinary Research, University of Coimbra, 3030-789 Coimbra, Portugal
Dr. Jeroen Leijten, Biomaterials Innovation Research Center, Division of Engineering in Medicine, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Cambridge, MA 02139, USA, Harvard-MIT Division of Health Sciences and Technology, Massachusetts Institute of Technology, Cambridge, MA 02139, USA. Department of Developmental BioEngineering, MIRA Institute for Biomedical Technology and Technical Medicine, University of Twente, Enschede, The Netherlands
Mario Miscuglio, Biomaterials Innovation Research Center, Division of Engineering in Medicine, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Cambridge, MA 02139, USA, Harvard-MIT Division of Health Sciences and Technology, Massachusetts Institute of Technology, Cambridge, MA 02139, USA.
Dr. Jeroen Rouwkema, Department of Biomechanical Engineering, MIRA Institute for Biomedical Technology and Technical Medicine, University of Twente, Enschede, The Netherlands
Dr. Mehmet Remzi Dokmeci, Biomaterials Innovation Research Center, Division of Engineering in Medicine, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Cambridge, MA 02139, USA, Harvard-MIT Division of Health Sciences and Technology, Massachusetts Institute of Technology, Cambridge, MA 02139, USA. Wyss Institute for Biologically Inspired Engineering, Harvard University, Boston, MA 02115, USA
Dr. Xavier Nissan, INSERM U861, I-STEM, AFM, Institute for Stem Cell Therapy and Exploration of Monogenic Diseases, Evry Cedex 91030, France
Dr. Lino Ferreira, CNC-Centre for Neuroscience and Cell Biology, University of Coimbra, Coimbra, Portugal, Institute for Interdisciplinary Research, University of Coimbra, 3030-789 Coimbra, Portugal.
Prof. Ali Khademhosseini, Biomaterials Innovation Research Center, Division of Engineering in Medicine, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Cambridge, MA 02139, USA, Harvard-MIT Division of Health Sciences and Technology, Massachusetts Institute of Technology, Cambridge, MA 02139, USA. Wyss Institute for Biologically Inspired Engineering, Harvard University, Boston, MA 02115, USA. Department of Physics, King Abdulaziz University, Jeddah 21569, Saudi Arabia, Department of Bioindustrial Technologies, College of Animal Bioscience and Technology, Konkuk University, Hwayang-dong, Gwangjin-gu, Seoul 143-701, Republic of Korea.
References
- 1.Davis MJ, Hill MA. Physiological reviews. 1999;79(2):387–423. doi: 10.1152/physrev.1999.79.2.387. [DOI] [PubMed] [Google Scholar]
- 2.Stefanadis C, Stratos C, Vlachopoulos C, Marakas S, Boudoulas H, Kallikazaros I, Tsiamis E, Toutouzas K, Sioros L, Toutouzas P. Circulation. 1995;92(8):2210–9. doi: 10.1161/01.cir.92.8.2210. [DOI] [PubMed] [Google Scholar]
- 3.Williams B. Journal of hypertension. 1998;16(12 Pt 2):1921–9. doi: 10.1097/00004872-199816121-00011. [DOI] [PubMed] [Google Scholar]
- 4.Anwar MA, Shalhoub J, Lim CS, Gohel MS, Davies AH. Journal of vascular research. 2012;49(6):463–78. doi: 10.1159/000339151. [DOI] [PubMed] [Google Scholar]
- 5.Heerkens EH, Izzard AS, Heagerty AM. Hypertension. 2007;49(1):1–4. doi: 10.1161/01.HYP.0000252753.63224.3b. [DOI] [PubMed] [Google Scholar]
- 6.Rensen SS, Doevendans PA, van Eys GJ. Netherlands heart journal : monthly journal of the Netherlands Society of Cardiology and the Netherlands Heart Foundation. 2007;15(3):100–8. doi: 10.1007/BF03085963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lu D, Kassab GS. Journal of the Royal Society, Interface / the Royal Society. 2011;8(63):1379–85. doi: 10.1098/rsif.2011.0177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Savoia C, Schiffrin EL. Current opinion in nephrology and hypertension. 2006;15(2):152–8. doi: 10.1097/01.mnh.0000203189.57513.76. [DOI] [PubMed] [Google Scholar]
- 9.Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. Cell. 2013;153(6):1194–217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maggio M, Guralnik JM, Longo DL, Ferrucci L. The journals of gerontology Series A, Biological sciences and medical sciences. 2006;61(6):575–84. doi: 10.1093/gerona/61.6.575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Polacheck WJ, Li R, Uzel SG, Kamm RD. Lab on a chip. 2013;13(12):2252–67. doi: 10.1039/c3lc41393d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bhatia SN, Ingber DE. Nature biotechnology. 2014;32(8):760–72. doi: 10.1038/nbt.2989. [DOI] [PubMed] [Google Scholar]
- 13.Ribas J, Sadeghi H, Manbachi A, Leijten J, Brinegar K, Zhang YS, Ferreira L, Khademhosseini A. Applied In Vitro Toxicology. 2016;2(2):82–96. doi: 10.1089/aivt.2016.0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huh D, Matthews BD, Mammoto A, Montoya-Zavala M, Hsin HY, Ingber DE. Science. 2010;328(5986):1662–8. doi: 10.1126/science.1188302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huh D, Leslie DC, Matthews BD, Fraser JP, Jurek S, Hamilton GA, Thorneloe KS, McAlexander MA, Ingber DE. Science translational medicine. 2012;4(159):159ra147. doi: 10.1126/scitranslmed.3004249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Douville NJ, Zamankhan P, Tung YC, Li R, Vaughan BL, Tai CF, White J, Christensen PJ, Grotberg JB, Takayama S. Lab on a chip. 2011;11(4):609–19. doi: 10.1039/c0lc00251h. [DOI] [PubMed] [Google Scholar]
- 17.Kim HJ, Huh D, Hamilton G, Ingber DE. Lab on a chip. 2012;12(12):2165–74. doi: 10.1039/c2lc40074j. [DOI] [PubMed] [Google Scholar]
- 18.Kim HJ, Ingber DE. Integrative biology : quantitative biosciences from nano to macro. 2013;5(9):1130–40. doi: 10.1039/c3ib40126j. [DOI] [PubMed] [Google Scholar]
- 19.Kim HJ, Li H, Collins JJ, Ingber DE. Proceedings of the National Academy of Sciences of the United States of America. 2016;113(1):E7–E15. doi: 10.1073/pnas.1522193112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee J, Wong M, Smith Q, Baker AB. Lab on a chip. 2013;13(23):4573–82. doi: 10.1039/c3lc50894c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shao Y, Tan X, Novitski R, Muqaddam M, List P, Williamson L, Fu J, Liu AP. The Review of scientific instruments. 2013;84(11):114304. doi: 10.1063/1.4832977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sinha R, Le Gac S, Verdonschot N, van den Berg A, Koopman B, Rouwkema J. Lab on a chip. 2015;15(2):429–39. doi: 10.1039/c4lc01259c. [DOI] [PubMed] [Google Scholar]
- 23.Meza D, Abejar L, Rubenstein DA, Yin W. Journal of biomechanical engineering. 2016;138(3) doi: 10.1115/1.4032550. [DOI] [PubMed] [Google Scholar]
- 24.Zhang W, Zhang YS, Bakht SM, Aleman J, Shin SR, Yue K, Sica M, Ribas J, Duchamp M, Ju J, Sadeghian RB, Kim D, Dokmeci MR, Atala A, Khademhosseini A. Lab on a chip. 2016;16(9):1579–86. doi: 10.1039/c6lc00001k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang J, Lian Q, Zhu G, Zhou F, Sui L, Tan C, Mutalif RA, Navasankari R, Zhang Y, Tse HF, Stewart CL, Colman A. Cell stem cell. 2011;8(1):31–45. doi: 10.1016/j.stem.2010.12.002. [DOI] [PubMed] [Google Scholar]
- 26.De Sandre-Giovannoli A, Bernard R, Cau P, Navarro C, Amiel J, Boccaccio I, Lyonnet S, Stewart CL, Munnich A, Le Merrer M, Levy N. Science. 2003;300(5628):2055. doi: 10.1126/science.1084125. [DOI] [PubMed] [Google Scholar]
- 27.Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, Scott L, Erdos MR, Robbins CM, Moses TY, Berglund P, Dutra A, Pak E, Durkin S, Csoka AB, Boehnke M, Glover TW, Collins FS. Nature. 2003;423(6937):293–8. doi: 10.1038/nature01629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blagosklonny MV. Aging. 2011;3(7):685–91. doi: 10.18632/aging.100352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Olive M, Harten I, Mitchell R, Beers JK, Djabali K, Cao K, Erdos MR, Blair C, Funke B, Smoot L, Gerhard-Herman M, Machan JT, Kutys R, Virmani R, Collins FS, Wight TN, Nabel EG, Gordon LB. Arteriosclerosis, thrombosis, and vascular biology. 2010;30(11):2301–9. doi: 10.1161/ATVBAHA.110.209460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burtner CR, Kennedy BK. Nature reviews. Molecular cell biology. 2010;11(8):567–78. doi: 10.1038/nrm2944. [DOI] [PubMed] [Google Scholar]
- 31.Merideth MA, Gordon LB, Clauss S, Sachdev V, Smith AC, Perry MB, Brewer CC, Zalewski C, Kim HJ, Solomon B, Brooks BP, Gerber LH, Turner ML, Domingo DL, Hart TC, Graf J, Reynolds JC, Gropman A, Yanovski JA, Gerhard-Herman M, Collins FS, Nabel EG, Cannon RO, 3rd, Gahl WA, Introne WJ. The New England journal of medicine. 2008;358(6):592–604. doi: 10.1056/NEJMoa0706898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McClintock D, Gordon LB, Djabali K. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(7):2154–9. doi: 10.1073/pnas.0511133103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu GH, Barkho BZ, Ruiz S, Diep D, Qu J, Yang SL, Panopoulos AD, Suzuki K, Kurian L, Walsh C, Thompson J, Boue S, Fung HL, Sancho-Martinez I, Zhang K, Yates J, 3rd, Izpisua Belmonte JC. Nature. 2011;472(7342):221–5. doi: 10.1038/nature09879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dreesen O, Stewart CL. Aging. 2011;3(9):889–95. doi: 10.18632/aging.100383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nissan X, Blondel S, Navarro C, Maury Y, Denis C, Girard M, Martinat C, De Sandre-Giovannoli A, Levy N, Peschanski M. Cell reports. 2012;2(1):1–9. doi: 10.1016/j.celrep.2012.05.015. [DOI] [PubMed] [Google Scholar]
- 36.Brassard JA, Fekete N, Garnier A, Hoesli CA. Biogerontology. 2016;17(1):129–45. doi: 10.1007/s10522-015-9602-z. [DOI] [PubMed] [Google Scholar]
- 37.Blondel S, Egesipe AL, Picardi P, Jaskowiak AL, Notarnicola M, Ragot J, Tournois J, Le Corf A, Brinon B, Poydenot P, Georges P, Navarro C, Pitrez PR, Ferreira L, Bollot G, Bauvais C, Laustriat D, Mejat A, De Sandre-Giovannoli A, Levy N, Bifulco M, Peschanski M, Nissan X. Cell death & disease. 2016;7:e2105. doi: 10.1038/cddis.2015.374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ugolini GS, Rasponi M, Pavesi A, Santoro R, Kamm R, Fiore GB, Pesce M, Soncini M. Biotechnology and bioengineering. 2016;113(4):859–69. doi: 10.1002/bit.25847. [DOI] [PubMed] [Google Scholar]
- 39.Zheng W, Huang R, Jiang B, Zhao Y, Zhang W, Jiang X. Small. 2016;12(15):2022–34. doi: 10.1002/smll.201503241. [DOI] [PubMed] [Google Scholar]
- 40.Aragona M, Panciera T, Manfrin A, Giulitti S, Michielin F, Elvassore N, Dupont S, Piccolo S. Cell. 2013;154(5):1047–59. doi: 10.1016/j.cell.2013.07.042. [DOI] [PubMed] [Google Scholar]
- 41.Zhou J, Niklason LE. Integrative biology : quantitative biosciences from nano to macro. 2012;4(12):1487–97. doi: 10.1039/c2ib00171c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wilson E, Sudhir K, Ives HE. The Journal of clinical investigation. 1995;96(5):2364–72. doi: 10.1172/JCI118293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Greiner AM, Chen H, Spatz JP, Kemkemer R. PloS one. 2013;8(10):e77328. doi: 10.1371/journal.pone.0077328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu B, Qu MJ, Qin KR, Li H, Li ZK, Shen BR, Jiang ZL. Biophysical journal. 2008;94(4):1497–507. doi: 10.1529/biophysj.106.098574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Albinsson S, Nordstrom I, Hellstrand P. The Journal of biological chemistry. 2004;279(33):34849–55. doi: 10.1074/jbc.M403370200. [DOI] [PubMed] [Google Scholar]
- 46.Bader M, Peters J, Baltatu O, Muller DN, Luft FC, Ganten D. Journal of molecular medicine. 2001;79(2–3):76–102. doi: 10.1007/s001090100210. [DOI] [PubMed] [Google Scholar]
- 47.Hardin CD, Vallejo J. Cardiovascular research. 2006;69(4):808–15. doi: 10.1016/j.cardiores.2005.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yu J, Bergaya S, Murata T, Alp IF, Bauer MP, Lin MI, Drab M, Kurzchalia TV, Stan RV, Sessa WC. The Journal of clinical investigation. 2006;116(5):1284–91. doi: 10.1172/JCI27100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Grayson TH, Ohms SJ, Brackenbury TD, Meaney KR, Peng K, Pittelkow YE, Wilson SR, Sandow SL, Hill CE. BMC genomics. 2007;8:404. doi: 10.1186/1471-2164-8-404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Patel HH, Zhang S, Murray F, Suda RY, Head BP, Yokoyama U, Swaney JS, Niesman IR, Schermuly RT, Pullamsetti SS, Thistlethwaite PA, Miyanohara A, Farquhar MG, Yuan JX, Insel PA. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2007;21(11):2970–9. doi: 10.1096/fj.07-8424com. [DOI] [PubMed] [Google Scholar]
- 51.Wang KY, Lee MF, Ho HC, Liang KW, Liu CC, Tsai WJ, Lin WW. BioMed research international. 2015;2015:173970. doi: 10.1155/2015/173970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bautista LE, Vera LM, Arenas IA, Gamarra G. Journal of human hypertension. 2005;19(2):149–54. doi: 10.1038/sj.jhh.1001785. [DOI] [PubMed] [Google Scholar]
- 53.Humbert M, Monti G, Brenot F, Sitbon O, Portier A, Grangeot-Keros L, Duroux P, Galanaud P, Simonneau G, Emilie D. American journal of respiratory and critical care medicine. 1995;151(5):1628–31. doi: 10.1164/ajrccm.151.5.7735624. [DOI] [PubMed] [Google Scholar]
- 54.Golembeski SM, West J, Tada Y, Fagan KA. Chest. 2005;128(6 Suppl):572S-573S. doi: 10.1378/chest.128.6_suppl.572S-a. [DOI] [PubMed] [Google Scholar]
- 55.Datla SR, Griendling KK. Hypertension. 2010;56(3):325–30. doi: 10.1161/HYPERTENSIONAHA.109.142422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Paravicini TM, Touyz RM. Diabetes care. 2008;31(Suppl 2):S170–80. doi: 10.2337/dc08-s247. [DOI] [PubMed] [Google Scholar]
- 57.Kojima M, Shiojima I, Yamazaki T, Komuro I, Zou Z, Wang Y, Mizuno T, Ueki K, Tobe K, Kadowaki T, et al. Circulation. 1994;89(5):2204–11. doi: 10.1161/01.cir.89.5.2204. [DOI] [PubMed] [Google Scholar]
- 58.Kranzhofer R, Schmidt J, Pfeiffer CA, Hagl S, Libby P, Kubler W. Arteriosclerosis, thrombosis, and vascular biology. 1999;19(7):1623–9. doi: 10.1161/01.atv.19.7.1623. [DOI] [PubMed] [Google Scholar]
- 59.ten Freyhaus H, Huntgeburth M, Wingler K, Schnitker J, Baumer AT, Vantler M, Bekhite MM, Wartenberg M, Sauer H, Rosenkranz S. Cardiovascular research. 2006;71(2):331–41. doi: 10.1016/j.cardiores.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 60.Kappert K, Schmidt G, Doerr G, Wollert-Wulf B, Fleck E, Graf K. Hypertension. 2000;35(1 Pt 2):255–61. doi: 10.1161/01.hyp.35.1.255. [DOI] [PubMed] [Google Scholar]
- 61.North BJ, Sinclair DA. Circulation research. 2012;110(8):1097–108. doi: 10.1161/CIRCRESAHA.111.246876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sterneckert JL, Reinhardt P, Scholer HR. Nature reviews. Genetics. 2014;15(9):625–39. doi: 10.1038/nrg3764. [DOI] [PubMed] [Google Scholar]
- 63.Cau P, Navarro C, Harhouri K, Roll P, Sigaudy S, Kaspi E, Perrin S, De Sandre-Giovannoli A, Levy N. Seminars in cell & developmental biology. 2014;29:125–47. doi: 10.1016/j.semcdb.2014.03.021. [DOI] [PubMed] [Google Scholar]
- 64.Vazao H, das Neves RP, Graos M, Ferreira L. PloS one. 2011;6(3):e17771. doi: 10.1371/journal.pone.0017771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dahl KN, Ribeiro AJ, Lammerding J. Circulation research. 2008;102(11):1307–18. doi: 10.1161/CIRCRESAHA.108.173989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Verstraeten VL, Ji JY, Cummings KS, Lee RT, Lammerding J. Aging cell. 2008;7(3):383–93. doi: 10.1111/j.1474-9726.2008.00382.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Goldman RD, Shumaker DK, Erdos MR, Eriksson M, Goldman AE, Gordon LB, Gruenbaum Y, Khuon S, Mendez M, Varga R, Collins FS. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(24):8963–8. doi: 10.1073/pnas.0402943101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cao K, Blair CD, Faddah DA, Kieckhaefer JE, Olive M, Erdos MR, Nabel EG, Collins FS. The Journal of clinical investigation. 2011;121(7):2833–44. doi: 10.1172/JCI43578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dahl KN, Scaffidi P, Islam MF, Yodh AG, Wilson KL, Misteli T. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(27):10271–6. doi: 10.1073/pnas.0601058103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Harvey A, Montezano AC, Touyz RM. Journal of molecular and cellular cardiology. 2015;83:112–21. doi: 10.1016/j.yjmcc.2015.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Austin ED, Ma L, LeDuc C, Berman Rosenzweig E, Borczuk A, Phillips JA, 3rd, Palomero T, Sumazin P, Kim HR, Talati MH, West J, Loyd JE, Chung WK. Circulation. Cardiovascular genetics. 2012;5(3):336–43. doi: 10.1161/CIRCGENETICS.111.961888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fahmy RG, Waldman A, Zhang G, Mitchell A, Tedla N, Cai H, Geczy CR, Chesterman CN, Perry M, Khachigian LM. Nature biotechnology. 2006;24(7):856–63. doi: 10.1038/nbt1225. [DOI] [PubMed] [Google Scholar]
- 73.Collado M, Blasco MA, Serrano M. Cell. 2007;130(2):223–33. doi: 10.1016/j.cell.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 74.Trigueros-Motos L, Gonzalez JM, Rivera J, Andres V. Frontiers in bioscience. 2011;3:1285–97. doi: 10.2741/226. [DOI] [PubMed] [Google Scholar]
- 75.Endisha H, Merrill-Schools J, Zhao M, Bristol M, Wang X, Kubben N, Elmore LW. Pathobiology : journal of immunopathology, molecular and cellular biology. 2015;82(1):9–20. doi: 10.1159/000368856. [DOI] [PubMed] [Google Scholar]
- 76.Acosta JC, O’Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, Fumagalli M, Da Costa M, Brown C, Popov N, Takatsu Y, Melamed J, d’Adda di Fagagna F, Bernard D, Hernando E, Gil J. Cell. 2008;133(6):1006–18. doi: 10.1016/j.cell.2008.03.038. [DOI] [PubMed] [Google Scholar]
- 77.Kuilman T, Michaloglou C, Vredeveld LC, Douma S, van Doorn R, Desmet CJ, Aarden LA, Mooi WJ, Peeper DS. Cell. 2008;133(6):1019–31. doi: 10.1016/j.cell.2008.03.039. [DOI] [PubMed] [Google Scholar]
- 78.Jurk D, Wilson C, Passos JF, Oakley F, Correia-Melo C, Greaves L, Saretzki G, Fox C, Lawless C, Anderson R, Hewitt G, Pender SL, Fullard N, Nelson G, Mann J, van de Sluis B, Mann DA, von Zglinicki T. Nature communications. 2014;2:4172. doi: 10.1038/ncomms5172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bifulco M, D’Alessandro A, Paladino S, Malfitano AM, Notarnicola M, Caruso MG, Laezza C. The FEBS journal. 2013;280(23):6223–32. doi: 10.1111/febs.12544. [DOI] [PubMed] [Google Scholar]
- 80.Varela I, Pereira S, Ugalde AP, Navarro CL, Suarez MF, Cau P, Cadinanos J, Osorio FG, Foray N, Cobo J, de Carlos F, Levy N, Freije JM, Lopez-Otin C. Nature medicine. 2008;14(7):767–72. doi: 10.1038/nm1786. [DOI] [PubMed] [Google Scholar]
- 81.Capell BC, Erdos MR, Madigan JP, Fiordalisi JJ, Varga R, Conneely KN, Gordon LB, Der CJ, Cox AD, Collins FS. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(36):12879–84. doi: 10.1073/pnas.0506001102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yang SH, Meta M, Qiao X, Frost D, Bauch J, Coffinier C, Majumdar S, Bergo MO, Young SG, Fong LG. The Journal of clinical investigation. 2006;116(8):2115–21. doi: 10.1172/JCI28968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Marcuzzi A, De Leo L, Decorti G, Crovella S, Tommasini A, Pontillo A. Pediatric research. 2011;70(1):78–82. doi: 10.1038/pr.2011.30310.1203/PDR.0b013e31821b581c. [DOI] [PubMed] [Google Scholar]
- 84.Cao K, Graziotto JJ, Blair CD, Mazzulli JR, Erdos MR, Krainc D, Collins FS. Science translational medicine. 2011;3(89):89ra58. doi: 10.1126/scitranslmed.3002346. [DOI] [PubMed] [Google Scholar]
- 85.Jain MK, Ridker PM. Nature reviews. Drug discovery. 2005;4(12):977–87. doi: 10.1038/nrd1901. [DOI] [PubMed] [Google Scholar]
- 86.Schonbeck U, Libby P. Circulation. 2004;109(21 Suppl 1):II18–26. doi: 10.1161/01.CIR.0000129505.34151.23. [DOI] [PubMed] [Google Scholar]
- 87.Weitz-Schmidt G. Trends in Pharmacological Sciences. 2002;23(10):482–487. doi: 10.1016/s0165-6147(02)02077-1. http://dx.doi.org/10.1016/S0165-6147(02)02077-1. [DOI] [PubMed] [Google Scholar]
- 88.Gordon LB, Kleinman ME, Miller DT, Neuberg DS, Giobbie-Hurder A, Gerhard-Herman M, Smoot LB, Gordon CM, Cleveland R, Snyder BD, Fligor B, Bishop WR, Statkevich P, Regen A, Sonis A, Riley S, Ploski C, Correia A, Quinn N, Ullrich NJ, Nazarian A, Liang MG, Huh SY, Schwartzman A, Kieran MW. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(41):16666–71. doi: 10.1073/pnas.1202529109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zampetaki A, Zhang Z, Hu Y, Xu Q. American journal of physiology. Heart and circulatory physiology. 2005;288(6):H2946–54. doi: 10.1152/ajpheart.00919.2004. [DOI] [PubMed] [Google Scholar]
- 90.Harrison DG, Guzik TJ, Lob HE, Madhur MS, Marvar PJ, Thabet SR, Vinh A, Weyand CM. Hypertension. 2011;57(2):132–40. doi: 10.1161/HYPERTENSIONAHA.110.163576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Osorio FG, Barcena C, Soria-Valles C, Ramsay AJ, de Carlos F, Cobo J, Fueyo A, Freije JM, Lopez-Otin C. Genes & development. 2012;26(20):2311–24. doi: 10.1101/gad.197954.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Libby P, O’Brien KV. Journal of cellular physiology. 1983;115(2):217–23. doi: 10.1002/jcp.1041150217. [DOI] [PubMed] [Google Scholar]
- 93.Pritchard RH, Lava P, Debruyne D, Terentjev EM. Soft Matter. 2013;9(26):6037–6045. doi: 10.1039/C3SM50901J. [DOI] [Google Scholar]
- 94.Miller BG, Gattone VH, 2nd, Overhage JM, Bohlen HG, Evan AP. The Anatomical record. 1986;216(1):95–103. doi: 10.1002/ar.1092160116. [DOI] [PubMed] [Google Scholar]
- 95.Costa KD. Disease markers. 2003;19(2–3):139–54. doi: 10.1155/2004/482680. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.