

Abstract
Increased renal reabsorption of sodium is a significant risk factor in hypertension. An established clinical marker for essential hypertension is elevated sodium lithium countertransport (SLC) activity. NHA2 is a newly identified Na+(Li+)/H+ antiporter with potential genetic links to hypertension, which has been shown to mediate SLC activity and H+-coupled Na+(Li+) efflux in kidney-derived MDCK cells. To evaluate a putative role in sodium homeostasis, we determined the effect of dietary salt on NHA2. In murine kidney sections, NHA2 localized apically to distal convoluted (both DCT1 and 2) and connecting tubules, partially overlapping in distribution with V-ATPase, AQP2, and NCC1 transporters. Mice fed a diet high in sodium chloride showed elevated transcripts and expression of NHA2 protein. We propose a model in which NHA2 plays a dual role in salt reabsorption or secretion, depending on the coupling ion (sodium or protons). The identified novel regulation of Na+/H+ antiporter in the kidney suggests new roles in salt homeostasis and disease.
Keywords: Hypertension, Na+/H+ antiport, NHEDC2, SLC, Sodium diet, Nephron
Introduction
NHA2 is a recently identified sodium proton (Na+/H+) antiporter belonging to a group of cation proton antiporters, CPA2, whose eukaryotic members have been poorly characterized [1]. Gene linkage analysis implicated NHA2 in essential hypertension, and heterologous expression in yeast provided the first evidence for a role in sodium homeostasis [29]. In kidney-derived MDCK cells, NHA2 was found to mediate phloretin-sensitive sodium lithium counter-transport (SLC) activity, an established marker for hypertension [14]. Elevation in SLC activity is a secondary biochemical indication of an abnormality in sodium homeostasis and has been extensively documented in erythrocytes and fibroblasts [3, 13, 30]. Numerous studies have reported a relationship between hypertension, SLC activity, and sodium levels in several ethnic groups, including Caucasians and Asians [4, 7, 8, 13]. In addition to the correlation with essential hypertension, SLC activity and intracellular Na+ concentrations are highly heritable [8, 13]. All monogenic forms of hypertension are mutations, which alter renal salt handling [15]. Thus, aberration in Na+-H+ transport could be an underlying contributor as well as useful predictor of the risk of developing hypertension.
The kidney plays a dominant role in sodium reabsorption, both in animal models and in human disease [18]. Fuster et al. demonstrated coexpression of NHA2 with calbindin-D28K, which is strongly expressed in the distal convoluted tubule (DCT) [11]. NHA2 expression was not detected in the glomerulus, proximal tubule and thick ascending limb, using RT-PCR [11]. Although the distal convoluted tubule reabsorbs only ~5 % of the filtered load of sodium, it is critical for fine adjustment of urinary Na+ and K+, extracellular fluid volume, and blood pressure regulation [9]. The key role of sodium transporters expressed in the DCT with respect to blood pressure regulation is highlighted not only by the frequent clinical use of thiazide diuretics (which target NCC, a transporter exclusively expressed in the DCT) as potent antihypertensives, but also by genetic mutations affecting NCC: for example, the hypotension seen in Gitelman’s syndrome and the hypertension seen in pseudohypoaldosteronism type II [6, 28]. Reabsorption from this distal site is dynamic, susceptible to change in response to dietary sodium intake with subsequent up- or down-regulation of Na+ transporters and channels [9]. Therefore, we undertook a more comprehensive analysis of NHA2 distribution and localization in the murine nephron and tested the hypothesis that NHA2 expression is regulated by dietary salt.
Methods
Quantitative real-time PCR
mRNA from kidney tissue was isolated using TRIzol Reagent (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. After DNAseI (AmpGrade; Invitrogen), treatment RNA was reverse transcribed with Random Primers (Invitrogen) and SuperScript II reverse transcriptase (Invitrogen). Gene expression levels were measured by quantitative real-time PCR (qPCR) using Taqman gene expression assays.
Immunolocalization in mouse nephron
Anesthetized C57BL/6 mice were fixed by perfusion with 4 % periodate-lysine-paraformaldehyde (PLP), and tissue sections were prepared and probed with the antibodies at the following dilutions: (a) anti-NHA2 antibody (1:100 dilution) raised in rabbit against a 15-aa peptide of HsNHA2 [29], (b) anti-AQP2 raised in chicken (gift from Dr. James Wade, Univ. of Maryland) at a dilution of 1:300, (c) anti-NCC antibody (Feldan, Qubec, Canada) was used at 1:100 dilution, (d) anti-α-NaKATPase antibody was used as previously described [24], (e) antibody Alexa Fluor 594-phalloidin (Molecular probes, Invitrogen) was used at 1:200 dilution to label actin, and (f) V-ATPase B1/2 (Santa Cruz Biotechnology, Inc.) at 1:50 dilution.
Animals
All animal procedures were approved by the Institutional Animal Care and Use Committees at the University of Alberta and Johns Hopkins University. Quantitative real-time PCR experiments were done on cDNA obtained from male FVB/N mice (8–10 weeks of age) that were fed compositionally matched diets consisting of either a control Na diet (0.4 %) or a Narich diet (8 %) for 7 days. Immunofluorescence experiments were done on kidney sections obtained from wild-type mice fed either a control Na diet (0.2 % NaCl, Teklad) or high-salt diet (8 % NaCl Teklad) for 3.5 weeks.
Results and discussion
NHA2 is localized apically to distal convoluted tubule and connecting tubules in the mouse nephron
We sought to establish the distribution of NHA2 to specific regions of the nephron (Fig. 1a) using a previously characterized polyclonal antibody [29]. Double-labeling of mouse kidney sections with the F-actin marker phalloidin, abundant in the brush border cells of the proximal tubule [22], showed a clear separation of NHA2 and phalloidin signals (Fig. 1b), consistent with the absence of NHA2 mRNA from proximal tubules in RT-PCR studies of microdissected rat tubules [11]. We observed apical localization of NHA2, distinct from the basal distribution of Na+, K+-ATPase α subunit (Fig. 1c). In the distal convoluted tubule, identified by staining for the thiazide-sensitive sodium-chloride cotransporter (NCC), co-expression with NHA2 was observed (Fig. 1d). In all the sections observed, NCC always localized with NHA2 but not vice versa. It has been shown that NCC expression levels vary along the distal nephron, with stronger expression in the early (DCT1) part of the distal convoluted tubule compared to the later (DCT2) [20, 25]. However, NHA2 expression may extend beyond the distal convoluted tubule. This is suggested by coexpression with calbindin-D28K, a calcium-binding protein that is highly expressed in DCT2 but is also expressed in the connecting tubules, albeit at lower levels [21]. Beginning at the connecting tubule (CNT), Aquaporin 2 (AQP2) expression levels increase further down the nephron with maximal expression in the principal cells of cortical collecting duct (CCD). When NHA2 and AQP2 were localized, three types of labeled tubule segments were identified: (1) segments where NHA2 co-stained with AQP2 (Fig. 2a, b), (2) segments containing only AQP2 (Fig. 2c), and (3) segments containing only NHA2 (Fig. 2c). Overall, NHA2 localized with AQP2 in ~3–5 % of the total tubules examined. Taken together, we conclude that NHA2 is distributed through DCT1, DCT2, and CNT sections of the nephron.
Fig. 1.
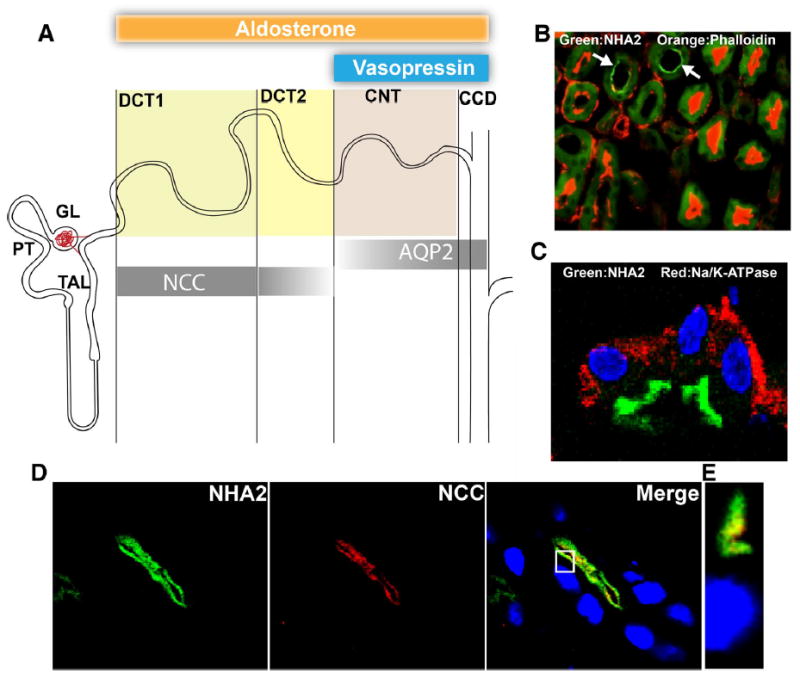
NHA2 is localized to the apical membranes of distal convoluted tubules in the mouse nephron. a Schematic representation of the segmentation of the mouse distal nephron and distribution of segment specific marker proteins. Shadings of bars indicate relative changes along the segments in immunohistochemical abundance. Target regions for aldosterone and vasopressin are shown on the top. Markers: NCC = Sodium chloride cotransporter, AQP2 = Aquaporin 2. b A merged microscopic image showing localization of NHA2 (green) and phalloidin (red) in tissue sections from mouse nephron. White arrows point to regions in tissue stained by NHA2 antibody. Green background observed in other areas is fixed-tissue auto-fluorescence. c A merged microscopic image showing subcellular distribution of NHA2 (green) and α-Na/K-ATPase (red) in tissue sections from mouse nephron. d Confocal microscopic image of a single tubule. Left panel NHA2 (green), middle panel DCT1 marker NCC (red), right panel merged image showing colocalization (yellow), nuclei are stained with DAPI (blue). e An orthogonal view of a slice from the confocal image (white box from the merged image in d)
Fig. 2.
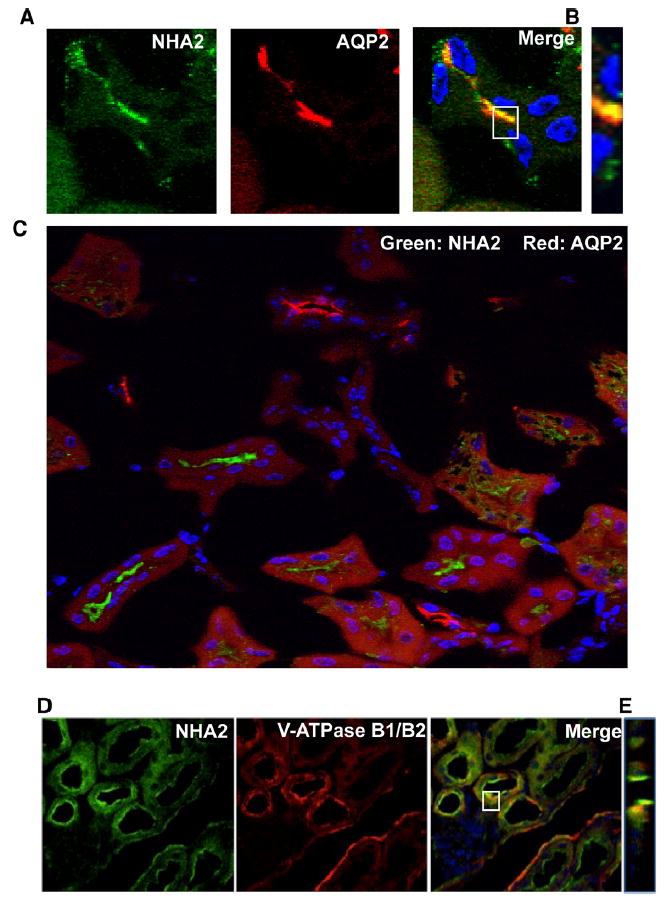
NHA2 partly localizes with Aquaporin 2 in the mouse nephron. a Confocal microscopic image of a single tubule. Left panel NHA2 (green), middle panel Aquaporin 2 (red), right panel merged image showing colocalization (yellow), nuclei are stained with DAPI (blue). b An orthogonal view of a slice from the confocal image (white box from the merged image in a). c A merged microscopic image showing NHA2 (green) and Aquaporin 2 (red) localized independently. d Microscopic images showing colocalization of NHA2 with V-ATPase in mouse nephron. Left panel indirect immunofluorescence of NHA2 (green), middle panel indirect immunofluorescence of V-ATPase B1/B2 (red), right panel merge of V-ATPase B1/B2 and NHA2 (yellow), Nuclei stained by DAPI (blue). e Orthogonal view of a slice from the merged confocal image (from d) showing colocalization of V-ATPase and NHA2
NHA2 partially overlaps in distribution with V-ATPase
Previously, we provided evidence for an unusual chemiosmotic coupling of NHA2 to the proton motive force generated by V-type H+-ATPase at the plasma membrane of MDCK cells [14]. Colocalization and co-immunoprecipitation of NHA2 and V-ATPase revealed a close functional interaction, resulting in a virtual Na+(Li+)-H+ efflux pump. While the majority of plasma membrane sodium-coupled secondary transporters move Na+ ions downhill and into the cytoplasm, we showed that NHA2 mediates H+-driven cation efflux in MDCK cells, recapitulating its phylogenetic origins with bacterial and fungal CPA2 orthologs. Here, we extend our observations to mouse kidney where we demonstrate colocalization of NHA2 with B1 subunit of V-ATPase (Fig. 2d, e). Although the V-ATPase is typically thought of as localizing to intercalated cells, this pump has also been localized to the apical membrane of early (DCT1) and late (DCT2) distal convoluted tubules [12], consistent with apical colocalization with NHA2 in these sections. It is worth noting that the close coupling between cation/proton exchange and the V-ATPase is a characteristic feature of insect epithelia to mediate salt efflux according to the Wieczorek model [27] but has not been comprehensively evaluated in mammalian models.
Dietary salt elevates NHA2 expression in the mouse nephron
Although the collecting duct has been much more well-studied in terms of sodium and water regulation, recent studies in both rodents and humans suggest that most of the transporters and other regulatory proteins localized in the collecting duct are also highly abundant in earlier nephron segments, DCT2 and CNT [20]. Furthermore, micropuncture studies have shown that the great majority of K+ secretion occurs prior to the CCD, and patch clamp studies have demonstrated that the Na+ reabsorptive capacity of the CNT is ten times higher than that of the CCD [16, 17, 23]. Recent findings further suggest that DCT2 and CNT, rather than the collecting duct, regulate sodium reabsorption and potassium excretion when sodium intake is high [20]. In contrast, the collecting duct has been implicated in salt homeostasis under conditions when upstream nephron segments are overwhelmed by genetic defects in specific transporters or by a diet that is very low in sodium [20]. As our immunohistochemistry studies highlighted the presence of NHA2 in the segments upstream of the collecting duct, we hypothesized that a diet rich in sodium would effect the expression of NHA2. We used quantitative RT-PCR of homogenized kidney extracts from mice placed on high-sodium diet. As shown in Fig. 3a, compared with animals on control sodium diet, the mice on high sodium diet had a 3.84-fold higher NHA2 mRNA levels. Consistent with this, immunohistochemistry of kidney sections from mice on a high sodium diet showed more intense NHA2 labeling (Fig. 3b) that was quantitatively evaluated to be elevated by ~5-fold (Fig. 3c). Taken together, we show that NHA2 expression responds to high salt diet. Similarly, the orthologous Nha antiporters in Drosophila were induced by a high salt (Na+ but not K+) diet [5].
Fig. 3.
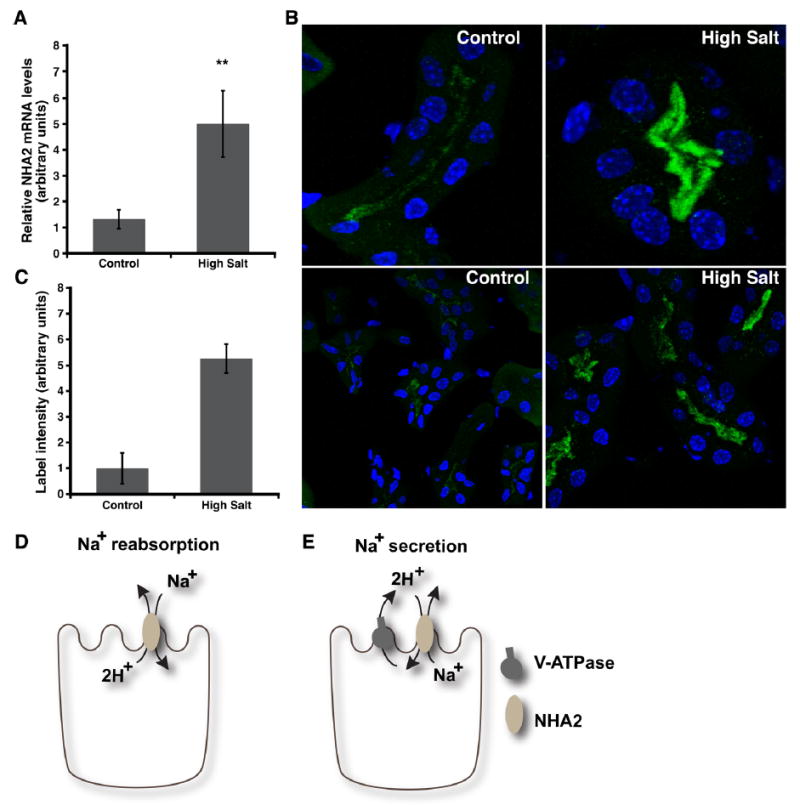
Dietary salt regulates NHA2 expression. a Real-time qPCR data showing NHA2 transcript levels in kidneys from mice fed either control or high salt diet (n = 8, error bars represent SD) as described under “Methods.” b Confocal microscopic images of NHA2 labeling in kidney sections from mice on control diet (left panel: single tubule (top), multiple tubules (bottom)) or high salt diet (right panel: single tubule (top), multiple tubules (bottom)). c Quantification of NHA2 labeling intensity from mice with either control or high salt diet. A minimum of three cells per tubule and at least 10 tubules were measured for each diet type (error bars represent SD). d Cartoon showing sodium gradient driven electrogenic transport by NHA2 on the apical membrane. This mode shows Na+ reabsorption under conditions of high Na+ concentrations in the filtrate that could overcome the unfavorable energetics of H+ extrusion under negative membrane potentials. Alternatively, electroneutral exchange (not shown) would also allow Na+ reabsorption. e Cartoon showing proton gradient coupled Na+ extrusion by electrogenic NHA2 transport on the apical membrane, as discussed in the text
Implications for function
The recognition of a new class of eukaryotic cation proton antiporters (CPA2), with distinct kinetic characteristics and mode of regulation, has opened the possibility of novel roles in physiology and human disease. Unlike electroneutral CPA1 antiporters, microbial NhaA transporters of the CPA2 subtype mediate electrogenic exchange of 2H+ for Na+, allowing them to extrude Na+ against an inwardly directed electrochemical gradient. Consistent with a physiological function in Na+ secretion, NhaA members exhibit alkaline pKa for transport, which favors periplasmic H+ binding [2]. Similar to NhaA, human NHA2 appears more active at neutral to alkaline pH, although evidence is still lacking for electrogenic transport [14]. Depending on the stoichiometry of transport, our current findings are consistent with roles in either sodium reabsorption and sodium secretion and could potentially accommodate both, not unlike the dual absorption or secretion of bicarbonate or protons [26]. In case of electroneutral transport, apical localization of NHA2 in segments of DCT could result in Na+ reabsorption, similar to NHE isoforms (Fig. 3d). Alternatively, electrogenic 2H+/Na+ transport, together with colocalization with V-ATPase in apical membranes of DCT, would favor a role in sodium excretion (Fig. 3e), similar to our previous observations in the basolateral membranes of MDCK cells [14]. This latter function is consistent with elevation of NHA2 expression in response to a high salt diet. We note that in contrast, salt reabsorbing mechanisms, including the apical sodium channel ENaC, are elevated in response to salt depletion [10].
The modern Western diet is high in sodium and is known to be associated with hypertension [19, 20]. We have noted that NHA2 lies within a limited region of human chromosome 4 previously linked to phloretin-sensitive and amiloride-insensitive SLC, an established marker of essential hypertension [14]. We have also shown that NHA2 mediates SLC activity with the same inhibitor characteristics in kidney-derived MDCK cells (3). What remains to be determined is whether NHA2 contributes to the dysregulation of sodium handling mechanisms underlying hypertension. The elevation of NHA2 expression in mice fed a high salt diet is consistent with a role in salt handling. Under these conditions, it is possible that uncoupling of NHA2 from V-ATPase at the apical membrane of nephron segments, when sodium levels are elevated in the filtrate, would result in influx of Na+ and excessive salt reabsorption that contributes to hypertension. Reabsorption of Na+ by a 2H+/Na+ electrogenic mechanism would generally be unfavorable given the interior negative membrane potential, unless Na+ concentrations in the filtrate are very high. Thus, a better understanding of the role of NHA2 in the kidney awaits mechanistic study of transport. As a first analysis of NHA2 expression, regulation, and localization in the kidney, this study provides a starting point for detailed investigation of NHA2 function and physiological role in renal function and disease.
Acknowledgments
This work was supported by grants from the National Institutes of Health R01 DK108304 (to R.R.), DK081610 (J.L.P.), Canadian Institute of Health Research (R.T.A.), and University of Michigan-Dearborn startup funds (KCK).
Abbreviations
- DCT
Distal convoluted tubule
- NCC
Sodium chloride cotransporter
- CNT
Connecting tubule (CNT)
- AQP2
Aquaporin 2
- CPA
Cation proton antiporter
- CCD
Cortical collecting duct
References
- 1.Brett CL, Donowitz M, Rao R. Evolutionary origins of eukaryotic sodium/proton exchangers. American journal of physiology Cell physiology. 2005;288:C223–C239. doi: 10.1152/ajpcell.00360.2004. [DOI] [PubMed] [Google Scholar]
- 2.Calinescu O, Fendler K. A universal mechanism for transport and regulation of CPA sodium proton exchangers. Biol Chem. 2015;396:1091–1096. doi: 10.1515/hsz-2014-0278. [DOI] [PubMed] [Google Scholar]
- 3.Canessa M. Red cell sodium-lithium countertransport and cardiovascular risk factors in essential hypertension. Trends in cardiovascular medicine. 1995;5:102–108. doi: 10.1016/1050-1738(95)00004-S. [DOI] [PubMed] [Google Scholar]
- 4.Canessa M, Adragna N, Solomon HS, Connolly TM, Tosteson DC. Increased sodium-lithium countertransport in red cells of patients with essential hypertension. N Engl J Med. 1980;302:772–776. doi: 10.1056/NEJM198004033021403. [DOI] [PubMed] [Google Scholar]
- 5.Chintapalli VR, Kato A, Henderson L, Hirata T, Woods DJ, Overend G, Davies SA, Romero MF, Dow JA. Transport proteins NHA1 and NHA2 are essential for survival, but have distinct transport modalities. Proc Natl Acad Sci U S A. 2015;112:11720–11725. doi: 10.1073/pnas.1508031112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cruz DN, Simon DB, Nelson-Williams C, Farhi A, Finberg K, Burleson L, Gill JR, Lifton RP. Mutations in the Na-Cl cotransporter reduce blood pressure in humans. Hypertension. 2001;37:1458–1464. doi: 10.1161/01.hyp.37.6.1458. [DOI] [PubMed] [Google Scholar]
- 7.de Wardener HE, He FJ, MacGregor GA. Plasma sodium and hypertension. Kidney Int. 2004;66:2454–2466. doi: 10.1111/j.1523-1755.2004.66018.x. [DOI] [PubMed] [Google Scholar]
- 8.Duhm J, Becker BF. Studies on lithium transport across the red cell membrane. V. On the nature of the Na + –dependent Li + countertransport system of mammalian erythrocytes. J Membr Biol. 1979;51:263–286. doi: 10.1007/BF01869087. [DOI] [PubMed] [Google Scholar]
- 9.Ecelbarger CA, Tiwari S. Sodium transporters in the distal nephron and disease implications. Curr Hypertens Rep. 2006;8:158–165. doi: 10.1007/s11906-006-0013-z. [DOI] [PubMed] [Google Scholar]
- 10.Ergonul Z, Frindt G, Palmer LG. Regulation of maturation and processing of ENaC subunits in the rat kidney. Am J Physiol Renal Physiol. 2006;291:F683–F693. doi: 10.1152/ajprenal.00422.2005. [DOI] [PubMed] [Google Scholar]
- 11.Fuster DG, Zhang J, Shi M, Bobulescu IA, Andersson S, Moe OW. Characterization of the sodium/hydrogen exchanger NHA2. J Am Soc Nephrol. 2008;19:1547–1556. doi: 10.1681/ASN.2007111245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gluck SL, Underhill DM, Iyori M, Holliday LS, Kostrominova TY, Lee BS. Physiology and biochemistry of the kidney vacuolar H + –ATPase. Annu Rev Physiol. 1996;58:427–445. doi: 10.1146/annurev.ph.58.030196.002235. [DOI] [PubMed] [Google Scholar]
- 13.Kammerer CM, Cox LA, Mahaney MC, Rogers J, Shade RE. Sodium-lithium countertransport activity is linked to chromosome 5 in baboons. Hypertension. 2001;37:398–402. doi: 10.1161/01.hyp.37.2.398. [DOI] [PubMed] [Google Scholar]
- 14.Kondapalli KC, Kallay LM, Muszelik M, Rao R. Unconventional chemiosmotic coupling of NHA2, a mammalian Na+/H+ antiporter, to a plasma membrane H+ gradient. J Biol Chem. 2012 doi: 10.1074/jbc.M112.403550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lifton RP, Gharavi AG, Geller DS. Molecular mechanisms of human hypertension. Cell. 2001;104:545–556. doi: 10.1016/s0092-8674(01)00241-0. [DOI] [PubMed] [Google Scholar]
- 16.Malnic G, Klose RM, Giebisch G. Micropuncture study of renal potassium excretion in the rat. Am J Phys. 1964;206:674–686. doi: 10.1152/ajplegacy.1964.206.4.674. [DOI] [PubMed] [Google Scholar]
- 17.Malnic G, Klose RM, Giebisch G. Microperfusion study of distal tubular potassium and sodium transfer in rat kidney. Am J Phys. 1966;211:548–559. doi: 10.1152/ajplegacy.1966.211.3.548. [DOI] [PubMed] [Google Scholar]
- 18.McCormick JA, Yang CL, Ellison DH. WNK kinases and renal sodium transport in health and disease: an integrated view. Hypertension. 2008;51:588–596. doi: 10.1161/HYPERTENSIONAHA.107.103788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meneton P, Jeunemaitre X, de Wardener HE, MacGregor GA. Links between dietary salt intake, renal salt handling, blood pressure, and cardiovascular diseases. Physiol Rev. 2005;85:679–715. doi: 10.1152/physrev.00056.2003. [DOI] [PubMed] [Google Scholar]
- 20.Meneton P, Loffing J, Warnock DG. Sodium and potassium handling by the aldosterone-sensitive distal nephron: the pivotal role of the distal and connecting tubule. Am J Physiol Renal Physiol. 2004;287:F593–F601. doi: 10.1152/ajprenal.00454.2003. [DOI] [PubMed] [Google Scholar]
- 21.Nijenhuis T, Hoenderop JG, van der Kemp AW, Bindels RJ. Localization and regulation of the epithelial Ca2+ channel TRPV6 in the kidney. J Am Soc Nephrol. 2003;14:2731–2740. doi: 10.1097/01.asn.0000094081.78893.e8. [DOI] [PubMed] [Google Scholar]
- 22.Nurnberger A, Rabiger M, Mack A, Diaz J, Sokoloff P, Muhlbauer B, Luippold G. Subapical localization of the dopamine D3 receptor in proximal tubules of the rat kidney. The journal of histochemistry and cytochemistry : official journal of the Histochemistry Society. 2004;52:1647–1655. doi: 10.1369/jhc.4A6359.2004. [DOI] [PubMed] [Google Scholar]
- 23.Reineck HJ, Osgood RW, Ferris TF, Stein JH. Potassium transport in the distal tubule and collecting duct of the rat. Am J Phys. 1975;229:1403–1409. doi: 10.1152/ajplegacy.1975.229.5.1403. [DOI] [PubMed] [Google Scholar]
- 24.Tamkun MM, Fambrough DM. The (Na+ + K+)-ATPase of chick sensory neurons. Studies on biosynthesis and intracellular transport. J Biol Chem. 1986;261:1009–1019. [PubMed] [Google Scholar]
- 25.Verlander JW. The thiazide-sensitive NaCl cotransporter: a new target for acute regulation of salt and water transport by angiotensin II. Am J Physiol Renal Physiol. 2007;293:F660–F661. doi: 10.1152/ajprenal.00260.2007. [DOI] [PubMed] [Google Scholar]
- 26.Wagner CA, Finberg KE, Stehberger PA, Lifton RP, Giebisch GH, Aronson PS, Geibel JP. Regulation of the expression of the Cl-/anion exchanger pendrin in mouse kidney by acid-base status. Kidney Int. 2002;62:2109–2117. doi: 10.1046/j.1523-1755.2002.00671.x. [DOI] [PubMed] [Google Scholar]
- 27.Wieczorek H, Putzenlechner M, Zeiske W, Klein U. A vacuolar-type proton pump energizes K+/H+ antiport in an animal plasma membrane. J Biol Chem. 1991;266:15340–15347. [PubMed] [Google Scholar]
- 28.Wilson FH, Disse-Nicodeme S, Choate KA, Ishikawa K, Nelson-Williams C, Desitter I, Gunel M, Milford DV, Lipkin GW, Achard JM, Feely MP, Dussol B, Berland Y, Unwin RJ, Mayan H, Simon DB, Farfel Z, Jeunemaitre X, Lifton RP. Human hypertension caused by mutations in WNK kinases. Science. 2001;293:1107–1112. doi: 10.1126/science.1062844. [DOI] [PubMed] [Google Scholar]
- 29.Xiang M, Feng M, Muend S, Rao R. A human Na+/ H+ antiporter sharing evolutionary origins with bacterial NhaA may be a candidate gene for essential hypertension. Proc Natl Acad Sci USA. 2007;104:18677–18681. doi: 10.1073/pnas.0707120104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zerbini G, Mangili R, Gabellini D, Pozza G. Modes of operation of an electroneutral Na+/Li + countertransport in human skin fibroblasts. Am J Phys. 1997;272:C1373–C1379. doi: 10.1152/ajpcell.1997.272.4.C1373. [DOI] [PubMed] [Google Scholar]