


Abstract
Vitamin E derivatives possess many essential features for drug-delivery applications, such as biocompatibility, stability, improvement of water solubility of hydrophobic compounds, anticancer activity, and the ability to overcome multidrug resistance (MDR). Herein, vitamin E derivatives are used to overcome MDR through a combined P-glycoprotein (P-gp) inhibition and mitochondrial impairment strategy. A novel nanomicellar drug-delivery system as a carrier for doxorubicin (DOX) was developed, in which d-α-tocopheryl polyethylene glycol 1000 succinate was used as a P-gp inhibitor, α-tocopheryl succinate was introduced as a mitochondrial disrupting agent, and d-α-tocopheryl polyethylene glycol 2000 succinate was used as the main building block of micelles. The optimal ratio between the components of the nanocarrier was determined. The resultant DOX-loaded mixed micelles exhibited a suitable size of 52.08 nm, high drug-loading encapsulation efficiency (>98%), high stability, and pH-dependent drug release. In vitro experiments demonstrated a significantly increased cytotoxic activity of DOX-loaded mixed micelles against resistant MCF-7/Adr cells (45-fold higher than DOX after 48 h of treatment). In vivo studies revealed superior antitumor efficiency with less cardio- and hepatotoxicities of DOX-loaded micelles compared with that of free DOX. These results highlight that the developed DOX-loaded mixed micelles have a promising potential to overcome MDR in chemotherapy for clinical usage.
Keywords: vitamin E derivatives, P-gp inhibition, multidrug resistance, mitochondrial impairment, drug delivery
Graphical abstract
1. INTRODUCTION
During the past few decades, nanomedicine has provided novel approaches to improve the delivery of chemotherapeutic drugs to tumors. However, multidrug resistance (MDR) still remains a serious obstacle to effective cancer treatment. The principle mechanisms of MDR may include active membrane transport of compounds from cells, decreased drug concentration due to overexpression of drug-metabolizing enzymes or glutathione, and increased DNA repair and failure of apoptosis.1 One of the major mechanisms in transport-associated MDR is the overexpression of drug efflux pumps, such as P-glycoprotein (P-gp), which extrude different agents out of the cells. P-gp, also named multidrug resistance protein 1 (MDR-1 protein), is a well-known adenosinetriphosphate (ATP)-dependent cell membrane transporter, which in normal cells plays a protective role by removing xenobiotics (toxins or drugs) from the cells into urine, bile, and intestinal lumen.2,3 However, cancer cells, which overexpress P-gp, present a clinical challenge for chemotherapy because a large number of structurally diverse anticancer agents, such as doxorubicin, paclitaxel, vinblastine, and so forth, are P-gp substrates. Reduced intracellular drug accumulation through active extracellular transport leads to decreased therapeutic efficacy and greatly complicates the treatment of drug-resistant types of cancer.3
To limit drug extrusion and hence overcome P-gp-associated MDR, numerous P-gp inhibitors belonging to diverse chemical groups have been identified.4 It has been widely observed that one of the most efficient strategies to improve chemotherapy of resistant malignancies is to combine a P-gp inhibitor with an anticancer drug. By impairing the P-gp efflux function with the inhibiting agents, the intratumoral distribution and the tissue pharmacokinetics of anticancer drugs can be improved. However, current P-gp inhibitors still have drawbacks, including high side effects and necessity of the use of high doses, resulting in high toxicity or unpredictable effects on drug metabolism.5
d-α-Tocopheryl polyethylene glycol 1000 succinate (TPGS 1k) is a nonionic amphiphilic molecule composed of hydrophobic α-tocopheryl succinate (α-TOS, a vitamin E derivative) and hydrophilic poly(ethylene glycol) (PEG) with a molecular weight of 1 kDa. TPGS 1k has been approved by the FDA as a safe pharmaceutical adjuvant.6 One of the major benefits of the TPGS 1k molecule is its ability to overcome MDR. It was demonstrated that TPGS 1k can act as an inhibitor of P-gp through a mechanism that could be due to the suppression of P-gp ATPase, the energy source that drives active transport by P-gp.7,8 Additionally, TPGS 1k containing a 1 kDa PEG chain has been shown to exhibit a higher P-gp inhibition activity than TPGS analogues containing PEG chains with other lengths.9
However, the hydrophilic part of TPGS 1k is not sufficiently long to prolong the circulation time of micelles in the blood stream. Moreover, the critical micelle concentration (CMC) of TPGS 1k is comparatively high (0.2 mg/mL), indicating unstable behavior during blood circulation.10 In this regard, TPGS including a PEG chain of 2 kDa (TPGS 2k), which possesses CMC = 0.02 mg/mL and hence better stability, can be considered as the main building block of mixed micelles. TPGS 2k can also confer a “stealth” capability on the drug carrier.6 Unfortunately, further experiments have demonstrated that the mixture of TPGS 1k and TPGS 2k has comparatively low drug encapsulation efficiency (EE). This might be ascribed to the insufficient hydrophobic interaction between the short hydrophobic part of the TPGS molecules and the drug. To overcome the aforementioned issues, hydrophobic molecules should be included in the micellar core.
The vitamin E derivative, α-TOS, exists as the alpha (α) isomer and represents the hydrophobic moiety of TPGS 1k and TPGS 2k. α-TOS is found to possess anticancer activity, which is associated with the production of intracellular reactive oxygen species (ROS) that decreases the mitochondrial membrane potential (MMP), leading to the release of cytochrome c into the cytosol and further activation of the apoptotic caspase cascade.11,12 Moreover, α-TOS has been demonstrated to have high selectivity against cancer cells while showing very limited or no toxicity toward normal cells.13–15 In addition, it was reported that because of its hydrophobicity, inclusion of α-TOS in the system’s hydrophobic portion can overcome the limitations of the nanocarriers associated with low drug EE and lack of stability in physiological environments.16
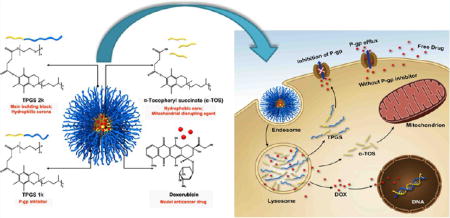
In this work, based on the rationale that vitamin E derivatives could improve the efficacy of anticancer drugs, micelles consisting of a mixture of TPGS 1k, TPGS 2k, and α-TOS have been developed. In this combined system, each component plays an essential role. Because of their amphiphilic properties, TPGS 1k and TPGS 2k form the micelles; TPGS 1k also inhibits P-gp, thus enhancing intracellular drug accumulation, and TPGS 2k, due to its longer PEG chains, forms the hydrophilic outer layer (corona) of the system to reduce protein adsorption.6 α-TOS was added to the system not only to improve the stability and drug EE but also to enhance the therapeutic efficacy of the drug-loaded micelles by targeting the mitochondria (Figure 1). The DOX-loaded mixed micelles exhibited 45-fold higher antitumor efficiency in comparison to DOX against multidrug-resistant breast cancer cells (MCF-7/ Adr) after treatment for 48 h. This excellent result from the drug-loaded system was attributed to the enhanced cellular uptake of the nanodelivery system, a markedly decreased drug efflux through P-gp inhibition, and activation of the mitochondria-specific apoptotic pathway in tumor cells as a result of mitochondrial impairment. In vivo results demonstrated superior antitumor activity with less cardiotoxicity and hepatotoxicity of DOX-loaded micelles as compared with that of the free DOX solution. The obtained results indicated the promising potential of the developed DOX-loaded mixed micelles based on vitamin E derivatives to overcome MDR in cancer therapy for clinical usage.
Figure 1.

Structural composition and the proposed mechanism of action of mixed micelles.
2. MATERIALS AND METHODS
2.1. Materials
TPGS 1k, PEG 2000 (PEG 2K, Mw = 2000 Da), verapamil hydrochloride, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT), pyrene, 2′,7′-dichlorodihydrofluorescin diacetate (DCFH-DA), N′-dicyclohexylcarbodiimide (DCC), Hoechst 33342, and 4-dimethylaminopyridine (DMAP) were all purchased from Sigma-Aldrich Co. (St. Louis, MO, USA). α-TOS was ordered from Tokyo Chemical Industry, Co., Ltd. (Tokyo, Japan). Doxorubicin hydrochloride (DOX·HCl) was provided by Dalian Meilun Biological Technology, Co., Ltd. (Dalian, China). Triethylamine (TEA) was purchased from Xilong Chemical Company Co., Ltd. (Shantou, China). Dry dichloromethane (DCM), and trichloromethane were purchased from Beijing Chemical Reagent Co., Ltd. (Beijing, China). Rhodamine 123 (Rho123) was purchased from Aladdin (Shanghai, China).
RPMI 1640 medium, Dulbecco’s modified Eagle’s medium (DMEM), and fetal bovine serum (FBS) were ordered from Wisent Inc (Multicell, Wisent Inc., St. Bruno, Quebec, Canada). Antibiotic solution (penicillin and streptomycin) and 0.25% trypsin−EDTA were bought from Life Technologies (USA). The MMP assay kit with 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide (JC-1) was provided by Invitrogen (Eugene, OR, USA). Caspase- 9 and caspase-3 kits were purchased from Beyotime Biotechnology Co., Ltd. (Jiangsu, China). All chemicals were of analytical grade and used directly without further purification.
2.2. Cell Lines
Human sensitive breast cancer cells (MCF-7), hepatic (L02) cells, normal rat kidney (NRK) cells, and human vein endothelial cells (HUVEC) were provided by the Cell Resource of Medical Sciences and Peking Union Medical College and were cultured in DMEM containing 10% (v/v) FBS and 1% (v/v) penicillin–streptomycin solution. Multidrug-resistant MCF-7/Adr cells were provided by Dr. Meng (Chinese Academy of Sciences, China). MCF-7/Adr cells were cultured in RPMI 1640 cell culture media containing FBS and antibiotics in the same proportion. The cells were cultivated in a humidified atmosphere at 37 °C and 5% CO2.
2.3. Synthesis of TPGS 2k
TPGS 2k was synthesized as described previously.17 Briefly, α-TOS, PEG 2K, DCC, and DMAP were dissolved in dry DCM at a molar ratio of 1:1:2:0.1. The solution was stirred overnight in a nitrogen atmosphere in the dark. After the reaction, the by-products were removed through filtration, and then, the solution was precipitated in excess cold diethyl ether. The precipitate was rinsed with diethyl ether again and dissolved in water, followed by dialyzing against water using a membrane with a molecular weight cutoff of 3500 Da. The final product TPGS 2k was obtained after lyophilization, as a white powder.17,18 The product was analyzed using proton nuclear magnetic resonance 1H NMR (AVANCE III, Bruker BioSpin, Germany) in CDCl3 and Fourier transform infrared (FTIR) spectroscopies (Spectrum One, PerkinElmer Instruments Co., Ltd., USA) in the range of 4000–400 cm−1.
2.4. Preparation of Blank and DOX-Loaded Mixed Micelles
The preparation of DOX-loaded mixed micelles (T1k2k-TOS-DOX), blank mixed micelles (T1k2k-TOS), and DOX-loaded mixed micelles without α-TOS (T1k2k-DOX) was done using the thin-film hydration technique.17 T1k2k-TOS-DOX micelles were prepared as follows: First, 6.0 mg of DOX·HCl was dissolved and stirred with TEA (molar ratio of DOX/TEA = 1:3) in 3 mL of methanol at room temperature to obtain hydrophobic DOX. Then, the DOX solution was mixed with chloroform solutions of TPGS 1k, TPGS 2k, and α-TOS in certain weight ratios. The chloroform was removed by rotary vacuum evaporation, and the formed thin film was hydrated in 15 mL of phosphate-buffered saline (1× PBS, pH 7.4) at 45 °C for 30 min, followed by sonication for 5 min. Then, the solution was filtered through a 0.22 µm syringe filter to get rid of nonloaded DOX aggregates. The T1k2k-TOS micelles, blank mixed micelles with various content of the components, T1k2k, or T1k2k-DOX micelles were prepared by dissolving certain weight ratios of TPGS 1k, TPGS 2k, α-TOS, or DOX in chloroform. The subsequent procedure was identical as that for T1k2k-TOS-DOX micelles.
2.5. Characterization of Mixed Micelles
The morphology of T1k2k-TOS and T1k2k-TOS-DOX micelles was investigated using a transmission electron microscope (TEM, HT7700, Hitachi, Japan). The micelles were prestained with 1% uranyl acetate. The particle size, polydispersity index (PDI), and zeta (ζ) potential of the mixed micelles were measured using dynamic light scattering (DLS) (Zetasizer 5000, Malvern Instrument, Inc., UK). To evaluate the stability of the micelles, their particle size, PDI, and ζ potential were recorded after storage at +4 °C for 2 months.
2.6. Determination of CMC
The CMC value of the mixed micelles was measured using pyrene as a fluorescent probe. Briefly, 10 µL of 1 × 10−5 M pyrene acetone solution was added to empty 1.5 mL tubes. After the acetone was removed, a series of aqueous solutions of mixed components, with concentrations ranging from 1 × 10−4 to 1 mg/mL, were added to each tube. The final pyrene concentration was kept as 1 × 10−7 M. The solution was sonicated for 30 min and left overnight at room temperature in the dark. The fluorescence intensity of each sample was measured using a fluorescence spectrophotometer (F-7000, Hitachi, JPN). The excitation wavelength was 335 nm, and the selected emission wavelengths were those corresponding to the maximum intensities of the first and third peaks (I1: 374 nm and I3: 384 nm, respectively) in the emission spectrum.17–19 The CMC value of the micelles was determined as the intersection point of two lines obtained by linear regression.
2.7. Drug EE% and Drug-Loading (DL%) Content
The EE% and DL% of the formulated micelles were determined using a fluorescence spectrophotometer with 480 nm excitation and 590 nm emission. The absorbance of unloaded DOX was determined after separation using an ultrafiltration membrane (Mw = 30 000). The total drug amount was measured after the micelles had been collapsed by dilution with methanol. The EE% and DL% were calculated as follows
2.8. In Vitro Drug Release
The release profile of DOX from the T1k2k-TOS-DOX micelles was examined by dialysis using a membrane with a molecular weight cutoff of 8000–12 000 Da. One mL of T1k2k-TOS-DOX micelles (1 mg/mL) was dialyzed against 30 mL of PBS (pH 5.0 and 7.4) at 37 °C under shaking at 100 rpm. At designated time points, 1 mL aliquots were collected and replaced by 1 mL of fresh PBS. The amount of released DOX was determined using a fluorescence spectrophotometer, with a calibration curve.
2.9. In Vitro Cell Cytotoxicity
To evaluate the toxicity of the mixed micelles against cancer cells, the MTT assay was performed. MCF-7 or MCF-7/Adr cells were seeded in 96-well plates (1 × 104 cells per well) and cultured for 24 h. Then, the culture medium was removed, and the cells were treated with the medium containing DOX, T1k2k, T1k2k-TOS, or T1k2k-TOS-DOX micelles at DOX equivalent concentrations ranging from 1 to 50 µg/mL. Untreated cells were used as the control. The incubation was performed for 48 h. After the incubation period had elapsed, the cell viability measurement was performed according to the standard protocol. The viability of the cells was presented as the percentage of the absorbance of the treated cells over that of the control group.
The viabilities of L02, HUVEC, and NRK cells exposed to blank mixed micelles with different TPGS 1k or α-TOS contents were measured using the same method, as described above.
2.10. Colony Formation Assay
MCF-7 and MCF-7/Adr were seeded in 6-well plates at a density of 300 cells per well and left in the incubator until cells formed sufficiently large colonies. Then, the cells were treated and cultured for another 7 days. After that, the cells were fixed with 4% paraformaldehyde, stained with crystal violet, and rinsed with tap water. Colonies of more than 50 cells were counted. The experiment was accomplished in triplicate.
2.11. In Vitro Cellular Uptake and Distribution
The cellular uptake of DOX and T1k2k-TOS-DOX micelles was examined using qualitative confocal microscopy and quantitative flow cytometry analysis. For confocal microscopy, MCR-7/Adr cells were seeded into 35 mm glass-bottom dishes (1 × 105 cells per well) and cultured for 24 h. Then, the cells were exposed to DOX or T1k2k-TOS-DOX micelles (final DOX concentration: 5 µg/mL). After 6 h, the cells were fixed with 4% paraformaldehyde for 20 min and rinsed with PBS, followed by nuclei staining with Hoechst 33342 for 20 min. The fixed cell monolayer was rinsed with PBS and imaged using confocal laser scanning microscopy (CLSM) (LSM710, Carl Zeiss Microscope, Co. Ltd., GER) with excitation at 488 nm. For flow cytometry, the cells were seeded into 6-well plates (3 × 105 cells per well) and incubated with DOX or T1k2k-TOS-DOX micelles, as described above. Subsequently, the cells were rinsed with PBS, collected, and investigated using an Attune acoustic focusing cytometer (Applied Biosystems, Life Technologies, Carlsbad, CA).
To show the localization of the mixed micelles in the cells, MCR-7/ Adr cells were seeded into 35 mm glass-bottom dishes (1 × 105 cells per well) and cultured for 24 h. Then, the cells were treated with DOX or T1k2k-TOS-DOX micelles (final DOX concentration: 5 µg/mL) for 30 min, washed with PBS, and stained with LysoTracker Deep Red for 15 min. After rinsing with PBS, the cells were stained with Hoechst 33342 for 20 min, followed by PBS washing and imaging using CLSM.
2.12. Evaluation of the Inhibition of P-gp Efflux
To determine the ability of the mixed micelles to suppress the P-gp function, the P-gp-mediated efflux of DOX was evaluated using CLSM and flow cytometry. Verapamil was used as the standard P-gp inhibitor. MCF-7/Adr cells were seeded into 35 mm glass-bottom dishes (1 × 105 cells per well) for CLSM and into 6-well plates (3 × 105 cells per well) for flow cytometry and cultured for 24 h. Then, the cells were pretreated with verapamil (100 µM), T1k2k-TOS, T1k2k, or a drug-free cell culture medium (control) for 12 h, and then, 5 µg/mL DOX was added and incubated for 6 h. After refreshing the culture medium, the cells were observed using CLSM. For flow cytometry, after refreshing the culture medium, the cells were further incubated, collected at designated time points (0, 3, 6, 18, and 24 h), and then analyzed. In the control group, the cells were preincubated with a medium for 12 h and treated with DOX.
2.13. ROS Production
The intracellular ROS level was estimated using a fluorogenic dye, DCFH-DA, which under oxidizing conditions converts into the highly fluorescent compound 2′,7′-dichlorofluorescein (DCF). Briefly, MCF-7/Adr cells were seeded into 6-well plates (3 × 105 cells per well) and incubated for 24 h. DOX, T1k2k, T1k2k-TOS, or T1k2k-TOS-DOX micelles were then added (final DOX concentration: 5 µg/mL). After 6 h, the drug-containing medium was replaced with DCFH-DA solution (6 µM) in a serum-free RPMI 1640 medium for 30 min. Then, the cells were rinsed with PBS, harvested, and assayed using a flow cytometer.
2.14. Mitochondrial Depolarization
Changes in the MMP were investigated using the JC-1 dye. In brief, MCF-7/Adr cells were seeded in 8-well coverglass chambers (1.5 × 104 cells per well) and incubated for 24 h. Then, the culture medium was replaced with the medium including T1k2k or T1k2k-TOS micelles for 6 h. Carbonyl cyanide 3-chlorophenyl hydrazone (CCCP) was used as the MMP disrupter in the positive control group. After the incubation period had elapsed, the cells were stained with the JC-1 dye for another 20 min at 37 °C, rinsed with PBS, and imaged using CLSM.
2.15. Caspase-9 and Caspase-3 Activity Assays
The caspase activities were analyzed according to the manufacturers’ protocol using caspase-9 and caspase-3 activity kits. In brief, MCF-7/Adr cells were treated with the DOX solution, T1k2k, T1k2k-TOS, and T1k2k-TOS-DOX micelles for 12 h (final DOX concentration: 5 µg/mL). Then, the cells were collected, lysed, and centrifuged at 20 000g for 15 min at 4 °C. The supernatant (50 µL) was incubated with 10 µL of caspase-9 and caspase-3 substrates separately for 1 h at 37 °C. The activity of caspase-9 and caspase-3 was assessed using a microplate reader at an absorbance of 405 nm. The concentration of the total cellular protein for each sample was normalized using the Bradford protein assay kit.
2.16. In Vivo Biodistribution
All in vivo experiments were performed according to guidelines set by the ethics committee of Peking University. Instead of DOX, DiR iodide 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindotricarbocyanine iodide was used to prepare T1k2k-TOS-DiR micelles according to the same procedure. For in vivo biodistribution, after the tumor volume reached approximately 400 mm3, MCF-7/Adr-bearing BALB/c nude mice were injected with 100 µL of PBS, free DiR, and T1k2k-TOS-DiR through the tail vein at a dose equal to 10 µg/mL of DiR. The real-time biodistribution was monitored in the near-infrared region at 0.5, 3, 8, 12, 24, and 48 h after injection, using a Maestro in vivo Spectrum Imaging System (Cambridge Research & Instrumentation, Woburn, MA). After 48 h, the mice were killed, and the tumors and the major organs were isolated for further ex vivo imaging.
2.17. In Vivo Antitumor Effect
To establish the tumor model, 1 × 107 MCF-7/Adr cells were subcutaneously inoculated in the right flank. After the tumor volume reached 100 mm3, the mice were divided into five groups (n = 6 per group) and treated intravenously with saline, DOX, T1k2k, T1k2k-TOS, and T1k2k-TOS-DOX solutions at a DOX-equivalent dose of 5 mg/kg. The drugs were administrated every 3 days, and the body weight and the tumor size were measured every 2 days. The following formula was used for calculating the tumor volume: V = 0.5 × L × W2, where L is the longest diameter and W is the shortest diameter. At the end of the treatment period, one mouse of each group was killed, and the tissues of heart, liver, and tumor were collected and sent to Wuhan Goodbio Technology Co. Ltd. for histological analysis.
2.18. Statistical Analysis
The data are reported as the mean ± standard deviation (SD). Statistical evaluation was performed using one-way analysis of variance (ANOVA). p < 0.05 was considered to indicate statistically significant differences.
3. RESULTS AND DISCUSSION
3.1. Synthesis and Characterization of TPGS 2k
TPGS 2k was synthesized by the linkage of α-TOS to PEG 2K via an ester bound (Figure S1). The chemical structure of TPGS 2k was confirmed using 1H NMR and FTIR (Figure S2) spectroscopies. The 1H NMR spectrum of TPGS 2k shows a characteristic peak at 3.65 ppm, which belongs to –CH2 protons of PEG, whereas the peak at 0.86 ppm corresponds to –CH3 protons of α-TOS. The other peaks in the aliphatic region are assigned to various portions of α-TOS (Figure S2A).
The FTIR spectra of TPGS 1k and TPGS 2k are shown in Figure S2B. Similar to TPGS 1k, the carbonyl band of TPGS 2k (C═O) with a reduced intensity appears at 1738 cm−1, the peak at 2889 cm−1 is assigned to –CH2, and the absorption band at 1110 cm−1 results from the stretching vibration of C– O of PEG. All of these results proved that the reaction between α-TOS and PEG 2K was successful.
3.2. Optimization of the Composition of the Carrier
The results in Figure S3 confirmed that the CMC value of TPGS 2k was 10-fold lower in comparison with TPGS 1k, which is consistent with previous reports. In this regard, the presence of TPGS 2k in the system is essential to keep appropriate stability. In addition, it was found out that the content of TPGS 1k in the mixed micelles is proportional to the CMC values of the mixed micelles.
It has been found that TPGS 1k exhibits better P-gp inhibition than TPGS 2k.9 To confirm this, MCF-7/Adr cells were exposed to DOX, or DOX was coincubated with TPGS 1k or TPGS 2k. The fluorescence intensity was then detected using flow cytometry. As shown in Figure S4, the highest fluorescence intensity was observed in cells treated with DOX coincubated with TPGS 1k, which is consistent with previous studies.
To determine the most optimal TPGS 1k and TPGS 2k content in the system and to achieve the best balance between safety and P-gp inhibition activity of the final mixed micelles, nanoparticles were prepared with various amounts of TPGS 1k. The TPGS 1k mass content (%) in the mixture of TPGS 1k/ TPGS 2k was varied in the range of 16.6–83.3%, whereas the amount of α-TOS was kept constant (10% of the total mass of TPGS 1k and TPGS 2k) (Table S1). The cytotoxicity of T1k2k-TOS micelles containing various levels of TPGS 1k against normal L02 cells was measured via the MTT assay. Data showed that the toxic effect increased with increasing TPGS 1k in the system (Figure S5A). Flow cytometry results from the cells coincubated with DOX and blank mixed micelles with various percentages of TPGS 1k in the system revealed that the highest fluorescence intensity of DOX was detected when the system contained 50% of TPGS 1k (Figure S5B). Further increasing the TPGS 1k content in the mixed micelles did not aid in the reduction of drug efflux; however, it increased the cytotoxicity against normal cells, thereby decreasing the safety of the nanocarrier. These data can be explained as follows: As the amount of TPGS 1k increases in the system, the P-gp inhibition becomes more significant. However, when the level of TPGS 1k reaches 50%, it leads to saturation; hence, higher levels of TPGS 1k will enhance the cytotoxicity but not the ability to inhibit P-gp. Taking into account the obtained results, it was concluded that the optimal content of TPGS 1k in the system is 50%, which provides the micelles with the highest Pgp inhibition activity in cancer cells and moderate cytotoxicity against normal cells. Thus, the mass ratio between TPGS 1k and TPGS 2k in the system was determined as 50/50.
The next step in the optimization of the system was the determination of the amount of α-TOS in the micelles. To obtain the desirable properties of the final nanocarrier, such as stability, high capacity, and cytotoxicity against tumor cells, drug-loaded micelles with different α-TOS contents were fabricated. During the calculations and the fabrication process, the total amount of TPGS 1k and TPGS 2k was considered as one component and specified as TPGS, the mass of which was kept constant, while the α-TOS content was changed. The particle size, PDI, DOX EE%, and DL% of T1k2k-TOS with various mass ratios of α-TOS were investigated. As shown in Table S2 and Figure S6, nanoparticles with a higher content of α-TOS, particularly with a α-TOS/TPGS ratio of 1.5:1 and 2:1, exhibited unstable behavior, which was confirmed by the formation of insoluble precipitates in solution. Therefore, it was not possible to measure their EE%. This instability might be attributed to the excessive amount of α-TOS, which cannot be embedded into the mixed micelles because the maximum capacity of the hydrophobic core had already been reached. Thus, the free hydrophobic molecules of α-TOS (and/or DOX) are precipitated in the aqueous environment, which leads to the formation of aggregates. When the α-TOS/TPGS ratio was 1:1, the instability was reduced but the PDI still remained high (0.349 ± 0.02), which indicates a broad particle size distribution. This was reflected in the transparency of the solution. Thus, based on the aforementioned data, mixed micelles with α-TOS/TPGS ratios of 1:1; 1.5:1, and 2:1 were excluded from further study. The DLS results obtained for micelles with α-TOS/TPGS ratios of 0.25:1 and 0.5:1 showed that these nanoparticles possess suitable mean diameters (17.39 ± 0.64 and 24.87 ± 2.39 nm, respectively) and PDIs (0.266 ± 0.03 and 0.217 ± 0.015, respectively). Meanwhile, the EE% value of micelles with ratios of 0.25:1 and 0.5:1 was up to 93.92 ± 2.25 and 98.90 ± 1.44%, respectively. Further cell viability assays confirmed that the higher α-TOS content in the system enhanced the cytotoxic effect against cancer cells while exhibiting limited or no toxicity toward normal cells (Table S3). Taking into account the observations that mixed micelles with a α-TOS/TPGS ratio of 0.5:1 have a suitable size, the highest EE%, and the lowest IC50 value for cancer cells, it is sensible to hypothesize that this proportion of components is optimal for achieving the best antitumor efficacy of the DOX-loaded system.
After the optimal mass ratio between α-TOS and TPGS was established as 0.5:1, which means that the ratio of α-TOS/TPGS 1k/TPGS 2k is 1:1:1, all further experiments were performed using mixed micelles that were fabricated using that proportion of the components, and the micelles were named as T1k2k, T1k2k-DOX, T1k2k-TOS, and T1k2k-TOS-DOX.
3.3. Characterization and Stability of the Micelles
The particle size, PDI, and ζ potential of T1k2k and T1k2k-TOS are shown in Table S4. The increase in the mean diameter indicated that α-TOS was successfully loaded into the mixed micelles. The particle size, ζ potential, PDI, EE%, and DL% of T1k2k-TOS (blank micelles) and T1k2k-TOS-DOX (drug-loaded micelles) are summarized in Table 1 and Figure 2. The mean diameter of T1k2k-TOS-DOX micelles is less than that of T1k2k-TOS micelles. This might be due to the hydrophobic interaction between the encapsulated DOX and the components of the nanocarrier’s core, leading to compression of the system and hence causing the drug-loaded micelles to be more compact than the blank micelles. The PDI of T1k2k-TOS-DOX micelles is 0.234 ± 0.006, indicating the uniform size distribution. It is well-established that the most effective particle size for drug-delivery applications is in the range of 10–200 nm. Nanoparticles with hydrodynamic diameters beyond this range can be cleared from circulation either through the mononuclear phagocyte system (MPS) or the renal pathway more rapidly.20 Moreover, the mean diameter of T1k2k-TOS-DOX micelles is in the desired size range to extravasate from the blood vessels into the tumor interstitium via the enhanced permeability and retention (EPR) effect.20,21 In TEM images, T1k2k-TOS and T1k2k-TOS-DOX micelles showed a generally spherical morphology with an average diameter of 40 and 20 nm, respectively (Figure 2A,D). The EE% of T1k2k-TOS-DOX micelles was up to 98.33 ± 1.37%, which is much higher than that of T1k2k-DOX micelles (43.68 ± 3.72%) (Table S5). This indicates that α-TOS exerts a great influence on the drug-encapsulating capacity of the micelles by increasing the lipophilic portion of the system, thus forming a strongly hydrophobic core.
Table 1.
Physicochemical Characterization of T1k2k-TOS and T1k2k-TOS-DOX Micelles
| formulation | average sizea (nm) | PDI | ζ potential (mV) | EE (%) | DL (%) |
|---|---|---|---|---|---|
| T1k2k-TOS | 83.13 ± 1.04 | 0.195 ± 0.004 | −14.6 ± 1.55 | ||
| T1k2k-TOS-DOX | 52.08 ± 3.34 | 0.234 ± 0.006 | −16.36 ± 1.53 | 98.33 ± 1.37 | 8.94 ± 0.10 |
Size distribution by volume.
Figure 2.

(A) TEM image and (B) DLS analysis of T1k2k-TOS micelles. (C) I1/I3 intensity ratio vs concentration of T1k2k micelles. (D) TEM image and (E) DLS analysis of T1k2k-TOS-DOX micelles. (F) Cumulative release profile of DOX from T1k2k-TOS-DOX micelles in solutions of pH = 5.5 and 7.4. Data are presented as mean ± SD (n = 3).
The main characteristic of micellar stability is the CMC, above which amphiphilic polymers self-assemble into nanosized structures.22 The CMC value of the fabricated micelles was determined using pyrene as a fluorescence probe. It was reported that the CMC value of TPGS 1k is relatively high (0.2 mg/mL), whereas the CMC of TPGS 2k is 10-fold lower (0.02 mg/mL), suggesting that the micelles fabricated from TPGS 2k are more stable.6,23 Mixed micelles comprising TPGS 1k and TPGS 2k (T1k2k) at an equal mass ratio (50/50) exhibited an intermediate value of CMC equal to 0.063 mg/mL (Figure 2C). These results are in accordance with the hypothesis that the addition of TPGS 2k to TPGS 1k can decrease the CMC value and hence improve the stability of the micelles. Practically, no significant changes were found in the particle size, size distribution, or ζ potential of the T1k2k-TOS-DOX micelles after 2 months storage at +4 °C (Table S6). However, under the same conditions, the solution containing T1k2k- DOX micelles clearly contained large insoluble aggregated precipitates, indicating that the micelles were unstable without α-TOS during the storage period (Figure S7). Thus, the data suggest that the presence of α-TOS in the system is crucially important as a stabilizing agent.
3.4. In Vitro Drug Release
Because the uptake of regular micelles by the cells is usually through the endocytic pathway, the release behavior of DOX from T1k2k-TOS-DOX was evaluated under different pH conditions: physiological pH (7.4), and pH of the lysosomal environment (5.5). As shown in Figure 2F, T1k2k-TOS-DOX micelles exhibited an obvious pH-dependent release behavior with an initial burst of drug release followed by sustained release. At 12 h, the cumulative drug release of DOX at pH = 5.5 was 54.8% compared with only 14.1% at pH = 7.4. After 48 h, the T1k2k-TOS-DOX micelles released 73% of the total amount of DOX at pH = 5.5 and 19.3% at pH = 7.4. The isoelectric point (pI) of DOX is 9.06; hence, decreasing the pH from 7.4 to 5.5 results in an increase in the positive charge of DOX molecules and hence in the electrostatic repulsion between them, which causes release of the drug from the micelles.24 This pH-sensitive feature is predicted to prevent drug release during circulation in the blood, keep the drug-loaded nanosystem intact before uptake by cancer cells, and induce sufficient release of DOX in the acidic lysosomes after endocytosis by the tumor cells.
3.5. In Vitro Cell Cytotoxicity
To evaluate the cytotoxic activities of T1k2k-TOS-DOX micelles, in vitro cell cytotoxicity experiments were performed with both sensitive MCF-7 and multidrug-resistant cells (MCF-7/Adr) using the MTT assay. The cells were incubated with different concentrations of DOX solution, T1k2k, T1k2k-TOS, and T1k2k-TOS-DOX micelles for 48 h. As shown in Figure 3A, both T1k2k-TOS-DOX micelles and DOX solution demonstrated marked cell cytotoxicity against sensitive MCF-7 cells after 48 h of treatment (IC50 of T1k2k-TOS-DOX micelles and DOX solution was 2.50 ± 0.15 and 3.44 ± 0.63, respectively; see Table 2). However, in resistant MCF/Adr cells (Figure 3B), DOX exhibited a negligible toxic effect (IC50 was 158.62 ± 4.68, Figure S8), whereas the formulated T1k2k-TOS-DOX micelles exhibited a much greater efficacy (IC50 was 3.52 ± 0.24). The developed T1k2k-TOS-DOX micelles increased the inhibitory potency of DOX in resistant cancer cells (45-fold) and reserved the cytotoxicity against sensitive cancer cells, indicating the ability of T1k2k-TOS-DOX micelles to overcome MDR. This might be largely determined by the following reasons: inhibition of the P-gp efflux pump by TPGS 1k, thereby increasing the concentration of intracellular DOX7,25 and mitochondrial impairment by α-TOS resulting in cell death. The second reason was supported by the data, which showed that the T1k2k-TOS micelles themselves also have a concentration-dependent cytotoxic effect, which is mainly caused by the activation of mitochondrial apoptosis by α-TOS.12,14 However, T1k2k-TOS micelles at a concentration of 50 µg/mL (a DOX-equivalent dose of 5.0 µg/mL) exhibited quite a low cytotoxic efficacy in comparison to T1k2k-TOS-DOX micelles. The data suggest that the success in the overcoming of MDR by T1k2k-TOS-DOX micelles was attained by the synergism between increased intracellular DOX accumulation due to P-gp inhibition provided by TPGS 1k and mitochondrial dysfunction caused by α-TOS.
Figure 3.

In vitro cytotoxicity of DOX, T1k2k, T1k2k-TOS, and T1k2k-TOS-DOX micelles against (A) MCF-7 and (B) MCF-7/Adr cells after 48 h of treatment. Data are presented as mean ± SD (n = 5). (C) Colony formation assays of MCF-7 and MCF-7/Adr cells. The histogram represents the number of colonies of MCF-7 and MCF-7/Adr cells after 7 days of treatment. Colonies of more than 50 cells were counted.
Table 2.
IC50 (µg/mL) of DOX, T1k2k, T1k2k-TOS, and T1k2k-TOS-DOX Micelles after 48 h of Incubation with MCF-7 and MCF-7/Adr Cells
| MCF-7 | MCF-7/Adr | |
|---|---|---|
| DOX | 3.44 ± 0.63 | 158.62 ± 4.68 |
| T1k2k | 7.14 ± 1.06 | 12.64 ± 1.54 |
| T1k2k-TOS | 4.98 ± 0.94 | 8.61 ± 1.12 |
| T1k2k-TOS-DOX | 2.50 ± 0.15 | 3.52 ± 0.24 |
3.6. Colony Formation
Colony formation assay was performed to analyze the ability of the cancer cells to proliferate and grow into colonies. MCF-7 and MCF-7/Adr cells were exposed to different formulations, and the number of colonies containing more than 50 cells was counted. As shown in Figure 3C, sensitive MCF-7 cells treated with T1k2k-TOS-DOX micelles or DOX had less number of colonies, whereas in resistant MCF-7/Adr cell lines, only the group exposed to T1k2k-TOS-DOX micelles exhibited a significantly reduced ability to colony formation. The results indicate that T1k2k- TOS-DOX micelles possess high antiproliferation capability not only against sensitive cells but also against resistant cell lines, which is in accordance with the cytotoxicity test that displayed the potency of T1k2k-TOS-DOX micelles to overcome MDR.
3.7. Cellular Uptake and Distribution
The internalization of T1k2k-TOS-DOX micelles into MCF-7/Adr cancer cells was visualized using CLSM. From Figure 4A, it can be seen that the cells which were incubated with T1k2k-TOS-DOX micelles displayed a much stronger red fluorescence compared with the cells treated with the DOX solution, indicating an increased cellular uptake of the mixed micelles. The red fluorescence emitted from DOX showed that the drug molecules were located throughout the cytoplasm and were concentrated around the nucleus, while a small amount of DOX penetrated into the nucleus. The results indicate that the encapsulation of the drug into the mixed micelle nanocarrier is able to significantly improve drug penetration and accumulation within the cells, leading to enhanced therapeutic potency against malignancies.
Figure 4.

(A) Fluorescence intensity of DOX in MCF-7/Adr cells visualized using CLSM. (B,C) Fluorescence intensity of DOX in MCF-7/Adr cells measured using flow cytometry. (D) Fluorescence intensity of DOX in MCF-7/Adr cells preincubated with verapamil, T1k2k-TOS, T1k2k, and cell culture medium (control), visualized using CLSM. (E) Fluorescence intensity of DOX in MCF-7/Adr cells preincubated with verapamil, T1k2k- TOS, T1k2k, and cell culture medium (control) at different time points, measured using flow cytometry. Data are presented as mean ± SD (n = 3). Notes: **, p < 0.01. The scale bar is 20 µm.
The enhanced uptake of T1k2k-TOS-DOX over DOX by MCF-7/Adr cells was further examined using flow cytometry (Figure 4B,C). The intracellular mean fluorescence intensity of T1k2k-TOS-DOX micelles was 5.3-fold higher than that of DOX (p < 0.01), confirming the superior concentration of micellar DOX in cells. These results were in accordance with the CLSM images described above.
The difference in the cellular uptake between the drug-loaded micelles and the free drug may be ascribed to their distinct uptake mechanisms.20,24,26 Moreover, the increased level of the intracellular drug might be due to the ability of TPGS 1k to inhibit drug efflux mediated by P-gp.
Subcellular localization results showed that the red fluorescence of micellar DOX was mainly colocalized with the green fluorescence of lysosomes at the early beginning of uptake (Figure S9). The data confirmed that T1k2k-TOS-DOX micelles were internalized principally through the endocytic pathway, which is consistent with previous reports about other nanoparticles.26
3.8. Evaluation of the Inhibition of P-gp Efflux
The micelle-induced inhibition of P-gp was investigated in comparison to the well-known P-gp inhibitor verapamil. To exclude the influence of the enhanced cellular uptake of micellar DOX, blank T1k2k-TOS and T1k2k micelles were used. The cells were pretreated with T1k2k, T1k2k-TOS micelles, or verapamil; then, DOX was added, and its fluorescence intensity was observed using CLSM (Figure 4D). The red fluorescence intensity of cells pretreated with verapamil, T1k2k-TOS, or T1k2k was more obvious than that of cells treated with the individual free DOX. The fluorescence intensities of verapamil, T1k2k-TOS, and T1k2k differ insignificantly, showing great ability of nanoformulations to decrease drug efflux on par with verapamil, leading to enhanced retention of drug in the cells. Later, flow cytometry results obtained at different time points confirmed that the fluorescence of DOX in cells pretreated with T1k2k-TOS and T1k2k was significantly higher compared with cells exposed only to DOX (p < 0.05) even after the culture medium was refreshed (Figure 4E). It is worth noting that, the difference between T1k2k-TOS and T1k2k is not obvious, revealing that the absence of α-TOS in the structure of T1k2k does not affect the P-gp inhibition ability. In addition, the intracellular drug concentration in the cells pretreated with T1k2k-TOS and T1k2k, as judged by the fluorescence intensity, was similar to the positive control group that was pretreated with verapamil. This confirmed that both T1k2k-TOS and T1k2k micelles have comparable effectiveness with verapamil at inhibiting the drug efflux associated with the P-gp function. These results, in conjunction with the cellular uptake studies, suggested that the higher uptake and the decreased efflux of the encapsulated drug contribute to the higher retention of drug in the cells treated with mixed micelles.
3.9. ROS Production
It is well-known that α-TOS can promote the ROS generation leading to cancer cell apoptosis. One of the main reasons for the generation of ROS is related to the ability of α-TOS to impair the mitochondria.12 The destabilization of the mitochondrial membrane by α-TOS results in cytosolic relocalization of pro-apoptotic factors, which in turn activates the caspase cascade causing programmed cell death.11,12 In this regard, it is reasonable to detect the ability of mixed micelles to induce intracellular ROS generation in MCF-7/Adr cells. The fluorescent dye, DCFH-DA, was used to probe the oxidative species. According to the results (Figure 5A), the ROS level was significantly enhanced in cells treated with T1k2k-TOS and T1k2k-TOS-DOX micelles, whereas no obvious changes were observed for the control and DOX groups, confirming the ability of mixed micelles to induce the intracellular accumulation of ROS. Free DOX did not display an ability to trigger ROS production compared with the control group, which was also confirmed by the insignificant effect between T1k2k-TOS and T1k2k-TOS-DOX. In addition, the cells treated with the T1k2k formulation showed an intermediate value of DCF fluorescence because of lesser amount of α-TOS, which can be produced by the hydrolysis of TPGS 1k and TPGS 2k. On the basis of the obtained results, it can be concluded that the mixed micelles have a great potency to trigger ROS generation.
Figure 5.

Mitochondria-specific apoptotic pathways. (A) Relative fluorescence intensity of DCF measured using flow cytometry after MCF-7/Adr cells were exposed to DOX, T1k2k, T1k2k-TOS, or T1k2k-TOS-DOX micelles. (B) Mitochondrial depolarization in MCF-7/Adr cells imaged using CLSM. Effect of DOX, T1k2k, T1k2k-TOS, and T1k2k-TOS-DOX micelles on (C) caspase-9 and (D) caspase-3 activity in MCF-7/Adr cells. Data are presented as mean ± SD (n = 3). Notes: *, p < 0.05; **, p < 0.01; ***, p < 0.001; ns, not significant. The scale bar is 20 µm.
3.10. Mitochondrial Depolarization
As mentioned above, the α-TOS molecule is able to disrupt mitochondrial function through the production of intracellular ROS, and as a result, causes decreased MMP. To investigate the changes in MMP in MCF-7/Adr cells treated with micelles containing α-TOS, a lipophilic cationic dye (JC-1) was used. JC-1 is present as a green monomer if the mitochondrial membrane is depolarized or as a red-emitting J-aggregate in the hyperpolarized mitochondria. The change in the fluorescence of the mitochondria of MCF-7/Adr cells was investigated using CLSM. For this assay, T1k2k-TOS-DOX micelles could not be used because of similar excitation (488 nm) and emission (maximum at 530, 590, and 560–590 nm) of J-monomers, J-aggregates, and DOX, respectively. This could lead to overlaps and inability to distinguish the different fluorescent signals. Therefore, the capability of T1k2k and T1k2k-TOS micelles to disrupt the mitochondrial function was investigated (Figure 5B). Control untreated cells exhibited both red and green fluorescence. However, the intensity of red fluorescence was obviously higher. Exposure of the cells to the uncoupling agent CCCP, a positive control, caused a dramatic decrease in the intensity of the red fluorescence and an enhanced intensity of the green fluorescence, illustrating mitochondrial membrane depolarization. Similar to the positive control, treatment with T1k2k-TOS micelles caused a shift from red to green in the fluorescence emission of JC-1, verifying that the mixed micelles have the potential to damage the membrane of mitochondria. For T1k2k group, the red–green shift was detected in a lesser degree, which might be attributed to a lesser amount of α-TOS produced by the hydrolysis of TPGS 1k and TPGS 2k. The low effect of T1k2k on mitochondria depolarization is consistent with its corresponding ability of ROS production.
3.11. Caspase-9 and Caspase-3 Activity Assays
Previous results demonstrated that α-TOS, which was present in the core of mixed micelles as a hydrophobic adjuvant, promoted increased generation of ROS, leading to mitochondria membrane depolarization that ultimately triggers caspase-9 and caspase-3 apoptotic pathways. The activities of caspase-9 and caspase-3 were evaluated by a colorimetric assay using peptide substrates that release the solvatochromic dye p-nitroaniline (pNA) upon cleavage by a caspase. As shown in Figure 5C,D, T1k2k-TOS and T1k2k-TOS-DOX possess a much higher ability, in comparison to control and T1k2k, to activate caspase-9 and caspase-3 apoptotic pathways. The effect of T1k2k-TOS and T1k2k-TOS-DOX is almost equal, concluding that the encapsulated DOX has negligible effect on caspase activation. This was confirmed by the inappreciable change in the caspase activity in cells treated with free DOX. It was also shown that T1k2k had a minor ability to activate caspases, which is consistent with the results of ROS generation and mitochondrial depolarization studies. The obtained results suggest that, owing to the presence of α-TOS in the system, T1k2k-TOS-DOX micelles have a high capability to activate caspase-9 and caspase-3.
In brief, the enhanced cytotoxicity of T1k2k-TOS-DOX micelles against MCF-7/Adr cells can be attributed to the significant increase in the cellular uptake and a substantial decrease in the DOX efflux. Moreover, the mixed micelles demonstrated an ability to disrupt mitochondrial function, thus contributing to the anticancer effect of the micelles.
3.12. In Vivo Biodistribution
To examine the distribution of the mixed micelles throughout the body, a real-time imaging assay was performed. DiR-loaded micelles were administrated intravenously, whereas PBS and free DiR were used as controls. The fluorescence of DiR was observed at designated time points after injection. The results demonstrated that, at 0.5 h, the micellar DiR fluorescence intensity was detected in the whole body. As time elapsed, the fluorescence signal was gradually intensified at the tumor site. The highest fluorescence level of DiR-loaded micelles was reached after 8 h, and the noticeable fluorescence intensity was maintained up to 48 h, whereas almost no signal was detected for free DiR (Figure 6A). Further ex vivo imaging confirmed high accumulation level of the micellar DiR dye in the tumor tissue, whereas no obvious florescence intensity was observed in the heart, kidney, and lung tissues at 48 h post injection (Figure 6B). The data suggest that the nanomicellar formulations have a desirable distribution and possess a high ability to improve the delivery and accumulation of payload at the tumor region, which can be achieved by a prolonged circulation time and the EPR effect. It also revealed that the nanomicellar system in a lesser degree can be uptaken by the reticuloendothelial system (RES), which was reflected in the fluorescence signal from the liver and spleen. However, further histological analysis did not reveal any significant damage of liver tissues.
Figure 6.

(A) In vivo imaging of tumor-bearing mice after 0.5, 3, 8, 12, 24, and 48 h of administration with PBS, DiR, and T1k2k-TOS-DiR. The tumor area of the mouse treated with T1k2k-TOS-DiR is indicated by oval. (B) Ex vivo fluorescence imaging of excised organs: tumor, liver, kidney, spleen, heart, and lung after 48 h of treatment.
In general, the biodistribution test demonstrated improved in vivo delivery and enhanced intratumoral accumulation of micellar cargo.
3.13. In Vivo Antitumor Effect
To verify the in vivo antitumor ability of the developed micelles, NSG mice bearing tumors derived from resistant breast cancer MCF-7/Adr cells were used. Five groups (n = 6) of mice were intravenously injected with saline, DOX, T1k2k, T1k2k-TOS, and T1k2k-TOS-DOX at a dose corresponding to 5 mg/kg of DOX. The amounts of T1k2k and T1k2k-TOS are equal to those contained in T1k2k-TOS-DOX. The drugs were administrated every 3 days, and the body weight and the tumor size were measured every 2 days (Figure 7). At the end of the treatment, DOX showed little inhibition of the tumor growth and T1k2k-TOS displayed moderate ability of tumor inhibition, whereas T1k2k-TOS-DOX exhibited obvious antitumor activity that was confirmed by the insignificant difference in the tumor volume at the beginning and end of treatment (Figure 7A). A much higher tumor inhibition level of T1k2k-TOS-DOX over DOX and T1k2k-TOS indicates the synergistic effect between them.
Figure 7.

(A) Tumor volume and (B) mice body weight changes after treatment with saline, DOX, T1k2k, T1k2k-TOS, and T1k2k-TOS-DOX. Data are presented as mean ± SD (n = 6) (C) survival curves of mice after different treatments (n = 5). (D) Histological analysis (H&E staining) of tumor, liver, and heart tissues after treatment with saline, DOX, T1k2k, T1k2k-TOS, and T1k2k-TOS-DOX.
During the treatment period, no significant alteration in the body weight was observed for mice treated with T1k2k, T1k2k-TOS, and T1k2k-TOS-DOX (Figure 7B). By contrast, mice injected with the DOX solution exhibited a considerable weight loss, and it is worth noting that all of the mice in this group were dead by day 13 of the treatment, suggesting high toxicity of the free drug at this dose. Later, it was revealed that the survival curve of the mice treated with T1k2k-TOS-DOX was much longer than that of the other groups (Figure 7C).
Further histological analysis was performed to evaluate the toxicity of different treatments. It is well-known that DOX possesses severe cardiotoxicity and hepatotoxicity.27–29 Hematoxylin and eosin (H&E) staining confirmed serious myocardial fiber necrosis, which is indicated by black arrows in the cardiac tissues and in the liver tissues of the group treated with free DOX (Figure 7D). By contrast, no visible tissue damage was detected for the T1k2k-TOS-DOX group, indicating that the cardiotoxicity and hepatotoxicity of DOX were circumvented by the T1k2k-TOS-DOX micelles. The components of the delivery system, T1k2k and T1k2k-TOS, did not exhibit any heart or liver tissue damage, which was understandable because vitamin E derivatives possess good biocompatibility. The analysis of tumor tissues revealed widespread necrosis after treatment with T1k2k-TOS-DOX, whereas no significant changes were registered for DOX and T1k2k. The blank T1k2k-TOS micelles demonstrated moderate tumor tissue necrosis due to antitumor activity of α-TOS.
In brief, in vivo antitumor experiments revealed that T1k2k- TOS-DOX micelles were endowed with significant tumor inhibition ability and negligible toxicity. It circumvented the cardiotoxicity and hepatotoxicity of DOX and prolonged the survival time of mice. This can be attributed to the enhanced accumulation of DOX at the tumor site, decreased damage of heart and liver, and synergism between DOX and α-TOS. The in vivo results support the possibility of T1k2k-TOS-DOX micelles as a potential antitumor drug to circumvent MDR in clinical usage.
4. CONCLUSIONS
This study presented mixed micelles as a delivery system based on vitamin E derivatives, which could efficiently enhance the antitumor activity of DOX and overcome drug resistance, while circumventing cardio- and hepatotoxicities. The drug-loaded micelles T1k2k-TOS-DOX demonstrated essential properties of nanocarriers, such as a suitable size for the EPR effect, enhanced drug loading ability, high stability behavior, and pH-dependent drug release. The prepared drug-encapsulated micelles resulted in an increased intracellular drug accumulation due to high cellular uptake and a reduced P-gp-mediated drug efflux. Furthermore, the presence of α-TOS in the system as a stabilizing agent resulted in a selective therapeutic effect against cancer cells and synergistic interaction with DOX. High accumulation of DOX and its synergism with α-TOS contribute to markedly improved cytotoxicity against the resistant cancer cell lines (45-fold higher than DOX). The simulative in vivo distribution experiment confirmed that the delivery system endowed the cargo with the preferable distribution into the tumor site. In vivo antitumor experiments verified a significantly enhanced MDR-tumor inhibition activity of T1k2k-TOS-DOX micelles with reduced toxicity. In brief, the developed mixed micelles could be considered as a promising and effective drug carrier in tumor therapy.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (grant no. 31600810), the Beijing Natural Science Foundation (grant no. 7174332), the Natural Science Foundation key project (31630027 and 31430031), and the National Distinguished Young Scholars grant (31225009). The authors also appreciate the support by the Strategic Priority Research Program (XDA09030301), the External Cooperation Program of BIC (121D11KYSB20130006) of the Chinese Academy of Sciences, and the CAS-TWAS President’s PhD Fellowship Program.
Footnotes
ASSOCIATED CONTENT
Supporting Information
Synthesis scheme of TPGS 2k, characterization of TPGS 1k and TPGS 2k, P-gp inhibition by TPGS 1k and TPGS 2k, results of optimization of the carrier’s composition, CMC values of micelles with different contents of TPGS 1k and TPGS 2k, stability data, cytotoxicity of DOX against MCF-7/Adr cells, and CLSM images of distribution of T1k2k-TOS-DOX or DOX in MCF-7/ Adr cells (PDF)
The authors declare no competing financial interest.
References
- 1.Chorawala M, Oza P, Shah G. Mechanisms of Anticancer Drugs Resistance: An Overview. Int. J. Pharm. Sci. Drug Res. 2012;4:1–9. [Google Scholar]
- 2.Ambudkar SV, Kimchi-Sarfaty C, Sauna ZE, Gottesman MM. P-Glycoprotein: From Genomics to Mechanism. Oncogene. 2003;22:7468–7485. doi: 10.1038/sj.onc.1206948. [DOI] [PubMed] [Google Scholar]
- 3.Kerb R, Hoffmeyer S, Brinkmann U. Abc Drug Transporters: Hereditary Polymorphisms and Pharmacological Impact in Mdr1, Mrp1 and Mrp2. Pharmacogenomics. 2001;2:51–64. doi: 10.1517/14622416.2.1.51. [DOI] [PubMed] [Google Scholar]
- 4.Abdallah HM, Al-Abd AM, El-Dine RS, El-Halawany AM. P-Glycoprotein Inhibitors of Natural Origin as Potential Tumor Chemo-Sensitizers: A Review. J. Adv. Res. 2015;6:45–62. doi: 10.1016/j.jare.2014.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ozben T. Mechanisms and Strategies to Overcome Multiple Drug Resistance in Cancer. FEBS Lett. 2006;580:2903–2909. doi: 10.1016/j.febslet.2006.02.020. [DOI] [PubMed] [Google Scholar]
- 6.Duhem N, Danhier F, Préat V. Vitamin E-Based Nanomedicines for Anti-Cancer Drug Delivery. J. Controlled Release. 2014;182:33–44. doi: 10.1016/j.jconrel.2014.03.009. [DOI] [PubMed] [Google Scholar]
- 7.Collnot E-M, Baldes C, Wempe MF, Kappl R, Hüttermann J, Hyatt JA, Edgar KJ, Schaefer UF, Lehr CM. Mechanism of Inhibition of P-Glycoprotein Mediated Efflux by Vitamin E Tpgs: Influence on Atpase Activity and Membrane Fluidity. Mol. Pharm. 2007;4:465–474. doi: 10.1021/mp060121r. [DOI] [PubMed] [Google Scholar]
- 8.Collnot E-M, Baldes C, Schaefer UF, Edgar KJ, Wempe MF, Lehr C-M. Vitamin E Tpgs P-Glycoprotein Inhibition Mechanism: Influence on Conformational Flexibility, Intracellular Atp Levels, and Role of Time and Site of Access. Mol. Pharm. 2010;7:642–651. doi: 10.1021/mp900191s. [DOI] [PubMed] [Google Scholar]
- 9.Collnot E-M, Baldes C, Wempe MF, Hyatt J, Navarro L, Edgar KJ, Schaefer UF, Lehr C-M. Influence of Vitamin E Tpgs Poly(Ethylene Glycol) Chain Length on Apical Efflux Transporters in Caco-2 Cell Monolayers. J. Controlled Release. 2006;111:35–40. doi: 10.1016/j.jconrel.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 10.Wu SH-W, Hopkins WK. Characteristics of D-α-Tocopheryl PEG 1000 Succinate for Applications as an Absorption Enhancer in Drug Delivery Systems. Pharm. Technol. 1999;23:52–68. [Google Scholar]
- 11.Liang D, Wang A-t, Yang Z-z, Liu Y-j, Qi X-r. Enhance Cancer Cell Recognition and Overcome Drug Resistance Using Hyaluronic Acid and α-Tocopheryl Succinate Based Multifunctional Nanoparticles. Mol. Pharm. 2015;12:2189–2202. doi: 10.1021/acs.molpharmaceut.5b00129. [DOI] [PubMed] [Google Scholar]
- 12.Neuzil J, Dong L-F, Ramanathapuram L, Hahn T, Chladova M, Wang X-F, Zobalova R, Prochazka L, Gold M, Freeman R, Turanek J, Akporiaye ET, Dyason JC, Ralph SJ. Vitamin E Analogues as a Novel Group of Mitocans: Anti-Cancer Agents That Act by Targeting Mitochondria. Mol. Aspects Med. 2007;28:607–645. doi: 10.1016/j.mam.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 13.Neuzil J, Weber T, Gellert N, Weber C. Selective Cancer Cell Killing by α-Tocopheryl Succinate. Br. J. Cancer. 2001;84:87. doi: 10.1054/bjoc.2000.1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Neuzil J. Vitamin E Succinate and Cancer Treatment: A Vitamin E Prototype for Selective Antitumour Activity. Br. J. Cancer. 2003;89:1822–1826. doi: 10.1038/sj.bjc.6601360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee K-Y, Chiang Y-T, Hsu N-Y, Yang C-Y, Lo C-L, Ku C-A. Vitamin E Containing Polymer Micelles for Reducing Normal Cell Cytotoxicity and Enhancing Chemotherapy Efficacy. Acta Biomater. 2015;24:286–296. doi: 10.1016/j.actbio.2015.06.014. [DOI] [PubMed] [Google Scholar]
- 16.Tao Y, Han J, Wang X, Dou H. Nano-Formulation of Paclitaxel by Vitamin E Succinate Functionalized Pluronic Micelles for Enhanced Encapsulation, Stability and Cytotoxicity. Colloids Surf., B. 2013;102:604–610. doi: 10.1016/j.colsurfb.2012.08.062. [DOI] [PubMed] [Google Scholar]
- 17.Mi Y, Liu Y, Feng S-S. Formulation of Docetaxel by Folic Acid-Conjugated D-α-Tocopheryl Polyethylene Glycol Succinate 2000 (Vitamin E Tpgs2k) Micelles for Targeted and Synergistic Chemotherapy. Biomaterials. 2011;32:4058–4066. doi: 10.1016/j.biomaterials.2011.02.022. [DOI] [PubMed] [Google Scholar]
- 18.Hao T, Chen D, Liu K, Qi Y, Tian Y, Sun P, Liu Y, Li Z. Micelles of D-α-Tocopheryl Polyethylene Glycol 2000 Succinate (Tpgs 2k) for Doxorubicin Delivery with Reversal of Multidrug Resistance. ACS Appl. Mater. Interfaces. 2015;7:18064–18075. doi: 10.1021/acsami.5b04995. [DOI] [PubMed] [Google Scholar]
- 19.Danhier F, Kouhé TTB, Duhem N, Ucakar B, Staub A, Draoui N, Feron O, Préat V. Vitamin E-Based Micelles Enhance the Anticancer Activity of Doxorubicin. Int. J. Pharm. 2014;476:9–15. doi: 10.1016/j.ijpharm.2014.09.028. [DOI] [PubMed] [Google Scholar]
- 20.Sun T, Zhang YS, Pang B, Hyun DC, Yang M, Xia Y. Engineered Nanoparticles for Drug Delivery in Cancer Therapy. Angew. Chem., Int. Ed. 2014;53:12320–12364. doi: 10.1002/anie.201403036. [DOI] [PubMed] [Google Scholar]
- 21.Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor Vascular Permeability and the Epr Effect in Macromolecular Therapeutics: A Review. J. Controlled Release. 2000;65:271–284. doi: 10.1016/s0168-3659(99)00248-5. [DOI] [PubMed] [Google Scholar]
- 22.Kataoka K, Harada A, Nagasaki Y. Block Copolymer Micelles for Drug Delivery: Design, Characterization and Biological Significance. Adv. Drug Delivery Rev. 2001;47:113–131. doi: 10.1016/s0169-409x(00)00124-1. [DOI] [PubMed] [Google Scholar]
- 23.Zhang Z, Tan S, Feng S-S. Vitamin E Tpgs as a Molecular Biomaterial for Drug Delivery. Biomaterials. 2012;33:4889–4906. doi: 10.1016/j.biomaterials.2012.03.046. [DOI] [PubMed] [Google Scholar]
- 24.Wei T, Liu J, Ma H, Cheng Q, Huang Y, Zhao J, Huo S, Xue X, Liang Z, Liang X-J. Functionalized Nanoscale Micelles Improve Drug Delivery for Cancer Therapy in Vitro and in Vivo. Nano Lett. 2013;13:2528–2534. doi: 10.1021/nl400586t. [DOI] [PubMed] [Google Scholar]
- 25.Dintaman JM, Silverman JA. Inhibition of P-Glycoprotein by D-α-Tocopheryl Polyethylene Glycol 1000 Succinate (TPGS) Pharm. Res. 1999;16:1550–1556. doi: 10.1023/a:1015000503629. [DOI] [PubMed] [Google Scholar]
- 26.Iversen T-G, Skotland T, Sandvig K. Endocytosis and Intracellular Transport of Nanoparticles: Present Knowledge and Need for Future Studies. Nano Today. 2011;6:176–185. [Google Scholar]
- 27.Ichikawa Y, Ghanefar M, Bayeva M, Wu R, Khechaduri A, Prasad SVN, Mutharasan RK, Naik TJ, Ardehali H. Cardiotoxicity of Doxorubicin Is Mediated through Mitochondrial Iron Accumulation. J. Clin. Invest. 2014;124:617–630. doi: 10.1172/JCI72931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Doroshow JH, Locker GY, Baldinger J, Myers CE. The Effect of Doxorubicin on Hepatic and Cardiac Glutathione. Res. Commun. Chem. Pathol. Pharmacol. 1979;26:285–295. [PubMed] [Google Scholar]
- 29.Mostafa MG, Mima T, Ohnishi ST, Mori K. S-Allylcysteine Ameliorates Doxorubicin Toxicity in the Heart and Liver in Mice. Planta Med. 2000;66:148–151. doi: 10.1055/s-2000-11124. [DOI] [PubMed] [Google Scholar]