

Abstract
Alterations in various developmental pathways are common themes in cancer. The early B-cell factors (EBFs) are a family of four highly conserved DNA-binding transcription factors with an atypical zinc finger and helix-loop-helix motif. They are involved in the differentiation and maturation of several cell lineages including B-progenitor lymphoblasts, neuronal precursors, and osteoblast progenitors. During B-cell development, EBF1 is required for the expression of Pax5, an essential factor for the production of antibody-secreting cells. Accumulating evidence indicates that genomic deletion of the EBF1 gene contributes to the pathogenesis, drug resistance and relapse of B-progenitor acute lymphoblastic leukemia (ALL). Epigenetic silencing and genomic deletion of the EBF3 locus in chromosome 10q are very frequent in glioblastoma (GBM). Strikingly, the frequency of EBF3 loss in GBM is similar to that of the loss of Pten, a key suppressor of gliomagenesis. Cancer-specific somatic mutations were detected in EBF3 in GBM and in both EBF1 and EBF3 in pancreatic ductal adenocarcinoma. These missense mutations occur in the DNA-binding domain or the conserved IPT/TIG domain, suggesting that they might disrupt the functions of these two proteins. Functional studies revealed that EBF3 represses the expression of genes required for cell proliferation (e.g., cyclins and CDKs) and survival (e.g., Mcl-1 and Daxx) but activates those involved in cell cycle arrest (e.g., p21 and p27), leading to growth suppression and apoptosis. Therefore, EBFs represent new tumor suppressors whose inactivation blocks normal development and contributes to tumorigenesis of diverse types of human cancer.
Introduction
Two key genetic events underlie the multi-step process of tumorigenesis. Tumor suppressor genes are inactivated through somatic point mutations, deletion and epigenetic silencing, and oncogenes are activated through gain-of-function somatic mutations, genomic amplification and transcriptional overexpression. It is now clear that a relatively small number of oncogenes and tumor suppressors such as Ras, PI3K, p53, Pten, Rb and p16INK4a are frequently mutated (1–4). Several recent large-scale sequencing studies have confirmed the high prevalence of mutations of these genes across diverse cancer types (1–6). These common mutations are believed to be the driving force in tumorigenesis. One surprising discovery from these studies is that, in addition to a few frequently mutated genes, specific tumors display low-frequency mutations in a diverse array of genes. The significance of these low-frequency mutations in tumorigenesis remains unclear. They might play a causal role, or they may be the so-called passenger mutations that do not contribute to cancer etiology or progression. Regardless, the diversity of genes with low-frequency mutations in different tumors reflects the notorious complexity of cancer.
Low frequency cancer-specific mutations have been observed in genes encoding two members of the early B-cell factors (EBF, or Olfactory neuronal transcription factor-1, Olf-1) in glioblastoma (GBM) (3) and pancreatic cancer (2). In a genome-wide screen for putative tumor suppressor genes, Zardo et al. found that the EBF3 locus on the human chromosome 10q is deleted or methylated in brain tumors (7). Thus, it is likely that EBF3 may function as a tumor suppressor in the brain. Furthermore, deletion of the EBF1 locus occurs in B-progenitor acute lymphoblastic leukemia (ALL) (8). Functional studies revealed that EBF3 activates genes involved in cell cycle arrest and apoptosis while repressing genes involved in cell survival and proliferation (9). Collectively, these studies suggest that EBF transcription factors act as tumor suppressors in several types of cancer. Biologically, these factors are involved in various developmental pathways including B-cell differentiation (10), neurogenesis (11–13) and bone development (14). Here, we introduce the structure and function of the EBF family of transcription factors, and will focus on the emerging evidence of their genetic and epigenetic inactivation in cancers as well as discuss the potential mechanisms by which these factors function in tumor suppression.
Structure and biochemical functions of EBFs
In 1993, Hagman et al. purified EBF1 from the nuclear extracts of a murine pre-B cell line through oligonucleotide affinity chromatography (15). The DNA sequence of the oligonucleotide was derived from the promoter of the Cd79a gene (also known as mb-1) encoding the Ig-α signaling molecule of the B cell receptor (also known as B-cell antigen receptor complex-associated protein alpha chain, or MB-1 membrane glycoprotein), an immunoglobulin-associated protein that is produced in the early stages of B-lymphocyte differentiation. EBF1 is essential for B-cell development, as mice lacking the Ebf1 gene do not produce functional B cells and immunoglobulins (16). In the same year, cDNA encoding a related factor called Olf-1 (orthologous to the human and mouse EBF1) was identified by screening rat cDNA clones that activated a reporter gene in yeast under the control of a synthetic promoter containing DNA elements derived from olfactory specific genes (17).
EBFs are found throughout the animal kingdom in organisms as diverse as Drosophila melanogaster, Caenorhabditis elegans, and humans (18). Invertebrate species carry a single EBF gene in their genomes (18). The ortholog in Drosophila is also known as collier. Together with their mammalian counterparts (Olf-1/EBF), these factors are also referred to as Olf-1/EBF (O/E) or Collier/Olf-1/EBF (COE). In the genomes of mice, humans and other vertebrates, there are four paralogous genes of the EBF family (EBF1-4). In humans, the EBF1-4 genes map to chromosomes 5q34, 8p21.2, 10q26.3 and 20p13, respectively. Similar to other DNA-binding transcription factors, EBF proteins contain well-defined modular domains with distinct structures and functions (Fig. 1). The DNA-binding domain (DBD) near the N-terminus consists of approximately 200 residues whose sequence is evolutionarily well conserved with >75% sequence identity between distant species. These proteins bind directly to DNA with a consensus sequence of 5′-CCCNNGGG-3′ as homo- or heterodimers (19) (Fig. 2). Within the DBD, there is a sequence motif that defines the signature of the EBF family (termed COE). This signature sequence is an atypical zinc finger (H-X3-C-X2-C-X5-C), and is absolutely required for DNA binding (20, 21). Mutation of any of the four Zn2+-binding residues to alanine abolishes the DNA-binding activity of EBF1 (21). Furthermore, the presence of Zn2+ or Cd2+ cations is required for EBF1 to bind to DNA in vitro (20). Thus, this motif is a bona fide Zn2+-binding structure. Because of the short length of this motif compared to other known zinc-binding structures, Fields et al. have designated this motif the ‘zinc knuckle’ (Zn-knuckle) of EBF (21).
FIGURE 1. The structural features of EBF family of transcription factors.

(A) Shown are the four paralogs (EBF1-4) and their corresponding accession numbers (GenBank or SwissProt databases). Specific domains are shown in shaded or striped boxes. Numbers refer to the position of amino acid residue in each protein. Cancer specific somatic mutations in EBF1 and EBF3 are indicated. The different domains are: DBD, DNA-binding domain; TAD, transactivation domain; COE, the signature sequence of EBF family; ZNF, zinc-finger motif; IPT/TIG (Immunoglobulin-like fold, Plexins, Transcription factors, or Transcription factor Immunoglobin); HLH, helix-loop-helix motif. Note that the ancestral EBF appears to have a typical HLH motif (H1LH2a) that lacks the H2b helix. The H2a and H2b appear to have arisen as a result of an exonic duplication in the vertebrates. (B) The signature zinc-finger motif of the EBF family of proteins in complex with zinc is depicted. The adjacent amino acid residues are also shown. This motif has been designated a zinc knuckle.
FIGURE 2. EBF mediates transcriptional activation and repression.

EBF1 binds to DNA as a dimer. Both the HLH motif and the DBD are involved in dimerization. EBF1 and EBF3 have been shown to interact with p300. This interaction is likely to underlie EBF-mediated gene activation through acetylation of core histones by p300 and related coactivator CBP. Several known EBF1 target genes are indicated.
EBF1 binds to the two related large zinc finger proteins Zfp423 and Zfp521. The COOH-terminal-most zinc fingers (28–30) interact with EBF1, and the N-terminal zinc fingers 2–8 bind to DNA. The Mi-2/NuRD (nucleosome remodeling and deacetylase) complex appears to bind to a conserved NH2-terminal motif in these two zinc finger proteins. Thus, EBF in conjunction with Zfp423/Zfp521 recruits the Mi-2/NuRD complex for EBF-mediated gene repression. The known components of the Mi-2/NuRD complex are depicted. ZF: zinc finger; MDB: the methyl CpG-binding domain; RbA: retinoblastoma associated protein A (also known as Rbbp4 (p48) and Rbbp7 (p46)); MTA, metastasis associated proteins (MTA1, 2 and 3).
The region C-terminal to the DBD shares sequence similarity with the conserved domains called IPT/TIG (Immunoglobulin-like fold, Plexins, Transcription factors, or Transcription factor Immunoglobin). Although the function of IPT/TIG has not been determined, it might function through protein interactions via homo- or heterodimerization with other proteins. An atypical helix-loop-helix (HLH) motif is found COOH-terminal to the IPT/TIG domain. In vertebrate EBFs, this motif consists of three α-helices. The first and second helices are similar to the typical HLH domains of v-myc and MyoD. Interestingly, the amino acid sequence of the third α-helix is nearly identical to that of the second one. Thus, this atypical structure of the vertebrate EBFs is better described as the HLHLH motif. Cross-species sequence analysis of the EBF family proteins suggests that the two similar helices probably arose due to an exonic duplication in vertebrates. Thus, the HLH motif in the EBF proteins of invertebrate species such as D. melanogaster still contains two dissimilar helices (18). Experimental data suggest that the HLH motif of the EBF proteins is involved in dimerization, as deletion of this motif abolishes stable dimer formation in solution (18, 20)(also see Fig. 2).
The less-conserved COOH-terminal domain has a major role in transcriptional activation. This domain is rich in serine, threonine and proline residues. Additional transactivation domain might exist, as COOH-terminal deletion mutants retain transactivation activity (20). Specifically, while the DBD alone did not promote transactivation, a construct encompassing amino acid residues 18–429 of the mouse EBF1 potently activated transcription of an artificial reporter gene construct containing two EBF-binding DNA sequences (20). This suggests that the ITP/TIG domain, the HLHLH domain, or both, could mediate transactivation.
EBF1 was found to interact with the coactivator p300 (22). Surprisingly, this appears to inhibit the histone acetyltransferase (HAT) activity of p300. EBF1 binds to both the HAT domain and the cysteine-histidine-rich region 3 (CH3) domain of p300 (22). Additionally, both EBF1 and EBF3 can interact with the COOH-terminal interferon-binding domain (IBiD) of p300 (our unpublished data). It has been shown that IBiD can bind promiscuously to diverse transcription factors that share no obvious sequence similarity, such as p53 and Ets-2 (23). Thus, it is likely that EBFs also recruit p300 and related cAMP response element binding protein-binding protein (CBP) to activate transcription (Fig. 2).
EBFs in development
EBF1 in B lymphocyte development
It has been extensively documented that EBF1 is a master regulator for B cell development (24–26). Ebf1-deficient mice produce only B-biased progenitor cells but not mature B cells (16). Ebf1-deficient cells fail to express B-cell markers such as the immunoglobulins. Mouse knock-out studies suggest a hierarchical model of early B cell commitment wherein E2A proteins (E47 and E12, two alternative splicing variants of the E2A gene, which homo- or heterodimerize via their helix–loop–helix domains) activate Ebf1, whose gene product in turn activates the production of Pax5, an essential regulator of B cell identity and function (10, 27) (Fig. 3). Interestingly, there appears to be a feed-forward loop between EBF1 and Pax5. Whereas EBF1 is required for activating Pax5 expression, the latter enhances EBF1 expression (28) (Fig. 3). In pre-pro–B cells (the first specified B cell progenitors), deficiency of the E2A-encoding gene (Tcfe2a) results in impaired expression of EBF1. Retroviral expression of Ebf1 rescues the expression of most early B lineage genes (24, 29–31). Thus, EBF1 has a critical role in inducing the specification of the B cell fate.
FIGURE 3. EBF1 is a critical component of the B cell pathway.
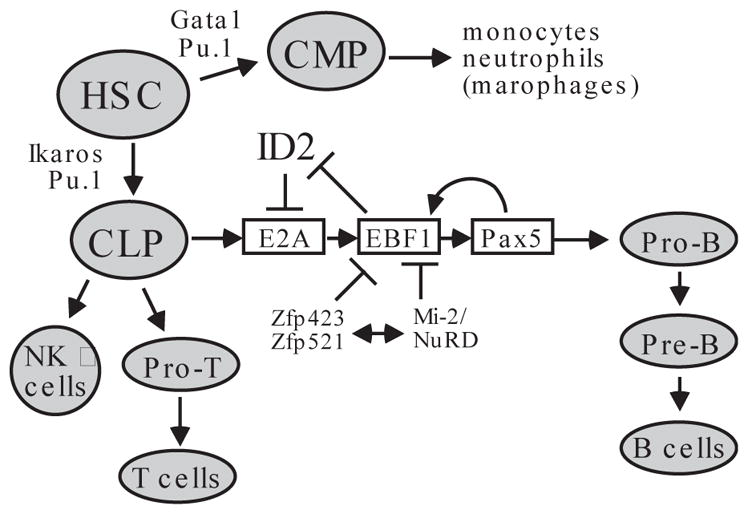
Hematopoietic stem cells (HSC) undergo differentiation mediated by PU.1 and Ikaros transcription factors to give rise to common lymphoid precursors (CLP). EBF1 acts in concert with transcription factors E2A and Pax5 to specify the pro-B cell pathway for the differentiation of CLP. There is extensive crosstalk among these factors to ensure commitment to the pro-B lineage. Factors that are known to suppress EBF1-dependent transcription include Zfp423/Zfp521 and the Mi-2/NuRD complex. For comparison, other lineages derived from CLP or HSC are also shown. CMP: common myeloid precursors; NK: natural killer cells.
At the onset of pro-B cell development from common lymphoid progenitor cells (CLP), EBF1 activates Pax5 through chromatin remodeling at the Pax5 locus (32) (Fig. 3). Pax5 expression controls the commitment of CLP to the B cell pathway by repressing genes inappropriate for the B-lineage and by activating genes required for B-cell maturation. In other cell types, the Pax5 promoter is repressed by the Polycomb group proteins (32). Independently of Pax5, EBF1 can also suppress alternative lineage options and promote B cell fate commitment. Retrovirus-mediated expression of Ebf1 in multipotential progenitor cells leads to the production of B cells. Furthermore, enforced Ebf1 expression in Pax5−/− hematopoietic progenitor cells inhibits their myeloid and T lineage potential in vivo (30).
EBF1-mediated DNA demethylation and chromatin remodeling appears to be a common mechanism by which it activates transcription of genes required for B-cell development. For example, EBF1, in conjunction with E2A and Runx1 (also known as AML1, CBFA2 and PEPB2αB), induces DNA demethylation and chromatin remodeling of the Cd79a and other B lineage target genes. This facilitates Pax5-mediated transactivation of these genes (32–34).
Like Pax5, EBF1 appears to repress the expression of genes that are inappropriate for the B cell lineage (30). Constitutive expression of EBF1 in Pax5-deficient Pro-B cells results in downregulation of numerous genes such as those encoding transcription factors C/EBPα, PU.1 and ID2 (differentiation proteins 2) (30). Likewise, Thal et al. showed that enforced expression of Ebf1 in IL-7Rα−/− bone marrow markedly suppresses the production of the Id2 and Id3 mRNA and restores B cell differentiation in vivo (35). Both ID2 and ID3 are inhibitory factors of E47, and thus EBF1-mediated downregulation of both factors is critical for B-cell development (35). Interestingly, siRNA-mediated knockdown of the Mi-2β subunit of the Mi-2/NuRD repression/remodeling complex appears to potentiate EBF1- and Pax5-mediated gene activation (34), suggesting the involvement of the Mi-2/NuRD silencing complex in EBF1-mediated gene repression (Fig. 2). Nonetheless, elucidation of the molecular mechanism by which EBF1 represses transcription requires further studies.
It is interesting to note that the D. melanogaster ortholog of EBF (Collier) was shown to play a role in immune response against parasitic wasps (36, 37). Analogous to vertebrate EBFs, Collier appears to specify the generation of lamellocytes, one of the three specific lineages of Drosophila hemocytes (blood cells). Lamellocytes devoted to encapsulation of large foreign bodies only differentiate from larval hematopoietic progenitors (prohemocytes) when the animal is challenged by specific immune threats such as parasitic invasion. Importantly, lamellocytes are not produced in parasitized collier mutant larvae (36). Thus, the involvement of EBFs in immunity predates the evolution of animals with more complex immune systems.
EBFs in bone development and adipogenesis
EBF2 is a regulator of osteoblast-dependent differentiation of osteoclasts. Targeted inactivation of Ebf2 results in reduced bone mass (14). In Ebf2-deficient cells, the osteoprotegerin (Opg) gene encoding a RANK decoy receptor is markedly downregulated. EBF2 can directly activate the Opg gene (14). In contrast, EBF1 appears to play an opposing role in regulating osteoblast development. Hesslein et al. showed that the Ebf1 mRNA and protein is produced in mesenchymal stem cells and in osteoblasts at most stages of differentiation (38). In Ebf1-deficient mice, osteoblastogenesis is increased (38).
EBF1 also regulates the production of adipocytes (39, 40). Ectopic expression of Ebf1 in the preadipocyte cell line 3T3-L1 fibroblasts or mouse embryonic fibroblasts induces adipogenesis (40). SiRNA-mediated downregulation of EBF1 or EBF2 blocks differentiation of 3T3-L1 cells, suggesting that both factors have a critical role and are not functionally redundant in driving the differentiation of fibroblasts to adipocytes (39). Additionally, in the bone marrow of Ebf1-deficient mice, the number of adipocytes is greatly increased, suggesting that EBF1 might block adipogenesis in this compartment (38).
EBFs in neurogenesis
The EBF orthologs in D. melanogaster (Collier), and C. elegans (Unc-3) are involved in neurogenesis, which might be mediated by interactions between EBF and the hedgehog or notch pathways (13). Studies based on these and other model organisms suggest that during neuronal differentiation the members of the EBF family have functionally distinct roles. For example, mouse EBF2 appears to act earlier than EBF1 or EBF3, and in Xenopus, EBF3 promotes differentiation of specific neuronal subtypes (13, 41). In mice all four members of the EBF family are expressed in olfactory receptor neurons, where they regulate the expression of olfactory genes (42, 43). During mouse embryogenesis EBF members are expressed in early post-mitotic neurons from the midbrain to the spinal cord and at specific sites in the embryonic forebrain, suggesting that they may regulate neuronal maturation in the central nervous system (CNS) (11–13). EBF2 is expressed in the embryonic CNS (44), and targeted inactivation of Ebf2 has revealed roles for EBF2 in peripheral nerve morphogenesis, migration of hormone-producing neurons, and projection of olfactory neurons (42, 45).
Interestingly, EBF1 is abundantly expressed in striatonigral medium spiny neurons (MSNs). Ebf1 deficiency in mice results in a markedly reduced number of striatonigral MSNs at postnatal day 14 (P14), although these neurons are properly specified in Ebf1−/− mice by P0. Thus, EBF1 appears to be a lineage-specific transcription factor essential for the differentiation of striatonigral MSNs (46). Recently Yin et al. showed that EBF1 has a critical role in terminal migration of mesodiencephalic dopaminergic (mDA) neurons that have multiple brain functions such as movement control, emotion and reward. EBF1 is primarily expressed in the immature mDA neurons and its depletion leads to retardation of mDA neurons in the marginal zone (47).
Non-redundancy of EBF family members
Although Ebf1 and Ebf2-deficient mice are viable (14, 16, 42, 45), homozygous Ebf3 knockout mice exhibit neonatal lethality by postnatal day two (42), suggesting that the EBF proteins are not functionally redundant. This is surprising, considering that the four paralogs of the EBF family are highly similar in amino acid sequence. The 50 NH2-terminal amino acid residues and the entire COOH-terminal transactivation domain are the most divergent regions among these paralogs. However, even in these regions the amino acid sequences are essentially identical among corresponding orthologs between mammalian species. Therefore, functional specificity of an individual EBF member might be determined by these slightly divergent domains, or more likely through specific regulation of their expression. Indeed, the promoter sequences of the four paralogs appear quite distinct (our unpublished analysis, but also see (28, 48)). The expression of EBF proteins in multiple tissues and their roles in diverse developmental processes suggest that they have fundamental cellular functions, and that their roles in lineage determination may involve cooperation with other tissue-restricted factors, as demonstrated in B lymphopoiesis (Fig. 3).
Potential roles of EBFs in tumor suppression
EBF3 is a putative tumor suppressor
Genetic analyses indicate that the loss of chromosome 10q is the most common genomic lesion of both primary and secondary GBMs and correlates with short patient survival. One consequence of the 10q loss is the deletion of the Pten locus in 10q22, a well-established genetic defect during gliomagenesis (49, 50). Pten encodes a phosphatase that dephosphorylates both lipids and proteins. It is mutated in about 25% of GBMs. Pten is also inactivated through other mechanisms. For example, promoter hypermethylation of the Pten gene occurs in 75–80% of all GBMs. However, hypermethylation of the Pten promoter occurs mostly in low-grade glioma and secondary GBMs (51).
The gene encoding the human EBF3 transcription factor also resides in 10q at 10q26.3. The EBF3 locus is close to the telomere of chromosome 10q, and is approximately 45 megabases (Mb) away from the Pten locus. A genome-wide screen using integrated genomic and epigenetic analyses revealed that the EBF3 locus at chromosome 10q26.3 is biallelically altered by methylation or deletion in the majority of high-grade brain tumors (7). The EBF3 gene was found to be inactivated in 50% of grade II, 83% of grade III, and 90% of grade IV brain tumors by deletion, or promoter hypermethylation of the EBF3 locus (7). It is noteworthy that the promoters of EBF3 gene in both mice and humans contain a very long CpG island spanning 1.6 kb, encompassing the proximal region of the promoter and the first two exons of the EBF3 gene. The long CpG island might render the promoters susceptible to DNA hypermethylation in tumors. Consistent with these observations, EBF3 is expressed in normal brain cells but is silenced in brain tumor cells (7). These data provided the first evidence that EBF3 is a potential tumor suppressor gene in the brain.
Consistent with previous findings (7), Maher et al. showed that genomic loss at 10q26.3 is as frequent as that at 10q22, occurring with a frequency of 85.2% in primary and secondary GBMs (52). These data suggest that both loci may be lost simultaneously, although it is also possible that each locus is deleted independently, as genomic loss of the regions between these two loci occurs at a slightly lower frequency (80%) (52). In a comprehensive analysis of the cancer genome in 206 GBMs, it was also found that the loss of the EBF3 genomic region is a frequent event (4). Since Pten inactivation alone is not sufficient to cause gliomas (49, 50, 53), and the loss of Pten is almost invariably associated with the loss of EBF3, it is tempting to speculate that genomic deletion of EBF3 might facilitate gliomagenesis when Pten is also lost. Similar genomic lesions also appear to occur in breast cancer. Quantitative comparative genomic hybridization analysis revealed that 10q26 was lost in 41 of 53 breast cancer cell lines (54). It is worth emphasizing that 10q loss would result in genetic deletion of many genes in this region. Therefore whether EBF3 loss in cancer is a specific contributing factor in tumorigenesis requires further investigation.
Remarkably, large-scale sequencing of all coding exons in tumors and their normal tissue counterparts has detected tumor-specific mutations of the EBF3 gene in GBM and pancreatic cancer. In a primary tumor of a 32-year-old male GBM patient, a missense mutation was found. It results in the change of amino acid residue from W to C at position 265 (W265C) of EBF3 (3). A missense mutation of EBF3 (R243W) was also discovered in a pancreatic ductal adenocarcinoma that metastasized to the lung from a 59-year-old male patient (2). Both cancer-specific somatic mutations occur within the highly conserved functional domains of EBF3 (Fig. 1). The former is in the ITP/TIG domain, whereas the latter is in the DBD. Although the functional impact of these mutations is currently unknown, they could affect DNA-binding or protein-protein interaction of EBF3.
There is also evidence suggesting that EBF3 has a tumor suppressive role in other types of cancer. For example, cDNA microarray-based gene expression analyses revealed significant downregulation of EBF3 expression in liver cancer compared to normal liver tissue (55). Epigenetic silencing of the EBF3 locus occurs in colon, liver, and head and neck squamous cell carcinoma (9, 56). Furthermore, EBF3 expression is reduced in metastatic prostate cancer compared with primary tumors (57). Collectively, these studies suggest that EBF3 acts as a tumor suppressor in multiple organs and that it can also inhibit metastasis.
Our recent studies have provided a plausible biochemical explanation regarding EBF3-mediated tumor suppression. We found that EBF3 regulates the expression of a variety of genes that have critical roles in cell growth, proliferation and apoptosis (9). Examples of EBF3-regulated genes include the Cip/Kip family of cyclin-dependent kinase inhibitors (CDKIs), and several cyclins and CDKs. In U87 glioma cells and Saos2 osteosarcoma cells, ectopic expression of wt EBF3, but not the H157A mutant defective for DNA-binding, causes initial activation and then significant repression of both p21cip1 (p21) and p27kip1 (p27). This downregulation coincides with cell detachment and apoptosis. Conversely, EBF3 can repress the expression of genes responsible for cell proliferation and survival such as cyclins A and B, CDK2, Daxx and Mcl-1. Therefore, EBF3 might act as a tumor suppressor by regulating the expression of specific set of genes, thereby preventing inappropriate proliferation (Fig. 4).
FIGURE 4. A potential anti-neoplastic pathway involving EBF3.

EBF3 regulates the expression of diverse genes involved in cell growth and proliferation. Specifically, ectopic expression of EBF3 activates the expression of p21 and p27 at early time points, which induce cell cycle arrest. At late time points following EBF3 expression, caspase-3 activation, PARP-1 (poly [ADP-ribose] polymerase 1) cleavage and apoptosis of tumor cells occur. Note that both p21 and p27 are downregulated at late time points. However, whether EBF3 directly mediates the repression of p21 and p27 gene is unclear. Nonetheless, enforced EBF3 expression also leads to marked downregulation of cyclins and CDKs as well as proteins involved in cell survival such as Daxx and Mcl-1 at earlier time points. Thus, EBF3 might directly repress these genes.
Genomic loss and somatic mutations of the EBF1 locus in cancer
As discussed above, EBF1 is an essential regulator of B-cell differentiation and maturation (10) (Fig. 3). It also has a critical role in neuronal differentiation (46, 47). Therefore, inactivation of EBF1 could block these developmental processes, resulting in neoplastic phenotypes. Recent genomewide analyses of diverse cancers provide comprehensive information regarding genetic abnormalities of cancer. Pediatric acute lymphoblastic leukemia (ALL) includes B-progenitor and T-lineage leukemias (T-ALL). These ALL subtypes have distinct genetic alterations. For example, the B-progenitor leukemias contain several well-known genomic translocations such as t(9;22)[BCR–ABL1], t(1;19)[TCF3–PBX1], t(12;21)[ETV6–RUNX1], and the rearrangements of MLL (myeloid/lymphoid or mixed-lineage leukemia). Mullighan et al. analyzed 242 cases of pediatric ALL using single nucleotide polymorphism (SNP) arrays that can detect loss or gain of precise genomic regions (8). Abnormalities at the Pax5 locus represent the most frequent genetic changes of ALL, occurring in 31.7% of cases. Translocation or deletion of Pax5 in B-cell precursor ALL results in reduced or abolished expression of EBF1 and other factors involved in B-lymphocyte development, thereby preventing B-cell maturation (58). These findings are perhaps not surprising given the key roles for Pax5 in B cell development (Fig. 3). Deletions of other key genes in the B-cell maturation pathway, such as TCF3 (encoding the human E2A), EBF1, LEF1, IKZF1 (IKAROS) and IKZF3 (AIOLOS), are also frequent events in ALL (8). It is striking that 82% of the patients with BCR-ABL1-like disease harbor deletions in these genes (59).
The studies by Mullighan et al. revealed eight cases of ALL that carry monoallelic deletions of EBF1 (8). It was shown previously that Ebf1+/− heterozygous mice have a 50% reduction of fraction B (now termed pro-B) cells. The pre-pro-B cells or fraction A were unaffected (16). Since the expression of EBF1 mRNA was detected in ALL cells with monoallelic deletion, EBF1 might be a haploinsufficient tumor suppressor (8). Detailed analysis of blasts from an ALL patient identified two distinct blast populations: one with monoallelic deletion and the other with homozygous deletion of the EBF1 gene. The latter population largely consists of immature B-cells with reduced or no expression of CD79a, a known EBF1 target gene (8). EBF1 deletion might also be an important factor in the relapse of ALL. Yang et al. found that 25% of relapsed ALL cases have deletions in the EBF1 locus (60). The fact that deletion of EBF1 was seen almost exclusively at relapse in some cases of pediatric ALL suggests that EBF1 loss confers drug resistance and might be a potential prognostic marker (60, 61). Taken together, these recent findings demonstrate that genomic lesions of the EBF1 gene contribute to the pathogenesis, drug resistance and disease relapse of ALL.
Implication of EBF-binding proteins and target genes in hematological and solid cancers
In mouse B-cell lymphomas, retroviral insertions occur frequently in two genetic loci: Evi3 (ecotropic viral integration site 3 encoding Evi3, also known as EHZF, Zfp521 or ZNF521), and Ebfaz (encoding EBF-associated zinc finger protein, EBFAZ, also known as OAZ, Zfp423, or ZNF423) (62–64). In normal B cells, Zfp521 is expressed at low levels and Zfp423 is not expressed (62, 63). Significantly, mice with retroviral insertion at Ebfaz (Zfp423) site develop lymphoblastic leukemia in most of the studied cases, although other types of lymphoid tumors including lymphoma are also diagnosed. Strikingly, the levels of Zfp423 expression were markedly higher in these tumors compared to their normal counterparts (64). Similarly, Zfp521 is also highly expressed following retroviral insertion at the Evi3 site (62). Interestingly, Viral integration at the Evi3 or Ebfaz sites appears mutually exclusive, suggesting that both Zfp521 and Zfp423 act in the same pathway during the development of B cell tumors (62, 63). Studies suggest that overexpression of Zfp423 and Zfp521 in B-cell leukemias and lymphomas represses EBF1 target genes that suppress leukemogenesis in B cells (65, 66). In support of this notion, Zfp521 is highly expressed in most human acute myelogenous leukemias. Similarly, its expression is particularly high in leukemias with rearrangements of the MLL gene. Consistent with these observations, the levels of Zfp521 expression are high in human hematopoietic progenitor cells and decline rapidly during cytokine-driven differentiation (65, 66). Furthermore, Zfp521 is also abundantly expressed in medulloblastomas and other brain tumors (65).
At the molecular level, Zfp521 and Zfp423 are highly similar to each other, each containing 30 Krüppel-type zinc fingers. Hata et al. showed that zinc fingers 28–30 of Zfp423 interact with EBF1, while zinc fingers 2–8 bind to the 5′-CCGCCC-3′ DNA sequence in concert with EBF1 binding to its cognate sequences (67) (Fig. 2). Experiments based on reporter gene assay suggest that human Zfp521 and mouse and rat orthologs of Zfp423 suppress EBF1-mediated transcriptional activation (64, 66, 68) (see Fig. 2), although their interactions with EBF1 may activate transcription in different tissue types (64, 67). Matsubara et al. reported that shRNA-mediated downregulation of Zfp521 relieved the repression of GATA-1 target genes and promoted erythrocyte differentiation (69). Intriguingly, Zfp521 and Zfp423 contain a conserved sequence motif near its N-terminus that is important for interacting with the Mi-2/NuRD remodeling and repression complex (65, 69–71). This evidence, along with the finding that siRNA-mediated down-regulation of Mi-2β subunit of the Mi-2/NuRD complex enhances EBF1-mediated transcriptional activation (34), suggests that Zfp521 and Zfp423 might recruit this corepressor complex to suppress EBF1 target genes (Fig. 2). In addition to regulatory roles in the hematopoietic progenitors, Zfp423 appears to have a critical role in the development of the CNS (72, 73). The interactions between Zfp521/Zfp423 and EBFs likely underpin these important developmental mechanisms whose disruption contributes to oncogenesis. Although the precise implications of the Zfp521/Zfp423-EBF pathway in oncogenesis remain to be determined, the suppression of EBF-mediated transcription by these two large zinc finger proteins inhibits several developmental pathways and can conceivably contribute to the etiology of leukemias, brain tumors and possibly other types of cancer.
As previously mentioned, EBF1 induces Pax5 expression by promoting epigenetic modifications and chromatin remodeling at the Pax5 promoter. It also represses the expression of the helix-loop-helix proteins ID2 and ID3, two repressors of Pax5 (30, 35). Overexpression of ID proteins can induce malignant transformation and was indeed observed in several types of cancer including colon (74), prostate (75), and lung cancer (76). Thus, EBF1-mediated downregulation of ID proteins may be a mechanism of tumor suppression in hematological and solid tumors. Additionally, genomic loss of the EBF1 locus or other EBF genes appears to occur in solid malignancies. Neve et al. detected genomic loss at 5q32 (encoding EBF1) or 20p13 (encoding EBF4) in 10 and 14 of 53 breast cancer lines, respectively (54). Although evidence supporting a tumor suppressive role for EBF1 in solid tumors is still preliminary, Jones et al. detected a cancer-specific somatic missense mutation in a primary ductal adenocarcinoma of the pancreas in a 60-year-old female patient (2). This mutation causes the amino acid substitution of arginine for glutamine at position 242, which lies within the DBD of EBF1 (R242Q, Fig. 1). It is noteworthy that the R242Q mutation of EBF1 and the R243W mutation of EBF3 affect two consecutive arginine residues within the well-conserved DBD of the EBF family. Thus, these mutations might impair the normal functions of EBFs. It will be important to determine how these mutations affect protein function of EBF1 and EBF3.
At present, little is known about potential inactivation of EBF2 or EBF4 in cancer. Our unpublished data indicate that EBF4 is expressed at higher levels in several tumor cell lines than EBF1 or EBF3. However, the biological functions of EBF4 remain a mystery.
Concluding remarks
EBFs are evolutionarily conserved transcription factors with specific roles in the differentiation and maturation of several cell lineages. Inactivation of EBF genes blocks normal development pathways, which results in the accumulation and expansion of undifferentiated progenitor cells, leading to tumorigenesis. Therefore, EBF inactivation due to genomic deletion, epigenetic silencing, or somatic point mutations might have causal roles in the tumorigenesis of several types of cancer including leukemia, GBM, and pancreatic cancer. Steadily accumulating evidence supports a tumor suppressive role for EBF1 and EBF3. Our understanding of their roles in human diseases such as cancer will depend on the illumination of their biological functions. Therefore, future studies should continue to focus on elucidating the biological roles of the EBF family members in development and other mechanisms. Genomewide analyses of diverse tumors and their normal counterparts will provide more evidence for the involvement of EBFs in cancer. Conventional approaches such as immunohistochemical staining and RT-PCR analysis of tumor specimens will be an important complement to large-scale studies in deepening our understanding of tumor suppressive roles of the EBF members. Tissue-specific inactivation of the Ebf genes in mice will allow the establishment of mouse tumor models to probe specific roles of EBFs in the development of different types of cancer. Alternatively, understanding how EBFs interact with their known binding partners such Zfp423 and Zfp521 and the functional impacts of these interactions will reveal the mechanisms of EBF pathways in development and tumorigenesis. Finally, investigation of potential genetic interactions between EBFs and other known tumor suppressors will provide further insight into their role in tumorigenesis. Given the fact that EBF3 is frequently lost together with Pten in GBMs, the simultaneous loss of both genes might facilitate gliomagenesis. Such studies will establish molecular basis for EBF proteins in tumor suppression and will provide clues for designing rational therapy for patients with specific types of cancer containing abnormalities in the EBF pathways.
Acknowledgments
I am grateful to two anonymous reviewers for insightful suggestions, and to Nicole T. Parker for critical reading and editing. I also apologize to colleagues whose publications could not be cited in this article due to space constraints. The research in the author’s laboratory was funded by the Bankhead-Coley Cancer Research Program, Florida Department of Health, Stop! Children’s Cancer Inc., and the National Cancer Institute.
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Wood LD, Parsons DW, Jones S, et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–13. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- 2.Jones S, Zhang X, Parsons DW, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–6. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Parsons DW, Jones S, Zhang X, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–12. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–8. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Greenman C, Stephens P, Smith R, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446:153–8. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sjöblom T, Jones S, Wood LD, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–74. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 7.Zardo G, Tiirikainen MI, Hong C, et al. Integrated genomic and epigenomic analyses pinpoint biallelic gene inactivation in tumors. Nat Genet. 2002;32:453–8. doi: 10.1038/ng1007. [DOI] [PubMed] [Google Scholar]
- 8.Mullighan CG, Goorha S, Radtke I, et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature. 2007;446:758–64. doi: 10.1038/nature05690. [DOI] [PubMed] [Google Scholar]
- 9.Zhao LY, Niu Y, Santiago A, et al. An EBF3-Mediated Transcriptional Program That Induces Cell Cycle Arrest and Apoptosis. Cancer Res. 2006;66:9445–52. doi: 10.1158/0008-5472.CAN-06-1713. [DOI] [PubMed] [Google Scholar]
- 10.Hagman J, Lukin K. Early B-cell factor ‘pioneers’ the way for B-cell development. Trends Immunol. 2005;26:455–61. doi: 10.1016/j.it.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 11.Garel S, Marin F, Mattei MG, Vesque C, Vincent A, Charnay P. Family of Ebf/Olf-1-related genes potentially involved in neuronal differentiation and regional specification in the central nervous system. Dev Dyn. 1997;210:191–205. doi: 10.1002/(SICI)1097-0177(199711)210:3<191::AID-AJA1>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 12.Garel S, Marin F, Grosschedl R, Charnay P. Ebf1 controls early cell differentiation in the embryonic striatum. Development. 1999;126:5285–94. doi: 10.1242/dev.126.23.5285. [DOI] [PubMed] [Google Scholar]
- 13.Dubois L, Vincent A. The COE--Collier/Olf1/EBF--transcription factors: structural conservation and diversity of developmental functions. Mech Dev. 2001;108:3–12. doi: 10.1016/s0925-4773(01)00486-5. [DOI] [PubMed] [Google Scholar]
- 14.Kieslinger M, Folberth S, Dobreva G, et al. EBF2 regulates osteoblast-dependent differentiation of osteoclasts. Dev Cell. 2005;9:757–67. doi: 10.1016/j.devcel.2005.10.009. [DOI] [PubMed] [Google Scholar]
- 15.Hagman J, Belanger C, Travis A, Turck CW, Grosschedl R. Cloning and functional characterization of early B-cell factor, a regulator of lymphocyte-specific gene expression. Genes Dev. 1993;7:760–73. doi: 10.1101/gad.7.5.760. [DOI] [PubMed] [Google Scholar]
- 16.Lin H, Grosschedl R. Failure of B-cell differentiation in mice lacking the transcription factor EBF. Nature. 1995;376:263–7. doi: 10.1038/376263a0. [DOI] [PubMed] [Google Scholar]
- 17.Wang MM, Reed RR. Molecular cloning of the olfactory neuronal transcription factor Olf-1 by genetic selection in yeast. Nature. 1993;364:121–6. doi: 10.1038/364121a0. [DOI] [PubMed] [Google Scholar]
- 18.Daburon V, Mella S, Plouhinec JL, Mazan S, Crozatier M, Vincent A. The metazoan history of the COE transcription factors. Selection of a variant HLH motif by mandatory inclusion of a duplicated exon in vertebrates. BMC Evol Biol. 2008;8:131. doi: 10.1186/1471-2148-8-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang SS, Betz AG, Reed RR. Cloning of a novel Olf-1/EBF-like gene, O/E-4, by degenerate oligo-based direct selection. Mol Cell Neurosci. 2002;20:404–14. doi: 10.1006/mcne.2002.1138. [DOI] [PubMed] [Google Scholar]
- 20.Hagman J, Gutch MJ, Lin H, Grosschedl R. EBF contains a novel zinc coordination motif and multiple dimerization and transcriptional activation domains. Embo J. 1995;14:2907–16. doi: 10.1002/j.1460-2075.1995.tb07290.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fields S, Ternyak K, Gao H, Ostraat R, Akerlund J, Hagman J. The ‘zinc knuckle’ motif of Early B cell Factor is required for transcriptional activation of B cell-specific genes. Mol Immunol. 2008;45:3786–96. doi: 10.1016/j.molimm.2008.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao F, McCarrick-Walmsley R, Akerblad P, Sigvardsson M, Kadesch T. Inhibition of p300/CBP by early B-cell factor. Mol Cell Biol. 2003;23:3837–46. doi: 10.1128/MCB.23.11.3837-3846.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin CH, Hare BJ, Wagner G, Harrison SC, Maniatis T, Fraenkel E. A small domain of CBP/p300 binds diverse proteins: solution structure and functional studies. Mol Cell. 2001;8:581–90. doi: 10.1016/s1097-2765(01)00333-1. [DOI] [PubMed] [Google Scholar]
- 24.Medina KL, Pongubala JM, Reddy KL, et al. Assembling a gene regulatory network for specification of the B cell fate. Dev Cell. 2004;7:607–17. doi: 10.1016/j.devcel.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 25.Hagman J, Lukin K. Transcription factors drive B cell development. Curr Opin Immunol. 2006;18:127–34. doi: 10.1016/j.coi.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 26.Busslinger M. Transcriptional control of early B cell development. Annu Rev Immunol. 2004;22:55–79. doi: 10.1146/annurev.immunol.22.012703.104807. [DOI] [PubMed] [Google Scholar]
- 27.Medina KL, Singh H. Genetic networks that regulate B lymphopoiesis. Curr Opin Hematol. 2005;12:203–9. doi: 10.1097/01.moh.0000160735.67596.a0. [DOI] [PubMed] [Google Scholar]
- 28.Roessler S, Gyory I, Imhof S, et al. Distinct promoters mediate the regulation of Ebf1 gene expression by interleukin-7 and Pax5. Mol Cell Biol. 2007;27:579–94. doi: 10.1128/MCB.01192-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seet CS, Brumbaugh RL, Kee BL. Early B cell factor promotes B lymphopoiesis with reduced interleukin 7 responsiveness in the absence of E2A. J Exp Med. 2004;199:1689–700. doi: 10.1084/jem.20032202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pongubala JM, Northrup DL, Lancki DW, et al. Transcription factor EBF restricts alternative lineage options and promotes B cell fate commitment independently of Pax5. Nat Immunol. 2008;9:203–15. doi: 10.1038/ni1555. [DOI] [PubMed] [Google Scholar]
- 31.Zhang Z, Cotta CV, Stephan RP, deGuzman CG, Klug CA. Enforced expression of EBF in hematopoietic stem cells restricts lymphopoiesis to the B cell lineage. Embo J. 2003;22:4759–69. doi: 10.1093/emboj/cdg464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Decker T, Pasca di Magliano M, McManus S, et al. Stepwise activation of enhancer and promoter regions of the B cell commitment gene Pax5 in early lymphopoiesis. Immunity. 2009;30:508–20. doi: 10.1016/j.immuni.2009.01.012. [DOI] [PubMed] [Google Scholar]
- 33.Maier H, Ostraat R, Gao H, et al. Early B cell factor cooperates with Runx1 and mediates epigenetic changes associated with mb-1 transcription. Nat Immunol. 2004;5:1069–77. doi: 10.1038/ni1119. [DOI] [PubMed] [Google Scholar]
- 34.Gao H, Lukin K, Ramirez J, Fields S, Lopez D, Hagman J. Opposing effects of SWI/SNF and Mi-2/NuRD chromatin remodeling complexes on epigenetic reprogramming by EBF and Pax5. Proc Natl Acad Sci U S A. 2009;106:11258–63. doi: 10.1073/pnas.0809485106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thal MA, Carvalho TL, He T, et al. Ebf1-mediated down-regulation of Id2 and Id3 is essential for specification of the B cell lineage. Proc Natl Acad Sci U S A. 2009;106:552–7. doi: 10.1073/pnas.0802550106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Crozatier M, Ubeda JM, Vincent A, Meister M. Cellular immune response to parasitization in Drosophila requires the EBF orthologue collier. PLoS Biol. 2004;2:E196. doi: 10.1371/journal.pbio.0020196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Krzemien J, Dubois L, Makki R, Meister M, Vincent A, Crozatier M. Control of blood cell homeostasis in Drosophila larvae by the posterior signalling centre. Nature. 2007;446:325–8. doi: 10.1038/nature05650. [DOI] [PubMed] [Google Scholar]
- 38.Hesslein DG, Fretz JA, Xi Y, et al. Ebf1-dependent control of the osteoblast and adipocyte lineages. Bone. 2009;44:537–46. doi: 10.1016/j.bone.2008.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jimenez MA, Akerblad P, Sigvardsson M, Rosen ED. Critical role for Ebf1 and Ebf2 in the adipogenic transcriptional cascade. Mol Cell Biol. 2007;27:743–57. doi: 10.1128/MCB.01557-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Akerblad P, Lind U, Liberg D, Bamberg K, Sigvardsson M. Early B-cell factor (O/E-1) is a promoter of adipogenesis and involved in control of genes important for terminal adipocyte differentiation. Mol Cell Biol. 2002;22:8015–25. doi: 10.1128/MCB.22.22.8015-8025.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liberg D, Sigvardsson M, Akerblad P. The EBF/Olf/Collier family of transcription factors: regulators of differentiation in cells originating from all three embryonal germ layers. Mol Cell Biol. 2002;22:8389–97. doi: 10.1128/MCB.22.24.8389-8397.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang SS, Lewcock JW, Feinstein P, Mombaerts P, Reed RR. Genetic disruptions of O/E2 and O/E3 genes reveal involvement in olfactory receptor neuron projection. Development. 2004;131:1377–88. doi: 10.1242/dev.01009. [DOI] [PubMed] [Google Scholar]
- 43.Wang SS, Tsai RY, Reed RR. The characterization of the Olf-1/EBF-like HLH transcription factor family: implications in olfactory gene regulation and neuronal development. J Neurosci. 1997;17:4149–58. doi: 10.1523/JNEUROSCI.17-11-04149.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Malgaretti N, Pozzoli O, Bosetti A, et al. Mmot1, a new helix-loop-helix transcription factor gene displaying a sharp expression boundary in the embryonic mouse brain. J Biol Chem. 1997;272:17632–9. doi: 10.1074/jbc.272.28.17632. [DOI] [PubMed] [Google Scholar]
- 45.Corradi A, Croci L, Broccoli V, et al. Hypogonadotropic hypogonadism and peripheral neuropathy in Ebf2-null mice. Development. 2003;130:401–10. doi: 10.1242/dev.00215. [DOI] [PubMed] [Google Scholar]
- 46.Lobo MK, Yeh C, Yang XW. Pivotal role of early B-cell factor 1 in development of striatonigral medium spiny neurons in the matrix compartment. J Neurosci Res. 2008;86:2134–46. doi: 10.1002/jnr.21666. [DOI] [PubMed] [Google Scholar]
- 47.Yin M, Liu S, Yin Y, et al. Ventral mesencephalon-enriched genes that regulate the development of dopaminergic neurons in vivo. J Neurosci. 2009;29:5170–82. doi: 10.1523/JNEUROSCI.5569-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Smith EM, Gisler R, Sigvardsson M. Cloning and characterization of a promoter flanking the early B cell factor (EBF) gene indicates roles for E-proteins and autoregulation in the control of EBF expression. J Immunol. 2002;169:261–70. doi: 10.4049/jimmunol.169.1.261. [DOI] [PubMed] [Google Scholar]
- 49.Xiao A, Wu H, Pandolfi PP, Louis DN, Van Dyke T. Astrocyte inactivation of the pRb pathway predisposes mice to malignant astrocytoma development that is accelerated by PTEN mutation. Cancer Cell. 2002;1:157–68. doi: 10.1016/s1535-6108(02)00029-6. [DOI] [PubMed] [Google Scholar]
- 50.Xiao A, Yin C, Yang C, Di Cristofano A, Pandolfi PP, Van Dyke T. Somatic induction of Pten loss in a preclinical astrocytoma model reveals major roles in disease progression and avenues for target discovery and validation. Cancer Res. 2005;65:5172–80. doi: 10.1158/0008-5472.CAN-04-3902. [DOI] [PubMed] [Google Scholar]
- 51.Wiencke JK, Zheng S, Jelluma N, et al. Methylation of the PTEN promoter defines low-grade gliomas and secondary glioblastoma. Neuro Oncol. 2007;9:271–9. doi: 10.1215/15228517-2007-003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Maher EA, Brennan C, Wen PY, et al. Marked genomic differences characterize primary and secondary glioblastoma subtypes and identify two distinct molecular and clinical secondary glioblastoma entities. Cancer Res. 2006;66:11502–13. doi: 10.1158/0008-5472.CAN-06-2072. [DOI] [PubMed] [Google Scholar]
- 53.Wei Q, Clarke L, Scheidenhelm DK, et al. High-grade glioma formation results from postnatal pten loss or mutant epidermal growth factor receptor expression in a transgenic mouse glioma model. Cancer Res. 2006;66:7429–37. doi: 10.1158/0008-5472.CAN-06-0712. [DOI] [PubMed] [Google Scholar]
- 54.Neve RM, Chin K, Fridlyand J, et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006;10:515–27. doi: 10.1016/j.ccr.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen X, Cheung ST, So S, et al. Gene expression patterns in human liver cancers. Mol Biol Cell. 2002;13:1929–39. doi: 10.1091/mbc.02-02-0023.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bennett KL, Karpenko M, Lin MT, et al. Frequently methylated tumor suppressor genes in head and neck squamous cell carcinoma. Cancer Res. 2008;68:4494–9. doi: 10.1158/0008-5472.CAN-07-6509. [DOI] [PubMed] [Google Scholar]
- 57.Dhanasekaran SM, Barrette TR, Ghosh D, et al. Delineation of prognostic biomarkers in prostate cancer. Nature. 2001;412:822–6. doi: 10.1038/35090585. [DOI] [PubMed] [Google Scholar]
- 58.An Q, Wright SL, Konn ZJ, et al. Variable breakpoints target PAX5 in patients with dicentric chromosomes: a model for the basis of unbalanced translocations in cancer. Proc Natl Acad Sci U S A. 2008;105:17050–4. doi: 10.1073/pnas.0803494105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Den Boer ML, van Slegtenhorst M, De Menezes RX, et al. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome-wide classification study. Lancet Oncol. 2009;10:125–34. doi: 10.1016/S1470-2045(08)70339-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yang JJ, Bhojwani D, Yang W, et al. Genome-wide copy number profiling reveals molecular evolution from diagnosis to relapse in childhood acute lymphoblastic leukemia. Blood. 2008;112:4178–83. doi: 10.1182/blood-2008-06-165027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mullighan CG, Su X, Zhang J, et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med. 2009;360:470–80. doi: 10.1056/NEJMoa0808253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Warming S, Liu P, Suzuki T, et al. Evi3, a common retroviral integration site in murine B-cell lymphoma, encodes an EBFAZ-related Kruppel-like zinc finger protein. Blood. 2003;101:1934–40. doi: 10.1182/blood-2002-08-2652. [DOI] [PubMed] [Google Scholar]
- 63.Warming S, Suzuki T, Yamaguchi TP, Jenkins NA, Copeland NG. Early B-cell factor-associated zinc-finger gene is a frequent target of retroviral integration in murine B-cell lymphomas. Oncogene. 2004;23:2727–31. doi: 10.1038/sj.onc.1207452. [DOI] [PubMed] [Google Scholar]
- 64.Hentges KE, Weiser KC, Schountz T, Woodward LS, Morse HC, Justice MJ. Evi3, a zinc-finger protein related to EBFAZ, regulates EBF activity in B-cell leukemia. Oncogene. 2005;24:1220–30. doi: 10.1038/sj.onc.1208243. [DOI] [PubMed] [Google Scholar]
- 65.Bond HM, Mesuraca M, Amodio N, et al. Early hematopoietic zinc finger protein-zinc finger protein 521: a candidate regulator of diverse immature cells. Int J Biochem Cell Biol. 2008;40:848–54. doi: 10.1016/j.biocel.2007.04.006. [DOI] [PubMed] [Google Scholar]
- 66.Bond HM, Mesuraca M, Carbone E, et al. Early hematopoietic zinc finger protein (EHZF), the human homolog to mouse Evi3, is highly expressed in primitive human hematopoietic cells. Blood. 2004;103:2062–70. doi: 10.1182/blood-2003-07-2388. [DOI] [PubMed] [Google Scholar]
- 67.Hata A, Seoane J, Lagna G, Montalvo E, Hemmati-Brivanlou A, Massague J. OAZ uses distinct DNA- and protein-binding zinc fingers in separate BMP-Smad and Olf signaling pathways. Cell. 2000;100:229–40. doi: 10.1016/s0092-8674(00)81561-5. [DOI] [PubMed] [Google Scholar]
- 68.Tsai RY, Reed RR. Cloning and functional characterization of Roaz, a zinc finger protein that interacts with O/E-1 to regulate gene expression: implications for olfactory neuronal development. J Neurosci. 1997;17:4159–69. doi: 10.1523/JNEUROSCI.17-11-04159.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Matsubara E, Sakai I, Yamanouchi J, et al. The role of zinc finger protein 521/early hematopoietic zinc finger protein in erythroid cell differentiation. J Biol Chem. 2009;284:3480–7. doi: 10.1074/jbc.M805874200. [DOI] [PubMed] [Google Scholar]
- 70.Lauberth SM, Rauchman M. A conserved 12-amino acid motif in Sall1 recruits the nucleosome remodeling and deacetylase corepressor complex. J Biol Chem. 2006;281:23922–31. doi: 10.1074/jbc.M513461200. [DOI] [PubMed] [Google Scholar]
- 71.Lin AC, Roche AE, Wilk J, Svensson EC. The N termini of Friend of GATA (FOG) proteins define a novel transcriptional repression motif and a superfamily of transcriptional repressors. J Biol Chem. 2004;279:55017–23. doi: 10.1074/jbc.M411240200. [DOI] [PubMed] [Google Scholar]
- 72.Cheng LE, Zhang J, Reed RR. The transcription factor Zfp423/OAZ is required for cerebellar development and CNS midline patterning. Dev Biol. 2007;307:43–52. doi: 10.1016/j.ydbio.2007.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Alcaraz WA, Gold DA, Raponi E, Gent PM, Concepcion D, Hamilton BA. Zfp423 controls proliferation and differentiation of neural precursors in cerebellar vermis formation. Proc Natl Acad Sci U S A. 2006;103:19424–9. doi: 10.1073/pnas.0609184103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wilson JW, Deed RW, Inoue T, et al. Expression of Id helix-loop-helix proteins in colorectal adenocarcinoma correlates with p53 expression and mitotic index. Cancer Res. 2001;61:8803–10. [PubMed] [Google Scholar]
- 75.Yuen HF, Chua CW, Chan YP, Wong YC, Wang X, Chan KW. Id proteins expression in prostate cancer: high-level expression of Id-4 in primary prostate cancer is associated with development of metastases. Mod Pathol. 2006;19:931–41. doi: 10.1038/modpathol.3800602. [DOI] [PubMed] [Google Scholar]
- 76.Rollin J, Blechet C, Regina S, Tenenhaus A, Guyetant S, Gidrol X. The intracellular localization of ID2 expression has a predictive value in non small cell lung cancer. PLoS ONE. 2009;4:e4158. doi: 10.1371/journal.pone.0004158. [DOI] [PMC free article] [PubMed] [Google Scholar]