

Abstract
Although ancillary pathways of glucose metabolism are critical for synthesizing cellular building blocks and modulating stress responses, how they are regulated remains unclear. In the present study, we used radiometric glycolysis assays, [13C6]-glucose isotope tracing, and extracellular flux analysis to understand how phosphofructokinase (PFK)-mediated changes in glycolysis regulate glucose carbon partitioning into catabolic and anabolic pathways. Expression of kinase-deficient or phosphatase-deficient 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase in rat neonatal cardiomyocytes co-ordinately regulated glycolytic rate and lactate production. Nevertheless, in all groups, >40% of glucose consumed by the cells was unaccounted for via catabolism to pyruvate, which suggests entry of glucose carbons into ancillary pathways branching from metabolites formed in the preparatory phase of glycolysis. Analysis of 13C fractional enrichment patterns suggests that PFK activity regulates glucose carbon incorporation directly into the ribose and the glycerol moieties of purines and phospholipids, respectively. Pyrimidines, UDP-N-acetylhexosamine, and the fatty acyl chains of phosphatidylinositol and triglycerides showed lower 13C incorporation under conditions of high PFK activity; the isotopologue 13C enrichment pattern of each metabolite indicated limitations in mitochondria-engendered aspartate, acetyl CoA and fatty acids. Consistent with this notion, high glycolytic rate diminished mitochondrial activity and the coupling of glycolysis to glucose oxidation. These findings suggest that a major portion of intracellular glucose in cardiac myocytes is apportioned for ancillary biosynthetic reactions and that PFK co-ordinates the activities of the pentose phosphate, hexosamine biosynthetic, and glycerolipid synthesis pathways by directly modulating glycolytic intermediate entry into auxiliary glucose metabolism pathways and by indirectly regulating mitochondrial cataplerosis.
Keywords: cardiac hypertrophy, glycolysis, heart failure, metabolomics, mitochondria, stable isotope
Introduction
Glycolysis is the central pathway of intermediary carbon metabolism. Not only does catabolism of glucose via glycolysis provide useable energy for the cell, but it also contributes glycolytic intermediates that serve as branch-point metabolites for the entry of glucose-derived carbon into ancillary pathways of glucose metabolism. These pathways are required for de novo synthesis of cellular components. For example, the activity of the pentose phosphate pathway (PPP) is required for nucleotide biosynthesis and the maintenance of cellular redox state, whereas the hexosamine biosynthetic pathway (HBP) regulates post-translational protein modifications, protein processing, and stress responses. The synthesis of glycero(phospho)lipids and serine is also dependent on the production of glycolytic intermediates [1]. In the heart, the PPP is activated during hypertrophy [2–4] and heart failure [5], and modulating the activity of this pathway regulates the severity of cardiac pathology [6–11]. Similarly, O-linked β-N-acetylglucosamine (O-GlcNAc)-modified proteins, which require synthesis of the sugar donor UDP-GlcNAc via the HBP [12,13], are elevated in hypertrophied and failing hearts [14–17]. Although less is known about the glycerolipid synthesis pathway (GLP), increased activity of this pathway may underlie pathologic cardiac hypertrophy [18]. In addition, numerous studies have identified aberrant ancillary pathway behavior in the context of diabetes (e.g. as reviewed in refs [19,20]).
The activities of ancillary pathways of glucose metabolism are regulated in part through expression of their rate-limiting or committed-step enzymes, cofactor availability, allosterism, and/or post-translational modifications [21–27]. Flux through these pathways may also be regulated by other means. Specific enzymatic reactions in the glycolytic pathway, especially the phosphofructokinase 1 (PFK1) reaction, can co-ordinate the activity of ancillary pathways in several cell types [28–31]; however, the effects of PFK activity and the glycolytic rate on biosynthetic pathway flux in the heart remain unclear. Understanding how PFK regulates ancillary biosynthetic pathway activity is important because its activity is modulated by increased work ex vivo [32,33], and catecholamines [34] and pressure overload in vivo [35].
In the present study, we examined how PFK activity regulates biosynthetic pathways in cardiomyocytes. For this, we altered PFK1 activity by expressing mutant forms of phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PFK2) in primary rat neonatal cardiomyocytes. This method of regulating glycolysis leverages the strong allosteric regulation of PFK1 by F-2,6-P2 [36–39] and is relevant to mechanisms regulating cardiovascular homeostasis. For example, kinases known to be important for cardiac responses to stress [40,41] are strong regulators of PFK2 activity [42–45]. By integrating radiometric assays, stable isotope tracing, and extracellular flux analyses, we demonstrate that the amount of glucose available for entry into ancillary pathways appears relatively high and that the PFK node of glycolysis exerts varying degrees of control over glucose incorporation in the end products of the PPP, the HBP, and the GLP by not only diverting glucose carbon into these pathways, but also by regulating mitochondrial activity and cataplerosis.
Experimental procedures
Materials
DMEM growth medium, containing l-glutamine, d-glucose, and pyruvate, was purchased from US Biological (Swampscott, MA, U.S.A.). Insulin, transferrin, and selenium (500×) were purchased from Lonza (Walkersville, MD, U.S.A.). Humulin R was from Eli Lilly (Indianapolis, IN, U.S.A.). All other reagents were from Sigma–Aldrich Corp. (St. Louis, MO, U.S.A.), unless indicated otherwise.
Rodent models
All procedures were approved by the University of Louisville Institutional Animal Care and Use Committee and were in accordance with the NIH guidelines. The euthanasia procedures were consistent with the AVMA Guidelines for the Euthanasia of Animals. Neonatal rat cardiomyocytes (NRCMs) were isolated from 1- to 2-day-old Sprague–Dawley rats as previously described [46–48]. Culture media were changed to serum-free DMEM 24 h prior to each experiment.
Gene transfer and expression analysis
Replication-deficient adenoviral vectors were constructed and purified by Vector BioLabs (Malvern, PA, U.S.A.) using cDNA for mutated forms of rat liver 6-phosphofructo-2-kinase/Fru-2,6-P2 bisphosphatase [PFKFB1 (6-phosphofructo-2-kinase/fructo-2,6-bisphosphatase isoform 1) isoform of PFK2]. The enzyme was engineered to have single amino acid point mutations to foster a kinase-deficient (S32D and T55V; GlycoLo) or phosphatase-deficient (S32A and H258A; GlycoHi) form of PFK2 [37,38,49–51]. A 1.4 kb BamHI/Nhe1 fragment of a pLenti6-3×FLAG-pd-PFK2 plasmid was subcloned into a Dual-CCM(+) shuttle vector, which has dual CMV promoters to drive both green fluorescent protein (GFP) and the insert. The backbone of the adenoviral vector is type 5 (dE1/E3). An Ad-GFP control virus, in which GFP is driven by the CMV promoter, was also purchased from Vector BioLabs.
Cardiac myocytes were transduced with adenovirus for 4 h in fresh media, followed by PBS wash and the addition of fresh media. For all viral treatments, a multiplicity of infection (MOI) of 50 was used. Viral transduction and functional expression of the target protein was confirmed by eGFP fluorescence, immunoblotting, and glycolysis assays. For measuring protein abundance, cells were lysed in a common lysis buffer containing 20 mM HEPES, 110 mM KCl, 1 mM EDTA, 1% NP-40, 0.1% SDS, and phosphatase and protease inhibitors. Cell lysates were then centrifuged at 13 000g for 20 min and the supernatant was collected. Protein was measured using the Lowry DC assay kit (Bio-Rad Laboratories, Hercules, CA, U.S.A.) and separated by SDS–PAGE, electroblotted onto PVDF membranes, and probed for PFKFB1 (Novus Biologicals, Littleton, CO, U.S.A.) according to the manufacturer's protocol. A horseradish peroxidase-linked secondary antibody (Cell Signaling Technology, Danvers, MA, U.S.A.) was used to detect and visualize the protein band with a Typhoon 9400 variable mode imager (GE Healthcare, Chicago, IL, U.S.A.).
Radiolabeled glycolysis assay
The rate of glucose utilization was determined using [5-3H]-glucose as previously described [52–54]. Briefly, after 48 h of transduction, the media were replaced with 2 ml of fresh, serum-free media containing 5.5 or 25 mM glucose with the addition of 2 µCi/ml [5-3H]-glucose (Moravek Biochemicals, Brea, CA, U.S.A.). Following incubation for 3 h, 100 µl of medium was collected and added to 100 µl of 0.2 M HCl in a microcentrifuge tube. This tube, with the tube cap removed, was placed in a scintillation vial containing 500 µl of dH2O to allow for evaporative diffusion of [3H]2O in the microcentrifuge tubes into the scintillation vials. To account for incomplete equilibration of [3H]2O and background, known amounts (µCi) of [5-3H]-glucose and [3H]2O (Moravek Biochemicals) were placed into microcentrifuge tubes, and these were placed into separate scintillation vials containing 500 µl of dH2O. After incubation at 37°C for 72 h, the microcentrifuge tube was removed from the vial, 10 ml of scintillation fluid was added, and scintillation counting was performed using a Tri-Carb 2900TR Liquid Scintillation Analyzer (Packard Bioscience Company, Meriden, CT, U.S.A.). Glucose utilization was then calculated as reported by Ashcroft et al. [52], with considerations for the specific activity of [5-3H]-glucose, incomplete equilibration and background, dilution of [5-3H]- to unlabeled glucose, and scintillation counter efficiency. To normalize glucose utilization to total protein, the cells were first washed with PBS (to remove adherent radioactivity) and then lysed in common lysis buffer. After centrifugation, protein concentration was measured using the Lowry assay kit (Bio-Rad Laboratories).
Stable isotope tracing
NRCMs were incubated in six-well plates for 18 h in glucose-free DMEM containing 1 mM pyruvate, 4 mM glutamine, and 25 mM [13C6]-glucose. Cell reactions were then quenched in cold acetonitrile and extracted in acetonitrile:water:chloroform (v/v/v, 2:1.5:1), as described previously [39,55–57], to obtain the polar, nonpolar, and insoluble proteinaceous fractions. The nonpolar (lipid) layer was collected, dried under a stream of nitrogen gas, and reconstituted in 0.1 ml of chloroform:methanol:butylated hydroxytoluene (2:1 + 1 mM) mixture. The extract was diluted 10× with 1 mM butylated hydroxytoluene solution in methanol and used for Fourier transform ion cyclotron resonance-mass spectrometry (FTICR-MS) analysis.
For stable isotope nucleotide analysis, the samples were prepared using a previously published protocol [39,57], with slight modifications. Briefly, lyophilized polar extracts were reconstituted in 50 μl of 5 mM aqueous hexylamine, adjusted to a pH of 6.3 with acetic acid (solvent A). Samples were then loaded onto a 100-μl capacity C18 tip (Thermo Fisher Scientific, Waltham, MA, U.S.A.) followed by washing twice with 50 μl of solvent A. The metabolites were eluted with 70% solvent A and 30% 1 mM ammonium acetate in 90% methanol (pH 8.5; solvent B). The resulting eluates were diluted 3× with methanol and analyzed via FTICR-MS.
[13C6]-glucose and [13C3]-lactate measurements in the media
For quantification of [13C6]-glucose and [13C3]-lactate in cell media, 10 μl of media were mixed with 9990 μl of 5 mM NH4OH in methanol containing 5 μM [D2]-glucose and 5 μM [D2]-lactate internal standard and used without any additional pretreatment for FTICR-MS analysis and quantification. Blank incubations without cells were run in parallel. The concentrations of [13C]-labeled glucose and lactate were assessed using seven-point calibration curves (0.3125–20 µM).
FTICR-MS analysis
Lipid and nucleotide spectra and [13C]-glucose/lactate spectra were acquired using a hybrid linear ion trap-FTICR mass spectrometer (Finnigan LTQ FT; Thermo Electron, Bremen, Germany), equipped with a TriVersa NanoMate ion source (Advion Bio-Sciences, Ithaca, NY, U.S.A.) with an A electrospray chip (nozzle inner diameter, 5.5 μm). The TriVersa NanoMate was operated by applying 1.5 kV with 0.5 psi head pressure in the positive ion mode and 1.6 kV and 0.7 psi in the negative ion mode. High mass accuracy data were collected using the FTICR analyzer over a mass range from 150 to 1600 Da (lipids; + and − mode) for 15 min at a target mass resolution of 400 000 at 400 m/z. For quantification of 13C6-glucose and 13C3-lactate in cell media, data were collected in the negative ion mode for 2 min over a mass range from 50 to 250 Da at a target mass resolution of 100 000 at 400 m/z. Prior to acquisition, the LTQ-FT was tuned and calibrated according to the manufacturer's default recommendations to achieve a mass accuracy of 2 ppm or less.
Stable isotope data analyses
The FTICR MS data were exported as exact mass lists into a spreadsheet file using Qual-Browser 2.0 (Thermo Electron). Isotopologue peak deconvolution, assignment, natural isotope abundance stripping, and quantification were performed using MetSign software, as described previously [39,58].
Extracellular flux analysis of cellular energetics
To measure cellular energetics in intact cardiomyocytes, the Seahorse Bioscience XF24 extracellular flux analyzer was used as described previously [59]. For this, 75 000 cells were plated into each well. An hour before each experiment, the media were replaced with 675 µl of assay media, i.e. unbuffered DMEM containing glucose (5.5 mM), glutamine (4 mM), and pyruvate (1 mM, pH 7.4). Following 1 h in a 37°C, CO2-free incubator, the cells were placed in the instrument for analysis. Basal oxygen consumption rates (OCRs) and extracellular acidification rates (ECARs) were measured using a programmed protocol: 3 cycles of 2 min mix, 2 min wait, and 3 min measure. To interrogate the effects of each treatment on cardiomyocyte bioenergetics, the following compounds were injected into each well with 2 cycles of 2 min mix, 2 min wait, 3 min measure following each injection: Port A, oligomycin (Oligo; 1 µM); Port B, carbonyl cyanide-p-(trifluoromethoxy)phenylhydrazone (FCCP; 1 µM); and Port C, antimycin A, and rotenone (AA/Rot; 10 µM/1 µM). The OCR and ECAR values were normalized to the total amount of protein in each well.
Statistical analysis
All values are mean ± SEM. All comparisons were made to the relevant control group and performed using one-way analysis of variance (ANOVA) with Dunnett's correction for multiple comparisons. The null hypothesis was rejected when P < 0.05.
Results
PFK regulates glycolysis and the distribution of glucose
To understand the role of PFK activity in cardiac myocyte metabolism, we expressed kinase-deficient (GlycoLo) or phosphatase-deficient (GlycoHi) PFK2 mutant constructs in NRCMs, which are conducive to well-controlled and detailed metabolic flux analyses. The mutant genes were driven by a CMV promoter (Figure 1A), and the bicistronic vector also contained GFP, which was used to determine relative transduction efficiency. As shown in Figure 1B, the vast majority of NRCMs were transduced with virus. These PFK2 mutants were intentionally derived from the liver-specific isoform of PFK2 (i.e. PFKFB1), so that expression of the proteins could be assessed by immunoblotting, without obfuscation by other isoforms (e.g. PFKFB2/PFKFB3). Myocytes transduced with the GlycoLo or the GlycoHi constructs showed strong bands at the predicted molecular mass for the mutant forms of PFK2, whereas cells transduced with Ad-GFP showed no expression of PFKFB1 (Figure 1C). Although GlycoHi-expressing cells routinely displayed a higher level of mutant protein expression relative to GlycoLo-expressing cells, the strength of the relative effect of each PFK2 mutant on the cellular glycolytic rate was equal, with a 2.1-fold increase and 2.2-fold decrease in glycolytic rate for the GlycoHi and GlycoLo constructs, respectively (Figure 1D). This effect on glycolytic rate was independent of glucose concentration, as rates were similar for groups incubated with either 5.5 or 25 mM glucose.
Figure 1. Phosphofructokinase activity regulates glucose metabolism in isolated cardiomyocytes.

Glucose utilization analyses in neonatal rat cardiomyocytes: (A) constructs of kinase-deficient and phosphatase-deficient PFK2 engineered to decrease (GlycoLo) or increase (GlycoHi) glycolytic rate, respectively; (B) GFP expression in cardiomyocytes 48 h after transduction (50 MOI) with adenoviruses expressing GFP only or the GlycoLo or GlycoHi adenoviruses; (C) immunoblot of mutant PFK2 protein expression; (D) glycolytic rate measured with [5-3H]-glucose under 5.5 or 25 mM glucose culture conditions. n = 3–5 independent isolations per group. (E) 13C6-glucose uptake, (F) 13C3-lactate production, and (G) the percentage of glucose converted into lactate following 18 h incubation with isotopic glucose. Data in E–G represent three replicates per group. *P < 0.05, **P < 0.01, ***P < 0.001.
To examine the effect of each mutant PFK2 enzyme on aerobic glycolysis, we replaced the normal growth media with media containing [13C6]-glucose and, after 18 h, measured the relative levels of [13C6]-glucose and [13C3]-lactate in the media. As shown in Figure 1E, the levels of [13C6]-glucose consumed by the cell were 1.8-fold higher in GlycoHi-expressing cells compared with those in Ad-GFP, with no difference in GlycoLo-expressing cells. Levels of [13C3]-lactate extruded into the media were 2.5-fold higher in GlycoHi cells compared with GFP control cells and >10-fold higher compared with GlycoLo virus-transduced cells (Figure 1F). As shown in Figure 1G, 29% of [13C6]-glucose was metabolized to [13C3]-lactate in GFP control cells, whereas in GlycoLo-expressing cells, 6% of labeled glucose was metabolized to lactate; in GlycoHi-expressing cells, 44% of labeled glucose was metabolized to lactate.
The assessment of [13C6]-glucose uptake, [13C3]-lactate production, and [5-3H]-glucose utilization provides estimates of not only how much glucose entered the cell and was later extruded as lactate, but also how much made it through the enolase step of glycolysis, where the tritiated label is eliminated as 3H2O. The mean values of glucose uptake and lactate production (provided by stable isotope tracing) and of radiometric measurements were used to calculate the amount of glucose unaccounted for by catabolism to pyruvate. In GFP control cardiomyocytes, only 44% of glucose was catabolized past the enolase step in glycolysis; of this fraction, the majority (65%) was metabolized to lactate (Figure 2A,B). These data show that, under control conditions, a substantial amount of glucose taken up by NRCMs is not catabolized to pyruvate, and that the majority of glucose that is catabolized to pyruvate is fated for lactate production.
Figure 2. Phosphofructokinase-mediated glucose distribution in isolated cardiomyocytes.
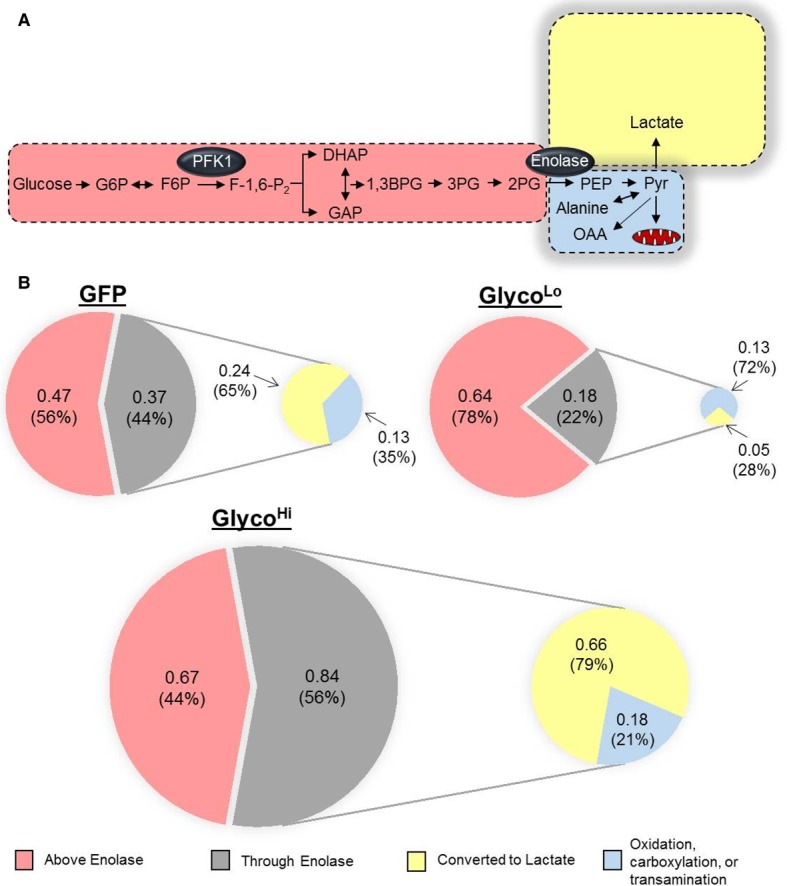
Schematics illustrating glucose utilization in myocytes transduced with GFP, GlycoLo, or GlycoHi adenoviruses. Measurements from radiometric and isotopic glucose utilization assays were used to delineate glucose distribution in neonatal rat cardiomyocytes transduced with each virus. (A) Schematic illustrating glucose distribution in control (GFP) myocytes. Distributions of glucose are drawn to relative scale. (B) Pie chart illustrating the distribution of glucose: numbers (in nmol/h/µg protein) and percentages (%) indicate the absolute and relative distribution of glucose ascribed to the portions of glucose metabolism: upstream of enolase (light red), through enolase (gray shading), converted into lactate (yellow), and available for transamination to alanine, anaplerotic formation of oxaloacetate (OAA), or glucose oxidation (light blue). Pie charts are drawn to scale. Both schematics consider glucose uptake and lactate production provided by [13C6]-glucose tracing and by glucose utilization measured by radiometric assays, respectively.
As shown in Figure 1E, glucose uptake was not different between GFP and GlycoLo cells, yet GlycoLo myocytes demonstrated 2-fold lower glucose utilization through the enolase step of glycolysis and 5-fold lower lactate production. In these cells, 78% of glucose-derived carbon was partitioned upstream of the enolase reaction, leaving 22% available for pathways downstream from enolase (Figure 2B). Although lactate production was markedly suppressed in GlycoLo cells, the absolute amount of glucose-derived carbon available for either mitochondrial oxidation, transamination to alanine, or carboxylation to oxaloacetate was identical with that of control cells. Interestingly, while GlycoHi cells showed 80% higher levels of glucose uptake compared with GFP control cells (see Figure 1E), 44% of glucose (by mass) was not catabolized via glycolysis. Nevertheless, a portion of glucose equivalent to that apportioned upstream of enolase (0.67 nmol/h/µg protein) taken into the cell could be accounted for as lactate (0.66 nmol/h/µg protein), and 21% of glucose carbon that was catabolized past the enolase reaction was available for fates other than lactate production (Figure 2B).
PFK activity regulates glucose carbon partitioning into the PPP in NRCMs
To determine how the activity of PFK regulates critical anabolic reactions derived from glycolysis, we modulated the glycolytic rate by expressing the GlycoLo or GlycoHi PFK2 mutants in cardiomyocytes and then assessed carbon incorporation into end products of ancillary biosynthetic pathways using stable isotope-resolved metabolomics [60]. Expression of the GlycoHi mutant modestly diminished incorporation of glucose-derived carbon into ATP. Lower fractional enrichment of 13C in GlycoHi-expressing cells is attributable to decreased carbon allocation into the phosphoribosyl ring of the nucleotide (m+5 isotopologue; Figure 3A,B), suggesting diminished flux of 13C-labeled glucose-6-phosphate (G6P) through the oxidative PPP (oxPPP). The higher level of the m+0 isotopologue of ATP (i.e. the unlabeled ATP pool) in GlycoHi cells further indicates that the flux of 13C6-G6P through the oxPPP was lower in GlycoHi cells. Similar changes were observed in other purines such as GTP (Figure 3C). Myocytes expressing the GlycoLo mutant showed 13C fractional enrichment values into ATP that were similar to GFP-transduced control cells (Figure 3A,B), suggesting that low PFK activity does not strongly affect purine synthesis in these cells.
Figure 3. Phosphofructokinase co-ordinates PPP activity.

Isolated cardiomyocytes were transduced with GFP, GlycoLo, or GlycoHi adenoviruses and incubated with [13C6]-glucose for 18 h. Stable isotope tracing of nucleotides in NRCMs incubated with media containing U-[13C6]-glucose: (A) atom-resolved map illustrating the biological and biochemical history of 13C incorporation into the purine ATP; fractional enrichment values of 13C in (B) ATP and (C) GTP after incubation with [13C6]-glucose; (D) atom-resolved map illustrating the biological and biochemical history of 13C incorporation into the pyrimidine UTP; fractional enrichment values of 13C in (E) UTP and (F) CTP after incubation with [13C6]-glucose. Colored x-axis labels correlate to the labeled molecule biosynthetic components indicated in the atom-resolved maps. Graph represents three replicates per group from one isolation. $P < 0.05, $$P < 0.01, ***,$$$P < 0.001, ****,$$$$P < 0.0001.
For the pyrimidines CTP and UTP, we found a higher m+0 isotopologue value in GlycoHi cells and a lower m+0 isotopologue value in GlycoLo cells compared with GFP controls. These data suggest that high rates of glycolysis diminish incorporation, and that low rates of glycolysis increase incorporation of glucose-derived carbon into pyrimidines (Figure 3D,E). Interestingly, both the GlycoLo and the GlycoHi NRCMs showed increased levels of CTP and UTP having only the ribose ring labeled (m+5 isotopologue). These data could indicate that either decreasing or increasing glycolysis augments oxPPP flux. This interpretation would be consistent with the observation that both GlycoLo and GlycoHi cells show more glucose available for entry into ancillary biosynthetic pathways (see Figure 2); however, it is also possible that higher levels of m+5 labeling could be due to an accumulation of these pyrimidine isotopologues, where the contribution of [13C6]-glucose to de novo synthesis of the uridine ring is diminished. Indeed, examination of the m+7 and m+8 isotopologues of UTP, which probably correspond to the addition of carbons from 13C-labeled aspartate to the molecule (see Figure 3D), shows that both the GlycoLo and GlycoHi cells have less incorporation of 13C, which is known to derive from Krebs cycle-(m+7) or pyruvate carboxylase-engendered (m+8) oxaloacetate. The m+6 isotopologue probably corresponds to complete 13C5-labeling of the ribose ring and 13C1 labeling of aspartate, occurring as a result of scrambling during the second turn of the Krebs cycle, as suggested previously [61]. Overall, these data suggest that high PFK activity modestly diminishes oxPPP flux and 13C incorporation into purines. Pyrimidine synthesis appears to be more robustly affected by PFK activity, with high rates of glycolysis diminishing incorporation of glucose-derived carbon into UTP and CTP molecules.
High PFK activity diminishes glucose-derived carbon incorporation into UDP-N-acetylhexosamine
The molecular synthesis of UDP-N-acetyl hexosamines requires end products or intermediates of several metabolic pathways, including those from the Krebs cycle, the pyrimidine biosynthetic pathway, glutamine, and glycolysis; hence, these pathways collectively contribute to UDP-N-acetylhexosamine (UDP-HexNAc) synthesis (Figure 4A). To determine how the glycolytic activity affects glucose-derived carbon allocation into the HBP, we measured fractional enrichment of 13C from [13C6]-glucose into UDP-HexNAc in NRCMs transduced with GlycoLo or GlycoHi virus. After 18 h, >90% of the UDP-HexNAc pool integrated 13C-glucose-derived carbon (Figure 4B), which suggests that UDP-HexNAc has a faster rate of glucose carbon incorporation relative to purines and pyrimidines. That the m+0 isotopologue was higher in GlycoHi cells indicates that high rates of glycolysis diminish glucose carbon incorporation into UDP-HexNAc. The lower 13C enrichment of many isotopologues (e.g. m+7, m+14, m+15, and m+16) appeared to be due to diminished input of metabolites that require mitochondrial transformation of 13C-glucose-derived carbon; however, the m+5, m+6, and m+11 isotopologues (which are the forms of UDP-HexNAc with the ribose, hexose, and ribose + hexose moieties constituted with 13C, respectively) demonstrate significantly higher fractional enrichment in GlycoHi cells. Given that both the GlycoLo and the GlycoHi cells had similar levels of glucose available for reactions upstream of enolase (see Figure 2), the simplest explanation for this result is that the mitochondrial cataplerotic metabolites (derived initially from [13C6]-glucose-derived) are limiting for the synthesis of UDP-HexNAc; as a result, the m+5 and m+6 isotopologues of the metabolite accumulate as 13C incorporation from cataplerotic metabolites slows down. This would be consistent with findings in pyrimidines, which suggest that cataplerotic generation of aspartate is limiting for the incorporation of glucose-derived carbon into CTP and UTP.
Figure 4. High phosphofructokinase activity diminishes glucose carbon incorporation into UDP-HexNAc.

Isolated cardiomyocytes were transduced with GFP, GlycoLo, or GlycoHi adenoviruses and incubated with [13C6]-glucose for 18 h. Stable isotope tracing of UDP-HexNAc in NRCMs incubated with media containing [13C6]-glucose: (A) atom-resolved map illustrating the biological and biochemical history of 13C incorporation into UDP-GlcNAc; (B) fractional enrichment values of 13C in UDP-HexNAc (which includes both UDP-GlcNAc and UDP-GalNAc) after incubation with [13C6]-glucose. Colored x-axis labels correlate to the labeled molecule biosynthetic components indicated in the atom-resolved maps. Graph represents three replicates per group from one isolation. *,$P < 0.05, **,$$P < 0.01, $$$P < 0.001, $$$$P < 0.0001.
Although low rates of glycolysis did not affect the overall level of incorporation of glucose-derived carbon into UDP-HexNAc, that the m+11 isotopologue in GlycoLo cells was higher than that in control cells suggests that low PFK activity promotes channeling of F6P (fructose-6-phosphate) not only into the HBP (contributing six 13C), but, because the ribose ring is included (contributing five 13C), also into the PPP (Figure 4A,B). Changes in the m+8 isotopologue are difficult to interpret because this isotopologue could consist of 13C-labeled N-acetylhexosamine and stably labeled UDP. Altogether, these results suggest that high PFK activity decreases glucose carbon allocation into UDP-HexNAc primarily via limiting the formation of cataplerotic intermediates (e.g. acetyl CoA, aspartate) derived initially from [13C6]-glucose.
PFK regulates glycerolipid synthesis in cardiomyocytes
In cardiac myocytes, glycerolipid synthesis requires free fatty acids (FFAs) and glycerol-3-phosphate (G3P). While FFAs can be derived from transporter-mediated uptake across the plasma membrane [62], myocytes have a relatively low capacity for free glycerol uptake; therefore, there is little contribution of glycerol kinase to G3P synthesis [63,64]. However, G3P can be generated from dihydroxyacetone phosphate (DHAP) by glycerol-3-phosphate dehydrogenase (Figure 5A). Nevertheless, it is unclear whether or how the rate of glycolysis affects glycerolipid synthesis. We found that in comparison with cells transduced with GFP virus, the GlycoLo cells showed diminished incorporation of 13C from 13C6-glucose into the glycerol (m+3) moiety of phosphatidylcholine (PC; Figure 5B), phosphatidylinositol (PI; Figure 5C), phosphatidylethanolamine (PE; Figure 5D), and phosphatidylserine (PS; Figure 5E). Incorporation of 13C6-glucose-derived carbon into fatty acids in PC, PI, PE, and PS was not different in GlycoLo cells from that in control cells, suggesting that only the synthesis of the glycerol backbone portion of these phospholipids is affected by low rates of glycolysis.
Figure 5. Phosphofructokinase regulates glycerolipid biosynthesis.

Stable isotope tracing of phospholipids and triacylglycerols in cardiomyocytes incubated with media containing 13C6-glucose for 18 h: (A) atom-resolved map illustrating the biological and biochemical history of 13C incorporation into glycerolipids; fractional enrichment values of 13C into: (B) PC; (C) PI; (D) PE; (E) PS; and (F) TAG. Graph represents three replicates per group from one isolation. *,$P < 0.05, **,$$P < 0.01, ****P < 0.0001.
High glycolytic rates affected 13C incorporation into phospholipids to a lesser extent. Although PE synthesis showed a slight increase in 13C fractional enrichment in glycerol (and a concomitant decrease in fractional enrichment of the unlabeled pool; Figure 5D), PI showed diminished labeling characterized by lower incorporation of 13C into fatty acids. Glycerol 13C labeling in PI from GlycoHi cells was identical with that of control cells. This dichotomy in labeling patterns in different phospholipids could suggest the presence of unique enzyme complexes that participate in metabolic channeling for the synthesis of distinct glycerolipids. Consistent with this idea, triacylglycerol (TAG) labeling was remarkably different from that of other glycerolipids. As shown in Figure 5F, TAG species collectively showed little 13C in the glycerol moiety. Furthermore, low rates of glycolysis had little effect on glucose carbon incorporation into TAGs; however, GlycoHi cells showed lower 13C incorporation into fatty acids. Collectively, these results suggest that, in isolated NRCMs, low rates of glycolysis diminish phospholipid synthesis by the DHAP → G3P pathway and that high rates of glycolysis decrease incorporation of glucose-derived carbon into the fatty acyl chains of PI and TAGs.
PFK activity regulates mitochondrial activity in NRCMs
Stable isotope labeling patterns of several end products of the PPP, HBP, and GLP suggest that high PFK activity diminishes the relative abundance of those 13C isotopologues that require metabolites derived from mitochondria. This suggests that cataplerosis [65] is diminished under conditions of high PFK activity. Because cataplerotic reactions depend on mitochondrial activity, we next measured respiration in cells expressing either the control construct (GFP) or the GlycoHi or GlycoLo construct. GlycoHi-expressing cells showed 50% lower basal oxygen consumption, whereas GlycoLo-expressing cells showed significantly higher basal OCR values (Figure 6A,C). These PFK-mediated changes in mitochondrial OCR are probably due to the Crabtree effect, which is characterized by decreases in mitochondrial activity under conditions of increased glucose availability or glycolytic activity [54,66–68]. Although ATP-linked OCR was not significantly different between the groups, proton leak was significantly diminished in GlycoHi cells. Upon the addition of a mitochondrial uncoupler, GlycoLo and GlycoHi cells showed relative changes in maximal respiratory rate and in reserve capacity that mirrored the pattern found for basal OCR and proton leak (Figure 6A,C). These data are consistent with the notion that high PFK activity diminishes mitochondrial activity, which limits the cataplerotic generation of metabolites required for the synthesis of end products of the PPP, HBP, and GLP.
Figure 6. Phosphofructokinase activity regulates mitochondrial activity in isolated cardiomyocytes.

Extracellular flux analyses of NRCMs: (A) oxygen consumption traces (OCR), (B) extracellular acidification rate (ECAR), and (C) calculated indices of respiration of cardiomyocytes transduced with GFP, GlycoLo, or GlycoHi adenoviruses. n = 3 independent isolations per group. *P < 0.05, **P < 0.01.
Although cells transduced with the GlycoHi virus had >2-fold elevation in basal ECAR compared with Ad-GFP controls, we observed no differences in ECAR in NRCMs expressing the GlycoLo PFK2 mutant (Figure 6B). The addition of oligomycin increased ECAR to similar values in all groups, suggesting that inhibition of mitochondrial ATP production overrides PFK2-mediated changes in glycolysis. Although extracellular acidification is a surrogate measure of lactate extrusion and aerobic glycolysis, protons are also generated during respiration via pyruvate dehydrogenase-mediated production of CO2 (i.e. CO2 → HCO3− + H+) [69,70]; thus, the inconsistency of ECAR with absolute measurements of the glycolytic rate and 13C3-lactate in GlycoLo cells (see. Figure 1) is probably due to an increase in mitochondrial activity. Taken together, measurements from radiometric assays, stable isotope tracing, and extracellular flux analyses indicate that PFK regulates ancillary pathways by not only direct partitioning of glucose (e.g. in the PPP and the GLP) but also regulating mitochondrial cataplerotic reactions.
Discussion
Most studies of cardiac metabolism have focused primarily on the catabolic reactions required for energy provision [71]. Considerably less effort has been devoted to understanding the regulation of ancillary pathways of glucose metabolism in the heart and how these pathways integrate with catabolic reactions. In the present study, we examined how PFK activity, which immediately precedes the ‘lysis’ step in glycolysis, regulates several 6-carbon- and 3-carbon-initiated anabolic pathways that require glycolytic intermediates. Our results demonstrate that, in cardiac myocytes, PFK regulates glucose carbon incorporation into ancillary biosynthetic pathways by: (1) directly regulating carbon flow into purines and phospholipids and (2) indirectly influencing mitochondrial cataplerotic activity required for the generation and subsequent incorporation of glucose-derived carbon into products of the PPP, HBP, and GLP. These studies also illustrate how radiometric measurements, stable isotope tracing, and extracellular flux analyses can be integrated to examine substrate fate and to develop detailed understanding of how interventions simultaneously affect anabolic and catabolic metabolism.
We chose to study the integration of anabolism and catabolism in the context of glycolytic regulation at the PFK node. This enzyme controls 65% of the flux through glycolysis in the heart [72], making it the major regulator of glycolytic activity. It is unlikely that, under the experimental conditions used, glucose entry was a major limiting step for its metabolism because NRCMs express both GLUT1 and GLUT4 [73]. Our stable and radiometric isotope tracing measurements show that, under control conditions, <50% of glucose taken into myocytes is catabolized past the enolase reaction. This suggests that a majority of the glucose entering the myocyte is not used for energy production, but is probably used by biosynthetic pathways. Although PFK activity was found to be a potent determinant of glycolysis-engendered lactate production, it did not have diametrically co-ordinate effects on the relative amounts of glucose found upstream of the enolase reaction. Surprisingly, both a decrease and an increase in glycolytic activity led to the accumulation of glucose upstream of the enolase reaction; however, the mechanisms by which low and high PFK activity augment the levels of glucose-derived carbon upstream of the enolase reaction appear to differ: when PFK activity was high, glucose uptake was increased, but glucose utilization was elevated; when PFK activity was low, glucose uptake was the same as that found in control cells, but glucose utilization was remarkably diminished. These results suggest that conditions of low PFK activity promote accumulation of metabolites (probably upstream of the PFK step), and that conditions of high glycolytic activity support levels of glucose-derived carbon upstream of the enolase reaction by augmenting glucose uptake. Additionally, our data suggest that the level of glucose available for glucose oxidation, alanine transamination, or pyruvate carboxylation was relatively stable and independent of PFK activity.
Although studies in some cell types such as adipocytes suggest that the HBP is a relatively minor pathway, utilizing only 2–3% of incoming glucose [74], our stable isotope tracing data suggest that the rate of entry of glucose-derived carbon into this pathway may be higher in cardiac myocytes. The unlabeled pool of UDP-HexNAc was below 10% in all groups, suggesting that sugar donors such as UDP-GlcNAc and UDP-GalNAc are synthesized and utilized at high rates. In contrast, >20% of the pool of nucleotides was unlabeled and 13C enrichment of the glycerol moiety of glycerolipids was below 20%. These findings appear to indicate that the HBP utilizes more glucose-derived carbon than either the PPP or the GLP. Given that up to 20% of glucose taken up by the heart could enter the PPP [75], our comparisons of 13C enrichment in end products of these pathways suggest that HBP flux in cardiomyocytes may be much higher than currently appreciated.
Our results are consistent with previous findings in epithelial cells [30], fibroblasts [76,77], and developing embryos [78], which show that glycolysis regulates glucose entry into the PPP. Our findings are also consistent with computational predictions of the role of PFK in regulating the PPP in the intact heart [72]. Similar to findings in neurons [79], in cardiac myocytes, high glycolytic rates driven by PFK decreased the incorporation of glucose carbon into the ribose ring of purines. This suggests that high rates of glycolysis decrease the flux of glucose through the oxPPP. Nevertheless, we identified remarkable differences in 13C labeling in ATP and GTP versus CTP and UTP. Although both classes of nucleotides use the metabolic precursor ribose-5-phosphate to form the ribose ring, purine ring carbon atoms originate from N10-formyltetrahydrofolate, CO2, and glycine, while pyrimidine ring carbons come from carbamoyl phosphate and glutamine, as well as aspartate generated by cataplerosis [80]. In the time frame examined, purines did not demonstrate incorporation of carbons that derive from metabolic products of the serine biosynthetic pathway and one-carbon metabolism. Hence, it is possible that glycine and N10-formyltetrahydrofolate derived from the serine biosynthetic pathway might be inadequate to participate in purine ring synthesis during the 18 h incubation time used in our experiments. Alternatively, unlabeled serine and folate present in the media could be used by these cells for de novo purine biosynthesis.
The decrease in mitochondrial activity detected by extracellular flux analysis in GlycoHi cells, paired with stable isotope tracing results in pyrimidines, suggests low cataplerotic generation of aspartate. Oxaloacetate generated in the Krebs cycle or via pyruvate carboxylation is transaminated to aspartate, which is subsequently used for pyrimidine biosynthesis. That the m+7 and m+8 isotopologues were lower in GlycoHi cells shows that these pathways were diminished under conditions of high PFK activity. From these data, we inferred that the decrease in de novo pyrimidine biosynthesis in the GlycoHi cells may be due to lower rates of cataplerosis. Because tricarboxylic acid anions are delicately balanced by the input and output of intermediates involved in mitochondrial substrate oxidation, anaplerosis, and cataplerosis [65], we reasoned that mitochondrial activity measurements could provide convergent evidence of PFK-mediated regulation of auxiliary glucose biosynthetic pathways. Indeed, the decrease in mitochondrial activity in GlycoHi cells, paired with the observation that 13C-aspartate entry into pyrimidines was low, suggests that inhibition of pyrimidine synthesis under conditions of high glycolytic activity is due to low rates of cataplerosis.
Similar reasoning as well as previous modeling, validation, and biochemical interpretation of 13C isotopologue data for UDP-GlcNAc [81] were used to determine how PFK affects the HBP, which is important given the known role of UDP-hexosamines in modulating N- and O-linked glycosylation [12,13]. The fact that the unlabeled pool of UDP-HexNAc was higher in GlycoHi cells suggests that the net effect of high PFK activity is a decrease in HBP flux, and, like that found for pyrimidines, this appears to be in part due to diminished mitochondrial activity. Nevertheless, there are limitations for interpreting current data on HBP flux. Because building UDP-HexNAc requires metabolites from many sources, it is possible that the contribution of carbon from non-labeled pools of metabolites may hinder interpretation. A potential solution to this problem would be to use a pulse-chase approach, where isotopic labeling in the system is first saturated (e.g. with both 13C and 15N) and then the stably labeled substrate(s) are replaced with unlabeled substrate(s). The disappearance of the stable label over time in isotopologues of UDP-HexNAc should, with higher confidence, reveal which intersecting metabolic pathways limit HBP activity and UDP-Glc(Gal)NAc synthesis under a given set of conditions.
To date, little evidence exists for whether or how glycolysis regulates GLP activity. Interestingly, G3P formed via glycerol-3-phosphate dehydrogenase 1 has been shown to inhibit PFK2 and activate fructose bisphosphatase 2 [82], suggesting feedback regulation of the GLP on both glycolytic and gluconeogenic activity. An indication that the glycolytic rate regulates GLP flux and glycerolipid synthesis is provided by studies, showing that genetic deletion or overexpression of PFKFB3, a highly active form of PFK2, in adipose tissue co-ordinately regulates fat deposition and obesity [83,84], which is consistent with the notion that glycolysis in adipocytes regulates TAG synthesis and storage. We found that low PFK activity directly limits glucose-derived carbon incorporation into the glycerol backbone of several phospholipids including PC, PI, PE, and PS; however, this was not the case with triacylglycerols, where 13C incorporation into glycerol was not different, but fatty acyl chain labeling was diminished in GlycoHi cells. These results suggest that the fatty acid pool derived from cataplerotic reactions is differentially channeled to route newly synthesized fatty acids for esterification into triacylglycerols rather than phospholipids.
There are several limitations to our study. Although NRCMs are beating cardiac myocytes, they rely on glycolysis for energy to a much greater extent than the adult heart [85]. Thus, findings regarding pathways that involve metabolites generated by cataplerosis may not entirely reflect metabolism in the adult heart. Glucose carbon fate in NRCMs appears to differ from the adult heart primarily at the level of coupling between glycolysis and glucose oxidation. In the adult human heart, previous studies show that 26% of extracted glucose undergoes oxidation and 12% is released as lactate [86]. In contrast, in NRCMs, we show that <13% of glucose carbon undergoes oxidation and ∼24% is released as lactate. Nevertheless, our finding that a large portion of exogenous glucose extracted by cardiac myocytes is apportioned for metabolic pathways other than glycolysis and glucose oxidation is supported by human in vivo isotope tracing studies in the myocardium, which demonstrate that 60% of exogenous glucose extracted by contracting myocytes enters a slow turnover pool amenable to entry into ancillary biosynthetic pathways [86]. Another consideration is that the NRCM culture medium is devoid of fatty acids, which could influence the coupling between glycolysis and glucose oxidation as well as biosynthetic pathway activity. Finally, it is important to consider that our stable isotope analyses did not include glycogen, glycine, serine, or sorbitol, which limits our understanding of how glycolysis regulates glycogen biosynthesis, serine biosynthesis, and the polyol pathway. Nevertheless, computational models created by Cortassa et al. [72] suggest that the polyol pathway is under strong control by activation or inhibition of PFK. Similarly, previous studies show that PFK activity regulates glycogen abundance in the heart [37,38]. Such a behavior would be consistent with co-ordinate regulation of ancillary biosynthetic pathway activity by PFK.
In summary, by using several methods to measure metabolic pathway activity, we found that >40% of glucose consumed by NRCMs is not catabolized to pyruvate, and that PFK has strong effects on the coupling between glucose oxidation and glycolysis. Pairing results from stable isotope-resolved metabolomics with extracellular flux analysis demonstrated that PFK activity regulates glucose carbon incorporation directly into the ribose and the glycerol moieties of purines and phospholipids, respectively, and that high PFK, by indirectly limiting mitochondrial activity, diminishes 13C incorporation into pyrimidines, UDP-N-acetylhexosamine(s), and the fatty acyl chains of phosphatidylinositol and triglycerides. These insights could help elucidate how pathological or physiological changes in glucose metabolism regulate the anabolic behavior and remodeling of the myocardium.
Abbreviations
- CMV
cytomegalovirus
- DHAP
dihydroxyacetone phosphate
- ECAR
extracellular acidification rate
- F6P
fructose-6-phosphate
- FFAs
fatty acids
- FTICR-MS
Fourier transform ion cyclotron resonance-mass spectrometry
- G3P
glycerol-3-phosphate
- G6P
glucose-6-phosphate
- GFP
green fluorescent protein
- GLP
glycerolipid synthesis pathway
- HBP
hexosamine biosynthetic pathway
- LTQ-FT
hybrid linear ion trap-FTICR mass spectrometer
- MOI
multiplicity of infection
- NRCM
neonatal rat cardiomyocyte
- OCR
oxygen consumption rate
- oxPPP
oxidative PPP
- PC
phosphatidylcholine
- PI
phosphatidylinositol
- PE
phosphatidylethanolamine
- PG
phosphoglycerol
- PS
phosphatidylserine
- pd-PFK2
phosphatase-deficient PFK2
- PFK1
phosphofructokinase 1
- PFK2
phosphofructokinase 2 (also denoted as 6-phosphofructo-2-kinase/fructo-2,6-bisphosphatase)
- PFKFB1
6-phosphofructo-2-kinase/fructo-2,6-bisphosphatase isoform 1
- PFKFB2
6-phosphofructo-2-kinase/fructo-2,6-bisphosphatase isoform 2
- PFKFB3
6-phosphofructo-2-kinase/fructo-2,6-bisphosphatase isoform 3
- PPP
pentose phosphate pathway
- TAG
triacylglycerol
- UDP-HexNAc
UDP-N-acetylhexosamine
Author Contribution
A.A.G. contributed to financial support, experimental studies, assembly of data, data analysis and interpretation, and manuscript writing, and gave final approval for publication of the manuscript. P.L. contributed to experimental studies, data analysis and interpretation, and manuscript writing, and gave final approval for publication of the manuscript. Y.Z. contributed to experimental studies and gave final approval for publication of the manuscript. X.Z. contributed to experimental studies and gave final approval for publication of the manuscript. A.B. contributed to financial support and gave final approval for publication of the manuscript. S.P.J. contributed to manuscript writing and gave final approval for publication of the manuscript. B.G.H. contributed to financial support, concept and design, assembly of data, data analysis and interpretation, and manuscript writing, and gave final approval for publication of the manuscript.
Funding
This work was supported by grants from the National Institutes of Health [HL122580 (to B.G.H.), HL130174 (to B.G.H.), HL55477 (to A.B.), GM103492 (to A.B.), HL131647 (to S.P.J.), and HL78825], a Predoctoral Fellowship from the American Heart Association [16PRE31010022 (to A.A.G.)], and the American Diabetes Association Pathway to Stop Diabetes Grant [1-16-JDF-041 (to B.G.H.)].
Competing Interests
The Authors declare that there are no competing interests associated with the manuscript.
References
- 1.Noor E., Eden E., Milo R. and Alon U. (2010) Central carbon metabolism as a minimal biochemical walk between precursors for biomass and energy. Mol. Cell 39, 809–820 doi: 10.1016/j.molcel.2010.08.031 [DOI] [PubMed] [Google Scholar]
- 2.Meerson F.Z., Spiritchev V.B., Pshennikova M.G. and Djachkova L.V. (1967) The role of the pentose-phosphate pathway in adjustment of the heart to a high load and the development of myocardial hypertrophy. Experientia 23, 530–532 doi: 10.1007/BF02137950 [DOI] [PubMed] [Google Scholar]
- 3.Zimmer H.G., Ibel H. and Steinkopff G. (1980) Studies on the hexose monophosphate shunt in the myocardium during development of hypertrophy. Adv. Myocardiol. 1, 487–492 PMID: [PubMed] [Google Scholar]
- 4.Zimmer H.G. and Peffer H. (1986) Metabolic aspects of the development of experimental cardiac hypertrophy. Basic Res. Cardiol. 81 (Suppl 1), 127–137 PMID: [DOI] [PubMed] [Google Scholar]
- 5.Gupte S.A., Levine R.J., Gupte R.S., Young M.E., Lionetti V., Labinskyy V. et al. (2006) Glucose-6-phosphate dehydrogenase-derived NADPH fuels superoxide production in the failing heart. J. Mol. Cell Cardiol. 41, 340–349 doi: 10.1016/j.yjmcc.2006.05.003 [DOI] [PubMed] [Google Scholar]
- 6.McCommis K.S., Douglas D.L., Krenz M. and Baines C.P. (2013) Cardiac-specific hexokinase 2 overexpression attenuates hypertrophy by increasing pentose phosphate pathway flux. J. Am. Heart Assoc. 2, e000355 doi: 10.1161/JAHA.113.000355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zimmer H.G. and Ibel H. (1984) Ribose accelerates the repletion of the ATP pool during recovery from reversible ischemia of the rat myocardium. J. Mol. Cell Cardiol. 16, 863–866 doi: 10.1016/S0022-2828(84)80010-3 [DOI] [PubMed] [Google Scholar]
- 8.Jain M., Cui L., Brenner D.A., Wang B., Handy D.E., Leopold J.A. et al. (2004) Increased myocardial dysfunction after ischemia-reperfusion in mice lacking glucose-6-phosphate dehydrogenase. Circulation 109, 898–903 doi: 10.1161/01.CIR.0000112605.43318.CA [DOI] [PubMed] [Google Scholar]
- 9.Zuurbier C.J., Eerbeek O., Goedhart P.T., Struys E.A., Verhoeven N.M., Jakobs C. et al. (2004) Inhibition of the pentose phosphate pathway decreases ischemia-reperfusion-induced creatine kinase release in the heart. Cardiovasc. Res. 62, 145–153 doi: 10.1016/j.cardiores.2004.01.010 [DOI] [PubMed] [Google Scholar]
- 10.Hecker P.A., Leopold J.A., Gupte S.A., Recchia F.A. and Stanley W.C. (2013) Impact of glucose-6-phosphate dehydrogenase deficiency on the pathophysiology of cardiovascular disease. Am. J. Physiol. Heart Circ. Physiol. 304, H491–H500 doi: 10.1152/ajpheart.00721.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Katare R., Caporali A., Emanueli C. and Madeddu P. (2010) Benfotiamine improves functional recovery of the infarcted heart via activation of pro-survival G6PD/Akt signaling pathway and modulation of neurohormonal response. J. Mol. Cell Cardiol. 49, 625–638 doi: 10.1016/j.yjmcc.2010.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Laczy B., Hill B.G., Wang K., Paterson A.J., White C.R., Xing D. et al. (2009) Protein O-GlcNAcylation: a new signaling paradigm for the cardiovascular system. Am. J. Physiol. Heart Circ. Physiol. 296, H13–H28 doi: 10.1152/ajpheart.01056.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dassanayaka S. and Jones S.P. (2014) O-GlcNAc and the cardiovascular system. Pharmacol. Ther. 142, 62–71 doi: 10.1016/j.pharmthera.2013.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lunde I.G., Aronsen J.M., Kvaloy H., Qvigstad E., Sjaastad I., Tonnessen T., et al. (2012) Cardiac O-GlcNAc signaling is increased in hypertrophy and heart failure. Physiol. Genomics 44, 162–172 doi: 10.1152/physiolgenomics.00016.2011 [DOI] [PubMed] [Google Scholar]
- 15.Facundo H.T., Brainard R.E., Watson L.J., Ngoh G.A., Hamid T., Prabhu S.D. et al. (2012) O-GlcNAc signaling is essential for NFAT-mediated transcriptional reprogramming during cardiomyocyte hypertrophy. Am. J. Physiol. Heart Circ. Physiol. 302, H2122–H2130 doi: 10.1152/ajpheart.00775.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Watson L.J., Facundo H.T., Ngoh G.A., Ameen M., Brainard R.E., Lemma K.M. et al. (2010) O-linked β-N-acetylglucosamine transferase is indispensable in the failing heart. Proc. Natl Acad. Sci. U.S.A. 107, 17797–17802 doi: 10.1073/pnas.1001907107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dassanayaka S., Brainard R.E., Watson L.J., Long B.W., Brittian K.R., DeMartino A.M. et al. (2017) Cardiomyocyte Ogt limits ventricular dysfunction in mice following pressure overload without affecting hypertrophy. Basic Res. Cardiol. 112, 23 doi: 10.1007/s00395-017-0612-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krishnan J., Suter M., Windak R., Krebs T., Felley A., Montessuit C. et al. (2009) Activation of a HIF1α-PPARγ axis underlies the integration of glycolytic and lipid anabolic pathways in pathologic cardiac hypertrophy. Cell Metab. 9, 512–524 doi: 10.1016/j.cmet.2009.05.005 [DOI] [PubMed] [Google Scholar]
- 19.Bouché C., Serdy S., Kahn C.R. and Goldfine A.B. (2004) The cellular fate of glucose and its relevance in type 2 diabetes. Endocr. Rev. 25, 807–830 doi: 10.1210/er.2003-0026 [DOI] [PubMed] [Google Scholar]
- 20.Brownlee M. (2001) Biochemistry and molecular cell biology of diabetic complications. Nature 414, 813–820 doi: 10.1038/414813a [DOI] [PubMed] [Google Scholar]
- 21.Sage A.T., Walter L.A., Shi Y., Khan M.I., Kaneto H., Capretta A. et al. (2010) Hexosamine biosynthesis pathway flux promotes endoplasmic reticulum stress, lipid accumulation, and inflammatory gene expression in hepatic cells. Am. J. Physiol. Endocrinol. Metab. 298, E499–E511 doi: 10.1152/ajpendo.00507.2009 [DOI] [PubMed] [Google Scholar]
- 22.James L.R., Tang D., Ingram A., Ly H., Thai K., Cai L. et al. (2002) Flux through the hexosamine pathway is a determinant of nuclear factor κB-dependent promoter activation. Diabetes 51, 1146–1156 doi: 10.2337/diabetes.51.4.1146 [DOI] [PubMed] [Google Scholar]
- 23.Zimmer H.-G. (1992) The oxidative pentose phosphate pathway in the heart: regulation, physiological significance, and clinical implications. Basic Res. Cardiol. 87, 303–316 doi: 10.1007/BF00796517 [DOI] [PubMed] [Google Scholar]
- 24.Palm D.C., Rohwer J.M. and Hofmeyr J.-H.S. (2013) Regulation of glycogen synthase from mammalian skeletal muscle—a unifying view of allosteric and covalent regulation. FEBS J. 280, 2–27 doi: 10.1111/febs.12059 [DOI] [PubMed] [Google Scholar]
- 25.Coleman R.A. and Lee D.P. (2004) Enzymes of triacylglycerol synthesis and their regulation. Prog. Lipid Res. 43, 134–176 doi: 10.1016/S0163-7827(03)00051-1 [DOI] [PubMed] [Google Scholar]
- 26.Kalhan S.C. and Hanson R.W. (2012) Resurgence of serine: an often neglected but indispensable amino acid. J. Biol. Chem. 287, 19786–19791 doi: 10.1074/jbc.R112.357194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grant G.A. (2012) Contrasting catalytic and allosteric mechanisms for phosphoglycerate dehydrogenases. Arch. Biochem. Biophys. 519, 175–185 doi: 10.1016/j.abb.2011.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yamamoto T., Takano N., Ishiwata K., Ohmura M., Nagahata Y., Matsuura T. et al. (2014) Reduced methylation of PFKFB3 in cancer cells shunts glucose towards the pentose phosphate pathway. Nat. Commun. 5, 3480 PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yi W., Clark P.M., Mason D.E., Keenan M.C., Hill C., Goddard W.A. III et al. (2012) Phosphofructokinase 1 glycosylation regulates cell growth and metabolism. Science 337, 975–980 doi: 10.1126/science.1222278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boada J., Roig T., Perez X., Gamez A., Bartrons R., Cascante M. et al. (2000) Cells overexpressing fructose-2,6-bisphosphatase showed enhanced pentose phosphate pathway flux and resistance to oxidative stress. FEBS Lett. 480, 261–264 doi: 10.1016/S0014-5793(00)01950-5 [DOI] [PubMed] [Google Scholar]
- 31.Blackmore P.F. and Shuman E.A. (1982) Regulation of hepatic altro heptulose 1,7-bisphosphate levels and control of flux through the pentose pathway by fructose 2,6-bisphosphate. FEBS Lett. 142, 255–259 doi: 10.1016/0014-5793(82)80147-6 [DOI] [PubMed] [Google Scholar]
- 32.Opie L.H., Mansford K.R.L. and Owen P. (1971) Effects of increased heart work on glycolysis and adenine nucleotides in the perfused heart of normal and diabetic rats. Biochem. J. 124, 475–490 doi: 10.1042/bj1240475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Neely J.R., Denton R.M., England P.J. and Randle P.J. (1972) The effects of increased heart work on the tricarboxylate cycle and its interactions with glycolysis in the perfused rat heart. Biochem. J. 128, 147–159 doi: 10.1042/bj1280147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Clark M.G. and Patten G.S. (1981) Epinephrine activation of phosphofructokinase in perfused rat heart independent of changes in effector concentrations. J. Biol. Chem. 256, 27–30 PMID: [PubMed] [Google Scholar]
- 35.Nascimben L., Ingwall J.S., Lorell B.H., Pinz I., Schultz V., Tornheim K. et al. (2004) Mechanisms for increased glycolysis in the hypertrophied rat heart. Hypertension 44, 662–667 doi: 10.1161/01.HYP.0000144292.69599.0c [DOI] [PubMed] [Google Scholar]
- 36.Van Schaftingen E., Jett M.F., Hue L. and Hers H.G. (1981) Control of liver 6-phosphofructokinase by fructose 2,6-bisphosphate and other effectors. Proc. Natl Acad. Sci. U.S.A. 78, 3483–3486 doi: 10.1073/pnas.78.6.3483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Donthi R.V., Ye G., Wu C., McClain D.A., Lange A.J. and Epstein P.N. (2004) Cardiac expression of kinase-deficient 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase inhibits glycolysis, promotes hypertrophy, impairs myocyte function, and reduces insulin sensitivity. J. Biol. Chem. 279, 48085–48090 doi: 10.1074/jbc.M405510200 [DOI] [PubMed] [Google Scholar]
- 38.Wang Q., Donthi R.V., Wang J., Lange A.J., Watson L.J., Jones S.P. et al. (2008) Cardiac phosphatase-deficient 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase increases glycolysis, hypertrophy, and myocyte resistance to hypoxia. Am. J. Physiol. Heart Circ. Physiol. 294, H2889–H2897 doi: 10.1152/ajpheart.91501.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Salabei J.K., Lorkiewicz P.K., Mehra P., Gibb A.A., Haberzettl P., Hong K.U. et al. (2016) Type 2 diabetes dysregulates glucose metabolism in cardiac progenitor cells. J. Biol. Chem. 291, 13634–13648 doi: 10.1074/jbc.M116.722496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.van Berlo J.H., Maillet M. and Molkentin J.D. (2013) Signaling effectors underlying pathologic growth and remodeling of the heart. J. Clin. Invest. 123, 37–45 doi: 10.1172/JCI62839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maillet M., van Berlo J.H. and Molkentin J.D. (2013) Molecular basis of physiological heart growth: fundamental concepts and new players. Nat. Rev. Mol. Cell Biol. 14, 38–48 doi: 10.1038/nrm3495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Deprez J., Vertommen D., Alessi D.R., Hue L. and Rider M.H. (1997) Phosphorylation and activation of heart 6-phosphofructo-2-kinase by protein kinase B and other protein kinases of the insulin signaling cascades. J. Biol. Chem. 272, 17269–17275 doi: 10.1074/jbc.272.28.17269 [DOI] [PubMed] [Google Scholar]
- 43.Almeida A., Moncada S. and Bolaños J.P. (2004) Nitric oxide switches on glycolysis through the AMP protein kinase and 6-phosphofructo-2-kinase pathway. Nat. Cell Biol. 6, 45–51 doi: 10.1038/ncb1080 [DOI] [PubMed] [Google Scholar]
- 44.Novellasdemunt L., Tato I., Navarro-Sabate A., Ruiz-Meana M., Méndez-Lucas A., Perales J.C. et al. (2013) Akt-dependent activation of the heart 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PFKFB2) isoenzyme by amino acids. J. Biol. Chem. 288, 10640–10651 doi: 10.1074/jbc.M113.455998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bockus L.B. and Humphries K.M. (2015) cAMP-dependent protein kinase (PKA) signaling is impaired in the diabetic heart. J. Biol. Chem. 290, 29250–29258 doi: 10.1074/jbc.M115.681767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jones S.P., Zachara N.E., Ngoh G.A., Hill B.G., Teshima Y., Bhatnagar A. et al. (2008) Cardioprotection by N-acetylglucosamine linkage to cellular proteins. Circulation 117, 1172–1182 doi: 10.1161/CIRCULATIONAHA.107.730515 [DOI] [PubMed] [Google Scholar]
- 47.Ngoh G.A., Facundo H.T., Hamid T., Dillmann W., Zachara N.E. and Jones S.P. (2009) Unique hexosaminidase reduces metabolic survival signal and sensitizes cardiac myocytes to hypoxia/reoxygenation injury. Circ. Res. 104, 41–49 doi: 10.1161/CIRCRESAHA.108.189431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sansbury B.E., Riggs D.W., Brainard R.E., Salabei J.K., Jones S.P. and Hill B.G. (2011) Responses of hypertrophied myocytes to reactive species: implications for glycolysis and electrophile metabolism. Biochem. J. 435, 519–528 doi: 10.1042/BJ20101390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Darville M.I., Crepin K.M., Hue L. and Rousseau G.G. (1989) 5' flanking sequence and structure of a gene encoding rat 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase. Proc. Natl Acad. Sci. U.S.A. 86, 6543–6547 doi: 10.1073/pnas.86.17.6543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kurland I.J., el-Maghrabi M.R., Correia J.J. and Pilkis S.J., (1992) Rat liver 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase properties of phospho- and dephospho-forms and of two mutants in which Ser32 has been changed by site-directed mutagenesis. J. Biol. Chem. 267, 4416–4423 PMID: [PubMed] [Google Scholar]
- 51.Vertommen D., Bertrand L., Sontag B., Di Pietro A., Louckx M.P., Vidal H. et al. (1996) The ATP-binding site in the 2-kinase domain of liver 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase. Study of the role of Lys-54 and Thr-55 by site-directed mutagenesis. J. Biol. Chem. 271, 17875–17880 doi: 10.1074/jbc.271.30.17875 [DOI] [PubMed] [Google Scholar]
- 52.Ashcroft S.J.H., Weerasinghe L.C.C., Bassett J.M. and Randle P.J. (1972) The pentose cycle and insulin release in mouse pancreatic islets. Biochem. J. 126, 525–532 doi: 10.1042/bj1260525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Radde B.N., Ivanova M.M., Mai H.X., Salabei J.K., Hill B.G. and Klinge C.M. (2015) Bioenergetic differences between MCF-7 and T47D breast cancer cells and their regulation by oestradiol and tamoxifen. Biochem. J. 465, 49–61 doi: 10.1042/BJ20131608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Salabei J.K., Lorkiewicz P.K., Holden C.R., Li Q., Hong K.U., Bolli R. et al. (2015) Glutamine regulates cardiac progenitor cell metabolism and proliferation. Stem Cells 33, 2613–2627 doi: 10.1002/stem.2047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lane A.N., Fan T.W.-M., Xie Z., Moseley H.N.B. and Higashi R.M. (2009) Isotopomer analysis of lipid biosynthesis by high resolution mass spectrometry and NMR. Anal. Chim. Acta 651, 201–208 doi: 10.1016/j.aca.2009.08.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Le A., Lane A.N., Hamaker M., Bose S., Gouw A., Barbi J. et al. (2012) Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 15, 110–121 doi: 10.1016/j.cmet.2011.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lorkiewicz P., Higashi R.M., Lane A.N. and Fan T.W.-M. (2012) High information throughput analysis of nucleotides and their isotopically enriched isotopologues by direct-infusion FTICR-MS. Metabolomics 8, 930–939 doi: 10.1007/s11306-011-0388-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wei X., Lorkiewicz P.K., Shi B., Salabei J.K., Hill B.G., Kim S. et al. (2017) Analysis of stable isotope assisted metabolomics data acquired by high resolution mass spectrometry. Anal. Methods 9, 2275–2283 doi: 10.1039/C7AY00291B [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hill B.G., Dranka B.P., Zou L., Chatham J.C. and Darley-Usmar V.M. (2009) Importance of the bioenergetic reserve capacity in response to cardiomyocyte stress induced by 4-hydroxynonenal. Biochem. J. 424, 99–107 doi: 10.1042/BJ20090934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lane A.N. and Fan, T.W.-M. (2015) Regulation of mammalian nucleotide metabolism and biosynthesis. Nucleic Acids Res. 43, 2466–2485 doi: 10.1093/nar/gkv047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sellers K., Fox M.P., Bousamra M. II, Slone S.P., Higashi R.M., Miller D.M. et al. (2015) Pyruvate carboxylase is critical for non-small-cell lung cancer proliferation. J. Clin. Invest. 125, 687–698 doi: 10.1172/JCI72873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kim T.T. and Dyck J.R.B. (2016) The role of CD36 in the regulation of myocardial lipid metabolism. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 1861, 1450–1460 doi: 10.1016/j.bbalip.2016.03.018 [DOI] [PubMed] [Google Scholar]
- 63.Tamboli A., Vander Maten M., O'Looney P. and Vahouny G.V. (1983) Metabolism of fatty acid, glycerol and a monoglyceride analogue by rat cardiac myocytes and perfused hearts. Lipids 18, 808–813 doi: 10.1007/BF02534640 [DOI] [PubMed] [Google Scholar]
- 64.Tamboli A., O'Looney P., Vander Maten M. and Vahouny G. V. (1983) Comparative metabolism of free and esterified fatty acids by the perfused rat heart and rat cardiac myocytes. Biochim. Biophys. Acta, Lipids Lipid Metab. 750, 404–410 doi: 10.1016/0005-2760(83)90046-2 [DOI] [PubMed] [Google Scholar]
- 65.Owen O.E., Kalhan S.C. and Hanson R.W. (2002) The key role of anaplerosis and cataplerosis for citric acid cycle function. J. Biol. Chem. 277, 30409–30412 doi: 10.1074/jbc.R200006200 [DOI] [PubMed] [Google Scholar]
- 66.Crabtree H.G. (1929) Observations on the carbohydrate metabolism of tumours. Biochem. J. 23, 536–545 doi: 10.1042/bj0230536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Diaz-Ruiz R., Rigoulet M. and Devin A. (2011) The Warburg and Crabtree effects: on the origin of cancer cell energy metabolism and of yeast glucose repression. Biochim. Biophys. Acta, Bioenergetics 1807, 568–576 doi: 10.1016/j.bbabio.2010.08.010 [DOI] [PubMed] [Google Scholar]
- 68.Sansbury B.E., Jones S.P., Riggs D.W., Darley-Usmar V.M. and Hill B.G. (2011) Bioenergetic function in cardiovascular cells: the importance of the reserve capacity and its biological regulation. Chem. Biol. Interact. 191, 288–295 doi: 10.1016/j.cbi.2010.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mookerjee S.A., Goncalves R.L.S., Gerencser A.A., Nicholls D.G. and Brand M.D. (2015) The contributions of respiration and glycolysis to extracellular acid production. Biochim. Biophys. Acta, Bioenergetics 1847, 171–181 doi: 10.1016/j.bbabio.2014.10.005 [DOI] [PubMed] [Google Scholar]
- 70.Mookerjee S.A., Gerencser A.A., Nicholls D.G. and Brand M.D. (2017) Quantifying intracellular rates of glycolytic and oxidative ATP production and consumption using extracellular flux measurements. J. Biol. Chem. 292, 7189–7207 doi: 10.1074/jbc.M116.774471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Taegtmeyer H., Lam T. and Davogustto G. (2016) Cardiac metabolism in perspective. Compr. Physiol. 6 (Suppl 1), 1675–1699 doi: 10.1002/cphy.c150056 [DOI] [PubMed] [Google Scholar]
- 72.Cortassa S., Caceres V., Bell L.N., O'Rourke B., Paolocci N. and Aon M.A. (2015) From metabolomics to fluxomics: a computational procedure to translate metabolite profiles into metabolic fluxes. Biophys. J. 108, 163–172 doi: 10.1016/j.bpj.2014.11.1857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shao D. and Tian R. (2015) Glucose transporters in cardiac metabolism and hypertrophy. Compr. Physiol. 6, 331–351 doi: 10.1002/cphy.c150016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Marshall S., Bacote V. and Traxinger R.R. (1991) Discovery of a metabolic pathway mediating glucose-induced desensitization of the glucose transport system. Role of hexosamine biosynthesis in the induction of insulin resistance. J. Biol. Chem. 266, 4706–4712 PMID: [PubMed] [Google Scholar]
- 75.Goodwin G.W., Cohen D.M. and Taegtmeyer H. (2001) [5-3H]glucose overestimates glycolytic flux in isolated working rat heart: role of the pentose phosphate pathway. Am. J. Physiol. Endocrinol. Metab. 280, E502–E508 PMID: [DOI] [PubMed] [Google Scholar]
- 76.Funato Y., Hayashi T., Irino Y., Takenawa T. and Miki H. (2013) Nucleoredoxin regulates glucose metabolism via phosphofructokinase 1. Biochem. Biophys. Res. Commun. 440, 737–742 doi: 10.1016/j.bbrc.2013.09.138 [DOI] [PubMed] [Google Scholar]
- 77.Franklin D.A., He Y., Leslie P.L., Tikunov A.P., Fenger N., Macdonald J.M. et al. (2016) p53 coordinates DNA repair with nucleotide synthesis by suppressing PFKFB3 expression and promoting the pentose phosphate pathway. Sci. Rep. 6, 38067 doi: 10.1038/srep38067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Miyazawa H., Yamaguchi Y., Sugiura Y., Honda K., Kondo K., Matsuda F. et al. (2017) Rewiring of embryonic glucose metabolism via suppression of PFK-1 and aldolase during mouse chorioallantoic branching. Development 144, 63–73 doi: 10.1242/dev.138545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rodriguez-Rodriguez P., Fernandez E., Almeida A. and Bolaños J. P. (2012) Excitotoxic stimulus stabilizes PFKFB3 causing pentose-phosphate pathway to glycolysis switch and neurodegeneration. Cell Death Differ. 19, 1582–1589 doi: 10.1038/cdd.2012.33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lehninger A.L., Nelson D.L. and Cox M.M. (2000) Lehninger Principles of Biochemistry, 3rd edn, Worth Publishers, New York [Google Scholar]
- 81.Moseley H.N., Lane A.N., Belshoff A.C., Higashi R.M. and Fan T.W.M. (2011) A novel deconvolution method for modeling UDP-N-acetyl-d-glucosamine biosynthetic pathways based on 13C mass isotopologue profiles under non-steady-state conditions. BMC Biol. 9, 37 doi: 10.1186/1741-7007-9-37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Frenzel J., Schellenberger W., Eschrich K. and Hofmann E. (1988) Control of the fructose 6-phosphate/fructose 2,6-bisphosphate cycle by sn-glycerol 3-phosphate. Biomed. Biochim. Acta 47, 461–470 PMID: [PubMed] [Google Scholar]
- 83.Huo Y., Guo X., Li H., Wang H., Zhang W., Wang Y. et al. (2010) Disruption of inducible 6-phosphofructo-2-kinase ameliorates diet-induced adiposity but exacerbates systemic insulin resistance and adipose tissue inflammatory response. J. Biol. Chem. 285, 3713–3721 doi: 10.1074/jbc.M109.058446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Huo Y., Guo X., Li H., Xu H., Halim V., Zhang W. et al. (2012) Targeted overexpression of inducible 6-phosphofructo-2-kinase in adipose tissue increases fat deposition but protects against diet-induced insulin resistance and inflammatory responses. J. Biol. Chem. 287, 21492–21500 doi: 10.1074/jbc.M112.370379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hue L. and Taegtmeyer H. (2009) The Randle cycle revisited: a new head for an old hat. Am. J. Physiol. Endocrinol. Metab. 297, E578–E591 doi: 10.1152/ajpendo.00093.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gertz E.W., Wisneski J.A., Stanley W.C. and Neese R.A. (1988) Myocardial substrate utilization during exercise in humans. Dual carbon-labeled carbohydrate isotope experiments. J. Clin. Invest. 82, 2017–2025 doi: 10.1172/JCI113822 [DOI] [PMC free article] [PubMed] [Google Scholar]