

SUMMARY
The mammalian imprinted Dlk1-Gtl2 locus produces multiple non-coding RNAs (ncRNAs) from the maternally inherited allele, including the largest miRNA cluster in the mammalian genome. This locus has characterized functions in some types of stem cell, but its role in hematopoietic stem cells (HSCs) is unknown. Here, we show that the Dlk1-Gtl2 locus plays a critical role in preserving long-term repopulating HSCs (LT-HSCs). Through transcriptome profiling in 17 hematopoietic cell types, we found that ncRNAs expressed from the Dlk1-Gtl2 locus are predominantly enriched in fetal liver HSCs and the adult LT-HSC population and sustain long-term HSC functionality. Mechanistically, the miRNA mega-cluster within the Dlk1-Gtl2 locus suppresses the entire PI3K-mTOR pathway. This regulation in turn inhibits mitochondrial biogenesis and metabolic activity and protects LT-HSCs from excessive reactive oxygen species (ROS) production. Our data therefore show that the imprinted Dlk1-Gtl2 locus preserves LT-HSC function by restricting mitochondrial metabolism.
In Brief
Qian and colleagues show that ncRNAs expressed from the imprinted locus Dlk1-Gtl2 maintain fetal liver and adult LT-HSCs through multiplexed inhibition of PI3K-mTOR signaling that in turn keeps mitochondrial biogenesis and metabolic activity in check.
INTR ODUCTION
Homeostasis in hematopoiesis requires a balance between stem cell maintenance and action to prevent bone marrow (BM) from exhaustion or overgrowth (Li and Clevers, 2010; Wilson et al., 2008). Cell-cycle status is essential for regulation of this balance, by which long-term (LT)-HSCs are preserved in a quiescent state for maintenance whereas short-term (ST)-HSCs and multipotent progenitor (MPP) cells are fast cycling for increasing cell mass and further differentiation (Yang et al., 2005). Previous studies reported that surface marker CD49b (Integrin α2) could further separate conventional LT-HSCs (CD34−Flk2−Lineage−Sca-1+c-Kit+ [LSK]) into CD49blo LT-HSCs that maintain permanent reconstituting ability and CD49bhi intermediate-term (IT)-HSCs that support only 6–8 months of multipotent hematopoiesis (Benveniste et al., 2010). Recent studies reported that metabolic properties are required for maintenance of different states of HSCs (Suda et al., 2011; Takubo et al., 2013), and a low mitochondrial potential correlates with HSC functionality (Simsek et al., 2010). How cell-cycle status and metabolic states are precisely controlled in HSCs remains largely unknown.
Epigenetic regulation, including ncRNAs, DNA methylation, histone modification, and chromatin remodeling, plays essential roles in orchestrating the balance between HSC maintenance and action (Cullen et al., 2014). Genomic imprinting, a unique epigenetic regulation resulting in a parent-of-origin-specific gene expression, is essential for normal mammalian development and growth (Bartolomei, 2009; Ferguson-Smith, 2011). We initiated our study on imprinting genes after seeing a differential expression of imprinted genes in HSCs (Haug et al., 2008). Systematic gene profiling conducted by several groups revealed predominant expression of imprinted genes in HSCs as well as other somatic stem cells (Berg et al., 2011; Ferrón et al., 2011; Zacharek et al., 2011). We further functionally proved that the imprinting at the H19-Igf2 locus is essential for maintaining HSC quiescence via suppression of Igf1r expression by H19-derived miR-675 (Venkatraman et al., 2013). In addition to miRNAs, long-length non-coding RNAs (lncRNAs) (>200 nt in length) have drawn attention for their potential regulatory roles in biological processes including stem cell property (Luo et al., 2015). These efforts have sparked interest in ncRNAs; however, whether and how ncRNAs preserve LT-HSCs remains poorly understood.
In this study, we performed transcriptome profiling in 17 sub-populations of hematopoietic stem, progenitor, and mature lineage cells, and we identified exclusively expressed “fingerprint” lncRNAs unique to each cell type. We found that lncRNAs in the Dlk1-Gtl2 locus are specifically enriched in the CD49blo LT-HSCs. The imprinted Dlk1-Gtl2 locus contains three protein-coding genes (Dlk1, Rtl1, and Dio3) on the paternally inherited allele and multiple lncRNAs and small ncRNAs on the maternally inherited allele, including Gtl2, Rian (containing 22 box C/D snoRNAs), and the largest miRNA mega-cluster in mammals (anti-Rtl1, which contains the miR-127/miR-136 cluster with 7 miRNAs, and Mirg, which contains the miR-379/miR-410 cluster with 39 miRNAs). Interestingly, all the ncRNAs are regulated by a common cis-element and epigenetic control, resulting in a large polycistronic transcription unit (da Rocha et al., 2008; Seitz et al., 2004; Tierling et al., 2006). The Dlk1-Gtl2 locus plays crucial roles in embryonic and adult stem cells (Ferrón et al., 2011; Lin et al., 2003; Snyder et al., 2013). However, whether the Dlk1-Gtl2 locus functions in HSCs and its underlying mechanism are largely uncharacterized. Here, we show how the Dlk1-Gtl2 locus preserves functionality of LT-HSCs by inhibiting the PI3K-mTOR pathway and restricting mitochondrial metabolism.
RESULTS
Unique lncRNA Fingerprints in 17 Hematopoietic Cell Types Revealed Enrichment of lncRNAs from the Dlk1-Gtl2 Locus in CD49blo LT-HSCs
To systematically identify lncRNAs that might play a role in LT-HSCs, we isolated four hematopoietic stem and progenitor cells (HSPCs) (CD49blo LT-HSCs, CD49bhi IT-HSCs, ST-HSCs, and MPPs), four committed progenitors (common lymphoid progenitor [CLP], common myeloid progenitor [CMP], granulocyte-macrophage progenitor [GMP], and megakaryocyte-erythroid progenitor [MEP]), and nine mature lineage cells (B cell, T cell, NK cell, dendritic cell, monocyte, macrophage, granulocyte, megakaryocyte, and nucleated erythrocyte) by fluorescence-activated cell sorting (FACS) from the BM of C57BL/6J mice (Figure 1A; Figures S1A–S1F; Table S1). Next, we conducted 100 bp paired-end high-throughput RNA-seq on poly-A+ RNA and identified reliable lncRNAs using the stringent filtering strategies as previously reported (Alvarez-Dominguez et al., 2014) (Table S2; see Supplemental Experimental Procedures). Several known HSC markers (CD150, EPCR, and ESAM) were well captured, and the markers used for FACS exhibited consistency between transcript and protein levels, supporting the high quality of our RNA-seq data (Figure S1G). Our strategies yielded 4,204 lncRNAs, 1,606 of which (38.2%) were distinct from those in Ensembl databases. We also found that 860 (20.5%) of these lncRNAs overlapped with HSC-related lncRNAs recently reported in an elegant study (Luo et al., 2015) (Figure S1H). We then removed the least expressed transcripts by setting the threshold of FPKM (fragments per kilobase of transcript per million mapped reads) at >1 in at least one sample, which yielded 1,693 expressed lncRNAs. Compared to protein-coding genes, these lncRNAs had lower expression levels (Figure S1I) but higher tissue specificity (Figure S1J, p = 2.2 × 10−16, Kolmogorov-Smirnov test), implying that they might play specific roles in different types of hematopoietic cells.
Figure 1. Unique lncRNA Fingerprints in 17 Hematopoietic Cell Types Revealed Enrichment of lncRNAs from the Dlk1-Gtl2 Locus in CD49blo LT-HSCs.

(A) Schematic representation of the hematopoietic hierarchy showing cell types used in this study and workflow for lncRNA discovery.
(B) A dendrogram displays hierarchical cluster analysis from 1,693 expressed lncRNAs based on Pearson correlations of log2 (FPKM + 2−11) values. The samples from HSPCs, progenitors, and lineage cells are surrounded by red, yellow, and purple rectangles, respectively.
(C) Population-distance analysis of 1,693 expressed lncRNAs shown in two principal components (PCs 1–2). Cell types are color-coded, labeled by name, and surrounded by different ellipses. HSPCs, red; committed progenitor cells, yellow; myeloid cells, violet; lymphoid cells, blue; nucleated erythrocytes, pink.
(D) Heat map of cell-type-specific lncRNA fingerprints based on cell specificity scores (color indicates specificity scores; darker corresponds to higher specificity scores). Representative fingerprint lncRNAs in different cell types are shown on the right.
(E) Schematic representation of the Dlk1-Gtl2 locus. Genes (rectangles) on the maternally inherited allele (in red) and the paternally inherited allele (in blue) are depicted. miRNA and C/D snoRNA genes are shown by hairpin loops and triangles, respectively. Arrows indicate the transcription directions, with the horizontal dash line indicating the hypothesis that Gtl2, anti-Rtl1, Rian, and Mirg may constitute a large polycistronic transcription unit. The IG-DMR and Gtl2-DMR are unmethylated (open circles) on the maternally inherited allele but methylated (filled circles) on the paternally inherited allele.
(F) UCSC Genome Browser images show RNA-seq signal as the density of mapped reads in the whole Dlk1-Gtl2 locus.
To investigate the ontogeny relatedness of hematopoietic cell types, we performed unsupervised hierarchical cluster analysis. The family tree analysis clearly showed that samples from HSPCs, progenitors, and lineages correlated positively, supporting the high specificity of lncRNAs (Figure 1B). We further performed principal components analysis (PCA) to measure and visualize differences among cell types. The PCA map showed CD49blo HSCs at the apex of the hematopoietic hierarchy (Figure 1C). We also observed the striking segregation of HSPCs, progenitor cells, and mature lineages, the latter of which further divided into three discrete clusters (lymphoid, myeloid, and erythroid) (Figure 1C). Together, both cluster and PCA analyses indicated that the lncRNA profiles distinguish CD49blo HSCs from other HSC subpopulations that eventually lost their functional capacity, supporting the previous report that permanently reconstituting CD49blo HSCs have the highest self-renewal ability (Benveniste et al., 2010).
We next determined the fingerprint lncRNAs in particular cell types. Based on specificity scores, we identified 461 (27.2% in 1693) fingerprint lncRNAs in 17 cell types that could be used as specific markers and might play essential roles during lineage commitment and differentiation (Figure 1D; Table S3). Only 954 of 13,775 (6.9%) protein-coding genes were identified as fingerprint genes, suggesting that lncRNAs have much higher percentages of lineage/stage-restricted gene expression. We specifically identified 12 lncRNAs enriched in CD49blo HSCs, three of which with the highest expression levels—Gtl2, Rian, and Gm26906—were derived from the imprinted Dlk1-Gtl2 locus on mouse chromosome 12qF1 (Figure 1E). Intriguingly, H19 was also enriched in CD49blo HSCs. Furthermore, our RNA-seq data revealed that all the ncRNAs expressed from the maternally inherited allele, starting from Gtl2 to Mirg, were exclusively enriched in CD49blo HSCs (Figure 1F), suggesting a potential role of ncRNAs from the Dlk1-Gtl2 locus in preserving CD49blo LT-HSCs.
Loss of Imprinting at the Dlk1-Gtl2 Locus Led to Deficiency in Fetal Liver HSCs
Both the intergenic germline-derived differentially methylated region (IG-DMR) and the Gtl2-DMR determine reciprocal expression of ncRNAs on the maternally inherited allele and protein-coding genes on the paternally inherited allele (Lin et al., 2003; Zhou et al., 2010). To determine the role of the Dlk1-Gtl2 locus in HSCs, we used the genetic mutant mice with straight knockout of IG-DMR (Lin et al., 2003). We bred the heterozygous IG-DMR mice with WT mice and then performed PCR to determine allele-specific mutants with deletion of IG-DMR from maternally or paternally inherited allele (mat ΔIG or pat ΔIG) and their control littermates (Figures 2A and S2A). Since mat ΔIG embryos were lethal after E16, we used E15 embryos, at which stage HSCs are mainly located in fetal livers. To test gene expression in the Dlk1-Gtl2 locus in fetal liver LSK cells and adult HSCs, we compared RNA-seq data from four adult HSPCs, adult LSK cells, and E15 fetal liver LSK cells. Interestingly, we found that E15 fetal liver LSK cells exhibited expression patterns of lncRNAs in the Dlk1-Gtl2 locus similar to those of CD49blo HSCs, but not similar to those of the other adult HSPCs (Figure 2B), suggesting that the CD49blo subset of HSCs are enriched with a feature of fetal HSCs. Thus we were able to study the behaviors of HSCs isolated from E15 fetal liver and transplanted into adult recipients to reveal potential roles of the Dlk1-Gtl2 locus in HSCs.
Figure 2. Loss of Imprinting at the Dlk1-Gtl2 Locus Led to Deficiency in Fetal Liver HSCs.

(A) Schematic representation of mating strategy to generate allele-specific mutant embyros.
(B) RNA-seq analysis of lncRNAs in the Dlk1-Gtl2 locus in E15 fetal liver LSK, adult BM LSK, and four adult HSPC populations.
(C) Absolute numbers of total fetal liver cells (n = 6).
(D–I) Flow cytometric analyses to show the gate, frequency (of total nucleated cells [TNCs]), and absolute numbers of LSK cells, CD93+ HSCs, and CD38 HSCs in WT and ΔIG fetal liver (n = 6).
(J) Tri-color heat map shows quantitative real-time PCR analysis of Dlk1-Gtl2 locus genes in HSPCs, progenitors, and lineage cells from WT and mat ΔIG fetal livers. β-Actin was used as an internal control.
(K and L) Frequency in TNCs and absolute numbers of HSPCs in WT and ΔGtl2 fetal liver (n = 3). Error bars, SEM; *p < 0.05; **p < 0.01. See also Figure S2 and Table S1, Table S2, and Table S6.
We next investigated the roles of the Dlk1-Gtl2 locus in fetal liver HSCs. Consistent with a previous study (Lin et al., 2007), we observed several symptoms of anemia, including pale skin and few vessels, in the E15 mat ΔIG embryos (Figure S2B). Although total cell numbers in WT and ΔIG mutant fetal livers remained similar (Figure 2C), we indeed observed a 2-fold decrease in both frequency and absolute number of LSK cells in mat ΔIG, but not in pat ΔIG, fetal liver (Figures 2D and 2E). To assess the effect on HSCs, we used CD93 (Morrison et al., 1995) and CD38 (Randall et al., 1996) as fetal liver HSC surface markers. Flow cytometry analysis showed that both the frequency and absolute numbers of CD93+ HSCs, CD38hi HSCs, and CD38lo HSCs declined 2-fold in mat ΔIG fetal livers (Figures 2F–2I). However, except for an increase in the frequency of MEP and erythroid, we did not observe any change in the other progenitors and lineage cells, reflecting a relatively specific reduction in HSCs caused by loss of mat ΔIG (Figures S2C–S2F). We next quantified the gene expression levels of the Dlk1-Gtl2 locus by quantitative real-time PCR in both WT and mat ΔIG fetal liver cells. Interestingly, we observed almost no expression in lineage cells and no change between WT and mat ΔIG progenitors (Figure 2J), supporting previous observations that most progenitors and lineages cells showed no difference in frequency and absolute numbers. In contrast, we observed a substantial decrease in ncRNAs expressed from the maternally inherited allele (Gtl2, Rian, and Mirg) and an increase in Dlk1 (but not Rtl1 or Dio3) from the paternally inherited allele in fetal liver HSCs (Figure 2J). To assess whether Dlk1 mediates HSC deficiency in the mat ΔIG mouse, we crossed mat ΔIG females with Tie2-cre+; Dlk1fl/fl males and found that deletion of Dlk1 could not fully rescue the decreased number of mat ΔIG fetal liver HSPCs, excluding the possibility that defective phenotype in mat ΔIG was due to increased Dlk1 expression (Figure S2G).
To assess whether loss of Gtl2 itself could lead to HSC deficiency, we used another genetic murine mutant of ΔGtl2, with a deletion of the first five exons of Gtl2 and Gtl2-DMR (Takahashi et al., 2009). Deletion of Gtl2 from the maternally inherited allele (mat ΔGtl2) caused perinatal death and repressed all the ncRNAs expressed from the maternally inherited allele, similar to the mat ΔIG mice (Zhou et al., 2010). Consistently, we found that HSPCs, but not progenitors and lineages, in the mat ΔGtl2 fetal livers exhibited decreased frequency and absolute numbers (Figures 2K, 2L, S2H, and S2I). Together, these data indicate that loss of either mat ΔIG or Gtl2 per se leads to a deficiency in fetal liver HSCs.
Loss of Imprinting at the Dlk1-Gtl2 Locus Impaired Fetal Liver HSC Long-Term Reconstitution Capacity
To characterize the functional roles of Dlk1-Gtl2 locus in HSCs, we conducted a competitive repopulation assay by transplanting 5 × 104 or 5 × 105 whole fetal liver cells from mutants or their control littermates as donor cells (CD45.2), together with 1 × 105 recipient BM cells (CD45.1 hereafter) derived from the Ptprc mutant strain, into lethally irradiated recipient mice. In the 5 × 104 group, we found that overall engraftment from mat ΔIG was reduced 1.4-fold, 1.8-fold, and 7.1-fold in primary (1st), secondary (2nd), and tertiary (3rd) recipients, respectively, compared to the control group (Figure 3A). Engraftment from pat ΔIG showed no difference (Figure S3A). Interestingly, we observed a significant myeloid lineage bias in the 2nd and 3rd mat ΔIG-transplant recipients (Figure 3A), consistent with a previous report of myeloid lineage bias associated with HSC aging and diminished total reconstitution potential (Beerman et al., 2010). In the 5 × 105 group, we observed a 2-fold decrease in the repopulation rate and a significant myeloid lineage bias from mat ΔIG only in the 3rd recipients, but no difference in the 1st or 2nd recipients (Figures S3B–S3D). We next analyzed donor HSCs in the recipient mice. In the 1st, 2nd, and 3rd recipients, the total cell numbers in BM were not affected; however, the absolute numbers of HSPCs, including CD49blo HSCs, declined significantly in mat ΔIG, but not in pat ΔIG, compared to control (Figures 3B and S3E–S3K). Intriguingly, we observed a significant decrease in the ratio of CD49blo/CD49bhi HSCs, from 30%/70% in control to 16%/84% in mat ΔIG, suggesting that deletion of IG-DMR from maternally inherited allele resulted in a reduction in LT-HSCs (Figure 3C). Further cell-cycle analyses in both fetal liver HSCs and donor CD49blo HSCs showed a decrease in the G0 phase fraction and a concomitant increase in the G1 phase fraction in mat ΔIG (Figures S3L and 3D). In addition, Kaplan-Meier survival analysis revealed that mice transplanted with mat ΔIG showed significantly reduced survival compared to controls (Figure 3E). To investigate whether impaired reconstitution capacity was due to functional defect in HSCs, we transplanted 150 sorted HSCs from WT and mat ΔIG fetal livers together with 2 × 105 rescue BM cells into recipient mice. We observed a striking reduction in reconstitution ability at 12 weeks posttransplantation in mat ΔIG compared to control (Figure 3F). Together, these results show that fetal liver cells from mat ΔIG mice have significantly reduced HSC numbers, accounting for reduced long-term reconstitution capacity.
Figure 3. Loss of Imprinting at the Dlk1-Gtl2 Locus Impaired Long-Term Reconstitution Capacity of Fetal Liver HSCs.

(A) 5 × 104 WT or mat ΔIG fetal liver cells were transplanted with 1 × 105 rescue cells into irradiated recipients. At 16 weeks posttransplant, BM isolated from 1st recipients was transplanted into 2nd recipients and, at 16 weeks post 2nd transplant, from 2nd into 3rd recipients at a dosage of 1 ×106 cells per mouse. Peripheral blood (PB) was analyzed for percent donor repopulation at the indicated number of weeks after transplants (top panels) and for percent mature donor-derived B, T, and myeloid cells (bottom panels) (n = 10).
(B) Absolute numbers of HSPCs in the BM from 3rd recipients (n = 5).
(C) Percentage of CD49blo and CD49bhi HSCs in the BM from 3rd recipients (n = 5).
(D) Cell-cycle analysis of CD49blo HSCs in BM from 1st recipients (n = 5).
(E) Kaplan-Meier survival curve for 1st, 2nd, and 3rd transplant recipients (n = 10).
(F) 150 sorted CD93+ HSCs from WT or mat ΔIG fetal livers were transplanted with 2 ×105 rescue cells into irradiated recipients. PB analysis for percent engrafted donor cells at the indicated number of weeks posttransplantation is shown.
(G–I) CRU frequency was determined in (H) using ELDA (Extreme Limiting Dilution Analysis). PB of the 5 ×104 group was analyzed for percent donor repopulation at the indicated number of weeks posttransplantation and for percent mature donor-derived B, T, and myeloid cells (I) (n = 10).
Error bars, SD; *p < 0.05; **p < 0.01. See also Figure S3.
To quantify functional HSCs, we further used ΔGtl2 mice to perform limiting dilution (5 × 103, 1.5 × 104, and 5 × 104) competitive repopulating unit (CRU) assays. We observed a 2.1-fold decrease in functional HSCs in mat ΔGtl2 mice compared to control (Figures 3G and 3H). In the 5 × 104 group, 20 weeks posttransplantation, we observed a 2.2-fold decrease in the overall engraftment and a significant myeloid lineage bias from mat ΔGtl2 (Figure 3I). These data reveal that loss of either mat ΔIG or Gtl2 per se compromises fetal liver HSC long-term reconstitution capacity.
Loss of Imprinting at the Dlk1-Gtl2 Locus Induced Hyperactivation of the PI3K-mTOR Pathway
We next sought to elucidate the molecular mechanisms underlying HSC defects from loss of imprinting at the Dlk1-Gtl2 locus. To this end, we performed RNA-seq in fetal liver LSK cells and identified 991 upregulated genes and 941 downregulated genes in mat ΔIG (Figures 4A and 4B). Gene Ontology (GO) enrichment analysis revealed that most of the upregulated GO terms enriched in the mat ΔIG LSK were related to proliferation, growth, development, and metabolism (Figure 4C; Table S4), suggesting that hyperactive proliferation and metabolism might be responsible for HSC deficiency in mat ΔIG mutants. Two terms (“regulation of cellular response to insulin stimulus” and “positive regulation of PI3K cascade”) were upregulated in mat ΔIG, leading us to hypothesize that the PI3K-mTOR pathway might be hyperactivated in mat ΔIG HSCs. The PI3K-mTOR pathway is known to contribute to hematopoiesis, with downregulation of mTORC1 being essential for HSC self-renewal and maintenance, whereas abnormal activation of mTOR signaling is usually associated with leukemogenesis (Chen et al., 2008; Hirao and Hoshii, 2013; Lee et al., 2010). Furthermore, our previous study showed that activation of PI3K-AKT by PTEN mutation led to HSC exhaustion (Zhang et al., 2006). To test our hypothesis, we conducted three individual sets of experiments. First, to detect phospho (p)-AktS473 and p-mTORS2448 activities in fetal liver HSCs, we performed immunostaining on sorted fetal liver LSK cells. Quantification of staining intensity revealed that, compared to control, mat ΔIG exhibited 1.2-fold, 2-fold, and 1.9-fold increased activity of p-AktS473, p-mTORS2448, and p-S6S235/236, respectively (Figures 4D–4G; Figures S4A and S4B). Second, to determine PI3K-mTOR activities, we performed intracellular flow cytometry and observed that median fluorescence intensities (MFIs) of p-AktS473, p-AktS308, and p-mTORS2448 were higher in LSK and HSCs, but not in progenitor and lineage cells, in mat ΔIG compared to WT (Figures 4H, 4I, and S4C). We also observed a significant increase in MFI of p-mTORS2481, indicating that both mTORC1 and mTORC2 were hyperactivated in mat ΔIG (Figure S4D). However, we observed no significant difference in MFIs of LKB1, p-AMPKαT172, p-FoxO1T24/FoxO3aT32, or p-SGK1S78 (Figures S4E–S4H). Third, to test PI3K-mTOR activities in adult HSCs, we performed flow cytometry in recipient BM cells. Interestingly, MFIs of p-AktS473 and p-mTORS2448 in donor CD49blo HSCs were 145% and 133% higher, respectively, in mat ΔIG than in WT (Figures 4J and 4K). Taken together, these data suggest that loss of imprinting at the Dlk1-Gtl2 locus induces hyperactivation of the PI3K-mTOR pathway in both fetal liver HSCs and adult donor HSCs.
Figure 4. Hyperactivation of the PI3K-mTOR Pathway in mat ΔIG Fetal Liver HSPCs.

(A) Poly-A+ RNA-seq workflow.
(B) MA plot (plot of mean log2 difference versus mean log2 intensity) for differential expression analysis between WT and mat ΔIG fetal liver LSK cells.
(C) GO enrichment analysis using −log10 of the uncorrected p value as x axis. The upregulated or downregulated GO terms in mat ΔIG are shown in red or blue, respectively. The numbers indicate the amount of enriched genes in each term.
(D–G) Representative image and quantification of staining intensity of LSK cells sorted from WT and ΔIG fetal livers for p-AktS473 (n ≥ 43) and p-mTORS2448 (n ≥ 152).
(H–K) MFIs of p-AktS473 and p-mTORS2448 in fetal liver HSCs (H and I, n = 4) and donor CD49blo HSCs from BM of 3rd recipients (J and K, n ≥ 4).
Error bars, SEM (H and I) or SD. Scale bars, 5 μm. *p < 0.05; **p < 0.01; ***p < 0.001. See also Figure S4, Table S4, and Table S7.
miRNAs in the Gtl2 Locus Suppressed Multiple Components of the PI3K-mTOR Pathway
Based on our RNA-seq data that the small ncRNAs were exclusively enriched in CD49blo HSCs together with Gtl2 (Figure 1F), we reasoned that these small ncRNAs might contribute to the hyperactivation of PI3K-mTOR signaling in the mutant HSCs. To test this idea, we performed small RNA-seq in both fetal liver HSCs and adult HSCs (Figure 5A). Intriguingly, we observed that small ncRNAs in the Gtl2 locus showed a widespread decrease in mat ΔIG fetal liver HSCs and exhibited an obvious enrichment in CD49blo HSCs relative to other adult HSPCs (Figures 5B and 5C). Moreover, ncRNAs in fetal liver HSCs and adult CD49blo HSCs showed striking similarity, suggesting that these ncRNAs might be specifically processed in HSCs. Combining the ncRNAs downregulated in mat ΔIG and upregulated in CD49blo, we identified 15 miRNAs and 10 snoRNAs that might be responsible for HSC deficiency and PI3K-mTOR pathway hyperactivation (Figure 5D; Table S5).
Figure 5. miRNAs in the Dlk1-Gtl2 Locus Repressed Multiple Components of the PI3K-mTOR Pathway.

(A) Small RNA-seq workflow.
(B and C) UCSC Genome Browser images show small RNA-seq signal as the density of mapped reads in the whole Dlk1-Gtl2 locus from either fetal liver HSCs (B) or adult HSPCs (C).
(D) The Venn diagram shows the overlap between ncRNAs downregulated in mat ΔIG and upregulated in CD49blo HSCs. The miRNAs are listed on the bottom.
(E) Schematic representation of the PI3K-mTOR pathway.
(F) Putative miRNAs and their binding sites in 21 genes of the PI3K-mTOR pathway.
(G) Luciferase reporter assays performed to determine functional miRNA binding sites (n = 2).
(H and I) Western blotting assays performed to detect expression of AKT1 and RPTOR in mouse MEF cells infected with miRNA overexpressing lentivirus. β-ACTIN was used as internal control.
(J) Scheme for transplantation assay rescued by overexpression of 10 miRNAs in Gtl2 locus.
(K) Quantification of functional HSCs by transplantation assay (n = 6).
(L) Absolute numbers of CD49blo HSCs in recipient mice at 16 weeks posttransplantation (n = 6).
Error bars, SEM (G) or SD; *p < 0.05; ns, not significant. See also Figure S5 and Table S5, Table S6, and Table S7.
Compared to snoRNAs, which function to methylate rRNA and snRNA (Cavaillé et al., 1996), miRNAs are widely reported to repress translation or trigger mRNA degradation by recognizing complementary target sites in the 3′ UTRs of cognate mRNAs (Bartel, 2009). We used TargetScan 6.2 to predict which components in the PI3K-mTOR pathway might be targeted by these miRNAs (Figure 5E). Unexpectedly, 21 genes distributed over the entire PI3K-mTOR pathway contain one or multiple conserved binding sites of Gtl2-derived miRNAs in their respective 3′ UTRs (Figure 5F). We then used luciferase reporter assays to determine whether the corresponding mRNAs involved in the mTOR pathway were direct targets of these miRNAs. We first cloned full-length fragments of their 3′ UTRs into the luciferase reporter plasmid psi-CHECK2. Then we transfected either a mature miRNA duplex or a negative control with cognate 3′ UTR constructs into HEK293T cells and found that most of these miRNAs significantly repressed the relative luciferase activities of the 3′ UTRs of IGF1, IGF1R, IRS1, PIK3R1, AKT1, RHEB, RPTOR, PGC-1α, RPS6KB1, and EIF4E (Figure 5G). Moreover, mutation of cognate miRNA binding sites in the 3′ UTRs of RHEB and AKT1 abrogated their repression (Figures S5A and S5B). Further, we infected mouse MEF cells with lentiviruses that overexpress individual miRNAs. Our western blot data showed that miRNAs in the Gtl2 locus could repress expression of AKT1 and RPTOR at protein levels (Figures 5H and 5I). We also measured expression levels of AKT1 and RPTOR in HSCs and found that these were markedly higher in both fetal liver HSCs and donor CD49blo HSCs of mat ΔIG compared to WT (Figures S5C–S5F). Together, these data indicate that multiple components of the PI3K-mTOR pathway are de novo and direct targets of miRNAs derived from the Gtl2 locus.
To assess whether the HSC deficiency in mat ΔIG was due to downregulation of these miRNAs, we individually cloned 10 highly expressed miRNAs within this locus into lentiviral vectors and infected sorted fetal liver LSK cells with a pool of lentiviruses (Figure 5J). 16 weeks posttransplantation, we found that the repopulation rate and absolute number of donor CD49blo HSCs in mat ΔIG HSCs were increased 2.9-fold and 5.7-fold, respectively, compared to WT HSCs after overexpression of these miRNAs. This implies that miRNAs in the Gtl2 locus are likely responsible for the HSC deficiency in mat ΔIG (Figures 5K and 5L).
Loss of Imprinting at the Dlk1-Gtl2 Locus Enhanced Mitochondrial Biogenesis, Metabolic Activity, and ROS Levels in HSCs
The PI3K-mTOR pathway serves as a sensor to integrate various environmental cues, such as nutrition and growth factors (Laplante and Sabatini, 2012). mTOR not only stimulates de novo synthesis of primary biosynthetic products such as proteins, nucleotides, and lipids, but also produces ATP by either enhancing mitochondrial activity through 4E-BP-dependent translational regulation (Morita et al., 2013) or activating PGC-1α, a master transcription regulator in the mitochondrial biogenic regulatory cascade (Cunningham et al., 2007). Since PGC-1α and EIF4E were directly targeted by miRNAs in the Gtl2 locus, we examined their expression in mat ΔIG HSCs. Indeed, we found MFIs of PGC-1α and p-4E-BP1T37/46 in both fetal liver and donor CD49blo HSCs to be significantly increased in mat ΔIG (Figures 6A–6D).
Figure 6. Loss of Imprinting at the Dlk1-Gtl2 Locus Enhanced Mitochondrial Biogenesis and Metabolic Activity and Increased ROS Levels in HSCs.

(A–D) MFI of PGC-1α and p-4E-BP1T37/46 in fetal liver HSCs (A and B, n = 3) and donor CD49blo HSCs from BM of 3rd recipients (C and D, n ≥ 4).
(E) Mitochondrial mass of WT and mat ΔIG cells assayed by MitoTracker Green staining (n = 3).
(F) Mitochondrial membrane potential of WT and mat ΔIG cells assayed by DilC5 staining (n = 3).
(G) Relative mitochondria DNA copy number of WT and mat ΔIG cells assayed by qRT-PCR (n = 2).
(H and I) Representative TEM images of mitochondrial morphology in WT and mat ΔIG LSK cells.
(J) Glucose uptake of WT and mat ΔIG cells assayed by 2-NBDG (n = 3).
(K) Relative basal ATP levels of WT and mat ΔIG cells (n = 2).
(L) ROS levels of WT and mat ΔIG cells assayed by H2-DCFDA (n = 3).
(M) Percentage of apoptotic cells in WT and mat ΔIG cells assayed by Annexin V and 7-AAD staining (n = 3).
Error bars, SEM or SD (C and D). *p < 0.05; **p < 0.01; ***p < 0.001. See also Figure S6 and Table S7.
We then measured mitochondrial properties in fetal liver HSCs. Using flow cytometry with MitoTracker Green and DilC5, we observed a significant increase in mitochondrial mass and mitochondrial membrane potential (Δψ) in HSPCs of mat ΔIG (Figures 6E and 6F). Also, the mtDNA copy number increased markedly in mat ΔIG LSK cells, but not in Lin+ cells, compared to control (Figure 6G). Moreover, using transmission electron microscopy (TEM) technology to measure the morphology of mitochondrion, we observed increased mitochondrial number and folds of cristae in mat ΔIG HSCs (Figures 6H and 6I). Mitochondria constitute the most prominent source of ATP production mainly through the Krebs cycle and oxidative phosphorylation (OXPHOS) (Fernie et al., 2004). Thus, we next investigated glucose metabolism in mat ΔIG mutants. By performing flow cytometry with 2-NBDG, a fluorescent glucose analog, we found increased glucose uptake in HSPCs of mat ΔIG (Figure 6J). Finally, we found greatly increased basal ATP levels in fetal liver LSK cells, but not in Lin+ cells, in mat ΔIG compared to contol (Figure 6K). In contrast, there was no difference in either mitochondrial activity or metabolism between pat ΔIG mutants and control (Figures S6A–S6D).
A hyperactive mTOR activity correlates with a high level of ROS (Jang and Sharkis, 2007). We next measured ROS levels using 2′,7′-dichlorodihydro-fluorescein diacetate (H2-DCFDA) and observed markedly increased ROS levels in HSPCs of mat ΔIG (Figure 6L), consitent with a previous study using a similar mouse model, TSC1−/− (Chen et al., 2008). We next measured apoptosis in fetal liver HSCs and found a significantly higher percentage of apoptotic cells in HSPCs of mat ΔIG than of WT, which to a certain extent explains the reduced HSPC number in mat ΔIG (Figure 6M).
To further investigate the role of the Dlk1-Gtl2 locus in mitochondrial metabolism and ROS levels, we performed metabolic assays in mat ΔGtl2 mutants. We found that compared to control, HSPCs in the mat ΔGtl2 fetal livers exhibited increased mitochondrial mass, Δψ, glucose uptake, oxygen consumption rate, ROS levels, and apoptosis (Figures S6E–S6J). These findings support the likelihood that loss of imprinting at the Dlk1-Gtl2 locus leads to enhanced mitochondrial biogenesis, metabolic activity, and increased ROS levels in HSCs.
Pharmacological Inhibition of the PI3K-mTOR Pathway or ROS Production Partially Rescued HSC Deficiency in mat ΔIG Mutants
Given that loss of imprinting at the Dlk1-Gtl2 locus activates the PI3K-mTOR pathway and enhances mitochondrial biogenesis, metabolic activity, and ROS production, we asked whether pharmacological inhibition of either the PI3K-mTOR pathway or ROS production could abrogate the HSC defect caused by the Dlk1-Gtl2 locus deletion. We first transplanted sorted HSCs from WT and mat ΔIG fetal livers into lethally irradiated recipient mice. After stabilization of donor BM cells, we administered vehicle control or rapamycin, which interacts with peptidylproline isomerase FKBP12 and specifically inhibits mTORC1 activity, to the recipients daily (Figure 7A). Indeed, we found that the repopulation rate after rapamycin treatment in mat ΔIG HSCs was 715% higher compared to only 45.9% higher in WT HSCs (Figure 7B). Likewise after rapamycin treatment, the absolute number of donor CD49blo HSCs in mat ΔIG was 311.5% higher compared to only 60.4% higher in WT mice (Figure 7C). Intracellular immunostaining revealed that rapamycin significantly reduced MFIs of p-mTORS2448, p-4E-BP1T37/46, and PGC-1α in donor CD49blo HSCs, supporting a decrease in mTORC1 activity following rapamycin administration (Figures 7D, S7A, and S7B). Furthermore, rapamycin clearly restored the mitochondrial mass, glucose uptake rate, percentage of G0 phase, and ROS levels in donor CD49blo HSCs (Figures 7E–7H). To further confirm that a hyperactivated PI3K-mTOR pathway led to HSC deficiency, we treated recipient mice with several different PI3K-mTOR pathway inhibitors, NVP-BEZ235 (dual PI3K and mTOR inhibitor), PP242 (mTOR inhibitor), or MK-2206 (pan AKT inhibitor). Our transplantation data revealed that the decreased repopulation rates in mat ΔIG were abrogated at varying levels by administration of these inhibitors (Figures S7C and S7D). We next tested whether inhibition of ROS production could abrogate the HSC defects. We transplanted whole fetal liver cells into lethally irradiated recipient mice and fed them with water containing N-acetyl cysteine (NAC), an antioxidant to scavenge free radicals (Figure 7I). Intriguingly, we found that NAC treatment not only reduced ROS levels, but also partially restored the repopulation rate and absolute numbers of donor CD49blo HSCs in mat ΔIG (Figures 7J–7L). Overall, these data show that inhibiting either the PI3K-mTOR pathway or ROS production partially restores the HSC deficiency in mat ΔIG mutants.
Figure 7. Rapamycin and NAC Partially Rescued Defective HSCs and Metabolic Effects Due to Loss of Imprinting at the Dlk1-Gtl2 Locus.

(A) Scheme for rapamycin administration.
(B) Repopulation assay for rapamycin administration at 12 weeks posttransplantation (n ≥ 4).
(C) Absolute numbers of CD49blo HSCs in recipient mice with rapamycin administration at 12 weeks posttransplantation (n ≥ 4).
(D) MFI of p-mTORS2448 in donor CD49blo HSCs from BM of mice treated with rapamycin (n ≥ 4).
(E–H) Mitochondrial mass, glucose uptake, cell-cycle analysis, and ROS levels in donor CD49blo HSCs from BM of mice treated with rapamycin (n ≥ 4).
(I) Scheme for NAC administration.
(J) Repopulation assay for NAC administration at 16 weeks posttransplantation (n = 6).
(K) Absolute numbers of CD49blo HSCs in recipient mice fed with NAC water at 16 weeks posttransplantation (n = 6).
(L) ROS levels in donor CD49blo HSCs from BM of mice fed with NAC water (n = 6).
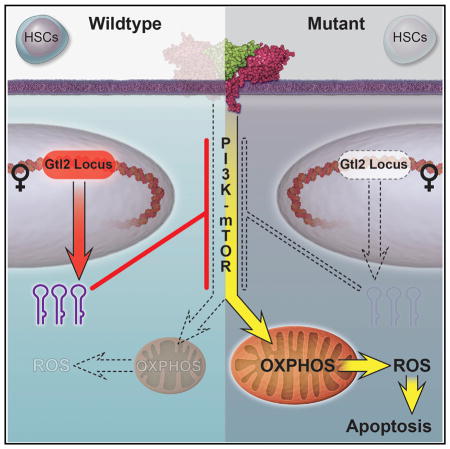
(M) Model depicting an essential role of the Dlk1-Gtl2 locus in preserving HSCs by inhibiting the PI3K-mTOR pathway and restricting mitochondrial metabolism.
Error bars, SD; *p < 0.05; **p < 0.01; ***p < 0.001. See also Figure S7 and Table S7.
DISCUSSION
In this study, by using global transcription profiling, we identified unique fingerprint lncRNAs in 17 hematopoietic cell types, in which lncRNAs in the Dlk1-Gtl2 locus were predominantly enriched in CD49blo LT-HSCs. Deletion of either IG-DMR or Gtl2 from the maternally inherited allele led to downregulation of Gtl2 and the downstream miRNA mega-cluster, activation of the PI3K-mTOR pathway, enhanced mitochondrial biogenesis, enhanced metabolic activity, and increased ROS levels, eventually leading to HSC apoptosis (Figure 7M).
Specific Expression of lncRNAs in Hematopietic Stem, Progenitor, and Lineage Cells
Recent studies have revealed critical roles of epigenetic regulation in HSCs, including ncRNAs, DNA methylation, and histone modification (Cullen et al., 2014; Luo et al., 2015). Our study, using 17 hematopoietic stem, progenitor, and lineage cells, systematically identified unique fingerprints of lncRNAs, opening the opportunity to investigate the roles and underlying molecular mechanisms of lncRNAs in each cell type. From this framework, using unsupervised hierarchical cluster analysis and PCA on lncRNAs, we revealed that CD49blo HSCs stand at the apex of the hematopoietic hierarchy, and lncRNAs from the Dlk1-Gtl2 locus were predominantly enriched in the CD49blo HSCs.
ncRNAs from the Gtl2 Imprinting Locus Link Fetal Liver HSCs to Adult LT-HSCs and Function to Maintain LT-HSCs
A known feature of fetal HSCs distinctive from adult HSCs is their cycling status. The reason why functionality of adult stem cells, but not fetal HSCs, is associated with quiescent status has been elusive. It is therefore striking to find a link between fetal liver HSCs and the adult CD49blo LT-HSCs in that they exhibit similar ncRNA expression patterns, particularly the ncRNAs from the maternally inherited allele in the Dlk1-Gtl2 locus (Figure 2B), suggesting that these Gtl2-derived ncRNAs play a role in maintaining both fetal and adult HSCs. Indeed, our study provides direct evidence that the Dlk1-Gtl2 locus is required for preservation of HSCs: loss of either mat ΔIG or Gtl2 per se in HSCs led to impaired HSC long-term reconstitution capacity. Consistent with previous studies that show that metabolic properties correlate with HSC functionality (Simsek et al., 2010; Suda et al., 2011; Takubo et al., 2013), our data further elucidate the role of the Dlk1-Gtl2 locus in preserving both fetal HSCs and adult LT-HSCs by inhibiting the PI3K-mTOR pathway and restricting mitochondrial metabolism.
An miRNA Mega-Cluster in the Gtl2 Locus Controls Mitochondrial Biogenesis and Metabolic Activity via Inhibiting the mTOR Pathway
The Dlk1-Gtl2 locus contains the largest cluster of miRNAs in the mammal genome and a large cluster of C/D box snoRNAs. A recent study revealed that Polycomb Repressive Complex 2 (PRC2) transcriptionally regulates the Gtl2 locus through DNA methylation at IG-DMR (Das et al., 2015). In this study, our RNA-seq data showed that these small ncRNAs were simultaneously expressed with Gtl2, supporting the hypothesis that these ncRNAs are transcribed as a large polycistronic transcription unit (Seitz et al., 2004; Tierling et al., 2006). Recent studies have linked the Dlk1-Gtl2 locus to energy metabolism. In one study, the Dlk1-Gtl2 locus was reported to be crucial for the transition to postnatal life by converging on the development of brown fat (Charalambous et al., 2012). In another study using a mouse model with the entire miR-379/miR-410 cluster deleted, researchers found that this cluster could control neonatal metabolic adaptation (Labialle et al., 2014). Consistently, our data reveal that deletion of either IG-DMR or Gtl2 in HSCs leads to downregulation of Gtl2 and the downstream miRNA mega-cluster and enhanced mitochondrial biogenesis and metabolic activity.
An interesting recent study showed that the mTOR pathway controls the transition from G0 to GAlert phase in quiescent stem cells, which functions as an “alerting” mechanism to enhance tissue regenerative function (Rodgers et al., 2014). Our study further reveals that the miRNA mega-cluster in the Gtl2 locus suppresses the entire PI3K-mTOR pathway and inhibits mitochondrial biogenesis and metabolic activity, suggesting the essential roles of metabolic states in preserving LT-HSCs.
EXPERIMENTAL PROCEDURES
Mice
All mice used in this study were housed in the AAALAC-accredited animal facility at the Stowers Institute for Medical Research (SIMR) and handled according to SIMR and National Institutes of Health guidelines. All procedures involving experimental animals were approved by the Institutional Animal Care and Use Committee (IACUC) of SIMR. C57BL/6, Ptprc, and Dlk1 floxed mice were purchased from Jackson Laboratory.
RNA-Seq
Total RNA was isolated using Trizol (Invitrogen). Sequencing libraries were prepared from 10–100 ng total RNA using the TruSeq RNA Sample Preparation Kit v2 (Illumina).
Intracellular Flow Cytometry
Cells from E15.0 fetal livers or recipient BM were initially stained with surface markers for 30–60 min at 4°C and washed with PBS (2% FBS). Cells were then fixed and permeabilized with Cytofix/Cytoperm buffer (BD Biosciences) for 30 min and stained on ice with individual primary antibody or isotype control for 30 min. Samples were then incubated with fluorescent dye conjugated secondary antibodies (Molecular Probes) for 30 min and analyzed by flow cytometry. FlowJo software was used to calculate the MFI for each sample.
Luciferase Reporter Assay
HEK293T cells (5 × 104/well) were seeded into 24-well plates 1 day before transfection. 200 ng of each 3′ UTR construct was co-transfected with 20 nM miRNA mimics or scramble control using Lipofectamine 2000 (Invitrogen) following the manufacturer’s instructions. Cells were harvested 48 hr after transfection, and then Firefly and Renilla luciferase activities were measured using the dual-luciferase reporter kit (Promega) on Victor3 Multilabel Plate Counter (Perkin Elmer).
Statistics
Values are shown as the mean ± SD or SEM as indicated. All statistical analyses were generated using GraphPad Prism 5 (GraphPad Software). Student’s t test was used for comparisons between two groups, whereas one-way ANOVA followed by Tukey’s post hoc tests and two-way ANOVA analysis followed by Bonferroni’s post hoc tests were used for comparisons among multiple groups. Log rank (Mantel-Cox) test was used for Kaplan-Meier survival. Statistical significance was defined as p < 0.05.
Supplementary Material
Figure S1. FACS Plots Showing the Strategy to Purify the 17 Hematopoietic Cell Populations Analyzed in This Study, Related to Figure 1. (A) Multipotent stem and progenitor cells, including CD49blo LT-HSCs, CD49bhi IT-HSCs, ST-HSC and MPP cells, from C57BL/6 mice. (B) Committed progenitor cells, including CMP, GMP, MEP and CLP cells, from C57BL/6 mice. (C) Mature T cells, B cells, NK cells and nucleated erythrocytes from C57BL/6 mice. (D) Mature dendritic cells, monocytes and granulocytes from C57BL/6 mice. (E) Mature megakaryocytes from Pf4-cre+;R26RtdTomato mice. (F) Mature macrophages from C57BL/6 mice. (G) RNA-seq analysis of known HSC markers and the markers used for FACS in 17 hematopoietic cell types. (H) Venn diagram shows the overlaps among lncRNAs annotated in Ensembl databases, reported in a recent study (Luo et al., 2015) and identified in this study. (I) LncRNAs have overall lower expression than protein-coding genes. Maximal FPKM counts (log2-normalized) of each lncRNA (green, n=1693) and coding (red, n=13775) transcript. (J) Cell specificity of RNAs according to type. Shown are distributions of maximal cell specificity scores calculated for each transcript from the RNA seq data for lncRNA (green, n=1693) and coding (red, n=13775).
Figure S2. Loss of Imprinting at Dlk1-Gtl2 Locus Did Not Affect Progenitors and Mature Lineage Cells in Fetal Liver, Related to Figure 2. (A) Electrophoresis patterns of PCR genotyping products from wt and ΔIG E15 embryo tails. (B) Gross phenotype of E15 wt littermate and ΔIG mutant embryos. (C and D) Frequency in TNC (C) and absolute numbers (D) of common progenitor cells in wt and ΔIG E15 fetal liver (n=5). (E and F) Frequency in TNC (E) and absolute numbers (F) of mature lineage cells in wt and ΔIG E15 fetal liver (n=4). (G) Absolute numbers of LSK cells and CD93+ HSCs in wt, mat ΔIG, pat Dlk1 KO and double mutant E15 fetal liver (n=2). (H and I) Frequency in TNC of common progenitor cells (H) and mature lineage cells (I) in wt and mat ΔGtl2 E15 fetal liver (n=3). Error bars, SEM. Scale bars, 5 mm. ns, not significant; *p < 0.05.
Figure S3. Loss of Maternal, but not Paternal, Imprinting at Dlk1-Gtl2 Locus Impaired Long-term Reconstitution Capacity of Fetal Liver HSCs, Related to Figure 3. (A) 5×104 wt or pat ΔIG fetal liver cells were transplanted with 1×105 rescue cells into irradiated recipients. PB was analyzed for percent donor repopulation at the indicated number of weeks posttransplantation (top panels) and for percent mature donor-derived B, T, and myeloid cells (bottom panels) (n=10). (B-D) 5×105 wt or mat ΔIG fetal liver cells were transplanted with 1×105 rescue cells into irradiated recipients. At 16 wks posttransplantation, BM isolated from 1st recipients was transplanted into 2nd recipients and, at 16 wks after 2nd transplant, from 2nd into 3rd recipients at a dosage of 1×106 cells per mouse. PB was analyzed for percent donor repopulation at the indicated number of weeks after 1st, 2nd, and 3rd transplant (top panels) and for percent mature donor-derived B, T, and myeloid cells (bottom panels) (n=10). (E and F) At 16 wks posttransplantation, absolute numbers of total BM cells (E) and HSPCs (F) in the BM from 1st recipients with 5×104 wt or mat ΔIG fetal liver cells (n=10). (G and H) At 16 wks posttransplantation, absolute numbers of total BM cells (G) and HSPCs (H) in the BM from 2nd recipients with 5×104 wt or mat ΔIG fetal liver cells (n≥8). (I) At 16 wks posttransplantation, absolute numbers of total BM cells from 3rd recipients with 5×104 wt or mat ΔIG fetal liver cells (n≥4). (J and K) At 16 wks posttransplantation, absolute numbers of total BM cells (J) and HSPCs (K) in the BM from 1st recipients with 5×104 wt or pat ΔIG fetal liver cells (n=10). (L) Cell cycle analysis of E15 wt or mat ΔIG fetal liver CD93+ HSCs (n≥3). Error bars, SD. *p < 0.05; **p < 0.01; ***p < 0.001.
Figure S4. Hyperactivation of PI3K-mTOR Pathway in mat ΔIG Fetal Liver HSPCs, Related to Figure 4 and Table S7. (A and B) Representative image (A) and quantification (B) of staining intensity of LSK cells sorted from wt and ΔIG fetal livers for p-S6S235/236 (n≥72). (C-H) MFI of p-AktS308 (C), p-mTORS2481 (D), LKB1 (E), p-AMPKαT172 (F), p-FoxO1T24/FoxO3aT32 (G) and p-SGK1S78 (H) in wt and mat ΔIG fetal liver HSCs (n=2–3). Error bars, SEM or SD (B). Scale bars, 5 μm. *p < 0.05; ***p < 0.001.
Figure S5. miRNAs in the Dlk1-Gtl2 Locus Repressed Multiple Components of PI3K-mTOR Pathway, Related to Figure 5 and Table S6, S7. (A) Schematic diagram shows mutations of miRNA binding sites on 3′UTRs of RHEB and AKT1. (B) Luciferase reporter assays shows repression of miRNAs was abrogated by mutations of cognate miRNA binding sites (n=2). (C and D) Expression of AKT1 (C) and RPTOR (D) determined by intracellular flow cytometry in wt and mat ΔIG fetal liver cells (n=2). (E and F) Expression of AKT1 (E) and RPTOR (F) determined by intracellular flow cytometry in donor CD49blo HSCs from BM of 3rd recipients (n≥4). Error bars, SEM or SD (E and F). *p < 0.05; **p < 0.01.
Figure S6. Loss of Gtl2 per se, whereas not pat ΔIG, Enhanced Mitochondrial Biogenesis, Metabolic Activity and Increased ROS Levels in HSCs, Related to Figure 6. (A) Mitochondrial mass of wt and pat ΔIG cells assayed by MitoTracker Green staining (n=2). (B) Mitochondrial membrane potential of wt and pat ΔIG cells assayed by DilC5 staining (n=2). (C) Glucose uptake of wt and pat ΔIG cells assayed by 2-NBDG (n=2). (D) The relative basal ATP levels of wt and pat ΔIG cells (n=2). (E) Mitochondrial mass of wt and mat ΔGtl2 cells assayed by MitoTracker Green staining (n=3). (F) Mitochondrial membrane potential of wt and mat ΔGtl2 cells assayed by DilC5 staining (n=3). (G) Glucose uptake of wt and mat ΔGtl2 cells assayed by 2-NBDG (n=3). (H) Oxygen consumption rate of wt and mat ΔGtl2 cells assayed by Seahorse assay. (I) ROS levels of wt and mat ΔGtl2 cells assayed by H2-DCFDA (n=3). (J) Percentage of apoptotic cells in wt and mat ΔGtl2 cells assayed by Annexin V and 7-AAD staining (n=3). Error bars, SEM. *p < 0.05; **p < 0.01.
Figure S7. Pharmacological Inhibition of PI3K-mTOR Pathway Partially Rescued HSC Deficiency in mat ΔIG Mutant, Related to Figure 7 and Table S7. (A and B) MFI of p-4E-BP1T37/46 (A) and PGC-1α (B) expression within donor CD49blo HSCs from BM of mice with rapamycin administration at 12 wks posttransplantation (n≥4). (C) Scheme for administration of PI3K-mTOR pathway inhibitors. (D) Repopulation assay for administration of PI3K-mTOR pathway inhibitors at 16 wks posttransplantation (n=6). Error bars, SD. *p < 0.05; **p < 0.01; ***p < 0.001.
Purification of Hematopoietic Cell Populations Used in This Study, Related to Figures 1, 2, 4, 5, Figure S1 and Supplemental Experimental Procedures.
FPKMs of All Transcripts Used in This Study by High-throughput RNA-seq, Related to Figures 1, 2, 4, 5, Figure S1 and Supplemental Experimental Procedures.
LncRNA Fingerprints Unique to Each Hematopoietic Cell Type, Related to Figure 1.
GO Analysis Using Slim-term List and Biological Process in the Mat ΔIG/Mat Wt, Related to Figure 4.
Small RNA Seq Identified 15 miRNAs and 10 snoRNAs in Both Fetal Liver HSC and Adult HSCs, Related to Figure 5.
Sequences of the Oligonucleotides for miRNAs, Plasmid Construction, and qRT-PCR Analysis, Related to Figures 2, 5, Figure S5 and Supplemental Experimental Procedures.
List of Proteins Tested by Antibodies and Characteristics of the Corresponding Antibodies Used, Related to Figures 4, 5, 6, 7, Figures S4, S5, S7 and Supplemental Experimental Procedures.
Highlights.
Transcriptome profiling reveals Gtl2-derived ncRNA enrichment in LT-HSCs
Loss of Dlk1-Gtl2 imprinting leads to functional defects in fetal liver HSCs
miRNAs of the Dlk1-Gtl2 locus suppress components of the entire PI3K-mTOR pathway
PI3K-mTOR inhibition restricts mitochondrial metabolism to preserve LT-HSC function
Acknowledgments
We thank M. Miller for model image modification; K. Tannen for proofreading and editing; and M. Hembree, H. Marshall, K. Zapien, D. Dukes, C. Semerad, A. Box, J. Park, L. Blunk, A. Perera, and A. Peak for technical support. We are grateful to Linheng Li laboratory members for scientific discussion and critical reading of the manuscript. This work was funded by the Stowers Institute for Medical Research and the American Society of Hematology (ASH) Scholar Award. L.Z. was supported by the National Natural Science Foundation of China (31200575) and the Natural Science Foundation of Tianjin (12JCQNJC07600).
Footnotes
ACCESSION NUMBERS
The accession numbers for the raw datasets reported in this paper are ArrayExpress: E-MTAB-2923, ArrayExpress: E-MTAB-3079 (mRNA-seq), and ArrayExpress: E-MTAB-3080 (small RNA-seq).
Supplemental Information for this article includes seven figures, seven tables, and Supplemental Experimental Procedures and can be found with this article online at http://dx.doi.org/10.1016/j.stem.2015.11.001.
AUTHOR CONTRIBUTIONS
P.Q. performed experiments, analyzed data, and wrote the manuscript. X.C.H. helped perform transplantations and RNA-seq. A.P. and H.L. performed bioinformatics analysis. Z.L., F.T., J.M.P., F.G., M.Z., L.Z., A.V., J.S.H., T.P., R.T.D., and W.X.D. helped perform part of the experiments. T.K. and A.C.F-S. contributed mouse lines. L.L. directed the overall project and co-wrote the manuscript. All authors contributed to reading and editing the manuscript.
References
- Alvarez-Dominguez JR, Hu W, Yuan B, Shi J, Park SS, Gromatzky AA, van Oudenaarden A, Lodish HF. Global discovery of erythroid long noncoding RNAs reveals novel regulators of red cell maturation. Blood. 2014;123:570–581. doi: 10.1182/blood-2013-10-530683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartolomei MS. Genomic imprinting: employing and avoiding epigenetic processes. Genes Dev. 2009;23:2124–2133. doi: 10.1101/gad.1841409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beerman I, Bhattacharya D, Zandi S, Sigvardsson M, Weissman IL, Bryder D, Rossi DJ. Functionally distinct hematopoietic stem cells modulate hematopoietic lineage potential during aging by a mechanism of clonal expansion. Proc Natl Acad Sci USA. 2010;107:5465–5470. doi: 10.1073/pnas.1000834107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benveniste P, Frelin C, Janmohamed S, Barbara M, Herrington R, Hyam D, Iscove NN. Intermediate-term hematopoietic stem cells with extended but time-limited reconstitution potential. Cell Stem Cell. 2010;6:48–58. doi: 10.1016/j.stem.2009.11.014. [DOI] [PubMed] [Google Scholar]
- Berg JS, Lin KK, Sonnet C, Boles NC, Weksberg DC, Nguyen H, Holt LJ, Rickwood D, Daly RJ, Goodell MA. Imprinted genes that regulate early mammalian growth are coexpressed in somatic stem cells. PLoS ONE. 2011;6:e26410. doi: 10.1371/journal.pone.0026410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavaillé J, Nicoloso M, Bachellerie JP. Targeted ribose methylation of RNA in vivo directed by tailored antisense RNA guides. Nature. 1996;383:732–735. doi: 10.1038/383732a0. [DOI] [PubMed] [Google Scholar]
- Charalambous M, Ferron SR, da Rocha ST, Murray AJ, Rowland T, Ito M, Schuster-Gossler K, Hernandez A, Ferguson-Smith AC. Imprinted gene dosage is critical for the transition to independent life. Cell Metab. 2012;15:209–221. doi: 10.1016/j.cmet.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Liu Y, Liu R, Ikenoue T, Guan KL, Liu Y, Zheng P. TSC-mTOR maintains quiescence and function of hematopoietic stem cells by repressing mitochondrial biogenesis and reactive oxygen species. J Exp Med. 2008;205:2397–2408. doi: 10.1084/jem.20081297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen SM, Mayle A, Rossi L, Goodell MA. Hematopoietic stem cell development: an epigenetic journey. Curr Top Dev Biol. 2014;107:39–75. doi: 10.1016/B978-0-12-416022-4.00002-0. [DOI] [PubMed] [Google Scholar]
- Cunningham JT, Rodgers JT, Arlow DH, Vazquez F, Mootha VK, Puigserver P. mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature. 2007;450:736–740. doi: 10.1038/nature06322. [DOI] [PubMed] [Google Scholar]
- da Rocha ST, Edwards CA, Ito M, Ogata T, Ferguson-Smith AC. Genomic imprinting at the mammalian Dlk1-Dio3 domain. Trends Genet. 2008;24:306–316. doi: 10.1016/j.tig.2008.03.011. [DOI] [PubMed] [Google Scholar]
- Das PP, Hendrix DA, Apostolou E, Buchner AH, Canver MC, Beyaz S, Ljuboja D, Kuintzle R, Kim W, Karnik R, et al. PRC2 Is Required to Maintain Expression of the Maternal Gtl2-Rian-Mirg Locus by Preventing De Novo DNA Methylation in Mouse Embryonic Stem Cells. Cell Rep. 2015;12:1456–1470. doi: 10.1016/j.celrep.2015.07.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson-Smith AC. Genomic imprinting: the emergence of an epigenetic paradigm. Nat Rev Genet. 2011;12:565–575. doi: 10.1038/nrg3032. [DOI] [PubMed] [Google Scholar]
- Fernie AR, Carrari F, Sweetlove LJ. Respiratory metabolism: glycolysis, the TCA cycle and mitochondrial electron transport. Curr Opin Plant Biol. 2004;7:254–261. doi: 10.1016/j.pbi.2004.03.007. [DOI] [PubMed] [Google Scholar]
- Ferrón SR, Charalambous M, Radford E, McEwen K, Wildner H, Hind E, Morante-Redolat JM, Laborda J, Guillemot F, Bauer SR, et al. Postnatal loss of Dlk1 imprinting in stem cells and niche astrocytes regulates neurogenesis. Nature. 2011;475:381–385. doi: 10.1038/nature10229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haug JS, He XC, Grindley JC, Wunderlich JP, Gaudenz K, Ross JT, Paulson A, Wagner KP, Xie Y, Zhu R, et al. N-cadherin expression level distinguishes reserved versus primed states of hematopoietic stem cells. Cell Stem Cell. 2008;2:367–379. doi: 10.1016/j.stem.2008.01.017. [DOI] [PubMed] [Google Scholar]
- Hirao A, Hoshii T. Mechanistic/mammalian target protein of rapamycin signaling in hematopoietic stem cells and leukemia. Cancer Sci. 2013;104:977–982. doi: 10.1111/cas.12189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang YY, Sharkis SJ. A low level of reactive oxygen species selects for primitive hematopoietic stem cells that may reside in the low-oxygenic niche. Blood. 2007;110:3056–3063. doi: 10.1182/blood-2007-05-087759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labialle S, Marty V, Bortolin-Cavaillé ML, Hoareau-Osman M, Pradère JP, Valet P, Martin PG, Cavaillé J. The miR-379/miR-410 cluster at the imprinted Dlk1-Dio3 domain controls neonatal metabolic adaptation. EMBO J. 2014;33:2216–2230. doi: 10.15252/embj.201387038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, Nakada D, Yilmaz OH, Tothova Z, Joseph NM, Lim MS, Gilliland DG, Morrison SJ. mTOR activation induces tumor suppressors that inhibit leukemogenesis and deplete hematopoietic stem cells after Pten deletion. Cell Stem Cell. 2010;7:593–605. doi: 10.1016/j.stem.2010.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Clevers H. Coexistence of quiescent and active adult stem cells in mammals. Science. 2010;327:542–545. doi: 10.1126/science.1180794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SP, Youngson N, Takada S, Seitz H, Reik W, Paulsen M, Cavaille J, Ferguson-Smith AC. Asymmetric regulation of imprinting on the maternal and paternal chromosomes at the Dlk1-Gtl2 imprinted cluster on mouse chromosome 12. Nat Genet. 2003;35:97–102. doi: 10.1038/ng1233. [DOI] [PubMed] [Google Scholar]
- Lin SP, Coan P, da Rocha ST, Seitz H, Cavaille J, Teng PW, Takada S, Ferguson-Smith AC. Differential regulation of imprinting in the murine embryo and placenta by the Dlk1-Dio3 imprinting control region. Development. 2007;134:417–426. doi: 10.1242/dev.02726. [DOI] [PubMed] [Google Scholar]
- Luo M, Jeong M, Sun D, Park HJ, Rodriguez BA, Xia Z, Yang L, Zhang X, Sheng K, Darlington GJ, et al. Long non-coding RNAs control hematopoietic stem cell function. Cell Stem Cell. 2015;16:426–438. doi: 10.1016/j.stem.2015.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita M, Gravel SP, Chénard V, Sikström K, Zheng L, Alain T, Gandin V, Avizonis D, Arguello M, Zakaria C, et al. mTORC1 controls mitochondrial activity and biogenesis through 4E-BP-dependent translational regulation. Cell Metab. 2013;18:698–711. doi: 10.1016/j.cmet.2013.10.001. [DOI] [PubMed] [Google Scholar]
- Morrison SJ, Hemmati HD, Wandycz AM, Weissman IL. The purification and characterization of fetal liver hematopoietic stem cells. Proc Natl Acad Sci USA. 1995;92:10302–10306. doi: 10.1073/pnas.92.22.10302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randall TD, Lund FE, Howard MC, Weissman IL. Expression of murine CD38 defines a population of long-term reconstituting hematopoietic stem cells. Blood. 1996;87:4057–4067. [PubMed] [Google Scholar]
- Rodgers JT, King KY, Brett JO, Cromie MJ, Charville GW, Maguire KK, Brunson C, Mastey N, Liu L, Tsai CR, et al. mTORC1 controls the adaptive transition of quiescent stem cells from G0 to G(Alert) Nature. 2014;510:393–396. doi: 10.1038/nature13255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seitz H, Royo H, Bortolin ML, Lin SP, Ferguson-Smith AC, Cavaillé J. A large imprinted microRNA gene cluster at the mouse Dlk1-Gtl2 domain. Genome Res. 2004;14:1741–1748. doi: 10.1101/gr.2743304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simsek T, Kocabas F, Zheng J, Deberardinis RJ, Mahmoud AI, Olson EN, Schneider JW, Zhang CC, Sadek HA. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell. 2010;7:380–390. doi: 10.1016/j.stem.2010.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder CM, Rice AL, Estrella NL, Held A, Kandarian SC, Naya FJ. MEF2A regulates the Gtl2-Dio3 microRNA mega-cluster to modulate WNT signaling in skeletal muscle regeneration. Development. 2013;140:31–42. doi: 10.1242/dev.081851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suda T, Takubo K, Semenza GL. Metabolic regulation of hematopoietic stem cells in the hypoxic niche. Cell Stem Cell. 2011;9:298–310. doi: 10.1016/j.stem.2011.09.010. [DOI] [PubMed] [Google Scholar]
- Takahashi N, Okamoto A, Kobayashi R, Shirai M, Obata Y, Ogawa H, Sotomaru Y, Kono T. Deletion of Gtl2, imprinted non-coding RNA, with its differentially methylated region induces lethal parent-origin-dependent defects in mice. Hum Mol Genet. 2009;18:1879–1888. doi: 10.1093/hmg/ddp108. [DOI] [PubMed] [Google Scholar]
- Takubo K, Nagamatsu G, Kobayashi CI, Nakamura-Ishizu A, Kobayashi H, Ikeda E, Goda N, Rahimi Y, Johnson RS, Soga T, et al. Regulation of glycolysis by Pdk functions as a metabolic checkpoint for cell cycle quiescence in hematopoietic stem cells. Cell Stem Cell. 2013;12:49–61. doi: 10.1016/j.stem.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tierling S, Dalbert S, Schoppenhorst S, Tsai CE, Oliger S, Ferguson-Smith AC, Paulsen M, Walter J. High-resolution map and imprinting analysis of the Gtl2-Dnchc1 domain on mouse chromosome 12. Genomics. 2006;87:225–235. doi: 10.1016/j.ygeno.2005.09.018. [DOI] [PubMed] [Google Scholar]
- Venkatraman A, He XC, Thorvaldsen JL, Sugimura R, Perry JM, Tao F, Zhao M, Christenson MK, Sanchez R, Yu JY, et al. Maternal imprinting at the H19-Igf2 locus maintains adult haematopoietic stem cell quiescence. Nature. 2013;500:345–349. doi: 10.1038/nature12303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson A, Laurenti E, Oser G, van der Wath RC, Blanco-Bose W, Jaworski M, Offner S, Dunant CF, Eshkind L, Bockamp E, et al. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell. 2008;135:1118–1129. doi: 10.1016/j.cell.2008.10.048. [DOI] [PubMed] [Google Scholar]
- Yang L, Bryder D, Adolfsson J, Nygren J, Månsson R, Sigvardsson M, Jacobsen SE. Identification of Lin(-)Sca1(+)kit(+)CD34(+)Flt3-short-term hematopoietic stem cells capable of rapidly reconstituting and rescuing myeloablated transplant recipients. Blood. 2005;105:2717–2723. doi: 10.1182/blood-2004-06-2159. [DOI] [PubMed] [Google Scholar]
- Zacharek SJ, Fillmore CM, Lau AN, Gludish DW, Chou A, Ho JW, Zamponi R, Gazit R, Bock C, Jäger N, et al. Lung stem cell self-renewal relies on BMI1-dependent control of expression at imprinted loci. Cell Stem Cell. 2011;9:272–281. doi: 10.1016/j.stem.2011.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Grindley JC, Yin T, Jayasinghe S, He XC, Ross JT, Haug JS, Rupp D, Porter-Westpfahl KS, Wiedemann LM, et al. PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature. 2006;441:518–522. doi: 10.1038/nature04747. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Cheunsuchon P, Nakayama Y, Lawlor MW, Zhong Y, Rice KA, Zhang L, Zhang X, Gordon FE, Lidov HG, et al. Activation of paternally expressed genes and perinatal death caused by deletion of the Gtl2 gene. Development. 2010;137:2643–2652. doi: 10.1242/dev.045724. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. FACS Plots Showing the Strategy to Purify the 17 Hematopoietic Cell Populations Analyzed in This Study, Related to Figure 1. (A) Multipotent stem and progenitor cells, including CD49blo LT-HSCs, CD49bhi IT-HSCs, ST-HSC and MPP cells, from C57BL/6 mice. (B) Committed progenitor cells, including CMP, GMP, MEP and CLP cells, from C57BL/6 mice. (C) Mature T cells, B cells, NK cells and nucleated erythrocytes from C57BL/6 mice. (D) Mature dendritic cells, monocytes and granulocytes from C57BL/6 mice. (E) Mature megakaryocytes from Pf4-cre+;R26RtdTomato mice. (F) Mature macrophages from C57BL/6 mice. (G) RNA-seq analysis of known HSC markers and the markers used for FACS in 17 hematopoietic cell types. (H) Venn diagram shows the overlaps among lncRNAs annotated in Ensembl databases, reported in a recent study (Luo et al., 2015) and identified in this study. (I) LncRNAs have overall lower expression than protein-coding genes. Maximal FPKM counts (log2-normalized) of each lncRNA (green, n=1693) and coding (red, n=13775) transcript. (J) Cell specificity of RNAs according to type. Shown are distributions of maximal cell specificity scores calculated for each transcript from the RNA seq data for lncRNA (green, n=1693) and coding (red, n=13775).
Figure S2. Loss of Imprinting at Dlk1-Gtl2 Locus Did Not Affect Progenitors and Mature Lineage Cells in Fetal Liver, Related to Figure 2. (A) Electrophoresis patterns of PCR genotyping products from wt and ΔIG E15 embryo tails. (B) Gross phenotype of E15 wt littermate and ΔIG mutant embryos. (C and D) Frequency in TNC (C) and absolute numbers (D) of common progenitor cells in wt and ΔIG E15 fetal liver (n=5). (E and F) Frequency in TNC (E) and absolute numbers (F) of mature lineage cells in wt and ΔIG E15 fetal liver (n=4). (G) Absolute numbers of LSK cells and CD93+ HSCs in wt, mat ΔIG, pat Dlk1 KO and double mutant E15 fetal liver (n=2). (H and I) Frequency in TNC of common progenitor cells (H) and mature lineage cells (I) in wt and mat ΔGtl2 E15 fetal liver (n=3). Error bars, SEM. Scale bars, 5 mm. ns, not significant; *p < 0.05.
Figure S3. Loss of Maternal, but not Paternal, Imprinting at Dlk1-Gtl2 Locus Impaired Long-term Reconstitution Capacity of Fetal Liver HSCs, Related to Figure 3. (A) 5×104 wt or pat ΔIG fetal liver cells were transplanted with 1×105 rescue cells into irradiated recipients. PB was analyzed for percent donor repopulation at the indicated number of weeks posttransplantation (top panels) and for percent mature donor-derived B, T, and myeloid cells (bottom panels) (n=10). (B-D) 5×105 wt or mat ΔIG fetal liver cells were transplanted with 1×105 rescue cells into irradiated recipients. At 16 wks posttransplantation, BM isolated from 1st recipients was transplanted into 2nd recipients and, at 16 wks after 2nd transplant, from 2nd into 3rd recipients at a dosage of 1×106 cells per mouse. PB was analyzed for percent donor repopulation at the indicated number of weeks after 1st, 2nd, and 3rd transplant (top panels) and for percent mature donor-derived B, T, and myeloid cells (bottom panels) (n=10). (E and F) At 16 wks posttransplantation, absolute numbers of total BM cells (E) and HSPCs (F) in the BM from 1st recipients with 5×104 wt or mat ΔIG fetal liver cells (n=10). (G and H) At 16 wks posttransplantation, absolute numbers of total BM cells (G) and HSPCs (H) in the BM from 2nd recipients with 5×104 wt or mat ΔIG fetal liver cells (n≥8). (I) At 16 wks posttransplantation, absolute numbers of total BM cells from 3rd recipients with 5×104 wt or mat ΔIG fetal liver cells (n≥4). (J and K) At 16 wks posttransplantation, absolute numbers of total BM cells (J) and HSPCs (K) in the BM from 1st recipients with 5×104 wt or pat ΔIG fetal liver cells (n=10). (L) Cell cycle analysis of E15 wt or mat ΔIG fetal liver CD93+ HSCs (n≥3). Error bars, SD. *p < 0.05; **p < 0.01; ***p < 0.001.
Figure S4. Hyperactivation of PI3K-mTOR Pathway in mat ΔIG Fetal Liver HSPCs, Related to Figure 4 and Table S7. (A and B) Representative image (A) and quantification (B) of staining intensity of LSK cells sorted from wt and ΔIG fetal livers for p-S6S235/236 (n≥72). (C-H) MFI of p-AktS308 (C), p-mTORS2481 (D), LKB1 (E), p-AMPKαT172 (F), p-FoxO1T24/FoxO3aT32 (G) and p-SGK1S78 (H) in wt and mat ΔIG fetal liver HSCs (n=2–3). Error bars, SEM or SD (B). Scale bars, 5 μm. *p < 0.05; ***p < 0.001.
Figure S5. miRNAs in the Dlk1-Gtl2 Locus Repressed Multiple Components of PI3K-mTOR Pathway, Related to Figure 5 and Table S6, S7. (A) Schematic diagram shows mutations of miRNA binding sites on 3′UTRs of RHEB and AKT1. (B) Luciferase reporter assays shows repression of miRNAs was abrogated by mutations of cognate miRNA binding sites (n=2). (C and D) Expression of AKT1 (C) and RPTOR (D) determined by intracellular flow cytometry in wt and mat ΔIG fetal liver cells (n=2). (E and F) Expression of AKT1 (E) and RPTOR (F) determined by intracellular flow cytometry in donor CD49blo HSCs from BM of 3rd recipients (n≥4). Error bars, SEM or SD (E and F). *p < 0.05; **p < 0.01.
Figure S6. Loss of Gtl2 per se, whereas not pat ΔIG, Enhanced Mitochondrial Biogenesis, Metabolic Activity and Increased ROS Levels in HSCs, Related to Figure 6. (A) Mitochondrial mass of wt and pat ΔIG cells assayed by MitoTracker Green staining (n=2). (B) Mitochondrial membrane potential of wt and pat ΔIG cells assayed by DilC5 staining (n=2). (C) Glucose uptake of wt and pat ΔIG cells assayed by 2-NBDG (n=2). (D) The relative basal ATP levels of wt and pat ΔIG cells (n=2). (E) Mitochondrial mass of wt and mat ΔGtl2 cells assayed by MitoTracker Green staining (n=3). (F) Mitochondrial membrane potential of wt and mat ΔGtl2 cells assayed by DilC5 staining (n=3). (G) Glucose uptake of wt and mat ΔGtl2 cells assayed by 2-NBDG (n=3). (H) Oxygen consumption rate of wt and mat ΔGtl2 cells assayed by Seahorse assay. (I) ROS levels of wt and mat ΔGtl2 cells assayed by H2-DCFDA (n=3). (J) Percentage of apoptotic cells in wt and mat ΔGtl2 cells assayed by Annexin V and 7-AAD staining (n=3). Error bars, SEM. *p < 0.05; **p < 0.01.
Figure S7. Pharmacological Inhibition of PI3K-mTOR Pathway Partially Rescued HSC Deficiency in mat ΔIG Mutant, Related to Figure 7 and Table S7. (A and B) MFI of p-4E-BP1T37/46 (A) and PGC-1α (B) expression within donor CD49blo HSCs from BM of mice with rapamycin administration at 12 wks posttransplantation (n≥4). (C) Scheme for administration of PI3K-mTOR pathway inhibitors. (D) Repopulation assay for administration of PI3K-mTOR pathway inhibitors at 16 wks posttransplantation (n=6). Error bars, SD. *p < 0.05; **p < 0.01; ***p < 0.001.
Purification of Hematopoietic Cell Populations Used in This Study, Related to Figures 1, 2, 4, 5, Figure S1 and Supplemental Experimental Procedures.
FPKMs of All Transcripts Used in This Study by High-throughput RNA-seq, Related to Figures 1, 2, 4, 5, Figure S1 and Supplemental Experimental Procedures.
LncRNA Fingerprints Unique to Each Hematopoietic Cell Type, Related to Figure 1.
GO Analysis Using Slim-term List and Biological Process in the Mat ΔIG/Mat Wt, Related to Figure 4.
Small RNA Seq Identified 15 miRNAs and 10 snoRNAs in Both Fetal Liver HSC and Adult HSCs, Related to Figure 5.
Sequences of the Oligonucleotides for miRNAs, Plasmid Construction, and qRT-PCR Analysis, Related to Figures 2, 5, Figure S5 and Supplemental Experimental Procedures.
List of Proteins Tested by Antibodies and Characteristics of the Corresponding Antibodies Used, Related to Figures 4, 5, 6, 7, Figures S4, S5, S7 and Supplemental Experimental Procedures.