

Abstract
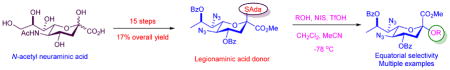
The synthesis of a legionaminic acid donor from N-acetyl neuraminic acid in fifteen steps and 17% overall yield is described. Activation of the adamantanyl thioglycoside in the donor with N-iodosuccinimide and trifluoromethanesulfonic acid in dichloromethane and acetonitrile at −78 °C in the presence of primary, secondary and tertiary alcohols affords the corresponding glycosides in excellent yield and good to excellent equatorial selectivity. In particular coupling to the 4-OH of a suitably protected neuraminic acid derivative affords a disaccharide that closely resembles the glycosidic linkage in the polylegionaminic acid from the lipopolysaccharide of the Legionella pneumophila virulence factor. A straightforward deprotection sequence enables conversion of the protected glycosides to the free N,N-diacetyl legionaminic acid glycosides.
Graphical Abstract
Introduction
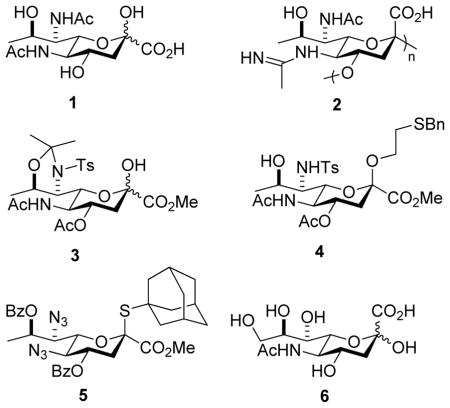
Legionaminic acid 1 is a member of the sialic acid family of nine carbon amino deoxy ulosonic acids that is only found in bacteria.1–3 It is the major component of the lipopolysaccharide virulence factor of Legionella pneumophila, where it occurs in the form of the α-(2→4)-linked (equatorial) homopolymer 2.4–7 Additionally, legionaminic acid glycosides are also found in the lipopolysaccharides of numerous other important Gram-negative pathogens1–2 including, for example, Campylobacter jejuni,8–10 Cronobacter turicensis,11 Enterobacter cloacae,12 and Acinetobacter baumannii.13C Consequently, legionamic acid glycosides are candidates for antibacterial vaccine development and for use in diagnostic tools.6,14 To facilitate such studies broader availability of legionaminic acid and its glycosides is required and, while recent biosynthetic work is promising in terms of accessing legionaminic acid15–16 itself and its glycosides,17–18 practical chemical syntheses are required. Initial synthetic work by Tsvetkov and coworkers was conducted with the aim of confirming the relative configuration but relied on the low yield condensation of oxaloacetic acid with a 2,4-diacetamido-2,4-deoxy-D-rhamnose derivative obtained in multiple steps from D-fucose.4,19 More recently a synthesis of a legionaminic acid glycosyl donor 3 has been reported in seventeen steps and 7% overall yield from the unnatural D-enantiomer of threonine by the Seeberger laboratory.14 This donor was used in the only chemical glycosylation of legionaminic acid reported; that of a simple primary alcohol by the Gin dehydrative method20 resulting in the isolation of the axial (β-) glycoside 4 in 63% yield after removal of the acetonide. It is reported that the equatorial (α-) anomer of 4 was also formed but could not be isolated pure, hence precluding any estimation of anomeric selectivity.14 Other glycosylation methods, including the use of anomeric chlorides, acetates and phosphites, thioglycosides, and N-phenyl trifluoroacetimidates, were stated to give irreproducible results.14 We report on the gram scale synthesis of the legionaminic acid thioglycoside 5 in fifteen steps and 17% overall yield from N-acetylneuraminic acid 6, and its use in the α-selective glycosylation of typical primary, secondary, and tertiary alcohols.
Results
Donor Synthesis
Synthesis of the glycosyl donor 5 (Scheme 1) began with N-acetylneuraminic acid 6, which was converted into the N-Boc adamantanyl thioglycoside 7 by a straightforward sequence of six known steps.21–22 Selective sulfonylation of the primary hydroxy group in 7 with 2,4,6-triisopropylbenzenesulfonyl chloride in pyridine then afforded 71% of the sulfonate ester 8, which on heating to reflux in acetone in the presence of excess sodium iodide gave the iodo derivative 9 in 94% yield. Selective diesterification of the triol 9 with benzoyl chloride in pyridine at 0 °C followed the established pattern in the N-acetylneuraminic acid series23 and gave the 4,8-di-O-benzoate 10 in 91% yield; acetylation of 9 with acetic anhydride in pyridine was less satisfactory owing to the formation of substantial amounts of triacetate. Deiodination to give 11 was best achieved with tris(trimethylsilyl)silane24 with initiation by azoisobutyronitrile in benzene at 60 °C, which left the thioglycoside intact but resulted in partial removal of the carbamate by the silyl iodide generated as byproduct of the radical reaction. As such, the crude reaction mixture was simply taken up in methanol and treated with hydrogen chloride in ether thereby completing removal of the Boc group and affording the amine 12 as the hydrochloride salt. This material was also not isolated but was converted to the corresponding azide by treatment with Stick’s reagent25–26 and triethylamine in the presence of catalytic copper sulfate in aqueous acetonitrile ultimately giving 13 in 78% overall yield for the three steps from iodide 10. The use of triethylamine as base for the introduction of the azide functionality arose from the need to suppress competing debenzoylation, while aqueous acetonitrile was employed as solvent owing to the poor solubility of 12 in the more typical aqueous methanol. With regard to the deiodination, hydrogenolytic methods were also successful but we were unable to fully suppress concomitant hydrogenolysis of the thioglycoside with a range of catalysts under both batch and flow conditions. In preparation for installation of the C-N bond at the 7-position, alcohol 13 was optimally converted to the ketone 14 in 85% yield using the Dess-Martin periodinane.27 Consistent with the precedent in the N-acetylneuraminic acid series28–29 reduction of 14 with Luche’s reagent30 in methanolic dichloromethane at −78 °C gave an 85:15 mixture of epimeric alcohols from which 15 was isolated in 82% yield. Finally, triflation of 15 with triflic anhydride in pyridine in dichloromethane at 0 °C gave 16, which was immediately stirred with excess sodium azide in DMF at 0 °C leading to the isolation of the desired donor 5 in 81% yield for the two steps. In summary, the stable thioglycoside 5 was prepared by the sequence outlined in Scheme 1 in fifteen straightforward steps and 17% overall yield from the readily available N-acetylneuraminic acid 6. This synthesis, albeit somewhat classical in nature, compares favorably with the previously reported seventeen step, 7% overall yield de novo synthesis of donor 3 from D-threonine14 and includes less steps requiring the tedious separation of diastereomeric mixtures.
Scheme 1.

Synthesis of the Donor 5.
Among the several variations explored on the sequence of steps outlined in Scheme 1, we draw attention only to the attempted reversal of the deiodination and regioselective benzoylation (9 → 10 → 13). Thus, hydrogenolysis of 9 over palladium hydroxide on charcoal in methanol gave the deiodo derivative 17, which was directly treated with HCl in methanol affording 18, and then with Stick’s reagent in the presence of catalytic copper sulfate potassium carbonate in aqueous methanol leading to triol 19 in 39% overall yield for the three steps. Treatment with benzoyl chloride in pyridine at 0 °C then afforded the 7,8-di-O-benzoate 20 in 46% isolated yield, rather than the anticipated 4,8-di-O-benzoate 15, along with 21% of the tri-O-benzoate 21 (Scheme 2). The typical selectivity sequence of preferential functionalization of the 4-OH in the presence of the 7-OH observed with neuraminic acid derivatives, and manifested here in the conversion of 9 to 10 (Scheme 1), therefore depends on the presence of a functional group at the 9-position, perhaps for reasons of steric buttressing, and is reversed when the 9-position is unfunctionalized.
Scheme 2.

Selective Formation of a 7,8-Di-O-benzoate from a 4,7,8-Triol.
Acceptor Synthesis
As a model acceptor for the eventual synthesis of polylegionaminic acid 2 a neuraminic acid derivative carrying a single free hydroxy group at the 4-position was prepared as outlined in Scheme 3. Thus, the azido triol 22, readily accessible from 6 by known methods,22 was converted with 2,2-dimethoxypropane to the 8,9-O-acetonide 23 in 83% yield, and then under standard conditions to the mono-4-O-silyl ether 24 in 91% yield. Subsequent acetylation of the remaining hydroxyl group gave 25 in 98% yield, and was followed by fluoride-mediated cleavage of the silyl ether to afford acceptor 26 in 98% yield. A series of three further acceptors 27–29 based on the galactopyranose framework were prepared by literature methods.31–33
Scheme 3.

Synthesis of Acceptor 26.
Glycosylation
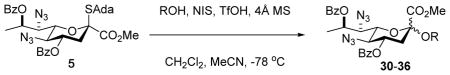
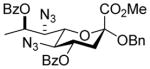
Glycosylation reactions (Table 1) were conducted at −78 °C in 1:2 acetonitrile:dichloromethane in the presence of acid-washed 4Å molecular sieves with activation by the N-iodosuccinimide and trifluoromethanesulfonic acid with quenching by addition of triethylamine at −78 °C. These conditions were selected as they were recently found to be highly satisfactory for the formation of equatorial glycosides of neuraminic acid from an adamantanyl thioglycoside carrying an azide at the 5-position.34 Anomeric selectivities were measured by integration of characteristic signals in the 1H NMR spectra of the crude reaction mixtures. Anomeric configurations were assigned following chromatographic purification by determination of the diagnostic heteronuclear coupling constants between the C1 carboxyl carbon and the axial hydrogen at C3 (Table 1). These coupling constants followed the well-established pattern in the neuraminic acid series.28,35–38
Table 1.
Glycosylation Reactions
| ||||
|---|---|---|---|---|
| Entry | Acceptor | Product | % Yield, α:β ratio | 3JCH (Hz) |
| 1 | BnOH |
 30 |
96%, α-only | 30: 7.2 |
| 2 |
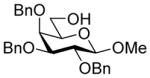 27 |
31αβ |
87%, 6.7:1 | 31α: 6.4 |
| 3 |
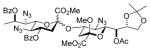
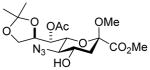 26 |
 32α,β |
82%, 4.4:1 | 32α: 6.9 |
| 4 |
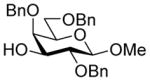 28 |
33α,β |
75%, a4.5:1 |
33α: 7.2 33β: 0 |
| 5 |
 29 |
34α,β |
88%, 34α:34β:35α= 4.7:1:0.9 | 34α: 7.0 |
 35 |
35α: 6,3 | |||
| 6 | 1-adamantanol |
 36α,β |
73%, 4.2:1 | 36α: 5.7 |
Glycosylation gave an 84% yield of an inseparable 4.5:1 α:β mixture of the disaccharides, which was saponified to give the products 33α and 33β.
Deprotection Reactions
The α-(2→6)-linked dibenzoyl disaccharide 31α was converted to the corresponding diol 37 with sodium methoxide in methanol, and then by hydrogenolysis and peracetylation to 38, before global hydrolysis of all esters with hot aqueous barium hydroxide to give the N,N-diacetyl legionaminic acid glycoside 39 in good overall yield (Scheme 4). The sequence of reactions in this deprotection protocol, with initial removal of the two benzoate esters, was selected to avoid possible O→N benzoate migration in the reverse sequence with prior hydrogenolysis of the azide groups. The α-(2→3)-linked disaccharide 33α from which the benzoate esters had already been stripped to facilitate separation of the anomers (Table 1, entry 4), was subjected to hydrogenolysis followed by acetylation to give 40. A second sample of 40 was obtained by removal of the benzoate esters from 34α, giving 41, and then hydrogenolysis and peracetylation. Finally, removal of all esters from 40 with barium hydroxide yielded the legionaminic acid glycoside 42 (Scheme 4). The conversion of both 33α and 34α to a common product 40 in this manner confirms the assignment of regiochemistry of the major product in the glycosylation of the galactosyl 3,4-diol 29 (Table 1, entry 5).
Scheme 4.

Deprotection Affording the Regioisomeric α-(2→6) and α-(2→3) Galactosyl Legionamic Acid Glycosides 39 and 41 and Proof of Regiochemistry of 34α.
Discussion
Glycosylation Selectivity
All glycosylation reactions proceeded with moderate to excellent yield and selectivity for the formation of the equatorial glycoside (Table 1). Not surprisingly, the optimal selectivity was observed with the reactive primary acceptor benzyl alcohol (Table 1, entry 1), followed by the primary carbohydrate-based acceptor 27 (Table 1, entry 2). Two standard carbohydrate-based secondary alcohols 26 and 28 (Table 1, entries 3 and 4) also performed satisfactorily. The galactose-based 3,4-diol 29 gave a comparable α,β-ratio for the 2→3-linked products 34α,β but in addition furnished a minor amount of the α-2→4-linked product 35 (Table 1, entry 5). We did not isolate the corresponding β-2→4-linked product and so are unable to comment on selectivity with respect to coupling to the 4-position of diol 29. Finally, coupling of the relatively reactive tertiary alcohol 1-adamantanol to donor 5 took place with only a minor loss of selectivity as compared to the secondary alcohols, giving a 4.2:1 α:β-ratio of the product 36 (Table 1, entry 6). These selectivities are moderately lower than those observed with the sialic acid donor 43 under comparable conditions.34
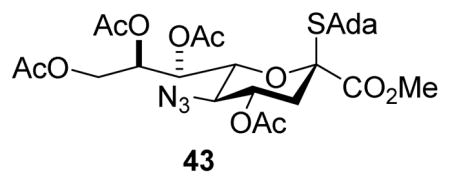
This change in selectivity may arise from the 9-deoxy nature of the side chain in 5 rendering it less disarming and so supportive of a greater degree of oxocarbenium-like character in the transition state for glycosylation. Alternatively, the 7-deoxy-7-azido substitution may be responsible for this change in selectivity albeit the acetoxy and azido groups have comparable electron-withdrawing ability as judged by their Hammett parameters.39 The side chain conformation of both donors 5 and 4334 are similar, as judged by the magnitude of their respective 3J6,7 coupling constants (Table 2), corresponding to the typical extended gg-conformation of the D-glycero-D-galacto-configured sialic acids,28,40–45 and suggesting that the selectivity difference does not arise from a simple conformational difference.
Table 2.
Key Spectral Parameters and Approximate Side Chain Conformations of 5, 43, 13, and 15.
| Cmpd | Key NOE Contacts | 3J6,7 (Hz) | C6-C7 Conformation |
|---|---|---|---|
| 5 | H8-H6 | 1.4 |
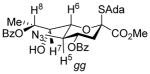
|
| 43 | H8-H6, H7-H5 | 2.2 |
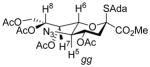
|
| 13 | H8-H6, H7-H5 | ≤1 |
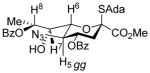
|
| 15 | H8-H5, H6-H9 | 2.9 |
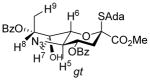
|
Influence of Configuration at C7 on Side Chain Conformation
The synthetic scheme adopted for the preparation of donor 5 (Scheme 1) provides a further opportunity to evaluate the influence of configuration at C7 on the side chain conformation. To this end, key spectral parameters and the proposed conformations of the C7 epimers 13 and 15, and of donor 5 are presented in Table 2. Thus, compounds 5 and 13 that retain the D-glycero-D-galacto configuration of N-acetyl neuraminic acid very predominantly adopt the gg-conformation, whereas 15, which differs from 13 only in configuration at C7 (overall D-glycero-L-altro configuration) adopts a predominantly gt conformation about the exocyclic bond consistent with earlier studies.19,28,46
Conclusion
A simple legionaminic acid thioglycoside in which both amines are protected in the form of azides was prepared in fifteen steps and good overall yield from readily available N-acetylneuraminic acid. This thioglycoside serves as an effective donor for coupling to a range of primary, secondary, and tertiary alcohols with which it gives moderate to excellent equatorial selectivity. Successful stereoselective coupling to the neuraminic acid-based acceptor 26 is especially felicitous as the linkage obtained closely resembles that in the α-(2→4)-polylegionaminic acid from the lipopolysaccharide of the Legionella pneumophila virulence factor. A simple deprotection sequence then affords the legionaminic acid glycosides themselves in the form of the diacetamides. Further work will explore the influence of alternative protecting group strategies on anomeric selectivity and the ability to differentially functionalize the two amines.
Experimental Part
General Experimental
Solvents, reagents and commercially available starting materials were used without further purification. All solvents were dried according to standard methods. All reactions were performed under an atmosphere of dry nitrogen or argon. Reactions were monitored using pre-coated glass TLC plates (Silica Gel HL TLC Plates w/UV254) with visualization with UV light (254 nm) and/or heating with cerium ammonium molybdate solution [(Ce(SO4)2 (5 g); (NH4)6Mo7O24·4H2O (25 g); 1N H2SO4 (50 mL); H2O (450 mL)]. Purifications were performed by column chromatography over silica gel (230–400 Mesh, Grade 60, 40–63 μm). HPLC purifications were performed using a ZORBAX RX-SIL column (5 μm, 9.4 x 250 mm) with a flow rate of 6.2 mL/min. 1H and 13CC NMR spectra were recorded at 600 MHz and 300 K. Residual solvent peaks were used as an internal reference. Assignments of the signals on the 1H and 13CC NMR spectra were made by first-order analysis using iNMR software and were verified by COSY, HSQC and HMBC experiments. Specific rotations were measured with an Automatic Polarimeter with a path length of 1 dm and have units of deg cm2 g−1. High resolution mass spectra were recorded with Micromass LCT Premier XE (Waters) instrument using an electrospray source coupled with a time-of-flight mass analyzer.
Methyl (1-adamantanyl 3,5-dideoxy-5-N-(1,1-dimethylethoxy)carbonyl-2-thio-D-glycero-β-D-galacto-non-2-ulopyranosid)onate (7)
Compound 7 was obtained by a literature procedure21 from N-acetylneuraminic acid in 65% overall yield as an off-white foam.
Methyl (1-adamantanyl 3,5-dideoxy-5-N-(1,1-dimethylethoxy)carbonyl-9-O-((2,4,6-triisopropylphenyl)sulfonyl)oxy-2-thio-D-glycero-β-D-galacto-non-2-ulopyranosid)onate (8)
Compound 7 (0.95 g, 1.78 mmol) was dissolved in anhydrous pyridine (15 mL) and 2,4,6-triisopropylbenzenesulfonyl chloride (3.23 g, 10.68 mmol, 6 eq) was added portion-wise. The mixture was stirred at room temperature for 20 h and the reaction was monitored by TLC (hexane/ethyl acetate 1:1). After completion, the reaction was quenched by addition of methanol (0.43 mL, 10.68 mmol) and the mixture was concentrated in vacuo to dryness. The residue was adsorbed on silica gel and purified by flash column chromatography (hexane/ethyl acetate 3:2) to give compound 8 (1.01 g, 71%) as a white foam. [α]25 D −50.0 (c 1.5, CHCl3); 1H NMR (600 MHz, CDCl3) δ 7.17 (s, 2H), 4.74 (d, J = 8.5 Hz, 1H), 4.50 (dd, J = 10.6, 2.0 Hz, 1H), 4.23-4.19 (m, 2H), 4.13-4.06 (m, 3H), 4.01 (s, 1H), 3.77 (s, 3H), 3.72 (d, J = 6.5 Hz, 1H), 3.61 (t, J = 8.1 Hz, 1H), 3.54 (q, J = 9.5 Hz, 1H), 2.93-2.86 (m, 2H), 2.63 (d, J = 4.9 Hz, 1H), 2.58 (dd, J = 13.7, 4.3 Hz, 1H,), 1.99-1.90 (m, 6H), 1.86 (d, J = 11.7 Hz, 1H), 1.81 (t, J = 12.6 Hz, 1H), 1.63 (s, 6H), 1.40 (s, 9H), 1.25 (m, 18H); 13CC NMR (151 MHz, CDCl3) δ 170.7, 157.6, 153.9, 150.9, 128.9, 123.8, 86.0, 81.0, 72.2, 71.6, 69.7, 68.7, 67.9, 54.2, 52.7, 50.2, 43.2, 42.9, 36.0, 34.2, 29.8, 29.6, 28.2, 24.7, 24.7, 23.5, 23.5; HRMS (ESI) m/z calcd for: C40H63NO11S2Na, [M+Na]+ 820.3740; found: 820.3735.
Methyl (1-adamantanyl 3,5,9-trideoxy-9-iodo-5-N-(1,1-dimethylethoxy)carbonyl-2-thio-D-glycero-β-D-galacto-non-2-ulopyranosid)onate (9)
Compound 8 (0.49 g, 0.62 mmol) was dissolved in anhydrous acetone (4 mL) and sodium iodide (0.93 g, 6.20 mmol, 10 eq) was added to the mixture. The reaction was heated at 40 °C with stirring for 17 h and the progress was monitored by TLC (hexane/ethyl acetate 2:3). After completion, the mixture was directly adsorbed on silica gel and purified by flash column chromatography (hexane/ethyl acetate 2:3) to give compound 9 (0.37 g, 94%) as an off-white foam. [α]25 D −89.1 (c 3.05, CHCl3); 1H NMR (600 MHz, CDCl3) δ 4.89 (d, J = 8.6 Hz, 1H, ), 4.17 (d, J = 10.2 Hz, 1H), 4.01 (d, J = 7.3 Hz, 1H), 3.99-3.96 (m, 1H), 3.79 (s, 3H), 3.76 (dd, J = 10.2, 1.9 Hz, 1H), 3.64-3.59 (m, 2H), 3.58-3.52 (m, 2H), 3.24 (br s, 1H), 2.56 (dd, J = 13.5, 4.0 Hz, 1H), 2.29 (d, J = 6.0 Hz, 1H), 1.98 (s, 3H), 1.94 (d, J = 11.9 Hz, 3H), 1.91-1.83 (m, 4H), 1.63 (s, 6H), 1.43 (s, 9H); 13CC NMR (151 MHz, CDCl3) δ 170.9, 157.4, 86.1, 81.0, 72.1, 72.0, 69.5, 67.7, 54.2, 52.8, 50.2, 43.3, 42.8, 36.0, 29.8, 28.4, 17.0; HRMS (ESI) m/z calcd for: C25H40INO8SNa, [M+Na]+ 664.1417; found: 664.1434.
Methyl (1-adamantanyl 4,8-di-O-benzoyl-3,5,9-trideoxy-9-iodo-5-N-(1,1-dimethylethoxy)carbonyl-2-thio-D-glycero-β-D-galacto-non-2-ulopyranosid)onate (10)
Compound 9 (0.60 g, 0.94 mmol) was dissolved in anhydrous pyridine (15 mL). The solution was cooled to 0 °C and benzoyl chloride (0.38 mL, 3.27 mmol, 3.5 eq) was added dropwise. The mixture was stirred at 0 °C for 1 h and the reaction was monitored by TLC (hexane/ethyl acetate 3:2). After completion, the mixture was diluted with ethyl acetate and washed with sat. NaHCO3, 1 N HCl, brine, dried over Na2SO4, filtered, concentrated, co-evaporated with toluene and dried. The crude material was purified by flash column chromatography (hexane/ethyl acetate 78:22) to give 10 (0.72 g, 91%) as a white foam. [α]23 D −25.8 (c 1.8, CHCl3); 1H NMR (600 MHz, CDCl3) δ 8.10 (d, J = 7.3 Hz, 2H), 8.00 (d, J = 7.4 Hz, 2H), 7.60-7.56 (m, 2H), 7.47 (t, J = 7.8 Hz, 2H), 7.43 (t, J = 7.7 Hz, 2H), 5.61 (td, J = 11.2, 4.5 Hz, 1H), 4.95 (d, J = 8.7 Hz, 1H), 4.93-4.91 (m, 1H), 4.36 (br s, 1H,), 4.32 (dd, J = 10.4, 1.0 Hz, 1H), 4.26 (dd, J = 11.0, 2.4 Hz, 1H), 4.10 (d, J = 4.2 Hz, 1H), 3.92 (q, J = 9.6 Hz, 1H), 3.85 (dd, J = 11.0, 6.4 Hz, 1H), 3.82 (s, 3H), 2.74 (dd, J = 13.2, 4.5 Hz, 1H), 2.12 (dd, J = 13.2, 12.2 Hz, 1H), 1.93-1.89 (m, 6H), 1.74 (d, J = 11.4 Hz, 3H), 1.54 (d, J = 12.3 Hz, 3H), 1.46 (d, J = 12.0 Hz, 3H), 1.29 (s, 9H); 13CC NMR (151 MHz, CDCl3) δ 170.0, 166.6, 165.8, 157.3, 133.4, 133.3, 130.1, 129.9, 129.8, 129.8, 129.3, 128.4, 128.4, 86.0, 81.1, 74.3, 73.7, 70.2, 68.7, 52.8, 52.5, 50.6, 43.3, 40.4, 35.8, 29.7, 28.1,17.2; HRMS (ESI) m/z calcd for: C39H48INO10SNa, [M+Na]+ 872.1941; found: 872.1907.
Methyl (1-adamantanyl 5-azido-4,8-di-O-benzoyl-3,5,9-trideoxy-2-thio-D-glycero-β-D-galacto-non-2-ulopyranosid)onate (13)
A stirred solution of compound 10 (0.46 g, 0.54 mmol) and and tris(trimethylsilyl)silane (0.25 mL, 0.81 mmol, 1.5 eq) in deoxygenated benzene (10 mL) was heated to 60 °C and treated dropwise with a 0.1 M 2,2′-azobis(2-methylpropionitrile) solution in benzene (0.54 mL, 0.1 eq). The reaction was monitored by TLC (hexane/ethyl acetate 3:2). After full consumption of the starting material (1 h), the mixture was cooled down and concentrated in vacuo to dryness to give a crude preparation of compound 11. The residue was dissolved in anhydrous methanol (2.5 mL) and 2 M hydrogen chloride solution in diethyl ether (5 mL) was added. The mixture was stirred at room temperature for 2 h and then concentrated. The crude residue was dissolved in acetonitrile and the solution was extracted with hexanes to remove any non-polar impurities from the previous step. The acetonitrile layer was separated and concentrated to give a crude preparation of compound 12 that was taken up in a mixture of acetonitrile and water (4:1, 10 mL) and the mixture was cooled to 0 °C. Then triethylamine (0.23 mL, 1.62 mmol, 3 eq), copper (II) sulfate (9 mg, 0.05 mmol, 0.1 eq) and imidazole-1-sulfonyl azide hydrochloride25 (0.17 g, 0.81 mmol, 1.5 eq) were added to the reaction mixture. The mixture was allowed to warm to room temperature and was stirred for 8 h with reaction progress being monitored by TLC (hexane/ethyl acetate 3:2). After full consumption of the starting material, the mixture was diluted with ethyl acetate and was washed with 1 N HCl. The aqueous phase was then washed with ethyl acetate and combined organic layers were dried over Na2SO4, filtered, concentrated and dried. The crude material was purified by flash column chromatography (hexane/ethyl acetate 7:3) to give 13 (0.28 g, 78% over 3 steps) as a white foam. [α]23 D −49.0 (c 1.6, CHCl3); 1H NMR (600 MHz, CDCl3) δ 8.05-8.01 (m, 4H), 7.59-7.54 (m, 2H), 7.48-7.42 (m, 4H), 5.65 (ddd, J = 11.7, 10.0, 4.7 Hz, 1H), 4.99 (dq, J = 8.6, 6.1 Hz, 1H), 4.34 (d, J = 10.1 Hz, 1H), 4.03 (d, J = 8.6 Hz, 1H), 3.91 (t, J = 10.1 Hz, 1H), 3.77 (s, 3H), 2.83 (dd, J = 13.4, 4.7 Hz, 1H), 2.77 (br s, 1H), 1.91 (dd, J = 13.4, 11.7 Hz, 1H), 1.84 (s, 3H), 1.76 (dd, J = 11.7, 1.5 Hz, 3H), 1.62 (dd, J = 11.7, 1.2 Hz, 3H), 1.59 (d, J = 6.1 Hz, 3H), 1.50 (d, J = 12.4 Hz, 3H), 1.36 (d, J = 11.8 Hz, 3H); 13CC NMR (151 MHz, CDCl3) δ 170.3, 165.4, 165.2, 133.4, 133.1, 130.2, 129.7, 129.6, 129.3, 128.5, 128.5, 85.6, 72.3, 71.2, 71.1, 70.5, 60.8, 52.8, 50.3, 43.2, 39.7, 35.7, 29.6, 17.2; HRMS (ESI) m/z calcd for: C34H39N3O8SNa, [M+Na]+ 672.2356; found: 672.2335.
Methyl (1-adamantanyl 5-azido-4,8-di-O-benzoyl-3,5,9-trideoxy-7-oxo-2-thio-D-glycero-β-D-galacto-non-2-ulopyranosid)onate (14)
Compound 13 (0.11 g, 0.17 mmol) was dissolved in anhydrous dichloromethane (2 mL)and the solution was cooled to 0 °C and Dess-Martin periodinane (108 mg, 0.26 mmol, 1.5 eq) was added. The mixture was stirred at room temperature for 4 h and the reaction was monitored by TLC (hexane/ethyl acetate 85:15). After completion, the mixture was diluted with diethyl ether and 20% aqueous Na2S2O3 solution was added. The organic layer was then washed with brine, dried over Na2SO4, filtered, concentrated and dried. The crude material was purified by flash column chromatography (hexane/ethyl acetate 9:1) to give 14 (0.094 g, 85%) as a white foam. [α]25 D −76.2 (c 2.05, CHCl3); 1H NMR (600 MHz, CDCl3) δ 8.09-8.07 (m, 2H), 8.02-8.00 (m, 2H), 7.58-7.55 (m, 2H), 7.46-7.42 (m, 4H), 5.57 (q, J = 6.8 Hz, 1H), 5.47 (ddd, J = 11.2, 9.7, 4.8 Hz, 1H), 5.03 (d, J = 9.8 Hz, 1H), 3.84 (t, J = 9.8 Hz, 1H), 3.79 (s, 3H), 2.80 (dd, J = 13.7, 4.8 Hz, 1H), 2.03 (d, J = 11.7 Hz, 3H), 1.99-1.94 (m, 4H), 1.91 (d, J = 11.7 Hz, 3H), 1.70 (d, J = 6.8 Hz, 3H), 1.63 (s, 6H); 13CC NMR (151 MHz, CDCl3) δ 202.6, 169.8, 166.0, 165.2, 133.4, 133.4, 129.9, 129.7, 129.3, 129.1, 128.5, 128.4, 86.2, 73.7, 73.5, 70.6, 60.9, 52.8, 50.7, 43.3, 39.1, 35.9, 29.8, 16.2; HRMS (ESI) m/z calcd for: C34H37N3O8SNa, [M+Na]+ 670.2199; found: 670.2187.
Methyl (1-adamantanyl 5-azido-4,8-di-O-benzoyl-3,5,9-trideoxy-2-thio-D-glycero-β-L-altro-non-2-ulopyranosid)onate (15)
A solution of compound 14 (0.172 g, 0.265 mmol) in dichloromethane (2.25 mL) was cooled to −78 °C and a solution of cerium(III) chloride heptahydrate (0.30 g, 0.79 mmol, 3 eq) in methanol (4.75 mL) was added. After 1 h sodium borohydride (0.015 g, 0.40 mmol) was added and the reaction was monitored by TLC (hexane/ethyl acetate 7:3). After completion, the mixture was quenched with sat aqueous NH4Cl, warmed to room temperature and concentrated. The residue was diluted with ethyl acetate, washed with water and brine dried over Na2SO4, filtered, concentrated and dried. The crude material was purified by flash column chromatography (hexane/ethyl acetate 3:1) to give 15 (0.14 g, 82%) as a white foam and the recovered epimer 13 (0.024 g; 14%). [7agr;]22 D −68.4 (c 3.5, CHCl3); 1H NMR (600 MHz, CDCl3) δ 8.06 (dd, J = 8.2, 1.1 Hz, 2H), 8.01 (dd, J = 8.2, 1.1 Hz, 2H), 7.58-7.53 (m, 2H), 7.45-7.42 (m, 4H), 5.57 (ddd, J = 11.2, 9.4, 4.8 Hz, 1H), 5.53 (dt, J = 12.3, 6.3 Hz, 1H), 4.38 (dd, J = 10.2, 2.9 Hz, 1H), 4.06 (t, J = 10.2 Hz, 1H), 4.04 (dd, J = 12.3, 2.9 Hz, 1H), 3.57 (s, 3H), 2.97 (br s, 1H), 2.72 (dd, J = 13.6, 4.8 Hz, 1H), 1.99-1.96 (m, 6H), 1.94 (dd, J = 13.6, 11.2 Hz, 1H), 1.83 (d, J = 10.3 Hz, 3H), 1.64-1.59 (m, 6H), 1.49 (d, J = 6.3 Hz, 3H); 13CC NMR (151 MHz, CDCl3) δ 170.2, 166.0, 165.2, 133.4, 133.0, 130.4, 129.7, 129.7, 129.3, 128.5, 128.3, 85.7, 75.2, 71.9, 71.8, 70.9, 60.3, 52.5, 50.4, 43.4, 39.1, 35.9, 29.8, 17.4; HRMS (ESI) m/z calcd for: C34H39N3O8SNa, [M+Na]+ 672.2356; found: 672.2347.
Methyl (1-adamantanyl 5,7-diazido-4,8-di-O-benzoyl-3,5,7,9-tetradeoxy-2-thio-D-glycero-β-D-galacto-non-2-ulopyranosid)onate (5)
Compound 15 (0.14 g, 0.21 mmol) was dissolved in anhydrous dichloromethane (2 mL), the solution was cooled to 0 °C and pyridine (170 μL, 2.10 mmol, 10 eq) was added followed by a drop-wise addition of triflic anhydride (106 μL, 0.63 mmol, 3 eq). The mixture was stirred at 0 °C for 1 h and the reaction was monitored by TLC (hexane/ethyl acetate 7:3). After completion, the mixture was diluted with dichloromethane and poured into ice-cold 1N HCl solution. The organic phase was washed with cold water, dried over Na2SO4, filtered and concentrated. The residue was dissolved in anhydrous dimethylformamide (4 mL) and the solution was cooled to 0 °C followed by addition of sodium azide (0.27 g, 4.20 mmol, 20 eq). The mixture was stirred at 0 °C for 16 h and the reaction was monitored by TLC (hexane/ethyl acetate 4:1). After completion, the mixture was diluted with ethyl acetate and washed with water. The organic phase was washed with brine, dried over Na2SO4, filtered, concentrated and dried. The crude material was purified by flash column chromatography (hexane/ethyl acetate 93:7) to give 5 (0.12 g, 81% over the two steps) as a white foam. [α]21 D −50.7 (c 2.9, CHCl3); 1H NMR (600 MHz, CDCl3) δ 8.06-8.02 (m, 4H), 7.59-7.56 (m, 2H), 7.47-7.43 (m, 4H), 5.65 (ddd, J = 11.7, 10.0, 4.7 Hz, 1H), 5.38 (quintet, J = 6.2 Hz, 1H), 4.47 (dd, J = 10.1, 1.4 Hz, 1H), 3.87 (t, J = 10.0 Hz, 1H), 3.85 (dd, J = 6.4, 1.4 Hz, 1H), 3.77 (s, 3H), 2.87 (dd, J = 13.4, 4.7 Hz, 1H), 1.98 (dd, J = 13.4, 11.7 Hz, 1H), 1.89-1.85 (m, 6H), 1.71 (d, J = 11.3 Hz, 3H), 1.68 (d, J = 6.2 Hz, 3H), 1.55 (d, J = 12.2 Hz, 3H), 1.44 (d, J = 12.2 Hz, 3H); 13CC NMR (151 MHz, CDCl3) δ 169.6, 165.3, 165.2, 133.5, 133.3, 129.8, 129.7, 129.7, 129.2, 128.5, 128.5, 85.8, 71.2, 71.0, 71.0, 64.1, 61.5, 52.7, 50.5, 43.2, 39.8, 35.8, 29.6, 17.2; HRMS (ESI) m/z calcd for: C34H38N6O7SNa, [M+Na]+ 697.2420; found: 697.2410.
Methyl (1-adamantanyl 5-azido-3,5,9-trideoxy-2-thio-D-glycero-β-D-galacto-non-2-ulopyranosid)onate (19)
Compound 9 (0.35 g, 0.55 mmol) was dissolved in methanol (15 mL) and Pearlman’s catalyst (1.55 g, 2.20 mmol) was added to the mixture. The flask was filled with hydrogen gas (45 psi) and the mixture was stirred at room temperature for 48 h until starting material was no longer present. The mixture was filtered through a pad of Celite and the filtrate was concentrated. The residue was dissolved in anhydrous methanol (7.5 mL) and 2 M hydrogen chloride solution in diethyl ether (5 mL) was added. The mixture was stirred at room temperature for 2 h and then concentrated. The residue was dissolved in a mixture of methanol and water (2:1, 6 mL). The mixture was cooled to 0 °C and potassium carbonate (0.23 g, 1.65 mmol, 3 eq), copper(II) sulfate (9 mg, 0.06 mmol, 0.1 eq) and imidazole-1-sulfonyl azide hydrochloride25 (0.19 g, 1.10 mmol, 2 eq) were added. The mixture was allowed to warm to room temperature and was stirred for 16 h with reaction progress being monitored by TLC. After consumption of the starting material, the mixture was slightly concentrated, acidified with 1 N HCl and diluted with ethyl acetate. The aqueous phase was then washed with ethyl acetate and the combined organic layers were dried over Na2SO4, filtered, concentrated and dried. The crude material was purified by flash column chromatography to give 19 (0.09 g, 39% over the three steps) as a slightly yellow oil. [α]22 D −185.5 (c 4.0, MeOH); 1H NMR (600 MHz, CD3OD) δ 4.22 (d, J = 10.4 Hz, 1H), 4.03 (ddd, J = 11.9, 9.6, 4.7 Hz, 1H), 3.81 (s, 3H), 3.80-3.76 (m, 1H), 3.42 (d, J = 8.9 Hz, 1H), 3.34 (t, J = 10.0 Hz, 1H), 2.42 (dd, J = 13.6, 4.8 Hz, 1H), 1.98-1.96 (m, 6H), 1.92 (d, J = 11.0 Hz, 3H), 1.74 (dd, J = 13.6, 11.9 Hz, 1H), 1.68 (s, 6H), 1.29 (d, J = 6.2 Hz, 3H); 13CC NMR (150 MHz, CD3OD) δ 172.4, 85.9, 74.2, 70.8, 67.9, 65.5, 64.1, 52.0, 49.5, 43.0, 42.6, 35.7, 29.9, 19.8; HRMS (ESI) m/z calcd for: C20H31N3O6SNa, [M+Na]+ 464.1831; found: 464.1836.
Methyl (1-adamantanyl 5-azido-7,8-di-O-benzoyl-3,5,9-trideoxy-2-thio-D-glycero-β-D-galacto-non-2-ulopyranosid)onate (20)
Compound 19 (0.08 g, 0.18 mmol) was dissolved in anhydrous pyridine (3 mL). The solution was cooled to 0 °C and benzoyl chloride (52 μL, 0.45 mmol, 2.5 eq) was added dropwise. The mixture was stirred at 0 °C for 1 h and the reaction was monitored by TLC. After completion, the mixture was diluted with dichloromethane and washed with sat. NaHCO3. The aqueous layer was extracted with dichloromethane and the combined organic layers were washed with water, brine, dried and concentrated. The crude material was purified by flash column chromatography to give 20 (0.05 g, 46%) as a slightly yellow oil. [α]23 D −27.2 (c 1.0, CHCl3); 1H NMR (600 MHz, CDCl3) δ 8.15 (dd, J = 8.2, 1.0 Hz, 2H), 8.02 (dd, J = 8.1, 0.9 Hz, 2H), 7.61-7.58 (m, 1H), 7.55-7.53 (m, 1H), 7.49-7.46 (m, 2H), 7.44-7.41 (m, 2H), 5.88 (dd, J = 6.0, 1.6 Hz, 1H), 5.43 (quintet, J = 6.0 Hz, 1H), 4.36 (dd, J = 9.8, 1.6 Hz, 1H), 4.23 (ddd, J = 11.8, 9.8, 4.7 Hz, 1H), 3.81 (s, 3H), 3.07 (t, J = 9.8 Hz, 1H), 2.52 (dd, J = 13.7, 4.7 Hz, 1H), 1.91-1.87 (m, 6H), 1.77-1.72 (m, 4H), 1.58-1.54 (m, 6H), 1.47 (d, J = 11.9 Hz, 3H); 13CC NMR (151 MHz, CDCl3) δ 170.2, 165.7, 165.6, 133.5, 133.1, 130.1, 130.1, 129.7, 129.4, 128.6, 128.4, 86.0, 72.8, 71.1, 70.9, 68.6, 64.2, 52.6, 50.3, 43.3, 42.5, 35.8, 29.7, 16.1; HRMS (ESI) m/z calcd for: C34H39N3O8SNa, [M+Na]+ 672.2356; found: 672.2387.
Methyl (methyl 5-azido-3,5-dideoxy-D-glycero-D-galacto-non-2-ulopyranosid)onate (22)
Methyl (methyl per-O-acetyl-N-acetyl-β-D-glycero-D-galacto-non-2-ulopyranosid)onate22 (5.05 g, 10 mmol) was dissolved in anhydrous THF (40 mL) and di-tert-butyl dicarbonate (23.0 mL, 100 mmol, 10 eq) and 4-(dimethylamino)pyridine (0.49 g, 4 mmol, 0.4 eq) were added to the mixture. The reaction mixture was heated to 60 °C with stirring for 22 h and the progress was monitored by TLC (hexane/ethyl acetate 3:2). After completion, the mixture was directly adsorbed on silica gel and purified by flash column chromatography (hexane/ethyl acetate 65:35) to give the N-acetyl-N-imide as an orange oil. The residue was dissolved in anhydrous methanol (65 mL) and a catalytic amount of sodium methoxide (0.27 g, 5 mmol, 0.5 eq) was added to the mixture. The mixture was stirred at room temperature for 3 h and then 2 M hydrogen chloride solution in diethyl ether (35 mL) was added. The mixture was stirred at room temperature for 5 h and then concentrated to give the free amine. The residue was dissolved in a mixture of acetonitrile and water (4:1, 150 mL), cooled to 0 °C and triethylamine (4.18 mL, 30 mmol, 3 eq), copper(II) sulfate (0.160 g, 1 mmol, 0.1 eq) and imidazole-1-sulfonyl azide hydrochloride25 (3.14 g, 15 mmol, 1.5 eq) were added. The mixture was allowed to warm to room temperature and was stirred for 15 h with reaction progress being monitored by TLC (chloroform/methanol 85:15). After full consumption of the starting material, the mixture was diluted with ethyl acetate and was washed with 1 N HCl. The aqueous phase was then washed with ethyl acetate and the combined organic layers were dried over Na2SO4, filtered, concentrated and dried. The crude material was purified by flash column chromatography (chloroform/methanol 9:1) to give 22 (0.98 g, 30% over the four steps) as a colorless oil. [α]22 D −61.1 (c 4.85, MeOH); 1H NMR (600 MHz, CD3OD) δ 3.97 (ddd, J = 11.3, 9.9, 5.0 Hz, 1H), 3.82 (dd, J = 11.3, 2.3 Hz, 1H), 3.80 (s, 3H), 3.76-3.73 (m, 1H), 3.73-3.70 (m, 2H), 3.66 (dd, J = 11.3, 5.0 Hz, 1H), 3.44 (t, J = 9.9 Hz, 1H), 3.22 (s, 3H), 2.28 (dd, J = 12.9, 5.0 Hz, 1H), 1.61 (dd, J = 12.9, 11.3 Hz, 1H); 13CC NMR (151 MHz, CD3OD) δ 169.8, 99.0, 70.5, 70.2, 69.0, 68.0, 63.8, 63.2, 52.0, 50.3, 39.9; HRMS (ESI) m/z calcd for: C11H19N3O8Na, [M+Na]+ 344.1070; found: 344.1067.
Methyl (methyl 5-azido-3,5-dideoxy-8,9-O-isopropylidene-D-glycero-D-galacto-non-2-ulopyranosid)onate (23)
Compound 22 (0.88 g, 2.75 mmol) was dissolved in anhydrous acetone (30 mL) and 2,2-dimethoxypropane (0.56 mL, 4.54 mmol, 1.65 eq) was added to the mixture followed by camphorsulfonic acid (22 mg, 0.1 mmol, 0.04 eq). The reaction was sturred at room temperature for 6 h and the progress was monitored by TLC (toluene/i-PrOH 9:1). Upon completion, the reaction was quenched by addition of triethylamine (0.2 mL). After 15 min the reaction mixture was concentrated and the residue was dissolved in ethyl acetate and washed with water and brine. The organic layer was dried over Na2SO4, concentrated and purified by flash column chromatography (toluene/i-PrOH 95:5) to give 23 (0.82 g, 83%) as a white foam.
[α]22 D −59.0 (c 4.2, CHCl3); 1H NMR (600 MHz, CDCl3) δ 4.14 (dt, J = 8.4, 5.4 Hz, 1H), 4.12-4.06 (m, 2H), 4.02 (dd, J = 8.4, 4.9 Hz, 1H), 3.96 (d, J = 3.5 Hz, 1H), 3.81 (s, 3H), 3.71-3.68 (m, 1H), 3.62 (d, J = 10.4 Hz, 2H), 3.48 (t, J = 10.0 Hz, 1H), 3.19 (s, 3H), 2.35 (dd, J = 12.9, 5.1 Hz, 1H), 1.63 (dd, J = 12.9, 11.6 Hz, 1H), 1.37 (s, 3H), 1.26 (s, 3H); 13CC NMR (151 MHz, CDCl3) δ 169.7, 109.3, 99.1, 74.5, 70.7, 70.7, 68.3, 67.7, 62.8, 53.1, 51.2, 39.9, 26.9, 25.3; HRMS (ESI) m/z calcd for: C14H23N3O8Na, [M+Na]+ 384.1383; found: 384.1389.
Methyl (methyl 5-azido-4-O-(tert-butyldimethylsilyl)-3,5-dideoxy-8,9-O-isopropylidene-D-glycero-D-galacto-non-2-ulopyranosid)onate (24)
Compound 23 (0.87 g, 2.4 mmol) and imidazole (0.36 mg, 5.28 mmol, 2.2 eq) were dissolved with stirring in anhydrous DMF (5 mL), the mixture was cooled to 0 °C and tert-butyldimethylsilyl chloride (0.40 mg, 2.64 mmol, 1.1 eq) was added. The mixture was allowed to warm to room temperature and the reaction progress was monitored by TLC (hexane/ethyl acetate 3:2). After 20 h DMF was removed in vacuo and the residue was dissolved in water and extracted with Et2O. The combined organic layers were dried over Na2SO4, concentrated and purified by flash column chromatography (hexane/ethyl acetate 7:3) to give 24 (1.04 g, 91%) as a white solid. [α]22 D −57.8 (c 2.25, CHCl3); 1H NMR (600 MHz, CDCl3) δ 4.13-4.09 (m, 2H), 4.06-4.01 (m, 2H), 3.79-3.75 (m, 4H), 3.59 (d, J = 10.2 Hz, 1H), 3.47 (t, J = 10.2 Hz, 1H), 3.20 (s, 3H), 2.48 (d, J = 10.3 Hz, 1H), 2.23 (dd, J = 13.1, 5.1 Hz, 1H), 1.65 (dd, J = 13.1, 11.1 Hz, 1H), 1.39 (s, 3H), 1.28 (s, 3H), 0.88 (s, 9H), 0.15 (s, 3H), 0.09 (s, 3H); 13CC NMR (151 MHz, CDCl3) δ 168.5, 109.2, 99.1, 74.9, 70.7, 70.5, 69.3, 67.5, 63.6, 52.7, 51.0, 40.7, 26.9, 25.6, 25.4, 17.8, -4.6, -5.1; HRMS (ESI) m/z calcd for: C20H37N3O8SiNa, [M+Na]+ 498.2248; found: 498.2254.
Methyl (methyl 7-O-acetyl-5-azido-4-O-(tert-butyldimethylsilyl)-3,5-dideoxy-8,9-O-isopropylidene-D-glycero-D-galacto-non-2-ulopyranosid)onate (25)
Compound 24 (1.04 g, 2.18 mmol) was dissolved in anhydrous DCM (10 mL), the mixture was cooled to 0°C and pyridine (3.53 mL, 44 mmol, 20 eq), acetic anhydride (2.06 mL, 22 mmol, 10 eq) and a catalytic amount of DMAP were added with stirring. The reaction mixture was allowed to warm to room temperature and the progress was monitored by TLC (hexane/ethyl acetate 4:1). After 7 h the mixture was concentrated in vacuo and the residue was purified by flash column chromatography (hexane/ethyl acetate 85:15) to give 25 (1.11 g, 98%) as a colorless oil. [α]20 D −22.1 (c 2.25, CHCl3); 1H NMR (600 MHz, CDCl3) δ 5.39 (dd, J = 7.3, 1.5 Hz, 1H), 4.32 (dt, J = 7.3, 6.1 Hz, 1H), 4.04-4.01 (m, 1H), 4.01-3.98 (m, 1H), 3.87 (dd, J = 8.7, 6.1 Hz, 1H), 3.79 (s, 3H), 3.62 (dd, J = 10.6, 1.5 Hz, 1H), 3.21 (s, 3H), 3.01 (dd, J = 10.6, 9.1 Hz, 1H), 2.24 (dd, J = 13.2, 5.1 Hz, 1H), 2.17 (s, 3H), 1.66 (dd, J = 13.1, 11.1 Hz, 1H), 1.37 (s, 3H), 1.29 (s, 3H), 0.88 (s, 9H), 0.14 (s, 3H), 0.09 (s, 3H); 13CC NMR (151 MHz, CDCl3) δ 170.0, 168.0, 109.2, 99.0, 73.7, 70.8, 70.1, 69.5, 66.6, 64.2, 52.6, 51.1, 40.5, 26.7, 25.6, 25.5, 20.9, 17.8, −4.7, −5.1; HRMS (ESI) m/z calcd for: C22H39N3O9SiNa, [M+Na]+ 540.2353; found: 540.2354.
Methyl (methyl 7-O-acetyl-5-azido-3,5-dideoxy-8,9-O-isopropylidene-D-glycero-D-galacto-non-2-ulopyranosid)onate (26)
Compound 25 (1.11 g, 2.14 mmol) was dissolved in anhydrous THF (25 mL) and 1.0 M tetrabutylammonium fluoride solution in THF (4.28 mL, 2 eq) was added. When TLC (hexane/ethyl acetate 1:1) showed complete conversion, the volatiles were removed in vacuo and the residue was purified by flash column chromatography (hexane/ethyl acetate 3:2) to give 26 (0.85 g, 98%) as a colorless oil. [α]21 D −27.5 (c 1.65, CHCl3); 1H NMR (600 MHz, CDCl3) δ 5.37 (dd, J = 7.2, 1.6 Hz, 1H), 4.33 (dt, J = 7.2, 6.1 Hz, 1H), 4.08 (ddd, J = 11.3, 9.4, 5.1 Hz, 1H), 4.03 (dd, J = 8.7, 6.1 Hz, 1H), 3.88 (dd, J = 8.7, 6.1 Hz, 1H), 3.79 (s, 3H), 3.66 (dd, J = 10.5, 1.6 Hz, 1H), 3.22 (s, 3H), 3.07 (dd, J = 10.5, 9.4 Hz, 1H), 2.55 (br s, 1H), 2.38 (dd, J = 13.1, 5.1 Hz, 1H), 2.16 (s, 3H), 1.70 (dd, J = 13.1, 11.3 Hz, 1H), 1.37 (s, 3H), 1.29 (s, 3H); 13CC NMR (151 MHz, CDCl3) δ 170.1, 167.9, 109.2, 98.8, 73.7, 70.6, 70.0, 68.8, 66.6, 63.4, 52.7, 51.2, 39.6, 26.6, 25.5, 20.8; HRMS (ESI) m/z calcd for: C16H25N3O9Na, [M+Na]+ 426.1488; found: 426.1494.
Acid-washed Molecular Sieves
Molecular sieves (4 Å, 30 g) were soaked in 2 N HCl (200 mL) for 12 h, then filtered and washed with de-ionized water (300 mL). The resulting solid was dried at 254 °C under vacuum for 24 h to give acid-washed molecular sieves (23 g), which were used directly for glycosylation.
General protocol for glycosylation (GP)
A mixture of donor 5 (101 mg, 0.15 mmol), acceptor (0.18 mmol, 1.2 eq) and activated 4Å acid-washed molecular sieves (300 mg) in anhydrous dichloromethane/acetonitrile (2:1) (3 mL) was stirred for 2 h at room temperature. Then the mixture was cooled to −78 °C and was treated with N-iodosuccinimide (41 mg, 0.18 mmol, 1.2 eq) and trifluoromethanesulfonic acid (2 μL, 0.023 mmol, 0.15 eq). The reaction mixture was stirred at −78 °C until completion and then quenched with triethylamine (25 μL). The mixture was diluted with dichloromethane, filtered through Celite, washed with 20% aqueous Na2S2O3, dried over Na2SO4 and concentrated under reduced pressure. The residue was adsorbed on silica gel and was purified by flash column chromatography to give the desired glycosylation products.
Methyl [1-benzyl (5,7-diazido-4,8-di-O-benzoyl-3,5,7,9-tetradeoxy-D-glycero-α-D-galacto-non-2-ulopyranosid)onate] (30)
Glycosylation of benzyl alcohol ((8.28 μL, 0.18 mmol, 1.2 eq) with 5 (100 mg, 0.15 mmol) was performed according to general procedure GP at −78 °C for 6 h to afford after flash column chromatography (hexane/ethyl acetate 80:20) compound 30 as the only product as colorless oil (87 mg, 96 %). [α]19 D −21.6 (c 1.0, CHCl3); 1H NMR (600 MHz, CDCl3) δ 8.05 (t, J = 7.8 Hz, 7H), 7.67-7.55 (m, 4H), 7.45 (t, J = 7.5 Hz, 4H), 5.63 (m, 1H), 5.17 (m, 1H), 4.82 (d, J = 11.4 Hz, 1H), 4.45 (d, J = 11.4 Hz, 1H), 4.05 (d, J = 10.3 Hz, 1H), 3.97 (t, J = 10.0 Hz, 1H), 3.71 (d, J = 8.5 Hz, 1H), 3.41 (s, 3H), 2.96 (dd, J = 13.0, 4.8 Hz, 1H), 2.02 (t, J = 12.4 Hz, 1H), 1.61 (d, J = 6.3 Hz, 3H); 13CC NMR (150 MHz, CDCl3) δ 167.9 (3JC1,H3ax = 7.2 Hz), 165.2, 165.1, 136.8, 133.6, 133.2, 133.1, 129.7, 129.1, 128.4, 128.3, 128.0, 127.8, 98.7, 72.9, 71.4, 68.6, 66.9, 63.4, 61.02, 52.57, 37.71, 18.5; HRMS (ESI) m/z calcd for: C31H30N6O8Na, [M+Na]+ 637.2023; found: 637.2047.
Methyl [methyl (5,7-diazido-4,8-di-O-benzoyl-3,5,7,9-tetradeoxy-D-glycero-α-D-galacto-non-2-ulopyranosid)onate]-(2→6)-2,3,4-tri-O-benzyl-β-D-galactopyranoside (31α) and Methyl [methyl (5,7-diazido-4,8-di-O-benzoyl-3,5,7,9-tetradeoxy-D-glycero-β-D-galacto-non-2-ulopyranosid)onate]-(2→6)-2,3,4-tri-O-benzyl-β-D-galactopyranoside (31β)
Glycosylation of acceptor 27 (42 mg, 0.09 mmol, 1.2 eq) with donor 5 (51 mg, 0.075 mmol) was performed according to general procedure GP at −78 °C for 6 h to afford after flash column chromatography (cyclohexane/ethyl acetate 85:15) 31α (major isomer) and 31β (minor isomer) as slightly yellow oils (63 mg, 87% overall, α:β 6.7:1, separated after column chromatography).
31α: [α]21 D −3.4 (c 2.5, CHCl3); 1H NMR (600 MHz, CDCl3) δ 8.07-8.03 (m, 4H), 7.62-7.57 (m, 2H), 7.49-7.45 (m, 4H), 7.37-7.32 (m, 7H), 7.31-7.29 (m, 3H), 7.28-7.25 (m, 4H), 7.23-7.20 (m, 1H), 5.52 (dq, J = 8.2, 6.3 Hz, 1H), 5.15 (ddd, J = 12.0, 9.9, 4.9 Hz, 1H), 4.93 (d, J = 11.4 Hz, 1H), 4.88 (d, J = 11.0 Hz, 1H), 4.74 (m, 2H), 4.70 (d, J = 11.8 Hz, 1H), 4.61 (d, J = 11.4 Hz, 1H), 4.29 (d, J = 7.7 Hz, 1H), 3.95 (dd, J = 9.9, 1.5 Hz, 1H), 3.90 (t, J = 9.9 Hz, 1H), 3.87-3.84 (m, 2H), 3.79 (dd, J = 9.7, 7.7 Hz, 1H), 3.72 (dd, J = 8.2, 1.5 Hz, 1H), 3.59 (dd, J = 9.2, 8.0 Hz, 1H), 3.55 (s, 3H), 3.53-3.51 (m, 2H), 3.34 (s, 3H), 2.89 (dd, J = 12.9, 4.9 Hz, 1H), 1.91 (dd, J = 12.9, 12.0 Hz, 1H), 1.59 (d, J = 6.3 Hz, 3H); 13CC NMR (151 MHz, CDCl3) δ 167.5 (3JC1,H3ax = 6.4 Hz), 165.2, 165.0, 138.8, 138.7, 138.5, 133.6, 133.2, 130.0, 129.8, 129.7, 129.1, 128.6, 128.5, 128.3, 128.2, 128.1, 128.1, 127.9, 127.5, 127.4, 127.3, 104.9, 99.0, 82.1, 79.5, 75.1, 74.3, 73.2, 72.9, 72.9, 72.8, 71.5, 68.8, 63.4, 63.0, 60.9, 57.1, 52.7, 37.3, 17.9; HRMS (ESI) m/z calcd for: C52H54N6O13Na, [M+Na]+ 993.3647; found: 993.3619.
31β: [α]19 D −9.6 (c 0.4, CHCl3); 1H NMR (600 MHz, CDCl3) δ 8.05 (d, J = 7.5 Hz, 2H), 7.99 (d, J = 7.5 Hz, 2H), 7.59 (t, J = 7.3 Hz, 1H), 7.53 (t, J = 7.4 Hz, 1H), 7.47 (t, J = 7.6 Hz, 2H), 7.39-7.26 (m, 16H), 7.18 (t, J = 7.0 Hz, 1H), 5.45 (ddd, J = 11.3, 10.1, 4.9 Hz, 1H), 5.35 (quintet, J = 6.2 Hz, 1H), 4.98 (d, J = 11.5 Hz, 1H), 4.87 (d, J = 11.0 Hz, 1H), 4.76-4.70 (m, 3H), 4.56 (d, J = 11.5 Hz, 1H), 4.17 (d, J = 7.6 Hz, 1H), 3.93 (t, J = 10.1 Hz, 1H), 3.77-3.74 (m, 2H), 3.73-3.68 (m, 5H), 3.59 (dd, J = 9.4, 6.5 Hz, 1H), 3.52 (s, 3H), 3.44-3.38 (m, 3H), 2.76 (dd, J = 13.0, 4.9 Hz, 1H), 1.89 (dd, J = 13.0, 11.3 Hz, 1H), 1.54 (d, J = 6.2 Hz, 3H); 13CC NMR (151 MHz, CDCl3) δ 167.1 (3JC1,H3ax = 0.0 Hz), 165.3, 165.3, 138.8, 138.7, 138.5, 133.5, 133.3, 129.7, 129.7, 129.6, 129.2, 128.6, 128.3, 128.2, 128.1, 128.1, 127.8, 127.5, 127.4, 127.3, 104.6, 98.9, 82.2, 79.3, 75.0, 74.6, 74.1, 73.0, 72.5, 71.2, 71.1, 70.8, 63.4, 63.2, 60.9, 56.9, 52.9, 36.9, 17.2; HRMS (ESI) m/z calcd for: C52H54N6O13Na, [M+Na]+ 993.3647; found: 993.3662.
Methyl [methyl (5,7-diazido-4,8-di-O-benzoyl-3,5,7,9-tetradeoxy-D-glycero-α-D-galacto-non-2-ulopyranosid)onate]-(2→4)-methyl 7-O-acetyl-5-azido-3,5-dideoxy-8,9-O-isopropylidene-D-glycero-D-galacto-non-2-ulopyranoside (32α) and Methyl [methyl (5,7-diazido-4,8-di-O-benzoyl-3,5,7,9-tetradeoxy-D-glycero-β-D-galacto-non-2-ulopyranosid)onate]-(2→4)-methyl 7-O-acetyl-5-azido-3,5-dideoxy-8,9-O-isopropylidene-D-glycero-D-galacto-non-2-ulopyranoside (32β)
Glycosylation of acceptor 26 (53.7 mg, 0.13 mmol, 1.2 eq) with 5 (75 mg, 0.11 mmol) was performed according to general procedure GP at −78 °C for 6 h to afford after flash column chromatography over silica gel (hexane/ethyl acetate 4:1) 32α (major isomer) and 32β (minor isomer) as colorless oils (82.8 mg, 82% overall, α:β 4.4 : 1, separated after column chromatography).
32α: [α]21 D 36.4 (c 1.0, CHCl3);1H NMR (600 MHz, CDCl3) δ 8.04 (dd, J = 12.8, 4.8 Hz, 4H), 7.58 (m, 2H), 7.46 (t, J = 7.7 Hz, 4H), 5.55 (dq, J = 12.8, 6.3 Hz, 1H), 5.46 (dd, J = 6.7, 1.4 Hz, 1H), 5.15 (ddd, J = 11.8, 10.0, 4.7 Hz, 1H), 4.49 (ddd, J = 11.2, 9.5, 5.0 Hz, 1H), 4.32 (q, J = 6.2 Hz, 1H), 4.04 (dd, J = 8.8, 6.1 Hz, 1H), 3.96 (t, J = 9.7 Hz, 1H), 3.90 (dd, J = 8.8, 6.1 Hz, 1H), 3.77 (d, J = 1.9 Hz, 1H), 3.76 (s, 3H), 3.70 (d, J = 6.6 Hz, 1H), 3.69 (dd, J = 10.6, 1.5 Hz, 1H), 3.42 (s, 3H), 3.27 (s, 3H), 3.11 (t, J = 9.8 Hz, 1H), 2.91 (dd, J = 12.6, 4.7 Hz, 1H), 2.16 (s, 3H), 2.11 (dd, J = 12.3, 5.1 Hz, 1H), 1.95 (t, J = 12.3 Hz, 1H), 1.64 (dd, J = 12.4, 4.2 Hz, 1H), 1.59 (d, J = 6.3 Hz, 1H), 1.39 (s, 3H), 1.31 (s, 3H); 13CC NMR (151 MHz, CDCl3) δ 170.0, 167.7, 167.6, 165.2, 164.9, 133.6, 133.1, 130.3, 129.8, 129.6, 129.0, 128.6, 128.4, 109.1, 98.6, 98.2, 74.1, 71.5, 71.4, 70.5, 70.3, 69.8, 66.4, 61.2, 60.8, 52.9, 52.5, 51.1, 38.0, 36.6, 26.6, 25.5, 20.8, 17.8; HRMS (ESI) m/z calcd for: C40H47N9O16Na, [M+Na]+ 932.3038; found: 932.3030.
32β: [α]21 D 18.6 (c 1.0, CHCl3);1H NMR (400 MHz, CDCl3) δ 8.07 (m, 4H), 7.59 (t, J = 7.3 Hz, 2H), 7.41 (m, 4H), 5.51 (m, 2H), 5.38 (dd, J = 7.3, 1.4 Hz, 1H), 4.33 (dd, J = 12.8, 6.2 Hz, 1H), 4.17 (m, 1H), 4.05 (dd, J = 8.7, 6.0 Hz, 1H), 3.90 (m, 3H), 3.86 (s, 3H), 3.80 (s, 3H), 3.61 (dd, J = 10.6, 1.5 Hz, 1H), 3.30 (d, J = 6.3 Hz, 1H), 3.20 ( t, J = 9.8 Hz, 1H), 3.13 (s, 3H), 2.94 (dd, J = 13.3, 4.8 Hz, 1H), 2.56 (dd, J = 12.5, 5.0 Hz, 1H), 2.23 (s, 3H), 1.94 (t, J = 12.6 Hz, 1H), 1.78 (dd, J = 12.4, 4.1 Hz, 1H), 1.68 (d, J = 6.4 Hz, 1H), 1.39 (s, 3H), 1.31 (s, 3H); 13CC NMR (101 MHz, CDCl3) δ 170.1, 167.0, 166.3 (3JC1,H3ax = 0.0 Hz), 165.5, 165.2, 133.6, 133.3, 129.9, 129.8, 129.6, 129.1, 128.5, 128.4, 109.2, 100.0, 98.1, 76.7, 73.8, 73.6, 73.5, 72.2, 70.6, 66.7, 61.1, 60.9, 52.9, 52.7, 51.1, 38.8, 37.6, 26.7, 25.5, 20.1, 15.9; HRMS (ESI) m/z calcd for: C40H47N9O16Na, [M+Na]+ 932.3038; found: 932.3021.
Methyl [methyl (5,7-diazido-3,4,5,7,8,9-hexadeoxy-D-glycero-α-D-galacto-non-2-ulopyranosid)onate]-(2→3)-2,4,6-tri-O-benzyl-β-D-galactopyranoside (33α) and Methyl [methyl (5,7-diazido-4,8-di-O-benzoyl-3,5,7,9-tetradeoxy-D-glycero-β-D-galacto-non-2-ulopyranosid)onate]-(2→3)-2,6-di-O-benzyl-β-D-galactopyranoside (33β)
Glycosylation of acceptor 28 (83.6 mg, 0.18 mmol, 1.2 eq) and donor 5 (100 mg, 0.15 mmol) and was performed according to general procedure GP at −78 °C for 6 h to afford after flash column chromatography (hexane/ethyl acetate 4:1) an inseparable mixture of isomers (120.7 mg; Yield: 84 %). A solution of this mixture of isomers (120 mg; 0.12 mmol) in methanol (10 mL) was was treated with NaOMe (6.67 mg; 0.12 mmol). The resulting mixture was stirred at room temperature until completion. The reaction mixture was neutralized with Amberlyst-15 H+ ion exchange resin, filtered and evaporated. The residue was subjected to column chromatography on silica gel (hexane/ethyl acetate 97:3) to obtain compound 33α (major isomer) and compound 33β (minor isomer) as colorless oils (80.3 mg; 88%, 75% overall for 2 steps, α:β 4.5:1, separated after column chromatography).
33α: [α]21 D 18.2 (c 1.0, CHCl3);1H NMR (600 MHz, CDCl3) δ 7.56-7.17 (m, 15H), 4.86 (d, J = 11.2 Hz, 1H), 4.79 (d, J = 11.6 Hz, 1H), 4.64 (d, J = 11.2 Hz, 1H), 4.49 (dd, J = 11.7, 5.4 Hz, 2H), 4.37 ( d, J = 11.8 Hz, 1H), 4.26 ( d, J = 7.7 Hz, 1H), 4.05 (m, 1H), 3.95 ( dd, J = 9.9, 3.0 Hz, 1H), 3.77 (s, 3H), 3.61 (m, 7H), 3.53 (s, 3H), 3.45 (t, J = 9.8 Hz, 1H), 3.04 (dd, J = 9.0, 2.3 Hz, 1H), 2.49 (dd, J = 13.7, 4.6 Hz, 1H), 2.05 ( dd, J = 13.3, 10.9 Hz, 1H), 1.35 (d, J = 6.2 Hz, 3H). 13CC NMR (151 MHz, CDCl3) δ 168.9 (3JC1,H3ax = 7.2 Hz), 138.5, 138.2, 128.3, 128.2, 128.1, 127.9, 127.6, 127.5, 127.4, 127.3, 105.0, 100.4, 77.4, 76.7, 75.9, 74.9, 74.7, 73.3, 72.9, 69.9, 68.9, 65.8, 65.3, 63.4, 63.3, 57.0, 53.6, 37.6, 20.3; HRMS (ESI) m/z calcd for: C38H46N6O11Na, [M+Na]+ 762.3225; found: 762.3234.
33β: [α]21 D 11.6 (c 1.0, CHCl3); 1H NMR (600 MHz, CDCl3) δ 7.32 (m, 15H), 4.77 (d, J = 11.9 Hz, 1H), 4.72 (d, J = 11.2 Hz, 1H), 4.62 (d, J = 11.2 Hz, 1H), 4.59 (d, J = 11.9 Hz, 1H), 4.43 (d, J = 11.2 Hz, 1H), 4.38 (d, J = 11.2 Hz, 1H), 4.22 (d, J = 7.6 Hz, 1H), 4.17 (d, J = 2.2 Hz, 1H), 4.08 (d, J = 7.6 Hz, 1H), 4.17 (d, J = 2.2 Hz, 1H), 4.08 (d, J = 10.4 Hz, 1H), 4.05 (d, J = 2.5 Hz, 1H), 4.03 (d, J = 2.1 Hz, 1H), 3.67 (m, 3H), 3.53 (m, 2H), 3.49 (s, 3H), 3.38 (t, J = 10.0 Hz, 1H), 3.35 (s, 3H), 3.01 (d, J = 9.1 Hz, 1H), 2.60 (dd, J = 13.8, 4.8 Hz, 1H), 1.78 (dd, J = 13.7, 11.7 Hz, 1H), 1.13 (d, J = 6.1 Hz, 3H); 13CC NMR (151 MHz, CDCl3) δ 166.8 (3JC1,H3ax = 0.0 Hz), 138.8, 138.5, 137.2, 128.8, 128.6, 128.2, 127.5, 127.2, 105.5, 99.2, 76.8, 76.2, 75.2, 74.6, 73.7, 72.9, 71.4, 68.6, 68.1, 64.6, 63.8, 57.0, 52.3, 39.9, 21.8; HRMS (ESI) m/z calcd for: C38H46N6O11Na, [M+Na]+ 762.3225; found: 762.3116.
Methyl [methyl (5,7-diazido-4,8-di-O-benzoyl-3,5,7,9-tetradeoxy-D-glycero-α-D-galacto-non-2-ulopyranosid)onate]-(2→3)-2,6-di-O-benzyl-β-D-galactopyranoside (34α) and Methyl [methyl (5,7-diazido-4,8-di-O-benzoyl-3,5,7,9-tetradeoxy-D-glycero-β-D-galacto-non-2-ulopyranosid)onate]-(2→3)-2,6-di-O-benzyl-β-D-galactopyranoside (34β)
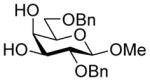
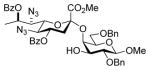
Glycosylation of acceptor 29 (67 mg, 0.18 mmol, 1.2 eq) with 5 (101 mg, 0.15 mmol) was performed according to general procedure GP at −78 °C for 6 h to afford after flash column chromatography (cyclohexane/ethyl acetate 4:1) 34α (major isomer) and a mixture separable by HPLC (gradient elution hexane/ethyl acetate from 95:5 to 7:3) of 34β (minor isomer) and 35 (α-glycoside (2→4)) as slightly yellow oils (116 mg, 88 %, α:β:(α(4-OH)) 4.7:1:(0.9), separated after column chromatography and HPLC).
34α: [α]22 D 15.3 (c 4.45, CHCl3); 1H NMR (600 MHz, CDCl3) δ 8.02-7.99 (m, 4H), 7.62-7.60 (m, 1H), 7.58-7.55 (m, 1H), 7.48-7.43 (m, 4H), 7.37-7.32 (m, 4H), 7.27-7.24 (m, 3H), 7.06-7.03 (m, 2H), 6.97-6.95 (m, 1H), 5.42 (dq, J = 8.6, 6.3 Hz, 1H), 5.27 (ddd, J = 11.8, 9.0, 5.0 Hz, 1H), 4.80 (d, J = 10.6 Hz, 1H), 4.64 (d, J = 11.9 Hz, 1H), 4.61 (d, J = 11.9 Hz, 1H), 4.51 (d, J = 10.6 Hz, 1H), 4.33 (d, J = 7.8 Hz, 1H), 4.05 (dd, J = 9.6, 3.3 Hz, 1H), 3.89 (s, 1H), 3.86-3.82 (m, 3H), 3.79 (dd, J = 10.2, 6.3 Hz, 1H), 3.72 (dd, J = 8.6, 1.6 Hz, 1H), 3.68 (t, J = 5.7 Hz, 1H), 3.58 (s, 3H), 3.53 (dd, J = 9.6, 7.8 Hz, 1H), 3.35 (s, 3H), 2.74 (dd, J = 13.3, 5.0 Hz, 1H), 2.61 (s, 1H), 2.05 (dd, J = 13.3, 11.8 Hz, 1H), 1.49 (d, J = 6.3 Hz, 3H); 13CC NMR (151 MHz, CDCl3) δ 167.8 (3JC1,H3ax = 7.0 Hz), 165.0, 164.8, 138.3, 138.1, 133.5, 133.3, 129.9, 129.8, 129.7, 129.1, 128.5, 128.5, 128.4, 128.2, 128.1, 127.6, 127.6, 127.5, 104.7, 99.2, 77.5, 75.3, 75.3, 73.6, 72.9, 72.7, 71.6, 69.6, 69.3, 68.4, 63.8, 60.7, 56.9, 53.0, 36.1, 18.1; HRMS (ESI) m/z calcd for: C45H48N6O13Na, [M+Na]+ 903.3177; found: 903.3167.
34β: [α]22 D 10.9 (c 0.65, CHCl3); 1H NMR (600 MHz, CDCl3) δ 8.05-8.04 (m, 4H), 7.60-7.55 (m, 2H), 7.47-7.43 (m, 4H), 7.38 (d, J = 7.5 Hz,), 7.30-7.27 (m, 6H), 7.25-7.20 (m, 2H), 5.62 (ddd, J = 11.4, 10.0, 4.5 Hz, 1H), 5.54-5.50 (m, 1H), 4.75 (d, J = 11.0 Hz, 1H), 4.66 (d, J = 11.0 Hz, 1H), 4.58-4.53 (m, 2H), 4.36 (dd, J = 10.0, 1.2 Hz, 1H), 4.24 (d, J = 7.6 Hz, 1H), 3.97-3.95 (m, 1H), 3.91-3.88 (m, 2H), 3.73 (dd, J = 9.5, 3.1 Hz, 1H), 3.70 (dd, J = 10.2, 5.4 Hz, 1H), 3.62 (dd, J = 10.2, 5.4 Hz, 1H), 3.57 (s, 3H), 3.52-3.50 (m, 4H), 3.45 (t, J = 5.4 Hz, 1H), 2.95 (dd, J = 13.2, 4.5 Hz, 1H), 2.80 (d, J = 5.1 Hz, 1H), 1.97 (dd, J = 13.2, 11.4 Hz, 1H), 1.54 (d, J = 6.3 Hz, 3H); 13CC NMR (151 MHz, CDCl3) δ 166.9 (3JC1,H3ax = 0 Hz), 165.4, 165.3, 138.6, 137.9, 133.5, 133.2, 129.9, 129.8, 129.7, 129.3, 128.5, 128.5, 128.3, 128.2, 128.1, 127.7, 127.6, 127.4, 104.7, 99.7, 78.5, 77.3, 75.1, 73.6, 72.8, 72.0, 71.6, 71.1, 69.4, 68.6, 64.0, 61.1, 57.0, 52.8, 36.8, 16.6; HRMS (ESI) m/z calcd for: C45H48N6O13Na, [M+Na]+ 903.3177; found: 903.3151.
Methyl [methyl (5,7-diazido-4,8-di-O-benzoyl-3,5,7,9-tetradeoxy-D-glycero-α-D-galacto-non-2-ulopyranosid)onate]-(2→4)-2,6-di-O-benzyl-β-D-galactopyranoside (35)
This compound was isolated as a minor byproduct from the glycosylation of 29 with 5 as described above. [α]21 D 22.2 (c 0.7, CHCl3); 1H NMR (600 MHz, CDCl3) δ 8.05 (d, J = 7.7 Hz, 2H), 8.00 (d, J = 7.7 Hz, 2H), 7.62-7.56 (m, 2H), 7.49-7.44 (m, 4H), 7.40 (d, J = 7.5 Hz, 2H), 7.33-7.30 (m, 6H), 7.27-7.23 (m, 2H), 5.45-5.41 (m), 5.14 (ddd, J = 11.9, 10.0, 4.7 Hz, 1H), 4.86 (d, J = 11.3 Hz, 1H), 4.77 (d, J = 11.3 Hz, 1H), 4.57 (d, J = 12.2 Hz, 1H), 4.52 (d, J = 12.2 Hz, 1H), 4.33 (d, J = 2.5 Hz, 1H), 4.25 (d, J = 7.6 Hz, 1H), 3.99 (d, J = 8.2 Hz, 1H), 3.90 (t, J = 10.0 Hz, 1H), 3.63 (d, J = 10.5 Hz, 1H), 3.61-3.58 (m, 2H), 3.52 (s, 3H), 3.51-3.45 (m, 2H), 3.40 (dd, J = 9.6, 7.6 Hz, 1H), 3.26 (s, 3H), 3.21 (d, J = 5.9 Hz, 1H), 3.05 (dd, J = 13.0, 4.7 Hz, 1H), 2.01 (dd, J = 13.0, 11.9 Hz, 1H), 1.58 (d, J = 6.2 Hz, 3H); 13CC NMR (150 MHz, CDCl3) δ 166.9 (3JC1,H3ax = 6.3 Hz), 165.2, 164.9, 138.8, 138.2, 133.6, 133.2, 130.0, 129.8, 129.6, 129.0, 128.6, 128.5, 128.4, 128.2, 128.1, 127.6, 127.5, 127.4, 104.7, 98.8, 79.8, 74.7, 73.5, 73.4, 73.1, 72.4, 72.2, 71.4, 69.8, 69.1, 64.4, 60.7, 57.1, 52.8, 37.8, 17.7; HRMS (ESI) m/z calcd for: C45H48N6O13Na, [M+Na]+ 903.3177; found: 903.3174.
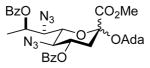
Methyl [1-adamantanyl (5,7-diazido-4,8-di-O-benzoyl-3,5,7,9-tetradeoxy-D-glycero-α-D-galacto-non-2-ulopyranosid)onate] (36α) and Methyl [1-adamantanyl (5,7-diazido-4,8-di-O-benzoyl-3,5,7,9-tetradeoxy-D-glycero-β-D-galacto-non-2-ulopyranosid)onate] (36β)
Glycosylation of 1-adamantanol (14 mg, 0.090 mmol, 1.2 eq) with 5 (51 mg, 0.075 mmol) was performed according to general procedure GP at −78 °C for 9 h to afford after flash column chromatography (hexane/acetone 95:5) compound 36α (major isomer) and compound 36β (minor isomer) as slightly yellow oils (36 mg, 73% overall, α:β 4.2:1, separated after column chromatography).
36α: [α]19 D −5.4 (c 1.3, CHCl3); 1H NMR (600 MHz, CDCl3) δ 8.04-8.02 (m, 4H), 7.59-7.56 (m, 2H), 7.47-7.44 (m, 4H), 5.51 (dq, J = 9.7, 6.2 Hz, 1H), 5.00 (ddd, J = 12.3, 10.2, 4.5 Hz, 1H), 4.29 (dd, J = 10.2, 1.7 Hz, 1H), 3.90 (t, J = 10.2 Hz, 1H), 3.63 (dd, J = 9.6, 1.7 Hz, 1H), 3.41 (s, 3H), 2.82 (dd, J = 12.7, 4.5 Hz, 1H), 2.10 (br s, 3H), 1.94 (t, J = 12.7 Hz, 1H), 1.79 (d, J = 2.4 Hz, 6H), 1.60 (d, J = 6.2 Hz, 3H), 1.59 (br s, 6H); 13CC NMR (151 MHz, CDCl3) δ 170.3 (3JC1,H3ax = 5.7 Hz), 165.2, 165.1, 133.5, 132.9, 130.3, 129.7, 129.6, 129.2, 128.5, 128.3, 97.9, 79.0, 72.7, 71.1, 68.3, 63.9, 61.2, 52.2, 43.2, 40.8, 36.0, 31.0, 18.5; HRMS (ESI) m/z calcd for: C34H38N6O8Na, [M+Na]+ 681.2649; found: 681.2632.
36β: [α]22 D −10.8 (c 0.4, CHCl3); 1H NMR (600 MHz, CDCl3) δ 8.05-8.03, 7.59-7.56 (m, 2H), 7.47-7.44 (m, 4H), 5.64 (ddd, J = 11.2, 10.1, 4.7 Hz, 1H), 5.42 (dq, J = 7.6, 6.2 Hz, 1H), 4.13 (dd, J = 10.1, 1.1 Hz, 1H), 3.88 (t, J = 10.1 Hz, 1H), 3.74 (s, 3H), 3.71 (dd, J = 7.6, 1.1 Hz, 1H), 2.78 (dd, J = 12.6, 4.7 Hz, 1H), 1.87 (br s, 3H), 1.72-1.67 (m, 7H), 1.65 (d, J = 6.2 Hz, 3H), 1.41 (d, J = 12.3 Hz, 3H), 1.29 (d, J = 11.8 Hz, 3H); 13CC NMR (150 MHz, CDCl3) δ 169.5 (3JC1,H3ax = 0.0 Hz), 165.4, 165.2, 133.4, 133.4, 129.7, 129.7, 129.4, 128.5, 128.5, 96.9, 78.2, 71.1, 71.0, 70.1, 63.7, 61.4, 52.4, 42.6, 40.3, 35.7, 30.7, 17.6; HRMS (ESI) m/z calcd for: C34H38N6O8Na, [M+Na]+ 681.2649; found: 681.2624.
Methyl [methyl (5,7-diazido-3,4,5,7,8,9-hexadeoxy-D-glycero-α-D-galacto-non-2-ulopyranosid)onate]-(2→6)-2,3,4-tri-O-benzyl-β-D-galactopyranoside (37)
To a solution of 31α (130 mg; 0.13 mmol) in methanol (15 mL) was added NaOMe (7.23 mg; 0.13 mmol). The resulting mixture was stirred at room temperature until complete conversion. The reaction mixture was neutralized with Amberlyst-15 H+ resin and filtered. The filtrate was evaporated and subjected to column chromatography over silica gel (Hexane/Ethyl acetate 6:4) to give 37 as colorless oil (89 mg; 87%).
[α]21 D 20.9 (c 1.0, CHCl3); 1H NMR: δ 7.28 (m, 15H), 4.97 (d, J = 11.6 Hz, 1H), 4.87 (d, J = 10.9 Hz, 1H), 4.75 (m, 2H), 4.71 ( d, J = 11.3 Hz, 1H), 4.58 (d, J = 11.6 Hz, 1H), 4.25 (d, J = 7.7 Hz, 1H), 4.12 (m, 1H), 3.83 (d, J = 2.7 Hz, 1H), 3.80 (m, 1H), 3.77 (t, J = 9.9 Hz, 1H), 3.61 (s, 3H), 3.59 (dd, J = 10.2, 5.4 Hz, 1H), 3.54 (s, 3H), 3.48 (m, 5H), 3.13 (dd, J = 9.1, 2.1 Hz, 1H), 2.68 (dd, J = 13.2, 4.7 Hz, 1H), 1.85 (dd, J = 12.8, 10.6 Hz, 1H), 1.40 (d, J = 6.2 Hz, 3H); 13CC NMR: δ 168.8, 138.7, 138.4, 128.3, 128.2, 128.1, 128.1, 127.6, 127.6, 127.5, 127.4, 127.3, 104.9, 98.8, 81.9, 79.5, 75.1, 74.2, 73.2, 73.1, 72.8, 72.6, 69.6, 65.8, 65.8, 63.8, 62.6, 57.1, 53.4, 39.4, 20.3; m/z calcd for: C38H46N6O11Na, [M+Na]+ 762.3225; found: 762.3241.
Methyl [methyl (5,7-diazido-3,4,5,7,8,9-hexadeoxy-D-glycero-α-D-galacto-non-2-ulopyranosid)onate]-(2→3)-2,6-di-O-benzyl-β-D-galactopyranoside (41)
To a solution of 34α (50 mg; 0.06 mmol) in methanol (10 mL) was added NaOMe (3 mg; 0.06 mmol). The resulting mixture was stirred at room temperature until complete conversion. Then the reaction mixture was quenched by Amberlyst-15 H+ resin and filtered. The filtrate was evaporated and subjected to column chromatography over silica gel (Hexane/Ethyl acetate 6:4) to give 41 as a colorless oil (27.1 mg; 72%). [α]21 D 30.4 (c 1.0, CHCl3); 1H NMR: δ 7.32 (m, 10H), 4.85 (d, J = 11.2 Hz, 1H), 4.62 (d, J = 11.2 Hz, 1H), 4.58 (m, 2H), 4.28 (d, J = 7.7 Hz, 1H), 4.06 (dd, J = 8.7, 6.2 Hz, 1H), 3.94 (dd, J = 9.6, 3.1 Hz, 1H), 3.81 (s, 3H), 3.75 (m, 4H), 3.59 (m, 5H), 3.44 (t, J = 10.1 Hz, H-5), 3.08 (dd, J = 8.9, 2.0 Hz, 1H), 2.52 (dd, J = 13.6, 4.6 Hz, 1H), 2.05 (t, J = 11.6 Hz, 1H), 1.35 (d, J = 6.2 Hz, 3H); 13CC NMR: δ 169.0, 138.4, 128.4, 128.2, 127.7, 127.6, 127.5, 104.8, 99.8, 77.3, 77.0, 76.7, 75.5, 74.9, 73.6, 73.1, 72.5, 69.8, 69.6, 65.4, 63.4, 57.0, 53.7, 38.0, 20.3; m/z calcd for: C31H40N6O11Na, [M+Na]+ 695.2653; found: 695.2661.
Methyl [methyl (4,8-di-O-acetyl-5,7-diacetamido-3,5,7,9-tetradeoxy-D-glycero-α-D-galacto-non-2-ulopyranosid)onate]-(2→6)-2,3,4-tri-O-benzyl-β-D-galactopyranoside (38)
To a solution of 37 (70 mg; 0.09 mmol) in 1:1 dioxane:water (8 mL) was added 10% Pd/C (70 mg) followed by glacial acetic acid (0.25 mL). The resulting mixture was stirred at room temperature under hydrogen gas (1 atm) for 16 h then filtered and was evaporated to dryness. Pyridine (5 mL) and acetic anhydride (5 mL) were added to the residue and the resulting mixture was stirred at room temperature for 10 h. The solvents were evaporated and the residue was subjected to column chromatography over silica gel (dichloromethane/methanol 94:6) to give 38 (45 mg; 68%). [α]21 D 26.2 (c 1.0, CHCl3); 1H NMR: δ 6.53 (d, J = 10.0 Hz, 1H), 5.71 (d, J = 7.9 Hz, 1H). 5.61 (d, J = 7.8 Hz, 1H), 5.36 (m, 1H), 5.20 (dd, J = 10.6, 7.9 Hz, 1H), 4.96 (dd, J = 10.6, 6.2 Hz, 1H), 4.94 (dd, J = 8.7 Hz, 3.5 Hz, 1H), 4.63 (d, J = 8.0 Hz, 1H), 4.30 (t, J = 9.8 Hz, 1H), 4.10 (dd, J = 9.3, 6.9 Hz, 1H), 3.84 (s, 3H), 3.60 (dd, J = 12.1, 9.4 Hz, 1H), 3.51 (s, 3H), 3.31 (m, 1H), 2.63 (dd, J = 12.6, 4.7 Hz, 1H), 2.15 (s, 3H), 2.13 (s, 3H), 2.12 (s, 3H), 2.07 (s, 3H), 1.99 (s, 3H), 1.98 (s, 3H), 1.92 (s, 3H), 1.67 (t, J = 12.4 Hz, 1H), 1.28 (d, J = 6.2 Hz, 3H); 13CC NMR: δ 171.9, 171.4, 170.7, 170.5, 170.4, 170.2, 169.6, 166.6, 101.7, 100.7, 72.3, 71.0, 70.0, 68.5, 67.3, 67.2, 66.8, 64.2, 57.1, 53.0, 52.0, 51.9, 38.7, 29.7, 23.6, 22.9, 21.4, 20.9, 20.8, 20.7, 17.7; m/z calcd for: C31H46N2O18Na, [M+Na]+ 757.2643; found: 757.2641.
Methyl [methyl (4,8-di-O-acetyl-5,7-diacetamido-3,5,7,9-tetradeoxy-D-glycero-α-D-galacto-non-2-ulopyranosid)onate]-(2→3)-2,4,6-tri-O-benzyl-β-D-galactopyranoside (40)
From 33α: To a solution of 33α (40 mg; 0.05 mmol) in 1:1 dioxane:water (6 mL) was added 10% Pd/C (70 mg) followed by glacial acetic acid (0.12 mL). The resulting mixture was stirred at room temperature under hydrogen gas (1 atm) for 16 h, then filtered off, and evaporated to dryness. Pyridine (5 mL) and acetic anhydride (5 mL) were added to the residue and the resulting mixture was stirred at room temperature for 10 h. Then the solvents were evaporated and the residue was subjected to column chromatography over silica gel (dichloromethane/methanol 94:6) to give 40 (28.1 mg; 73%).
From 41: To a solution of 41 (20 mg; 0.03 mmol) in 1:1 dioxane:water (3 mL) was added 10% Pd/C (20 mg) followed by glacial acetic acid (0.06 mL). The resulting mixture was stirred at room temperature under hydrogen gas (1 atm) for 16 h, then filtered off, and evaporated to dryness. Pyridine (3 mL) and acetic anhydride (3 mL) were added to the residue and the resulting mixture was stirred at room temperature for 10 h. Then the solvents were evaporated and the residue was subjected to column chromatography over silica gel (dichloromethane/methanol 94:6) to give 40 (14.3 mg; 66%).
[α]21 D 37.5 (c 1.0, CHCl3); 1H NMR (600 MHz, CDCl3) δ 6.35 (s, 1H), 5.34 (d, J = 8.9 Hz, 1H), 5.28 (s, 1H), 5.21 (m, 1H), 5.10 (dd, J = 12.3, 6.1 Hz, 1H), 5.03 (d, J = 2.9 Hz, 1H), 4.64 (d, J = 8.3 Hz, 1H), 4.55 (dd, J = 10.0, 3.2 Hz, 1H), 4.51 (d, J = 7.6 Hz, 1H), 4.33 (t, J = 10.2 Hz, 1H), 4.08 (d, J = 6.3 Hz, 1H), 3.83 (s, 3H), 3.52 (s, 3H), 2.64 (dd, J = 12.4, 4.6 Hz, 1H), 2.17 (s, 3H), 2.12 (s, 3H), 2.09 (s, 3H), 2.05 (s, 3H), 2.04 (s, 3H), 2.00 (s, 3H), 1.90 (s, 3H), 1.56 (t, J = 12.2 Hz, 1H), 1.24 (d, J = 6.2 Hz, 3H); 13CC NMR (151 MHz, CDCl3) δ 170.8, 170.8, 170.3, 170.3, 170.3, 170.21, 170.19, 167.55, 101.48, 96.79, 71.03, 71.03, 70.82, 68.36, 68.36, 67.89, 67.46, 62.05, 56.97, 53.10, 51.37, 51.05, 37.51, 23.40, 23.02, 21.73, 20.84, 20.82, 20.66, 20.66, 17.79; m/z calcd for: C31H46N2O18Na, [M+Na]+ 757.2643; found: 757.2662.
Methyl [5,7-Diacetamido-3,5,7,9-tetradeoxy-D-glycero-α-D-galacto-non-2-ulopyranosid)onic acid]-(2→6)-2,3,4-tri-O-benzyl-β-D-galactopyranoside (39)
To a solution of 38 (30 mg; 0.04 mmol) in H2O (3.0 mL) was added saturated aq Ba(OH)2 (1.0 mL). The resulting solution was stirred at 60 °C for 2 h. Then the reaction mixture was brought to room temperature and saturated with CO2. The precipitate was filtered off and the filtrate was frozen using a dry ice-acetone bath and lyophilized to obtain the white foam 39 (18.1 mg, 91 %). [α]21 D 3.3 (c 0.5, H2O); 1H NMR (600 MHz, D2O) δ 4.15 (d, J = 8.0 Hz, 1H), 3.82 (m, 2H), 3.76 (m, 3H), 3.68 (dd, J = 9.3, 2.9 Hz, 1H), 3.61 (dd, J = 8.1, 4.2 Hz, 1H), 3.49 (m, 2H), 3.43 (m, 4H) 3.33 (dd, J = 9.9, 8.0 Hz, 1H), 2.58 (dd, J = 12.4, 4.5 Hz, 1H), 1.83 (s, 3H), 1.78 (s, 3H), 1.52 (t, J = 12.1 Hz, 1H), 1.00 (d, J = 6.3 Hz, 3H); 13CC NMR (151 MHz, D2O) δ 173.9, 173.6, 173.3, 103.8, 100.4, 73.4, 72.5, 71.6, 70.6, 68.7, 68.6, 67.1, 63.7, 57.3, 53.9, 52.1, 40.2, 23.2, 21.9, 18.0; m/z calcd for: C20H33N2O13, [M-H]− 509.1983; found: 509.1962.
Methyl [5,7-Diacetamido-3,5,7,9-tetradeoxy-D-glycero-α-D-galacto-non-2-ulopyranosid)onic acid]-(2→3)-2,4,6-tri-O-benzyl-β-D-galactopyranoside (42)
To a solution of 40 (15 mg; 0.02 mmol) in H2O (1.5 mL) was added saturated aq Ba(OH)2 (1.0 mL). The resulting solution was stirred at 60 °C for 2 h. Then the reaction mixture was brought to room temperature and saturated with CO2. The precipitate was filtered off and the filtrate was frozen using a dry ice-acetone bath and lyophilized to obtain the white foam 42 (9.2 mg, 92 %). [α]21 D 1.8 (c 0.5, H2O); 1H NMR (600 MHz, D2O) δ 4.19 (d, J = 7.9 Hz, 1H), 3.91 (m, 1H), 3.79 (d, J = 6.4 Hz, 1H), 3.75 (s, 1H), 3.65 (m, 2H), 3.56 (m, 3H), 3.48 (t, J = 9.8 Hz, 1H), 3.40 (s, 3H), 3.37 (d, J = 8.0 Hz, 1H), 2.58 (dd, J = 12.8, 4.3 Hz, 1H), 1.81 (s, 3H), 1.77 (s, 3H), 0.99 (d, J = 6.2 Hz, 3H); 13CC NMR (151 MHz, D2O) δ 173.9, 173.8, 173.7, 103.5, 99.5, 75.8, 74.8, 71.7, 69.0, 68.6, 67.2, 67.0, 60.9, 57.0, 53.8, 51.9, 40.0, 22.4, 21.9, 18.0; m/z calcd for: C20H33N2O13, [M-H]− 509.1983; found: 509.1996.
Supplementary Material
Acknowledgments
We thank the NIH (GM62160) for support of this work, and acknowledge support from the NSF (MRI-084043) for the purchase of the 600 MHz NMR spectrometer in the Lumigen Instrument Center at Wayne State University.
Footnotes
Supporting Information Available. Copies of the 1H and 13CC NMR spectra of all new compounds. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.joc.******
References
- 1.Knirel YA, Shashkov AS, Tsvetkov YE, Jansson PE, Zaehringer U. Adv Carbohydr Chem Biochem. 2003;58:371–417. doi: 10.1016/s0065-2318(03)58007-6. [DOI] [PubMed] [Google Scholar]
- 2.Zunk M, Kiefel MJ. RSC Adv. 2014;4:3413–3421. [Google Scholar]
- 3.Severi E, Hood DW, Thomas GH. Microbiology. 2007;153:2817–2822. doi: 10.1099/mic.0.2007/009480-0. [DOI] [PubMed] [Google Scholar]
- 4.Tsvetkov YE, Shashkov AS, Knirel YA, Zähringer U. Carbohydr Res. 2001;331:233–237. doi: 10.1016/s0008-6215(01)00041-6. [DOI] [PubMed] [Google Scholar]
- 5.Kooistra O, Herfurth L, Lüneberg E, Frosch M, Peters T, Zähringer U. 2002;269:573–582. doi: 10.1046/j.0014-2956.2001.02684.x. [DOI] [PubMed] [Google Scholar]
- 6.Mas Pons J, Dumont A, Sautejeau G, Fugier E, Baron A, Dukan S, Vauzeilles B. Angew Chem Int Ed. 2014;53:1275–1278. doi: 10.1002/anie.201309072. [DOI] [PubMed] [Google Scholar]
- 7.Cazalet C, Jarraud S, Ghavi-Helm Y, Kunst F, Glaser P, Etienne J, Buchrieser C. Genome Res. 2008;18:431–441. doi: 10.1101/gr.7229808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McNally DJ, Aubry AJ, Hui JPM, Khieu NH, Whitfield D, Ewing CP, Guerry P, Brisson JR, Logan SM, Soo EC. J Biol Chem. 2007;282:14463–14475. doi: 10.1074/jbc.M611027200. [DOI] [PubMed] [Google Scholar]
- 9.Logan SM, Hui JPM, Vinogradov E, Aubry AJ, Melanson JE, Kelly JF, Nothaft H, Soo EC. FEBS J. 2009;276:1014–1023. doi: 10.1111/j.1742-4658.2008.06840.x. [DOI] [PubMed] [Google Scholar]
- 10.Schoenhofen IC, Vinogradov E, Whitfield DM, Brisson JR, Logan SM. Glycobiology. 2009;19:715–725. doi: 10.1093/glycob/cwp039. [DOI] [PubMed] [Google Scholar]
- 11.MacLean LL, Vinogradov E, Pagotto F, Perry MB. Carbohydr Res. 2011;346:2589–2594. doi: 10.1016/j.carres.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 12.Filatov AV, Wang M, Wang W, Perepelov AV, Shashkov AS, Wang L, Knirel YA. Carbohydr Res. 2014;392:21–24. doi: 10.1016/j.carres.2014.01.012. [DOI] [PubMed] [Google Scholar]
- 13.Vinogradov E, MacLean L, Xu HH, Chen W. Carbohydr Res. 2014;390:42–45. doi: 10.1016/j.carres.2014.03.001. [DOI] [PubMed] [Google Scholar]
- 14.Matthies S, Stallforth P, Seeberger PH. J Am Chem Soc. 2015;137:2848–2851. doi: 10.1021/jacs.5b00455. [DOI] [PubMed] [Google Scholar]
- 15.Glaze PA, Watson DC, Young NM, Tanner ME. Biochemistry. 2008;47:3272–3282. doi: 10.1021/bi702364s. [DOI] [PubMed] [Google Scholar]
- 16.Hassan MI, Lundgren BR, Chaumun M, Whitfield DM, Clark B, Schoenhofen IC, Boddy CN. Angew Chem Int Ed. 2016;55:12018–12021. doi: 10.1002/anie.201606006. [DOI] [PubMed] [Google Scholar]
- 17.Watson DC, Leclerc S, Wakarchuk WW, Young NM. Glycobiology. 2011;21:99–108. doi: 10.1093/glycob/cwq135. [DOI] [PubMed] [Google Scholar]
- 18.Watson DC, Wakarchuk WW, Gervais C, Durocher Y, Robotham A, Fernanades SM, Schnaar RL, Young NM, Gilbert M. Glycoconj J. 2015;32:729–734. doi: 10.1007/s10719-015-9624-4. [DOI] [PubMed] [Google Scholar]
- 19.Tsvetkov YE, Shashkov AS, Knirel YA, Zähringer U. Carbohydr Res. 2001;335:221–243. doi: 10.1016/s0008-6215(01)00235-x. [DOI] [PubMed] [Google Scholar]
- 20.Haberman JM, Gin DY. Org Lett. 2003;5:2539–2541. doi: 10.1021/ol034815z. [DOI] [PubMed] [Google Scholar]
- 21.Crich D, Li W. J Org Chem. 2007;72:7794–7797. doi: 10.1021/jo7012912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Buda S, Crich D. J Am Chem Soc. 2016;138:1084–1092. doi: 10.1021/jacs.5b13015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Furuhata K. Trends Glycosci Glycobiol. 2004;16:143–169. [Google Scholar]
- 24.Chatgilialoglu C. Acc Chem Res. 1992;25:188–194. [Google Scholar]
- 25.Goddard-Borger ED, Stick RV. Org Lett. 2007;9:3797–3800. doi: 10.1021/ol701581g. [DOI] [PubMed] [Google Scholar]
- 26.Potter GT, Jayson GC, Miller GJ, Gardiner JM. J Org Chem. 2016;81:3443–3446. doi: 10.1021/acs.joc.6b00177. [DOI] [PubMed] [Google Scholar]
- 27.Dess PB, Martin JC. J Org Chem. 1983;48:4155–4156. [Google Scholar]
- 28.Kancharla PK, Crich D. J Am Chem Soc. 2013;135:18999–19007. doi: 10.1021/ja410683y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Price NPJ, Furukawa T, Cheng F, Qi J, Chen W, Crich D. J Antibiotics. 2014;67:405–414. doi: 10.1038/ja.2014.15. [DOI] [PubMed] [Google Scholar]
- 30.Luche JL, Rodriguez-Hahn L, Crabbe P. J Chem Soc, Chem Commun. 1978;601 [Google Scholar]
- 31.Spijker NM, Zuurmond H, Westerduin P, Van Boeckel CA. A Rec Trav Chim Pays Bas. 1989;108:360–368. [Google Scholar]
- 32.Kohata K, Abbas SA, Matta KL. Carbohydr Res. 1984;132:127–135. doi: 10.1016/0008-6215(84)85070-3. [DOI] [PubMed] [Google Scholar]
- 33.Paulsen H, Hasenkamp T, Paal M. Carbohydr Res. 1985;144:45–55. [Google Scholar]
- 34.Dhakal B, Buda S, Crich D. J Org Chem. 2016;81:10617–10630. doi: 10.1021/acs.joc.6b02221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Czarniecki MF, Thornton ER. J Am Chem Soc. 1977;99:8273–8279. [Google Scholar]
- 36.Hori H, Nakajima T, Nishida Y, Ohrui H, Meguro H. Tetrahedron Lett. 1988;29:6317–6320. [Google Scholar]
- 37.Haverkamp J, Spoormaker T, Dorland L, Vliegenthart JFG, Schauer R. J Am Chem Soc. 1979;101:4851–4853. [Google Scholar]
- 38.Prytulla S, Lauterwein J, Klessinger M, Thiem J. Carbohydr Res. 1991;215:345–349. [Google Scholar]
- 39.Hansch C, Leo A, Taft RW. Chem Rev. 1991;91:165–195. [Google Scholar]
- 40.Rao VSR, Qasba PK, Balaji PV, Chandrasekaran R. Conformation of Carbohydrates. Harwood Academic Publishers; Amsterdam: 1998. [Google Scholar]
- 41.Christian R, Schulz G, Brandstetter HH, Zbiral E. Carbohydr Res. 1987;162:1–11. doi: 10.1016/0008-6215(87)80195-7. [DOI] [PubMed] [Google Scholar]
- 42.Brown EB, Brey WS, Jr, Weltner W., Jr Biochim Biophys Acta. 1975;399:124–130. doi: 10.1016/0304-4165(75)90218-4. [DOI] [PubMed] [Google Scholar]
- 43.Flippen JL. Acta Cryst. 1973;B29:1881–1886. [Google Scholar]
- 44.Veluraja K, Rao VSR. Biochim Biophys Acta. 1980;630:442–446. doi: 10.1016/0304-4165(80)90293-7. [DOI] [PubMed] [Google Scholar]
- 45.Sabesan S, Bock K, Lemieux RU. Can J Chem. 1984;62:1034–1045. [Google Scholar]
- 46.Bandgar BP, Zbiral E. Monat für Chemie. 1991;122:1075–1088. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.