

Abstract
Rationale
Mitochondrial changes occur during cell differentiation and cardiovascular disease. Dynamin-related protein 1 (DRP1) is a key regulator of mitochondrial fission. We hypothesized that DRP1 plays a role in cardiovascular calcification, a process involving cell differentiation and a major clinical problem with high unmet needs.
Objective
To examine the effects of osteogenic promoting conditions on DRP1, and whether DRP1 inhibition alters the development of cardiovascular calcification.
Methods and Results
DRP1 was enriched in calcified regions of human carotid arteries, examined by immunohistochemistry. Osteogenic differentiation of primary human vascular smooth muscle cells (SMCs) increased DRP1 expression. DRP1 inhibition in human SMCs undergoing osteogenic differentiation attenuated matrix mineralization, cytoskeletal rearrangement, mitochondrial dysfunction, and reduced type 1 collagen secretion and alkaline phosphatase activity. DRP1 protein was observed in calcified human aortic valves, and DRP1 RNA interference reduced primary human valve interstitial cell calcification. Mice heterozygous for Drp1 deletion did not exhibit altered vascular pathology in a PCSK9 gain-of-function atherosclerosis model. However, when mineralization was induced via oxidative stress, DRP1 inhibition attenuated mouse and human SMC calcification. Femur bone density was unchanged in mice heterozygous for Drp1 deletion, and DRP1 inhibition attenuated oxidative stress-mediated dysfunction in human bone osteoblasts.
Conclusions
We demonstrate a new function of DRP1 in regulating collagen secretion and cardiovascular calcification, a novel area of exploration for the potential development of new therapies to modify cellular fibrocalcific response in cardiovascular diseases. Our data also support a role of mitochondrial dynamics in regulating oxidative stress-mediated arterial calcium accrual and bone loss.
Keywords: Mitochondria, dynamin-related protein 1, DNM1L, calcification, collagen, oxidative stress, aortic valve calcification, atherosclerosis
Subject Terms: Atherosclerosis, Basic Science Research, Mechanisms, Oxidant Stress, Vascular Biology
INTRODUCTION
Mitochondria exist in dynamic networks where they are continuously generated, joined through fusion, and divided by fission, with dynamin-related protein 1 (DRP1) being a key protein allowing mitochondria to multiply or be degraded through mitochondrial fission1. Mitochondrial activity, shape, and localization have been suggested to regulate cell differentiation by influencing programs that control cell fate2. Differentiation is a characteristic phenotypic feature of vascular smooth muscle cells (SMCs)3, with phenotypic modulation of vascular cells allowing for the vasculature to efficiently perform and respond to different physiological and pathological conditions4. One such condition with high unmet needs involves SMCs and valve interstitial cells (VICs) undergoing differentiation into osteoblast-like cells. The resulting arterial and aortic valve accumulation of calcium mineral deposits promotes major adverse clinical events5.
Mitochondrial dynamics are associated with many biological processes related to cardiovascular calcification including: calcium homeostasis, cellular differentiation, apoptosis, oxidative stress, autophagy, and exocytosis6–10. Mice with a Drp1 middle domain C425F dominant-negative mutation develop cardiomyopathy along with spotty calcifications in heart tissue11. No mechanism has been suggested for the presence of cardiac calcification in these mice, and whether DRP1 regulates calcification of other tissues, including the vasculature is unknown. DRP1inhibition prevents rat vascular neointima formation in a balloon injury model12, suppresses lesion formation in diabetic ApoE-deficient mice13, and oxidized low density lipoprotein induces DRP1 mediated mitochondrial fragmentation in human SMCs14, suggesting a possible role of DRP1 in vascular disease. Reduced mitochondrial fusion protein, Mitofusin 2 (MFN2) that acts in an opposite function to that of DRP1 driven mitochondrial fission, is associated with atherosclerosis pathology in mice15, 16, rabbits17, and humans16. Overexpression of Mfn2 in atherosclerotic rabbits reduces lesion formation17. Taken together these studies suggest a role of mitochondrial dynamics in atherosclerosis, a disease highly associated with calcification. Mutations in the dynamin 1 like gene (DNM1L) encoding the DRP1 protein, including nonsense and dominant negative mutations, have been reported in a small number of human patients18–24. While these patients exhibited a wide range of pathologies, from normal early development to neuronal development defects, cardiovascular calcification has not been reported. As such, an involvement of DRP1 in human soft tissue calcification remains to be demonstrated. Therefore, we sought to examine whether DRP1 plays a role in human cardiovascular calcification, and if its inhibition alters the calcification process.
METHODS
Detailed methodology is included in the Online Supplemental Material.
RESULTS
DRP1 localized to calcified areas of human carotid arteries
To analyze DRP1 expression in human vascular disease conditions, we performed DRP1 immunostaining on human carotid atherosclerotic plaques (N=5). DRP1 immunoreactivity was high in tissues with calcification, and more specifically around calcified areas (Figure 1A and Online Figure I). DRP1 was observed in SMCs (HHF-35, αSMA) and some macrophages (CD68) in calcified human arteries (Figure 1C and D). SMC mitochondrial area was enlarged in calcified compared to non-calcified regions of carotid artery tissue from patients undergoing endarterectomy (N=3), examined by electron microscopy (Figure 1E).
Figure 1. DRP1 was enriched in calcified regions of human carotid arteries.
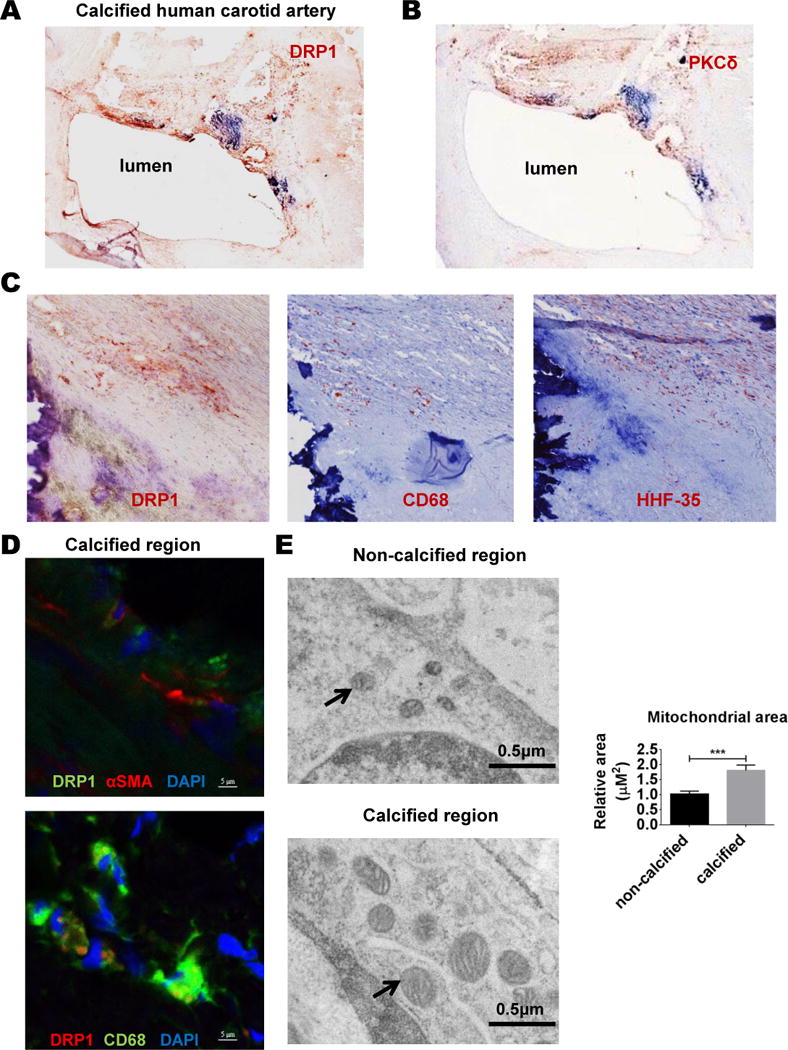
Representative human carotid artery tissue immunohistochemistry for (A) DRP1, (B) PKCδ, and adjacent sections for DRP1, CD68, and HHF-35 (smooth muscle cell marker) in calcified tissue (C); N=5 donors. (D) Representative immunofluorescence images showing DRP1 staining in both αSMA and CD68 positive cells in calcified areas of human carotid artery; N=5 donors. (E) Representative electron microscopy images of human carotid artery smooth muscle cells (SMCs) in non-calcified and calcified tissue regions. Arrows indicate examples of mitochondria. Relative quantification of SMC mitochondrial area (N=32–41 mitochondria from 3 donors); error bars indicate SEM; *** P<0.001.
Increased DRP1 in calcifying human SMCs associated with mitochondrial alterations
An established cell culture vascular calcification model25, promotes the transition of SMCs to an osteoblast-like cell phenotype. αSMA positive (Online Figure IIA) SMCs cultured under osteogenic conditions have increased activity of a key enzyme, tissue non-specific alkaline phosphatase (TNAP), peaking at two weeks in culture, and develop a mineralized matrix at three weeks in culture. Osteogenic transition of primary human coronary artery SMCs increased (approximately two-fold) DRP1 protein and mRNA (Figure 2A). To assess mitochondrial morphology in this model, mitochondria were labeled using MitoTracker Red and live cell mitochondrial imaging analysis was performed with confocal microscopy. Human SMC mitochondria appeared to be more fragmented when cultured in osteogenic media (OM) compared to control media (CM) (Figure 2B). We confirmed these observations through quantifying mitochondrial aspect ratios, a measurement of the major mitochondrial axis to the minor axis.
Figure 2. DRP1 and mitochondrial dysfunction increased in calcifying human SMCs.
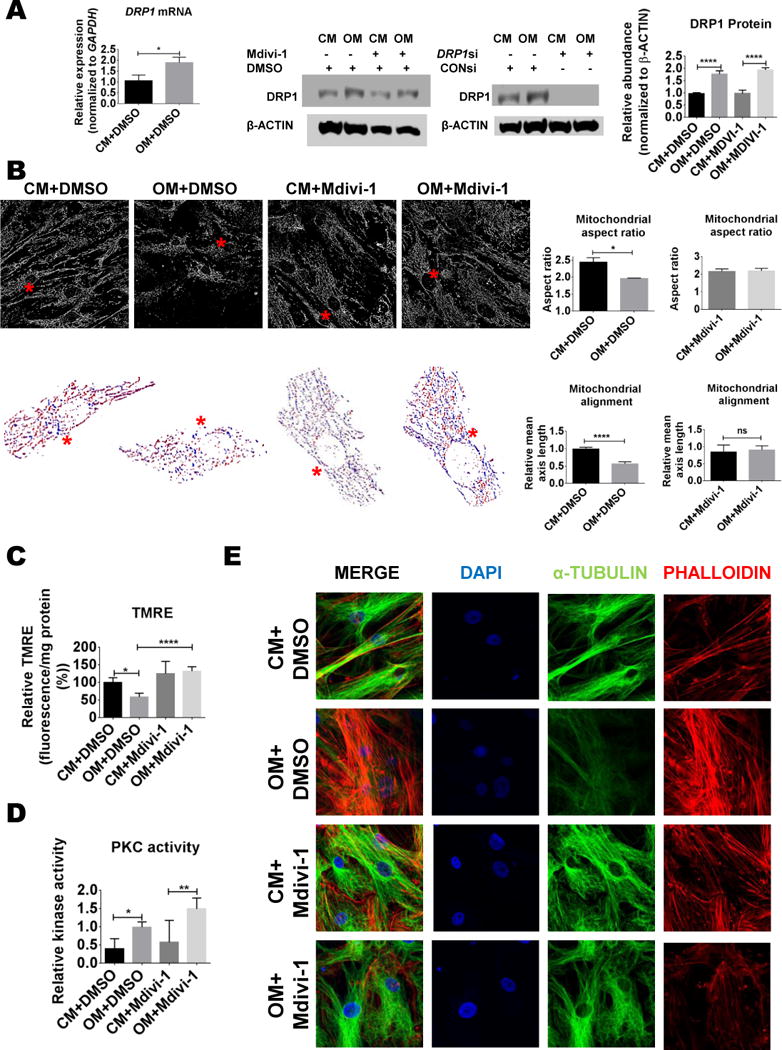
(A) DRP1 mRNA and protein from human smooth muscle cells (SMCs) cultured in control (CM) or osteogenic media (OM) for two weeks with vehicle (0.01% DMSO), Mdivi-1 (50 μmol/L), control siRNA (CONsi), or DRP1 siRNA (DRP1si). (B) Representative MitoTracker Red (mitochondria) human SMC processed binary confocal images from human SMCs cultured in CM or OM for two weeks. Red asterix indicates the location of a representative cell with mitochondrial alignment shown in the corresponding images below (Mitochondria color coded by major axis orientation). Quantified mitochondrial aspect ratio and alignment from SMCs cultured in CM or OM with vehicle or Mdivi-1 treatment for two weeks. (C) Mitochondrial membrane potential assayed by TMRE fluorescence and (D) PKC activity in human SMCs treated for two weeks. (E) Representative confocal microscopy images of DAPI, α-TUBULIN, and phalloidin from human SMCs treated for two weeks. Error bars indicate STDEV; ns= not significant, * P <0.05, **<0.01, ****<0.0001; N=3–4 donors.
Mitochondrial aspect ratios were reduced in two-week-OM cultured cells compared to CM conditions (Figure 2B). To test whether this change was due to DRP1 activity or protein content, the selective small molecule DRP1 inhibitor, mitochondrial division inhibitor 1 (Mdivi-1) was added at a concentration (50 μmol/L) previously reported to fully inhibit DRP1 activity in cells26. Mdivi-1 treatment attenuated OM-mediated mitochondrial aspect ratio reductions (Figure 2B) without altering OM-mediated increases in DRP1 protein (Figure 2A). To assess whether the changes in DRP1 and mitochondrial morphology we observed in human SMCs resulted in mitochondrial functional deficits, we utilized the tetramethylrhodamine ethyl ester (TMRE) reagent to measure mitochondrial membrane potential, a general marker of mitochondrial function27. TMRE fluorescence was reduced in cells cultured in OM for two weeks, and this decrease was attenuated by DRP1 inhibition (Figure 2C).
Previous studies link DRP1 and Protein Kinase C (PKC), with PKCδ inducing DRP1 driven fission activity in SMCs12, 28. Therefore we examined PKC in relation to DRP1 changes under calcifying conditions. Immunohistochemistry showed increased PKCδ in the same calcified regions of human carotid arteries that stained for DRP1 (Figure 1B). Dexamethasone, a component of OM, increases fatty acid synthesis29. Fatty acids increase DRP130, and have been associated with PKC activation31. To test whether altered mitochondrial fission observed in OM conditions involved PKC, we measured PKC activity in cells cultured for two weeks in CM and OM. In human SMCs, PKC enzyme activity was elevated in OM with and without the addition of Mdivi-1 (Figure 2D).
DRP1 inhibition attenuated OM induced cytoskeletal changes, TNAP activation, COL1A1 secretion, and human SMC calcification
The OM-mediated alterations in mitochondrial alignment and aspect ratio that were attenuated by DRP1 inhibition, along with reports of cytoskeleton involvement in DRP1 mediated fission32, could indicate cytoskeletal alterations33. As microtubule stability and PKC activity associate with SMC calcification suppression34, we assessed the tubulin and filamentous actin cytoskeleton of human SMCs undergoing osteogenic differentiation. Two-week OM treatment resulted in a modestly diffuse tubulin cytoskeleton and a more apparent actin cytoskeleton; a difference attenuated by Mdivi-1 (Figure 2E). Mdivi-1 increased α-TUBULIN acetylation, a modification associated with tubulin stability (Online Figure IIB).
We next assessed whether the effects of Mdivi-1 treatment on cytoskeleton reflect the ability of DRP1 inhibition to block the transition of human SMCs to osteoblast-like calcifying SMCs. Intracellular calcium levels analyzed by Fluo-4 did not change in cells treated in OM for two weeks, but slightly increased in both CM and OM conditions with the addition of Mdivi-1 (Figure 3A). DRP1 inhibition reduced OM-induced cellular TNAP mRNA, protein, and activity (Figure 3B–D). OM reduced the calcification inhibitor and TNAP substrate, pyrophosphate (PPi), which was partially restored by DRP1 inhibition in human SMCs (Figure 3E). DRP1 inhibition attenuated OM induced calcification, visualized by Alizarin Red and Osteosense staining (Figure 3F and 3J). To confirm the specificity of DRP1 inhibition in driving the anti-calcification effects of Mdivi-1, we reduced DRP1 in human SMCs with small interfering RNA oligonucleotides (siRNA; DRP1 western blot siRNA confirmation shown in Figure 2A). In agreement with the inhibitor results, DRP1 siRNA attenuated the formation of a mineralized matrix in human SMCs cultured in OM for three weeks (Figure 3F). Mdivi-1 reduced the expression of the TNAP regulator, RUNX2 in human SMCs (Online Figure IIIA). Additionally, DRP1 inhibition attenuated oxidative stress induced mineralization in human SMCs (Figure 3G) treated with a low concentration of hydrogen peroxide (H2O2; 0.3 mmol/L). This concentration of H2O2 induces SMC calcification without altering cell viability35, 36. As DRP1 inhibition did not fully inhibit OM induced TNAP activity, we looked at additional mechanistic pathways through which it may act to inhibit calcification. In our OM experimental conditions, DRP1 inhibition effects on SMC calcification were not mediated by the calcification inhibitors osteoprotegerin and matrix gla protein, ER stress, or oxidative stress without the addition of H2O2 (Online Figures IIC, IID, IIIB). As calcifying vesicles utilize a collagen matrix to attach and mineralize37, 38, we assessed type 1 collagen (COL1A1) content. Mdivi-1 reduced human SMC type 1 collagen secretion from a 7.5-fold induction to a 2.5-fold induction in OM (Figure 3H) without altering COL1A1 mRNA (Figure 3I). Collagen staining using CNA probe38 validated the increased collagen matrix produced in human OM treated SMCs that was attenuated by DRP1 inhibition (Figure 3J).
Figure 3. DRP1 inhibition attenuated human SMC osteogenic media-induced fibrocalcific response.
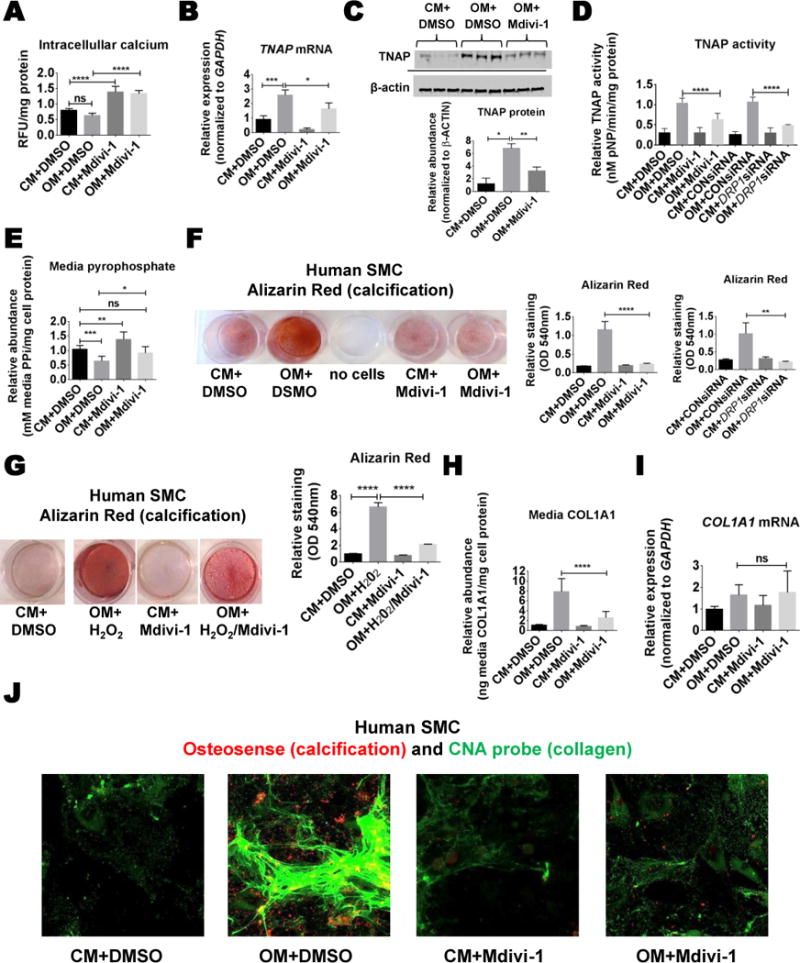
(A) Live cell intracellular calcium concentrations, and (B) TNAP mRNA, (C) protein, and (D) relative enzyme activity from human coronary smooth muscle cells (SMCs) cultured in control media (CM) or osteogenic media (OM) for two weeks with DMSO vehicle (0.01%) or Mdivi-1 (50 μmol/L). (E) Relative media pyrophosphate (PPi) from human coronary SMCs treated for two weeks. (F) Representative Alizarin Red stain image and quantification from human coronary artery SMCs treated with Mdivi-1 or DRP1 siRNA for three weeks. (G) Representative Alizarin Red stain and quantification for human aortic SMCs treated for three weeks in OM with H2O2 (0.3mmol/L), with or without Mdivi-1. (H) Human coronary artery SMC media COL1A1 ELISA from cells treated for two weeks. (I) COL1A1 mRNA from human coronary artery SMCs treated for two weeks. (J) Representative Osteosense (red; calcification) and CNA immunofluorescence (green; collagen) from human coronary artery SMCs treated for three weeks. N=3–6 donors; ns not significant, * P <0.05, **<0.01, ***<0.001, ****<0.0001; error bars indicate STDEV.
DRP1 siRNA reduced human valve interstitial cell calcification
To analyze DRP1 in human valve calcification, we performed DRP1 immunostaining on calcified human aortic valves obtained from patients undergoing aortic valve replacement surgery (N=5). DRP1 immunoreactivity was detected in calcified valve tissue (Figure 4A). We next tested the role of DRP1 in human valve calcification using an established cell culture calcification condition39, in which primary human valve interstitial cells (VICs) were treated with OM containing inorganic phosphate and L-ascorbic acid for three weeks. DRP1 mRNA was significantly increased in OM (Figure 4B). DRP1 siRNA reduced human VIC calcification (Figure 4C). TNAP mRNA and activity were not induced during VIC calcification in our experiments, which was reflected by no changes in VIC PPi levels (Figure 4D). Similarly, RUNX2 mRNA was not strongly induced under these experimental conditions (Online Figure IIIC). VIC COL1A1 secretion was increased by ~1.5-fold in OM. This modest induction in VICs was not altered by DRP1 knockdown (Figure 4E). Examination of calcification related transcription factors, MSX2 and SOX9, revealed no changes in MSX2 expression, whereas SOX9 was elevated by DRP1 siRNA in VICs, and by DRP1 inhibition in human SMCs (Online Figure IIIA and IIIC).
Figure 4. DRP1 knockdown attenuated human valve interstitial cell calcification.
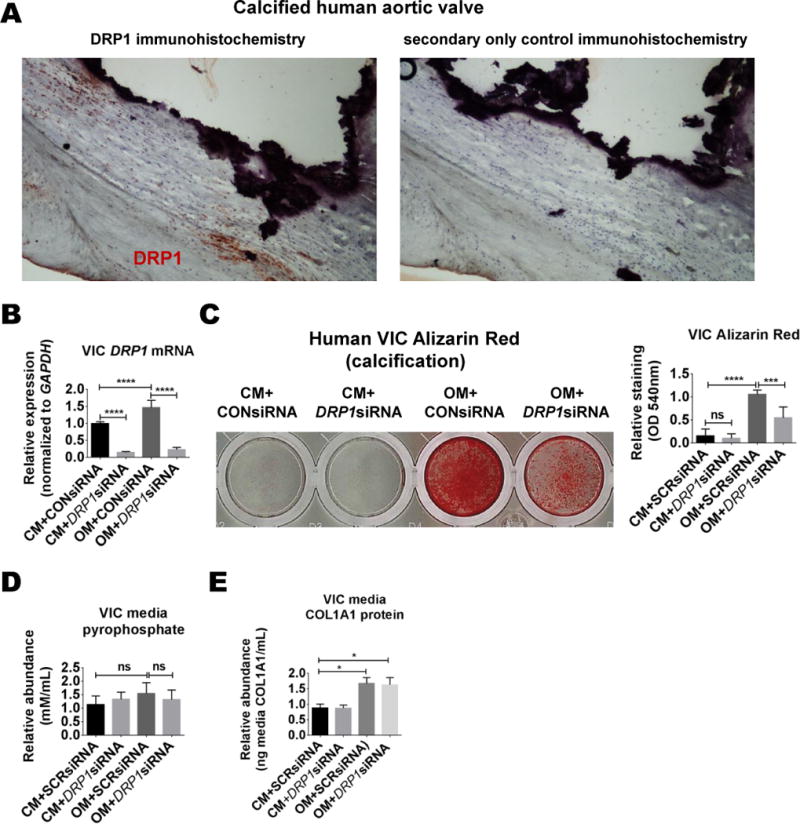
(A) Representative DRP1 immunohistochemistry image from calcified human aortic valve and adjacent section control (secondary antibody only) (N=5 donors). (B) DRP1 mRNA in primary human valve interstitial cells (VICs) treated in control media (CM) or osteogenic media (OM) with control or DRP1 siRNA for two weeks. (C) Representative Alizarin Red staining and quantification from human VICs treated for three weeks. (D) Media pyrophosphate and (E) media COL1A1 ELISA from VICs treated for two weeks. N=3 donors; ns= not significant, * P <0.05, **<0.01, ***<0.001, **** <0.0001; error bars indicate STDEV.
Mdivi-1 reduced human PBMC-derived osteoclastogenesis
We analyzed the in vitro effects of DRP1 inhibition on human vascular osteoclastogenesis through the use of primary human peripheral blood mononuclear cells (PBMCs). PBMCs were cultured in the presence of M-CSF and RANKL to drive osteoclastogenesis. 25 and 50 μmol/L Mdivi-1 reduced tartrate-resistant acid phosphatase (TRAP) activity in PBMCs by 68% and 72%, respectively (Online Figure IVA). 50 μmol/L Mdivi-1 disrupted large actin ring formation in human PBMCs, while actin rings could be seen similar to that observed in untreated cells with 25 μmol/L Mdivi-1, but large multinucleated TRAP-positive cells were still less apparent (Online Figure IVA). Given the similar dose effects observed in the PBMCs to reduce TRAP activity, we used 25 μmol/L Mdivi-1 to test the effects of DRP1 inhibition on human PBMC derived osteoclast bone resorption. Time course analysis involving adding Mdivi-1 at the start of the two-week osteoclast differentiation procedure, or starting at four days or seven days into the process, showed that Mdivi-1 alters human PBMC-derived osteoclasts through inhibition of differentiation, but not through regulating the bone resorption activity of fully formed osteoclast-like cells (Online Figure IVB).
Mdivi-1 attenuated oxidative stress mediated mouse SMC calcification
Immunohistochemistry was used to examine DRP1 in calcified Ldlr-deficient, ApoE-deficient, and PCSK9 gain-of-function atherosclerosis mouse models; however, DRP1 was not enriched in calcified mouse plaque (Online Figure I). In agreement, DRP1 protein and PKC activity were not increased in primary mouse SMCs cultured in OM (Online Figure VA and VB), and DRP1 inhibition did not suppress mouse SMC mineralization in OM alone (Online Figure VC). To test if the stress level required to elicit DRP1 changes was not reached in mouse SMC under the examined experimental conditions, we cultured mouse SMCs in OM with the addition of low levels of H2O2 (0.3 mmol/L). H2O2 increased oxidative stress (Online Figure VD) and DRP1 translocation to mitochondria (Figure 5A). Similar to human SMCs in OM, H2O2 addition to mouse SMCs induced Tnap expression (Figure 5B), the production of a calcified collagen-rich matrix (Figure 5C and 5D), and mitochondrial fragmentation (Figure 5E), all of which were attenuated by Mdivi-1 in addition to increasing Sox9 expression (Online Figure IIID).
Figure 5. Mdivi-1 attenuated H2O2 induced calcification in mouse SMCs.
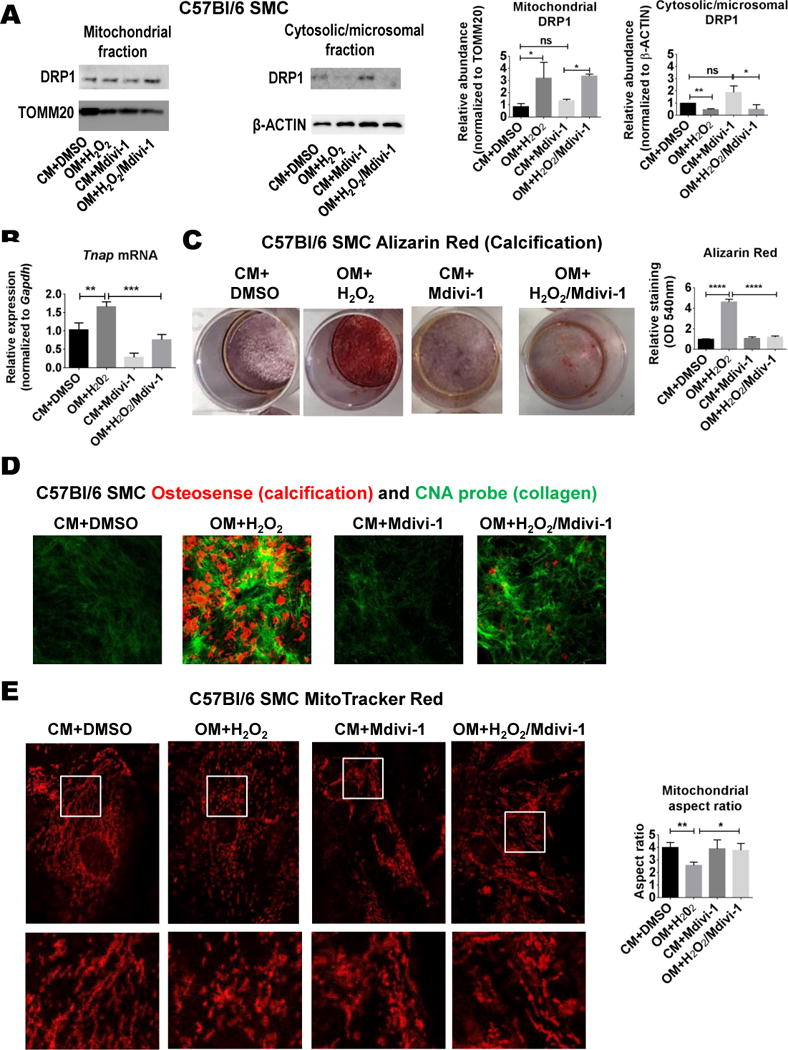
(A) Western blot analysis of DRP1 translocation to the mitochondria five hours post-treatment in control (CM) and osteogenic media (OM) with H2O2 (0.3 mmol/L), with DMSO vehicle (0.01%) or Mdivi-1 (50 μmol/L); TOMM20 served as mitochondrial fraction loading control. (B) Tnap mRNA from mouse SMCs cultured for two weeks. (C) Alizarin Red stain with quantification, and (D) Osteosense (red; calcification) and CNA (green; collagen) immunofluorescence from mouse aortic SMCs treated for three weeks. (E) MitoTracker Red stain immunofluorescence (white boxes indicate higher magnification area of corresponding lower panels) and mitochondrial aspect ratio quantification. N=3; ns=not significant, * P <0.05, **<0.01, ***<0.001, ****<0.0001; error bars indicate STDEV.
Drp1-deficiency is embryonic lethal in mice, therefore to further examine DRP1 inhibition in a mouse vascular calcification model, we utilized Drp1 heterozygous mice (Drp1+/−), which maintain about 75% of wild type DRP1 protein and are largely indistinguishable from wild type mice40, 41. Mice were given a single tail vein injection of murine PCSK9 gain-of-function adeno-associated virus (AAV; which mimics Ldlr-deficiency pathology in mice), fed an atherogenic diet for twenty weeks, and then analyzed in a similar manner to that which we and others have previously reported42–44. Drp1+/− male mice (N=5) did not have significantly altered body weight, serum triglycerides, or total cholesterol, compared to Drp1+/+ male siblings (Figure 6A). Serum total PCSK9 (endogenous + gain-of-function), trended lower but was not significantly reduced in Drp1+/− mice (Figure 6A). Additionally, aortic root Oil Red O, MAC3 (macrophage), and TRAP activity staining was not altered in Drp1+/− mice (Figure 6B). Picrosirius red stain visualized under polarized light (fibrillar collagen accumulation) trended lower, but was not significantly altered in Drp1+/− aortic roots (Figure 6B). In agreement with our primary mouse SMCs in OM without the addition of H2O2 data and the lack of change observed in the mouse vascular DRP1 immunohistochemistry, TNAP activity and vascular calcification measured by Osteosense fluorescence reflectance imaging45 in mouse aortas were not significantly different in Drp1+/− mice under these experimental conditions (Figure 7).
Figure 6. Drp1+/− did not significantly alter atherosclerosis pathology in a PCSK9 gain-of-function AAV mouse model.
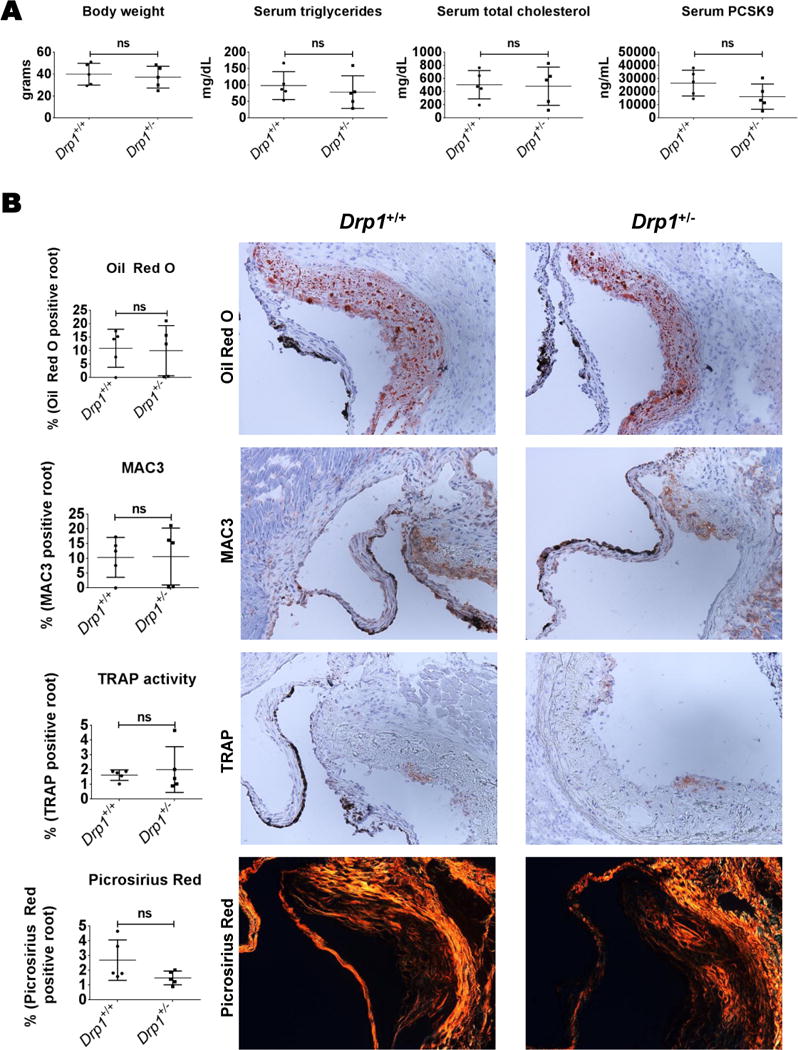
(A) Body weight and serum triglycerides, total cholesterol, and PCSK9 from mice twenty weeks post-PCSK9 gain-of-function AAV tail vein injection and maintained on an atherogenic diet. (B) Representative images of aortic root Oil Red O, MAC3 (macrophage), TRAP activity, and Picrosirius Red staining. N=5 C57Bl/6;129 male mice per group, with at least two sections per mouse analyzed; ns= not significant; error bars indicate STDEV.
Figure 7. Drp1+/− did not significantly alter vascular calcification pathology in a PCSK9 gain-of-function AAV mouse model.
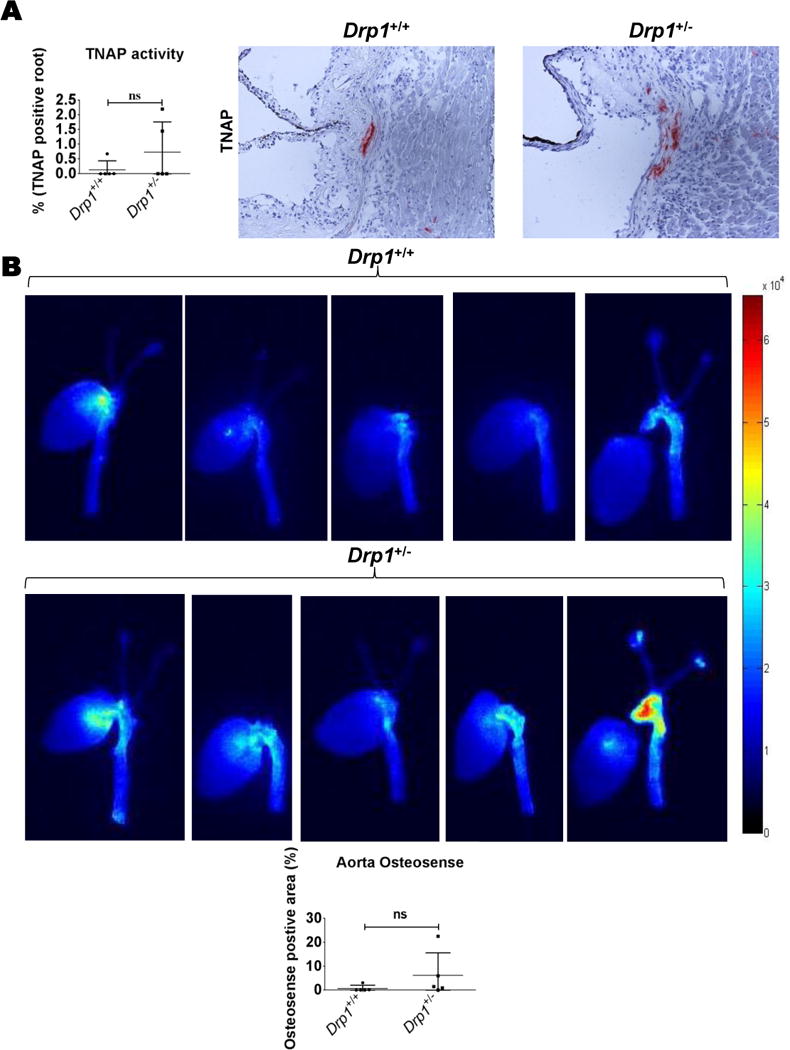
(A) TNAP activity representative images and quantification. (B) Calcification in intact mouse aortas (fluorescence reflectance imaging with Osteosense) 20 weeks post-PCSK9 gain-of-function AAV tail vein injection and maintained on an atherogenic diet. N=5 C57Bl/6;129 male mice per group, with at least two sections per mouse analyzed for immunohistochemistry; ns=not significant; error bars indicate STDEV.
DRP1 heterozygous deficiency did not alter mouse bone density and Mdivi-1 attenuated oxidative stress-mediated dysfunction in primary human bone osteoblasts
Given the connection between cardiovascular and bone calcification46, we examined bone density in Drp1+/− mice. Micro-CT analysis of mouse femurs revealed no changes in cortical thickness, bone volume/trabecular volume ratio, trabecular thickness, trabecular number, or trabecular spacing (Figure 8A). Human osteoblast mitochondria appeared more fragmented with the addition of H2O2; a difference that was attenuated with Mdivi-1 (Figure 8B). Additionally, Mdivi-1 suppressed human osteoblast mineralization dysfunction following H2O2 treatment, analyzed by Alizarin Red staining (Figure 8C and Online Figure VI).
Figure 8. DRP1 inhibition did not reduce bone mineralization.
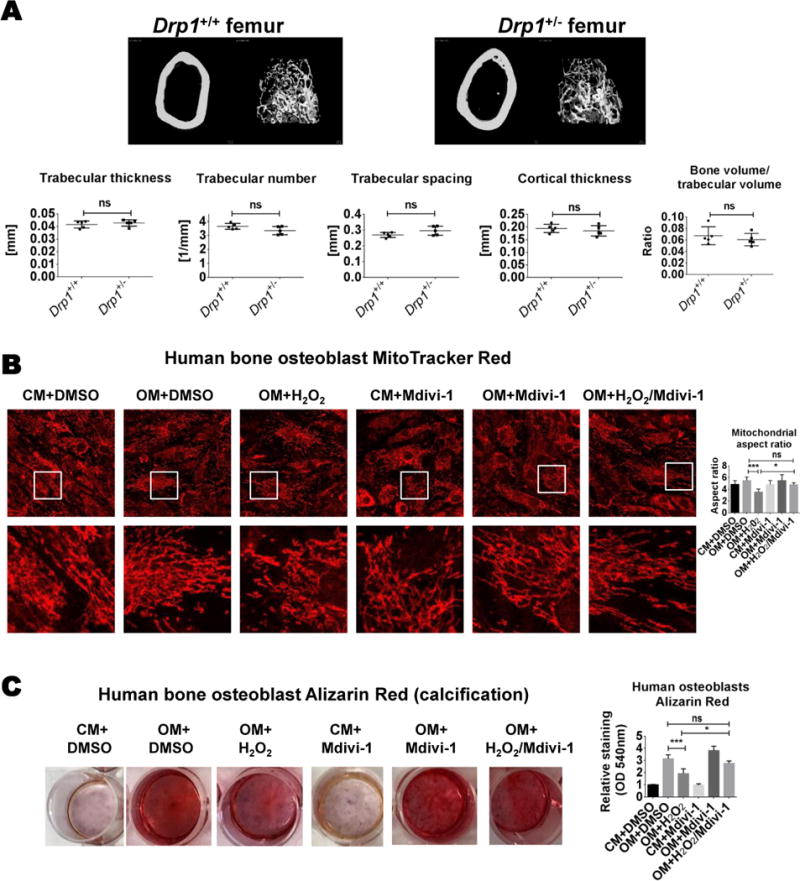
(A) Representative femur cortical and trabecular bone micro-CT images and trabecular thickness, trabecular number, trabecular spacing, cortical thickness, and bone volume/trabecular volume quantification (N=5 C57Bl/6;129 male mice/group). (B) Two week cultured MitoTracker Red stain (white boxes indicate higher magnification area of corresponding images below) and mitochondrial aspect ratio quantification, and (C) three-week cultured Alizarin Red staining and quantification from primary human bone osteoblasts treated in control media (CM) or osteogenic media (OM) with H2O2 (0.3 mmol/L), and with DMSO vehicle (0.01%) or Mdivi-1 (50 μmol/L); N=3; ns= not significant, * P <0.05, ***<0.001; error bars indicate STDEV.
DISCUSSION
We report the following novel findings: 1) DRP1 associates with calcified human cardiovascular tissue and cells; 2) DRP1 inhibition attenuates primary human SMC and VIC calcification; 3) Mdivi-1 reduces profibrotic COL1A1 secretion induced during human SMC osteogenic differentiation; and 4) Mdivi-1 attenuates oxidative stress-mediated human and mouse SMC calcification, in addition to suppressing oxidative stress mediated dysfunction in human osteoblasts. These results support our hypothesis that DRP1 promotes human cardiovascular calcification via regulating osteogenic differentiation. DRP1 associates with cell differentiation, with its inhibition blocking mouse myoblast47 and Drosophila follicle cell48 differentiation. In our calcification model, OM-reduced mitochondrial membrane potential and increased mitochondrial fragmentation could be attenuated by DRP1 inhibition, signifying a role of mitochondrial dynamics in osteogenic differentiation.
Along with its role in osteogenesis, TNAP is generally recognized as a marker of cell differentiation, and DRP1 inhibition reduces TNAP in pluripotent stem cells49. As DRP1 inhibition reduced TNAP activity in OM treated human SMCs and H2O2 treated mouse SMCs, TNAP reduction is a mechanistic component through which DRP1 regulates the reprogramming network that specifies SMC osteogenic differentiation. In contrast to the reduced TNAP activity we observed in human SMCs, DRP1 inhibition rescues oxidative stress driven TNAP reductions in human bone osteoblast-like cells50. Similarly, we found that Mdivi-1 suppressed oxidative stress mediated mineralization loss in primary human osteoblasts. No defects in long bones have been reported in patients with mutations in the DNM1L gene encoding DRP1, and we did not observe changes in mouse bone density in Drp1+/− femurs; suggesting cell type specificity in DRP1 regulation of mineralization. In agreement, the transcriptional program of calcifying SMCs and osteoblasts has been reported to not overlap51, suggesting cell type specific mechanistic differences between bone and ectopic calcification, although cell heterogeneity may account for some expression pattern differences. Cell stress can produce cell type specific calcification responses, exemplified by oxidative stress driving SMC but inhibiting osteoblast mineralization35. Mfn2 but not Drp1 is elevated during mouse osteoblast differentiation, and Mfn2 knockdown reduces mitochondrial elongation and osteoblast differentiation52. Cells from humans that do not express any wild type DRP1 are resistant to H2O2 induced mitochondrial fragmentation24. In agreement, we observed reduced mitochondrial aspect ratio and mineralization in human osteoblasts treated with H2O2, which was suppressed by Mdivi-1. While H2O2 similarly decreased SMC mitochondrial aspect ratio, it increased SMC calcification, which was attenuated by DRP1 inhibition. As such, calcifying SMCs and bone osteoblasts may have different metabolic needs that are better supported by fatty acid oxidation and mitochondrial respiration or glycolysis to supply energy, along with corresponding mitochondrial morphology changes during differentiation.
As our VIC calcification condition did not involve TNAP induction, DRP1 inhibition attenuates cardiovascular cell calcification through additional mechanisms. Along with RUNX236, the transcription factor SOX9 is associated with cardiovascular calcification53. Regulation of SOX9 by DRP1 requires further in-depth analysis beyond the scope of the present study. Our data indicate DRP1 inhibition increased SOX9 expression in human SMCs, VICs, and H2O2 treated mouse SMCs. SOX9 inhibits valve calcification54. High variability of SOX9 has been reported in non-calcified vessels while calcified carotid lesions contain higher SOX953, making the role of SOX9 in cardiovascular calcification unclear. However, increased SOX9 may act in a compensatory means to attenuate disease progression54.
Our previous study associated macrophages with vascular calcification through the production of extracellular vesicles contributing to mineralization55. Whether macrophages play an additional role in calcification via mineral resorption by differentiating into osteoclast-like cells in the vasculature, however, remains unclear. Mdivi-1 suppressed PBMC osteoclastic differentiation, but not mineral resorption in fully formed osteoclast in vitro and we did not observe changes in MAC3 staining or TRAP activity in the aorta of heterozygous Drp1-deficient mice. Future studies may address the role of mitochondrial dynamics in monocytes and macrophages during in vivo calcification including conditions of enhanced oxidative stress, as H2O2 enhances osteoclastic differentiation56. Collagen is trafficked in vesicles from the endoplasmic reticulum (ER)57,58 prior to being secreted from cells, but the molecular mechanisms of how this is accomplished are not clear. As the human SMC increase in OM-induced COL1A1 secretion exceeded the transcriptional increase, one intriguing possibility is that these cells may activate auxiliary mechanisms to increase the rate of collagen secretion involving DRP1. Alternatively, Mdivi-1 may have indirectly altered collagen transport resulting in less COL1A1 secretion in human SMCs undergoing osteogenic differentiation. In rat liver, DRP1 has been observed at the ER, and in fractions containing secretory proteins59. In mice in which Drp1 is deleted in the liver, VLDL secretion, serum cholesterol, and triglycerides significantly decrease on a high fat diet without altering mitochondrial respiratory activity, liver ATP, or lipid content60, further supporting a role of DRP1 in cellular trafficking. In the present study, DRP1 inhibition in SMCs modestly increased intracellular calcium, while blocking extracellular matrix mineralization. Intracellular calcium and calcium binding proteins such as annexins have been associated with extracellular vesicles involved in the calcification process61. DRP1 interacts with Annexin A662. Annexin protein may maintain the architectural and functional features of ER exit sites63. Whether calcium regulation and annexins participate in DRP1 regulation of protein secretion and calcification requires further exploration. DRP1 is an ER resident protein in mouse β-cells, and dominant-negative Drp1 expression helps maintain β-cell ER structure under stressed conditions64. Together, these studies suggest a role of DRP1 in ER maintenance, which may explain our observation of decreased OM-induced COL1A1 secretion in Mdivi-1 treated human SMCs.
DRP1inhibition suppresses vascular disease pathology in diabetic ApoE-deficient mice13, and balloon12 and wire injury65 rodent models. In contrast, heterozygous Drp1-deficient mice did not have altered vascular pathology in a PCSK9-gain-of-function model, and DRP1 was not elevated in calcified plaque of the two most commonly used atherosclerosis mouse models, ApoE-deficient and Ldlr-deficient mice. We found that attenuation of mouse SMC calcification by Mdivi-1 required the addition of oxidative stress to the osteogenic differentiation condition. As oxidative stress is elevated in diabetic and injury models, DRP1 inhibition may require the presence of certain stress levels in order to attenuate disease pathology in murine cardiovascular models. Oxidized low density lipoprotein enhances SMC but suppresses osteoblast differentiation, increases cellular oxidative stress35, and induces DRP1 mediated mitochondrial fission in SMCs14. A recent study found that treating rat SMCs with the antioxidant flavonoid Quercetin reduced DRP1 abundance and phosphate induced calcification in an oxidative stress dependent manner in vitro, along with suppressing calcification in adenine fed rats66, a model of chronic kidney disease. While DRP1 abundance and direct inhibition of DRP1 remain to be examined in chronic kidney disease, mitochondrial diameter is increased in the kidneys of diabetic mice, and this increase is attenuated by dominant-negative DRP167. The diabetes medication metformin suppresses diabetes-accelerated atherosclerosis in mice through DRP1 inhibition13, and attenuates progression of arterial calcification in HIV-infected metabolic syndrome patients68. Given the connection between metabolic disease and cardiovascular calcification, conditions with enhanced oxidative stress, like diabetes and chronic kidney disease, may provide clinically relevant in vivo models in which to further assess DRP1 inhibition effects on cardiovascular calcification. Humans heterozygous for DRP1 deletion or missense mutations do not have associated medical issues24, and Mdivi-1 does not produce organ toxicity in rats69; however, several concerns need to be addressed before assessing DRP1 inhibition in a clinical setting70, particularly under prolonged treatment conditions. Despite high unmet clinical needs of cardiovascular calcification, the underlying mechanisms remain obscure. Our current study identifies DRP1 as a novel regulator of cellular fibrocalcific response, which may lead to further research supporting the development of new therapies to combat cardiovascular calcification, a global health burden.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
Dynamin-related protein 1 (DRP1) regulates mitochondrial fission.
Mitochondrial morphology changes during cell differentiation, and DRP1 has been shown to promote the differentiation of some cell types.
What New Information Does This Article Contribute?
A novel association of DRP1 to cardiovascular calcification, with increased DRP1 observed in calcified human cardiovascular tissue and cells.
DRP1 inhibition attenuated primary human smooth muscle cell and valve interstitial cell calcification, inhibited oxidative stress-mediated smooth muscle cell calcification, and prevented oxidative stress-mediated dysfunction in bone osteoblasts.
DRP1 is a promising therapeutic target for multiple diseases; however, the role of DRP1 in cardiovascular disease is not clearly defined. Here we show that DRP1 associates with calcified human plaques and regulates osteogenic differentiation of cardiovascular cells. Additionally, we report a novel function of DRP1 in regulating osteogenic differentiation-induced type 1 collagen secretion. These results support a role of mitochondrial dynamics in the opposite mineralization response of vascular and bone cells to oxidative stress, and better define the role of DRP1 and mitochondrial dynamics in cardiovascular disease.
Acknowledgments
We thank Andrew Mlynarchik, Hengmin Zhang, Michael Creager, Brett Pieper, Eugenia Shvartz, Jose Luiz Figueiredo, Jung Choi, Alex Mojcher, and Tan Pham for technical assistance. We also thank Ken Mizuno and Jiro Matsumoto for their administrative support, and Carlijn Bouten for providing CNA probe.
SOURCES OF FUNDING
This study was supported by a research grant from Kowa Company, Ltd. (Tokyo, Japan, to M. A.) and the National Institutes of Health grants (R01HL114805 and R01HL136431 to E. A.)
Nonstandard Abbreviations and Acronyms
- AAV
adeno associate virus
- ApoE−/−
apolipoprotein E-deficient
- COL1A1
type 1 collagen
- CM
control media
- DNM1L
dynamin 1 like
- DRP1
dynamin-related protein 1
- ER
endoplasmic reticulum
- H2O2
hydrogen peroxide
- Ldlr−/−
low-density lipoprotein receptor-deficient
- M-CSF
macrophage colony-stimulating factor
- Mdivi-1
mitochondria division inhibitor-1
- Mfn2
mitofusin 2
- MSX2
Msh homeobox 2
- OM
osteogenic media
- PBMCs
peripheral blood mononuclear cells
- PCSK9
proprotein convertase subtilisin/kexin type 9
- PPi
pyrophosphate
- PKC
protein kinase c
- RANKL
receptor activator of nuclear factor kappa-B ligand
- RUNX2
runt-related transcription factor 2
- SMCs
smooth muscle cells
- SOX9
SOX gene family member 9
- TNAP
tissue non-specific alkaline phosphatase
- TMRE
tetramethylrhodamine ethyl ester
- TRAP
tartrate-resistant acid phosphatase
- VICs
valve interstitial cells
Footnotes
DISCLOSURES
None.
References
- 1.Smirnova E, Griparic L, Shurland DL, van der Bliek AM. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell. 2001;12:2245–2256. doi: 10.1091/mbc.12.8.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kasahara A, Scorrano L. Mitochondria: from cell death executioners to regulators of cell differentiation. Trends Cell Biol. 2014;24:761–770. doi: 10.1016/j.tcb.2014.08.005. [DOI] [PubMed] [Google Scholar]
- 3.Johnson RC, Leopold JA, Loscalzo J. Vascular calcification: pathobiological mechanisms and clinical implications. Circ Res. 2006;99:1044–1059. doi: 10.1161/01.RES.0000249379.55535.21. [DOI] [PubMed] [Google Scholar]
- 4.Rensen SS, Doevendans PA, van Eys GJ. Regulation and characteristics of vascular smooth muscle cell phenotypic diversity. Neth Heart J. 2007;15:100–108. doi: 10.1007/BF03085963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Demer LL, Tintut Y. Inflammatory, Metabolic, and Genetic Mechanisms of Vascular Calcification. Arterioscler Thromb Vasc Biol. 2014;34:715–723. doi: 10.1161/ATVBAHA.113.302070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Duchen M. Mitochondria and calcium: from cell signaling to cell death. J Physiol. 2000;1:57–68. doi: 10.1111/j.1469-7793.2000.00057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Madal S, Lindgren AG, Srivastava AS, Clark AT, Banerjee U. Mitochondrial function controls proliferation and early differentiation potential of embryonic stem cell. Stem Cells. 2011;3:486–495. doi: 10.1002/stem.590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang C, Youle RJ. The role of mitochondria in apoptosis. Annu Rev Genet. 2009;43:95–118. doi: 10.1146/annurev-genet-102108-134850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Giwimani S, Munjal C, Tyagi N, Sen U, Metreveli N, Tyagi SC. Miochondrial division/mitophagy inhibitor (Mdivi) amerliorates pressure overload induced heart failure. PLoS One. 2012;3:e32388. doi: 10.1371/journal.pone.0032388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koseoglu S, Dilks JR, Peters CG, Fitch-Tewfik JL, Fadel NA, Jasuja R, Italiano JE, Jr, Haynes CL, Flaumenhaft R. Dynamin-related protein-1 controls fusion pore dynamics during platelet granule exocytosis. Arterioscler Thromb Vasc Biol. 2013;33:481–488. doi: 10.1161/ATVBAHA.112.255737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ashrafian H, Docherty L, Leo V, et al. A mutation in the mitochondrial fission gene Dnm1l leads to cardiomyopathy. PLoS Genet. 2010;6:e1001000. doi: 10.1371/journal.pgen.1001000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lim S, Lee SY, Seo HH, Ham O, Lee C, Park JH, Lee J, Seung M, Yun I, Han SM, Lee S, Choi E, Hwang KC. Regulation of mitochondrial morphology by positive feedback interaction between PKCδ and Drp1 in vascular smooth muscle cell. J Cell Biochem. 2015;116:648–660. doi: 10.1002/jcb.25016. [DOI] [PubMed] [Google Scholar]
- 13.Wang Q, Zhang M, Torres G, Wu S, Ouyang C, Xie Z, Zou MH. Metformin suppreses diabetes-accelerated atherosclerosis via the inhibition of Drp1-mediated mitochondrial fission. Diabetes. 2017;66:193–205. doi: 10.2337/db16-0915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Swiader A, Nahapetyan H, Faccin J, D’Angelo R, Mucher E, Elbaz M, Boya P, Vindis C. Mitophagy acts as a safeguard mechanism against human vascular smooth muscle cell apoptosis induced by atherogenic lipids. Oncotarget. 2016;7:28821–28835. doi: 10.18632/oncotarget.8936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen KH, Guo X, Ma D, Guo Y, Li Q, Yang D, Li P, Qiu X, Wen S, Xiao RP, Tang J. Dysregulation of HSG triggers vascular proliferative disorders. Nat Cell Biol. 2004;6:872–883. doi: 10.1038/ncb1161. [DOI] [PubMed] [Google Scholar]
- 16.Liu C, Ge B, He C, Zhang Y, Liu X, Lui K, Qian C, Zhang Y, Peng W, Guo X. Mitofusin 2 decreases intracellular lipids in macrophages by regulating peroxisome proliferator-activeated receptor-γ. Biochem Biophys Res Commun. 2014;450:500–506. doi: 10.1016/j.bbrc.2014.06.005. [DOI] [PubMed] [Google Scholar]
- 17.Guo YH, Chen K, Gao W, Li Q, Chen L, Wang GS, Tang J. Overexpression of Mitofusin 2 inhibited oxidized low-density lipoprotein induced vascular smooth muscle cell proliferation and reduced atherosclerotic lesion formation in rabbit. Biochem Biophys Res Commun. 2007;363:411–417. doi: 10.1016/j.bbrc.2007.08.191. [DOI] [PubMed] [Google Scholar]
- 18.Waterham HR, Koster J, van Roermund CW, Mooyer PA, Wander RJ, Leonard JV. A lethal defect of mitochondrial and peroxisomal fission. N Engl J Med. 2007;356:1736–1741. doi: 10.1056/NEJMoa064436. [DOI] [PubMed] [Google Scholar]
- 19.Vanstone JR, Smith AM, McBride S, Naas T, Holcik M, Antoun G, Harper ME, Michaud J, Sell E, Chakraborty P, Tetreault, Care4Rare Consortium. Majewski J, Baird S, Boycott KM, Dyment DA, MacKenzie A, Lines MA. DNM1L-related mitochondrial fission defect presenting as refractory epilepsy. Eur J Hum Gent. 2016;24:1084–1088. doi: 10.1038/ejhg.2015.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chao YH, Robak LA, Xia F, Koenig MK, Adesina A, Bacino CA, Scaqlia F, Bellen HJ, Wangler MF. Missense variants in the middle domain of DNM1L in cases of infantile encephalopathy alter peroxisomes and mitochondria when assayed in Drosophila. Hum Mol Genet. 2016;25:1846–1856. doi: 10.1093/hmg/ddw059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sheffer R, Douiev L, Edvardson S, Shaaq A, Tamimi K, Soiferman D, Meiner V, Saada A. Postnatal microcephaly and pain insensitivity due to a de novo heterozygous DMN1L mutation causing impaired mitochondrial fission and function. Am J Med Genet A. 2016;170:1603–1607. doi: 10.1002/ajmg.a.37624. [DOI] [PubMed] [Google Scholar]
- 22.Yoon G, Malam Z, Paton T, Marshall CR, Hyatt E, Ivakine Z, Scherer SW, Lee KS, Hawkins C, Cohn RD, Findings of Rare Disease Genes (Forge) in Canada Consortium Steering Committee Lethal disorder of mitochondrial fission caused by mutations in DNM1L. J Pediatr. 2016;171:313–316. doi: 10.1016/j.jpeds.2015.12.060. [DOI] [PubMed] [Google Scholar]
- 23.Fahrner JA, Liu R, Perry MS, Klein J, Chan DC. A novel de novo dominant negative mutation in DNM1L impairs mitochondrial fission and presents as childhood epileptic encephalopathy. Am J Med Genet A. 2016;170:2002–2011. doi: 10.1002/ajmg.a.37721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nasca A, Legati A, Baruffini E, Nolli C, Moroni I, Ardissone A, Goffrini P, Ghezzi D. Biallelic mutations in DNM1L are associated with a slowly progressive infantile encephalopathy. Hum Mutat. 2016;37:898–903. doi: 10.1002/humu.23033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mori K, Shioi A, Jono S, Nishizawa Y, Morii H. Dexamethasone enhances In vitro vascular calcification by promoting osteoblastic differentiation of vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 1999;19:2112–2118. doi: 10.1161/01.atv.19.9.2112. [DOI] [PubMed] [Google Scholar]
- 26.Cassidy-Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T, Kurth MJ, Shaw JT, Hinshaw JE, Green DR, Nunnari J. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev Cell. 2008;14:193–204. doi: 10.1016/j.devcel.2007.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rottenberg H, Wu S. Quantitative assay by flow cytometry of the mitochondrial membrane potential in intact cells. Biochim Biophys Acta. 1998;1404:393–404. doi: 10.1016/s0167-4889(98)00088-3. [DOI] [PubMed] [Google Scholar]
- 28.Qi X, Disatnik MH, Shen N, Sobel RA, Mochly-Rosen D. Aberrant mitochondrial fission in neurons induced by protein kinase C{delta} under oxidative stress conditions in vivo. Mol Biol Cell. 2011;22:256–265. doi: 10.1091/mbc.E10-06-0551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maniscalco WM, Finkelstein JN, Parkhurst AB. Dexamethasone increases de novo fatty acid synthesis in fetal rabbit lung explants. Pediatr Res. 1985;19:1272–7. doi: 10.1203/00006450-198512000-00010. [DOI] [PubMed] [Google Scholar]
- 30.Peng L, Men X, Zhang W, Wang H, Xu S, Fang Q, Liu H, Yang W, Lou J. Invovlment of dynamin-related protein 1 in free fatty acid-induced INS-1 derived cell apoptosis. PLoS One. 2012;7:e49258. doi: 10.1371/journal.pone.0049258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nishizuka Y. Intracellular signaling by hydrolysis of phospholipids and activation of protein kinase C. Science. 1992;258:607–614. doi: 10.1126/science.1411571. [DOI] [PubMed] [Google Scholar]
- 32.Korobova F, Ramabhadran V, Higgs HN. An actin-dependent step in mitochondrial fission mediated by the ER-associated formin INF2. Science. 2013;339:464–467. doi: 10.1126/science.1228360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Anesti V, Scorrano L. The relationship between mitochondrial shape and function and the cytoskeleton. Biochim Biophys Acta. 2006;1757:692–699. doi: 10.1016/j.bbabio.2006.04.013. [DOI] [PubMed] [Google Scholar]
- 34.Lee K, Kim H, Jeong D. Microtubule stabilization attenuates vascular calcification through the inhibition of osteogenic signaling and matrix vesicle release. Biochem Biophys Res Commun. 2014;451:436–441. doi: 10.1016/j.bbrc.2014.08.007. [DOI] [PubMed] [Google Scholar]
- 35.Mody N, Parhami F, Sarafian Ta, Demer LL. Oxidative stress modulates osteoblastic differentiation of vascular and bone cells. Free Radic Biol Med. 2001;31:509–519. doi: 10.1016/s0891-5849(01)00610-4. [DOI] [PubMed] [Google Scholar]
- 36.Byon CH, Javed A, Dai Q, Kappes JC, Clemens TL, Darley-Usmar VM, McDonald JM, Chen Y. Oxidative stress induces vascular calcification through modulation of the osteogenic transcription factor Runx2 by AKT signaling. J Biol Chem. 2008;283:15319–15327. doi: 10.1074/jbc.M800021200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goettsch C, Hutcheson JD, Aikawa M, Iwata H, Pham T, Nykjaer A, Kjolby M, Rogers M, Shibasaki M, Hagita S, Kramann R, Rader DJ, Libby P, Singh SA, Aikawa E. Sortilin mediates vascular calcification via its recruitment into extracellular vesicles. J Clin Invest. 2016;126:1323–1326. doi: 10.1172/JCI80851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hutcheson JD, Goettsch C, Bertazzo S, Maldonado N, Ruiz JL, Goh W, Yakusaki K, Faits T, Bouten C, Franck G, Quillard T, Libby P, Aikawa M, Weinbaum S, Aikawa E. Genesis and growth of extracellular-vesicle-derived microcalcification in atherosclerotic plaques. Nat Mater. 2016;15:335–343. doi: 10.1038/nmat4519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bouchareb R, Mahmut A, Nsaibia MJ, Boulanger MC, Dahou A, Lépine JL, Laflamme MH, Hadji F, Couture C, Trahan S, Pagé S, Bossé Y, Pibarot P, Scipione CA, Romagnuolo R, Koschinsky ML, Arsenault BJ, Marette A, Mathieu P. Autotaxin derived from lipoprotein(a) and vlave interstitial cells promotes inflammation and mineralization of the aortic valve. Circulation. 2015;132:677–690. doi: 10.1161/CIRCULATIONAHA.115.016757. [DOI] [PubMed] [Google Scholar]
- 40.Wakabayashi J, Zhang Z, Wakabayashi N, Tamura Y, Fukaya M, Kensler TW, Lijima M, Sesaki H. The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice. J Cell Biol. 2009;186:805–816. doi: 10.1083/jcb.200903065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Manczak M, Sesaki H, Kageyama Y, Reddy PH. Dynamin-related protein 1 heterozygote knockout mice do not have synaptic and mitochondrial deficiencies. Biochim Biophys Acta. 2012;1822:862–874. doi: 10.1016/j.bbadis.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bjørklund MM, Hollensen AK, Hagensen MK, Dagnaes-Hansen F, Christoffersen C, Mikkelsen JG, Bentzon JF. Induction of atherosclerosis in mice and hamsters without germline genetic engineering. Circ Res. 2014;114:1684–1689. doi: 10.1161/CIRCRESAHA.114.302937. [DOI] [PubMed] [Google Scholar]
- 43.Roche-Molina, Sanz-Rosa D, Cruz FM, Garcia-Prieto J, Lopez S, Abia R, Muriana FJ, Fuster V, Ibanez B, Bernal JA. Induction of sustained hypercholesterolemia by single adeno-associated virus-mediated gene transfer of mutant hPCSK9. Arterioscler Thromb Vasc Biol. 2015;35:50–59. doi: 10.1161/ATVBAHA.114.303617. [DOI] [PubMed] [Google Scholar]
- 44.Goettsch C, Hutcheson JD, Hagita S, Rogers MA, Creager MD, Pham T, Choi J, Mylnarchik AK, Pieper B, Kjolby M, Aikawa M, Aikawa E. A single injection of gain-of-function mutant PCSK9 adeno-associated virus vector induces cardiovascular calcification in mice with no genetic modification. Atherosclerosis. 2016;251:109–118. doi: 10.1016/j.atherosclerosis.2016.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Aikawa E, Nahrendorf M, Figueiredo JL, Swirski FK, Shtatland T, Kohler RH, Jaffer FA, Aikawa M, Weissleder R. Osteogenesis associates with inflammation in early-stage atherosclerosis evaluated by molecular imaging in vivo. Circulation. 2007;116:2841–2850. doi: 10.1161/CIRCULATIONAHA.107.732867. [DOI] [PubMed] [Google Scholar]
- 46.Doherty TM, Asotra K, Fitzpatrick LA, Qiao JH, Wilkin DJ, Detrano RC, Dunstan CR, Shah PK, Rajavashisth TB. Calcification in atherosclerosis: bone biology and chronic inflammation at the arterial crossroads. Proc Natl Acad Sci U S A. 2003;100:11201–11206. doi: 10.1073/pnas.1932554100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim B, Kim JS, Yoon Y, Santiago MC, Brown MD, Park JY. Inhibition of Drp1-depenent mitochondrial division impairs myogenic differentiation. Am J Physiol Regul Integr Comp Physiol. 2013;305:R927–R938. doi: 10.1152/ajpregu.00502.2012. [DOI] [PubMed] [Google Scholar]
- 48.Mitra K, Rikhy R, Lilly M, Lippincott-Schwartz J. DRP1-dependent mitochondrial fission initiates follicle cell differentiation during Drosophila oogenesis. J Cell Biol. 2012;197:487–497. doi: 10.1083/jcb.201110058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vazquez-Martin A, Cufi S, Corominas-Faja B, Oliveras-Ferraros C, Vellon L, Menendez JA. Mitochondrial fusion by pharmacological manipulation impedes somatic cell reprogramming to pluripotency: new insight into the role of mitophagy in cell stemness. Aging. 2012;4:393–401. doi: 10.18632/aging.100465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gan X, Huang S, Yu Q, Yu H, Yan SS. Blockade of Drp1 rescues oxidative stress-induced osteoblast dysfunction. Biochem Biophys Res Commun. 2015;468:719–725. doi: 10.1016/j.bbrc.2015.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Alves RD, Eijen M, van de Peppel J, van Leeuwen JP. Calcifying vascular smooth muscle cells and osteoblasts: independent cell types exhiting extracellular matrix and biomineralization-related mimicries. BMC Genomics. 2014;15:965. doi: 10.1186/1471-2164-15-965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Forni MF, Peloggia J, Trudeau K, Shirihai O, Kowaltowski AJ. Murine mesenchymal stem cell commitment to differentiation is regulated by mitochondrial dynamics. Stem Cells. 2016;34:743–755. doi: 10.1002/stem.2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tyson KL, Reynolds JL, McNair R, Zhang Q, Weissber PL, Shanahan CM. Osteo/chondrocytic transcription factors and their target genes exhibit distinct patterns of expression in human arterial calcification. Aterioscler Thromb Vasc Biol. 2003;23:489–494. doi: 10.1161/01.ATV.0000059406.92165.31. [DOI] [PubMed] [Google Scholar]
- 54.Peacock JD, Levay AK, Gillaspie DB, Tao G, Lincoln J. Reduced sox9 function promotes heart valve calcification phenotypes in vivo. Circ Res. 2010;106:712–719. doi: 10.1161/CIRCRESAHA.109.213702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.New SE, Goettsch C, Aikawa M, Marchini JF, Shibasaki M, Yabusaki K, Libby P, Shanahan CM, Croce K, Aikawa E. Macrophage‐derived matrix vesicles: an alternative novel mechanism for microcalcification in atherosclerotic plaques. Circ Res. 2013;113:72–77. doi: 10.1161/CIRCRESAHA.113.301036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fraser JHE, Helfrich MH, Wallace HM, Ralston SH. Hydrogen peroxide, but not superoxide, stimulates bone resorption in mouse calvariae. Bone. 1996;19:223–226. doi: 10.1016/8756-3282(96)00177-9. [DOI] [PubMed] [Google Scholar]
- 57.Saito K, Chen M, Bard F, Chen S, Zhou H, Woodley D, Polischuk R, Schekman R, Malhotra V. TANGO1 facilitates cargo loading at endoplasmic reticulum exit sites. Cell. 2009;136:891–902. doi: 10.1016/j.cell.2008.12.025. [DOI] [PubMed] [Google Scholar]
- 58.Jin L, Pahuja KB, Wicklifee KE, Gorur A, Baumgärtel C, Schekman R, Rape M. Ubiquitin-dependent regulation of COPII coat size and function. Nature. 2012;482:495–500. doi: 10.1038/nature10822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yoon Y, Pitts KR, Dahan S, McNiven MA. A novel dynamin-like protein associates with cytoplasmic vesicles and tubules of the endoplasmic reticulum in mammalian cells. J Cell Biol. 1998;140:779–793. doi: 10.1083/jcb.140.4.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang L, Ishihara T, Ibayashi Y, Tatsushima K, Setoyama D, Hanada Y, Takeichi Y, Sakamoto S, Yokota S, Mihara K, Kang D, Ishihara N, Takayanagi R, Nomura M. Disruption of mitochondrial fission in the liver protects mice from diet-induced obesity and metabolic deterioration. Diabetologia. 2015;58:2371–2380. doi: 10.1007/s00125-015-3704-7. [DOI] [PubMed] [Google Scholar]
- 61.Kapustin AN, Davies JD, Reynolds JL, McNair R, Jones GT, Sidibe A, Schurgers LJ, Skepper JN, Proudfoot D, Mayr M, Shanahan CM. Calcium regulates key components of vascular smooth muscle cell-derived matrix vesicles to enhance mineralization. Circ Res. 2011;109:e1–12. doi: 10.1161/CIRCRESAHA.110.238808. [DOI] [PubMed] [Google Scholar]
- 62.Chlystun M, Campanella M, Law AL, Duchen MR, Fatimathas L, Levine TP, Gerke V, Moss SE. Regulation of mitochondrial morphogenesis by Annexin A6. PLoS One. 2013;8:e53774. doi: 10.1371/journal.pone.0053774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shibata H, Kanadome T, Sugiura H, Yokoyama T, Yamamuro M, Moss SE, Maki M. A new role for annexin A11 in the early secretory pathway via stabilizing Sec31A protein at the endoplasmic reticulum exit sites (ERES) J Biol Chem. 2015;290:4981–4993. doi: 10.1074/jbc.M114.592089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wikstrom JD, Israeli T, Bachar-Wikstrom E, Swisa A, Ariav Y, Waiss M, Kaganovich D, Dor Y, Cerasi E, Leibowitz G. AMPK regulates ER morphology and function in stressed pancreatic β-cells via phosphorylation of DRP1. Mol Endocrinol. 2013;27:1706–1723. doi: 10.1210/me.2013-1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang L, Yu T, Lee H, O’Brien DK, Sesaki H, Yoon Y. Decreasing mitochondrial fission diminishes vascular smooth muscle cell migration and ameliorates intimal hyperplasia. Cardiovasc Res. 2015;106:272–283. doi: 10.1093/cvr/cvv005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cui L, Li Z, Chang X, Cong G, Hao L. Quercetin attenuates vascular calcification by inhibiting oxidative stress and mitochondrial fission. Vascul Pharmacol. 2017;88:21–29. doi: 10.1016/j.vph.2016.11.006. [DOI] [PubMed] [Google Scholar]
- 67.Galloway CA, Lee H, Nejjar S, Jhun BS, Yu T, Hsu W, Yoon Y. Transgenic control of mitochondrial fission induces mitochondrial uncoupling and relieves diabetic oxidative stress. Diabetes. 2012;61:2093–2104. doi: 10.2337/db11-1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fitch K, Abbara S, Lee H, Stavrou E, Sacks R, Michel T, Hemphill L, Torriani M, Grinspoon S. Effects of lifestyle modification and metformin on atherosclerotic indices among HIV-infected patients with the metabolic syndrome. AIDS. 2012;26:587–597. doi: 10.1097/QAD.0b013e32834f33cc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liu JM, Yi Z, Liu SZ, Chang JH, Dang XB, Li QY, Zhang YL. The mitochondrial division inhibitor mdivi-1 attenuates spinal cord ischemia-reperfusion injury both in vitro and in vivo: involvement of BK channels. Brain Res. 2015;1619:155–165. doi: 10.1016/j.brainres.2015.03.033. [DOI] [PubMed] [Google Scholar]
- 70.Rosdah AA, Holien KJ, Delbridge LM, Dusting GJ, Lim SY. Mitochondria fission – a drug target for cytoprotection of cytodestruction? Pharmacol Res Perspect. 2016;4:e00235. doi: 10.1002/prp2.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.