

Abstract
Transcriptional deregulation is one of the core tenets of cancer biology and is underpinned by alterations in both protein-coding genes and noncoding regulatory elements. Large regulatory elements, so-called super-enhancers (SEs), are central to the maintenance of cancer cell identity, and promote oncogenic transcription to which cancer cells become highly addicted. Such dependence on SE-driven transcription for proliferation and survival offers an Achilles’ heel for the therapeutic targeting of cancer cells. Indeed, inhibition of the cellular machinery required for the assembly and maintenance of SEs dampens oncogenic transcription and inhibits tumor growth. In this article, we review the organization, function, and regulation of oncogenic SEs and their contribution to the cancer cell state.
Transcriptional Deregulation in Cancer
The maintenance of a specific cell state is reliant on precisely regulated gene expression programs. Such regulation involves highly orchestrated interactions between transcription factors (TFs) and the general transcriptional machinery that control transcription initiation and elongation, as well as chromatin-dependent mechanisms that govern DNA accessibility and transcriptional activation. The hallmarks of cancer cells -sustained proliferation, replicative immortality, apoptotic evasion and metastasis — are supported by aberrant gene expression programs [1, 2]. Thus, not surprisingly, transcriptional deregulation driven by changes in the genetic and epigenetic landscape is a fundamental mechanism of cancer [3, 4]. The intimate relationship between transcriptional deregulation and oncogenesis is underscored by the observation that many oncogenes and tumor suppressor genes encode TFs, strongly implicating altered gene regulation as a key oncogenic mechanism [1–4]. Moreover, in a wide range of cancers, recurrent chromosomal translocations result in chimeric TFs with oncogenic function [5]. The argument for a central role of transcriptional deregulation in oncogenesis became more persuasive with the identification of recurrent mutations in DNA-binding TFs and chromatin remodelers that disrupt the cancer genome and the epigenome, with consequent adverse effects on gene regulation [4, 6].
In metazoans, yet another level of gene regulation is provided by c/s-regulatory elements or enhancers, which drive tissue-specific gene expression necessary for the development of complex multicellular systems [7–9]. Given their function in specifying cell fate, enhancers are ideally positioned to contribute to inappropriate cell fate choices such as hyperproliferation and prolonged survival, both of which are recurrent themes in cancer [10, 11]. Enhancers are capable of activating gene expression over both short and long distances, independent of their position and orientation with respect to transcription start sites [12]. These sequences are enriched in binding sites for TFs that specify cell lineage and/or signaling pathways that impinge on them, thereby acting as a platform to integrate the environmental and developmental cues necessary to orchestrate spatio-temporally controlled gene expression [13]. TF binding to enhancers results in the recruitment of the Mediator complex, which facilitates enhancer interaction with the basal transcription machinery and RNA polymerase II (Pol II) at promoters in a gene-specific manner, a process mediated by “looping” of the loaded enhancer to the cognate promoter [11, 14] (Figure 1). Distinct functional states of enhancers are characterized by a unique combination of histone modifications: active enhancers are marked by monomethylation at lysine 4 (K4me1), acetylation at lysine 27 (K27ac), and the absence of trimethylation at lysine 4 (K4me3) of the histone H3 protein [13]. H3K4me1 and H3K27ac marks are conferred by the mixed lineage leukemia (MLL) family of methyltransferases (MLL2/3/4) and the CREB-binding protein (CBP)/p300 acetyltransferases, respectively; whereas the H3K4me3 mark is erased by the H3K4me3-specific demethylase KDM5C [13, 15]. Thus, the enzymatic machinery that confers and removes these histone modifications play a crucial role in the establishment and maintenance of the enhancer landscape, and its dysregulation is often associated with cancer [6]. Enhancer activity is also characterized by Pol II mediated transcription of noncoding RNA (enhancer RNA or eRNA) that contributes to enhancer function and gene regulation [16].
Figure 1. Organization and function of super-enhancers (SEs).
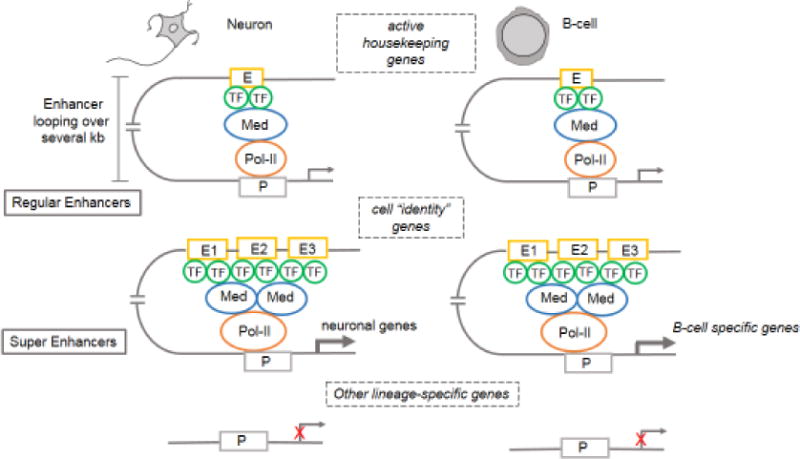
Transcription factor (TF) binding to enhancers (E) results in the recruitment of the Mediator (Med) complex, which facilitates enhancer interaction with the basal transcription machinery and RNA polymerase II (Pol II) at promoters (P) in a gene-specific manner, a process mediated by “looping” of the loaded enhancer to the cognate promoter. Enhancer looping can span large genomic distances (several kb). SEs are characterized by clustering of multiple constituent enhancers (E1–E3) located in close genomic proximity, and distinguished by the presence of multiple TF binding sites and increased density of the Mediator complex. Regular enhancers in neurons and hematopoietic cells are involved in the transcription of housekeeping genes, whereas, SEs regulate only cell-type-specific genes that confer “identity”. Non-lineage-specific genes are silenced in both cell types.
The recent identification of a subset of enhancers, called super-enhancers (SEs), has been instrumental in refining our understanding of cell-type-specific gene regulation [17–19]. Most cancer cells rely on aberrant transcription propelled by SEs, and such dependence offers productive targets for cancer therapy [18, 20–22]. Here we review the fundamental principles of SE organization and function, their formation in cancer cells and promising avenues for their selective therapeutic targeting, as well as open questions regarding their functional significance.
Super-enhancers: Organizing Principles
Super-enhancers are defined as clusters of enhancers in close genomic proximity that regulate the expression of genes that specify cell identity and functionally conform to cell type-specific biological processes. Regular enhancers and the genes they regulate, on the other hand, are common to a variety of cell types [17, 19, 23] (Figure 1). Some cell type-specific SEs overlap with other previously annotated regulatory regions implicated in tissue-specific gene expression [17], such as locus control regions [24], DNA methylation valleys [25], and transcription initiation platforms [26], suggesting that these diverse regulatory elements may typify distinct classes of SEs. SEs are frequently flanked by CTCF (CCCTC-binding factor) binding sites, suggesting that their activity may be constrained by boundary elements [27]. The notion of SEs stemmed from work in mouse embryonic stem cells (mESCs), in which large enhancer domains were involved in regulating the expression of key genes mediating pluripotency [19]. Subsequently, several studies not only extended the concept of SEs to diverse cell types, but also provided compelling evidence that they possess unique functional properties distinguishing them from regular enhancers [17, 18, 28]. These reports laid out the following iterative themes pertaining to SE-driven transcription: (i) SEs are densely occupied by H3K27ac and H3K4me1 enhancer marks, the Mediator complex (MED1) and bromodomain-containing protein 4 (BRD4); (ii) cell type-specific TFs and constituent enhancers within a SE exhibit an order-of-magnitude higher abundance of enhancer-associated chromatin marks and TFs as compared to the composition of regular enhancers [17–19, 28]; (iii) SE-driven genes are expressed at significantly higher levels than are genes under the control of regular enhancers [17–19, 28]; (iv) individual constituent enhancers of a SE are capable of increased transcriptional activation as compared to regular enhancers [19]; (v) SEs are characterized by differential binding of tissue-specific TFs. In mESCs, for example, both regular enhancers and SEs are equally enriched for Oct4, Sox2, and Nanog pluripotency marker binding sites, whereas, Klf4 and Esrrb binding sites are selectively localized to SEs [17, 19]. How the differences in TF binding at these enhancers contribute to differential gene regulation remains to be explored. Finally, (vi) SE-associated genes are much more sensitive to perturbation of associated enhancer-binding transcriptional regulator genes [18–21, 28, 29]. For example, knockdown of the mESC-specific TF Oct4 and the Mediator subunit Med12, leads to a more striking decrease in levels of SE-associated genes than of genes related to regular enhancers [19]. These observations support the hypothesis that enhancer function depends on cooperative and synergistic interactions among TFs, and that transcriptional output from enhancers with larger numbers of TF-binding sites will be more susceptible to changes in TF concentration. Intriguingly, master TFs enriched at SEs are themselves regulated by SE-driven transcription, suggesting a cooperative feedforward loop directed towards the maintenance of SEs at key target genes [19–22, 30]. Thus, the ability of SEs to regulate cell identity together with their vulnerability to perturbation could form the basis of cell state transitions associated with development, reprogramming and disease [17, 31, 32].
A fundamental question germane to SE biology lies in whether these enhancer clusters have unique functional significance compared with regular enhancers [33]. Several lines of evidence support the notion that SEs function as regulatory ensembles in which cooperative interactions between the constituent enhancers affect gene expression [18, 28, 34–37]. At certain well-studied loci, constituent enhancers within a SE physically interact with each other, exhibit interdependencies, and have nonredundant functions in gene regulation [28, 37]. Hnisz et al, for example, demonstrated cooperative behavior between enhancer elements at the Pou5f1 and Prdm14 SE loci as well as the concentration of signaling factors compared with typical enhancers [28]. Such cooperative interdependence between constituent enhancers is also evident in vivo, for example, at the mammary gland-specific Wap SE locus, where the establishment of a “seed enhancer” is essential to instituting additional enhancers required for optimal gene expression [37]. SE cooperativity is further highlighted by the observation that single TF binding promotes the assembly of large oncogenic and inducible SE domains [34, 35, 38]. Not all SEs conform to cooperative principles, however. Indeed, the constituent enhancers at the developmentally regulated alpha globin SE function independently and additively with respect to hematologic phenotype, gene expression, chromatin structure and chromosome conformation [39]. Similarly, cooperativity at the chromatin level is not always reflected at the transcriptional level. Proudhon et al for example, demonstrated that individual enhancer elements within the B-cell-specific Igk SE act synergistically, such that depletion of one element led to dissolution of the SE. Yet, this did not completely abolish transcription at the lgk locus, indicating an additive relationship among the remaining elements [36]. Together, these studies suggest that SE mapping based on chromatin marks is a good starting point for characterizing cell-type-specific gene regulation; however, the modus operandi of each SE could be highly context-specific, requiring rigorous genetic analysis to unravel the underlying regulatory grammar.
Super-enhancers: Critical Roles in Cancer
SEs maintain cancer cell identity
Oncogenic SEs were first identified in multiple myeloma cells, distinguished from regular enhancers by their higher densities of MED1 and BRD4 binding [18]. Functionally, most SEs were associated with genes that confer hematopoietic identity and contributed to the pathogenesis of multiple myeloma (MYC, IRF4, PRDM1 XBP1), as well as general oncogenes (CYCLIN D, MCL1, BCL-XL). These observations suggested that, similar to normal cells, cancer-associated SE domains mark lineage-restricted genes as well as putative oncogenes that normally serve as critical regulators of cell proliferation and apoptosis. Indeed, SEs mark lineage-specific TFs and oncogenes in a broad spectrum of cancers, including neuroblastoma [20], small-cell lung cancer (SCLC) [21], medulloblastoma [40], breast [41], esophageal [42] and gastric [43] cancers and melanoma [44]. The SEs associated with key oncogenes are unique to cancer cells and conspicuously absent in normal untransformed cells of identical lineage, suggesting that they are acquired during the course of tumorigenesis and underlie the oncogenic state in fundamental ways [17]. This prediction is further supported by the observation that most SEs acquired de novo segregate into functional categories recognized as hallmarks of cancer [1, 2, 17]. Together, these observations suggest that SEs facilitate the transition to a transformed state and contribute to its maintenance.
Although unique to cancer cells, the same oncogene can have a different SE architecture in diverse tumor types, further underscoring the role of SEs in maintaining cell identity. This scenario is best exemplified by tumor-specific SE profiles observed at the MYC and MYCN loci [11, 20, 21]; however, the significance of such diversely arranged SEs in gene regulation remains unclear. Thus far, the SE landscapes in established cancer cell lines have recapitulated the SE profiles identified in primary human tumors, supporting the hypothesis that SEs are key to the maintenance of tumor identity in vivo [20, 45].
SE-mediated regulation of cellular identity also provides a means to distinguish cancer subtypes. Medulloblastoma, for example, consists of four main subgroups based on underlying biochemical and genetic signatures. Mapping of the enhancer landscape in each sub-group revealed a unique set of SEs that correlated with tumor heterogeneity, independently predicted subgroup status and identified therapeutic vulnerabilities [40]. Importantly, analysis of SE-regulated TFs in these tumors facilitated reconstruction of the cell type-specific core regulatory circuitry, enabling the identification of LMX1A as a master TF in group 4 medulloblastoma. Interestingly, other genetically heterogeneous cancers, such as triple-negative breast cancer rely on a specific gene cluster driven by SEs to sustain proliferation and survival, thereby offering a uniform platform for therapeutically targeting this group of diverse tumors [41].
Cellular identity mediated by SEs also plays an important role during cell-fate transitions associated with cancer initiation. This contribution is illustrated by the association of melanoma initiation in BRAFV600E/p53 mutant zebrafish with reactivation of a neural crest cell state in a single melanocyte precursor cell [32]. Interestingly, this transition to a primitive cell state was linked to the establishment of SEs at sox10 and dlx2, two key genes involved in regulating neural crest cell specification. Indeed, human melanoma cells are associated with SEs at these gene loci, further emphasizing the marking of key cell identity genes as an essential feature of SEs. Together, these findings suggest a general strategy in which the discovery of unique SE domains in cancer cells could be used to predict the presence of candidate oncogenes and, on an entirely different level, their unique therapeutic vulnerabilities.
Convergence of oncogenic signaling pathways on SEs
Early studies in mESCs revealed that, as compared with regular enhancers, developmentally regulated SEs are enriched in one or more binding sites for the terminal TFs of the Wnt, Lif and Tgfβ pathways, signaling modules that function in the maintenance of stemness and pluripotency [28]. Interestingly, manipulation of these pathways resulted in larger expression changes in SE-associated genes compared with genes driven by regular enhancers [28]. These observations support a model in which SEs function as a platform to integrate developmentally regulated signaling to trigger appropriate gene expression. As an extension of this organizing principle, oncogenic SEs are enriched in TF binding sites pertinent to specific signaling pathways upon which cancer cells depend. In colorectal cancer cells driven by the oncogenic WNT pathway, for example, associated SEs are enriched in binding sites for TCF4, the terminal TF of the WNT pathway [28]. Similarly, SE-associated genes in SCLC are enriched for E2F1 binding sites, consistent with loss of the retinoblastoma tumor suppressor pathway and unrestrained E2F1 activity in this disease [21, 46]. This link between oncogenic signaling and transcription also operates in distinguishing cancer subtypes that rely on distinct signaling pathways. In estrogen receptor (ER)-positive breast cancer cells, for example, SE-associated genes are enriched for ERα binding, whereas in triple-negative breast cancer cells that lack steroid hormone expression, the associated SEs are enriched for a different repertoire of oncogenic TFs [17, 41].
Oncogenic signaling has been shown to regulate SE function through multiple mechanisms [34, 47–49]. In one example, unrestrained RAS activity promotes the formation of oncogenic SEs, and pharmacologic inhibition of aberrant RAS signaling leads to the loss of active enhancer marks, decommissioning of SEs, and concomitant reduction in gene expression [47]. Consistent with these observations, the introduction of oncogenic RAS and RAF (HrasG12V, BRAFV600E) stimulates the deposition of active enhancer marks, resulting in a gain of SE domains and increased oncogenic transcription [47]. In addition, oncogenic signaling can directly modulate SE function by directing the engagement of the transcriptional machinery at enhancers, for example, through the regulation of transcriptional pause-release. Transcriptional pausing refers to promoter-proximal stalling of active RNA Pol II, which serves to regulate gene expression by restricting elongation [50]. In normal liver cells, active Hippo signaling restricts both regular enhancer and SE-mediated gene expression by facilitating Pol II pausing [48]. However, in liver cancer cells, the loss of Hippo signaling results in increased expression of YAP, which encodes the terminal TF of the Hippo pathway [51]. Consequential binding of YAP to cognate motifs within enhancer domains leads to recruitment of the Mediator complex and the cyclin-dependent kinase CDK9, to allow productive elongation and aberrant expression of growth-promoting genes. Thus, YAP-induced SE activation serves as an oncogenic mechanism in liver cancer [48].
Another intriguing aspect of the impact of multiple signaling cues on DNA regulatory sequences pertains to the dynamic loading and unloading of signaling TFs at SEs, as illustrated in T-cell acute lymphoblastic leukemia (T-ALL) driven by aberrant NOTCH1. Although oncogenic NOTCH1 exhibits pervasive binding to the T-ALL genome, only a subset of these target sites respond to perturbations in NOTCH levels and thus qualify as bona fide NOTCH targets. Intriguingly, these dynamically responsive sites are contained within SEs, and loss of NOTCH1 binding at these sites results in the depletion of active enhancer marks and extensive remodeling of the associated SE domains [52]. Together, these studies suggest that one of the general functions of SEs may be to channel oncogenic signaling pathways into gene expression programs that are required to sustain cancer cells.
Establishment and Regulation of Oncogenic Super-enhancers
Understanding how oncogenic SEs are formed during tumorigenesis is a rapidly emerging area of scientific inquiry. Broadly speaking, inappropriate SE formation and function may stem from (i) alterations in cis-regulatory elements (cis-REs) [38, 53, 54], (ii) focal and large-scale chromosomal rearrangements [20, 30, 55–59], and (iii) rewiring of the cellular TF network by viral oncogenes [60–62].
SEs acquired through alterations in cis-REs
Germline and somatic mutations in the noncoding genome result in the deregulated expression of key oncogenes and tumor suppressors through altered promoter and enhancer function. In the context of cancer cells, a causal relationship between altered cis-REs and aberrant SE formation was first demonstrated in T-ALL where short insertions ~8 kb upstream of the TAL1 oncogene introduced binding sites for the transcription factor MYB, resulting in the formation of a SE driving TAL1 expression [38]. MYB binding to these de novo sites facilitates SE formation through recruitment of CBP/p300 acetyltransferase and the TAL1 transcription factor complex, which drive key genes involved in leukemogenesis (Figure 2A). In addition to insertional mutations, single nucleotide polymorphisms (SNPs) associated with certain cancers have a direct bearing on the regulation of oncogenic SEs. In neuroblastoma, for example, SE formation at the LMO1 oncogene locus is contingent upon GATA3 binding at a conserved intronic GATA binding site conferred by the tumorigenic “G” allele [54]. Interestingly, in cells harboring the protective “T” allele (TATA) at this site, LMO1 lacks a SE and shows greatly reduced expression [54] (Figure 2B). Such alterations in cis-REs can also disrupt SEs associated with tumor suppressor genes. In chronic lymphocytic leukemia associated with the 15q15.1 risk locus, a SNP within the intronic SE of the pro-apoptotic gene BMF, disrupts a binding site for the transcription factor RELA, resulting in compromised enhancer activity, reduced BMF expression, and unrestrained anti-apoptotic BCL2 function [53]. Together, these examples illustrate how alterations in cis-REs modulate SE formation by routing tissue-specific TFs to key drivers and repressors of oncogenesis.
Figure 2. Mechanisms of oncogenic super-enhancer formation.
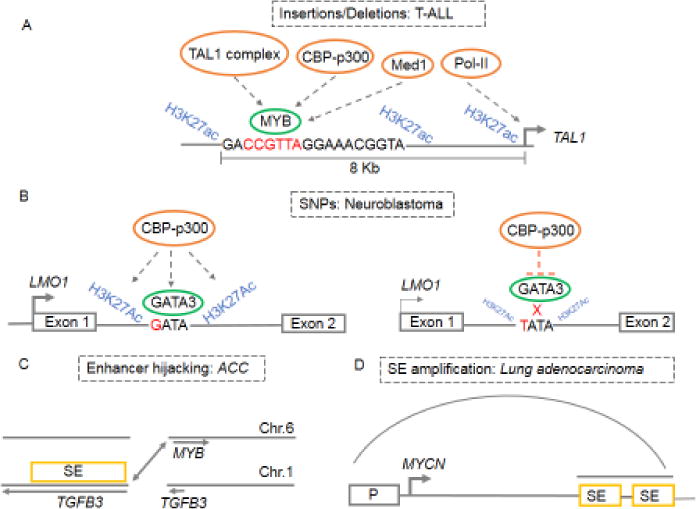
(A) Short insertion mutations that introduce novel MYB binding sites upstream of the TAL1 oncogene, resulting in the formation of a SE that drives TAL1 expression in T-ALL. MYB binding to these de novo sites facilitates SE formation through recruitment of CBP-p300 acetyl-transferase and the TAL1 transcription factor complex, which drive key genes involved in leukemogenesis [38]. (B) SE formation at the LMO1 oncogene in neuroblastoma is contingent upon GATA3 binding at a conserved intronic GATA binding site conferred by the tumorigenic “G” allele. Cells harboring the protective “T” allele (TATA) at this locus have impaired GATA3 binding, leading to diminished recruitment of H3K27Ac, resulting in decreased enhancer activity and greatly reduced LMO1 expression [54]. (C) A chromosomal translocation as found in adenoid cystic carcinoma (ACC) that repositions an unrelated SE in proximity to the MYB oncogene, resulting in its high expression [30]. (D) Focal amplification of SE regions as in lung adenocarcinoma, where a focal amplification ~450s kb downstream of the MYCN locus leads to SE formation and drives high expression of the oncogene [59]. Grey arcs represent physical interactions between SEs and the gene promoter (P).
SEs acquired through focal and large scale chromosomal rearrangements
Chromosomal rearrangements result in the juxtaposition of unrelated SEs to oncogenes, leading to their high-level expression. In adenoid cystic carcinoma, a chromosomal translocation repositions a distant SE in proximity to the MYB gene, resulting in its high expression [30] (Figure 2C). Moreover, MYB binding to the newly acquired SE generates a positive feedback loop that reinforces its own expression and activates a MYB-dependent oncogenic transcriptional program. Such “enhancer hijacking” is also evident in acute myeloid leukemia (AML), neuroblastoma, medulloblastoma, and colorectal cancer where structural rearrangements position the key oncogenic drivers EVI1, TERT, GFI1 and IGF2 under the regulatory control of SEs, leading to their sustained expression [55–58]. Such repositioning of a single enhancer can result in concomitant deregulation of multiple genes that may cooperatively contribute to tumorigenesis. This concept is illustrated in AML, where overexpression of EVI1 caused by hijacking of the GATA2 SE leads to the concomitant loss of GATA2 transcription [56]. Considering that GATA2 is a key regulator of hematopoiesis and that its loss is associated with AML, GATA2 haploinsufficiency might provide the precise context for EVI1-mediated oncogenic transformation [56, 63–65]. In addition to enhancer hijacking, aberrant oncogene expression is also driven by focal amplification of SEs, as seen with several cancer-related genes such as KLF5, USP12, PARD6B and MYC [59] (Figure 2D). In certain contexts, these amplified oncogenic SEs exhibit cell-type specific localization. For example, focally amplified 3’-SEs at the MYC locus exhibit distinct localization in T-ALL, AML, lung adenocarcinomas and uterine cancer, where they modulate MYC expression presumably through lineage-specific chromatin loops [49, 59]. A variation to this theme is observed in neuroblastoma, where a 350–2000 kb genomic region harboring the MYCN oncogene and its cognate SE is aberrantly amplified and contributes to high MYCN levels that drive tumorigenesis [20, 66].
SEs mediated by viral oncogenes
Virally mediated cellular transformation relies heavily on transcriptional output from the host cell, and is partly achieved through high levels of transcription driven by virus-induced SE formation at key genes involved in proliferation and survival. For example, during infection of human B cells by Epstein-Barr virus (EBV), the TFs encoded (EBNA2, 3A, 3C and EBNALP) or activated (RelA, RelB) by EBV induce the formation of SEs at key pro-survival and anti-apoptotic genes — MYC, MIR155, IKZF3 and BCL2 — resulting in massively upregulated transcription [60, 61]. This dependency on oncogenic transcription is highlighted by the observation that inhibiting viral oncoproteins and BRD4 result in the collapse of SE domains and the inhibition of cell growth [61]. Intriguingly, the expression of viral oncogenes per se can also be regulated by SEs. As depicted by human papillomavirus virus (HPV)-transformed cervical cancer cells, tandemly integrated copies of the HPV16 genome assemble SE-like elements that drive transcription of the E6/E7 oncogenes required to sustain unrestrained proliferation [62]. These lines of evidence suggest that the establishment of SE domains through cis and trans regulatory mechanisms, at key target genes, is a fundamental step toward tumorigenesis.
Targeting SE-driven Transcriptional Dependencies in Cancers
Cancer cells, unlike most normal healthy cells, generally possess increased levels of oncogenic transcriptional activity that facilitate growth-promoting pathways [3, 67]. Thus, inhibiting oncogenic transcription is an attractive therapeutic option, but presents significant challenges, considering that transcription is a fundamental biological process shared by all living cells and hence its targeting could have dire consequences on global gene expression [3, 68]. Thus, any clinically useful transcriptional inhibitor should selectively target oncogenic transcription with only minimal toxicity in normal cells. Recent studies have demonstrated that JQ1 (a competitive inhibitor of BRD4), and (a covalent inhibitor of CDKs 7 and 12), selectively kill cancer cells by inhibiting SE-driven oncogenic transcription, with both agents lacking systemic toxic effects in vivo [20–22, 29]. The targeting rationales for JQ1 and THZ1 and their mechanisms of action are discussed below.
Transcription initiation, pausing and elongation proceed through an ordered exchange of regulatory and enzymatic cofactors. SE-associated transcription depends, at the very least, on the cooperative binding of BRD4, the Mediator complex, and the stepwise recruitment of the CDK7-containing TFIIH initiation complex and the CDK9-containing p-TEFb elongation complex [50]. CDK7 phosphorylates RNA Pol II to initiate transcription, and plays an active role in promoter proximal pausing, by enabling the loading of the pause-inducing negative elongation factor (NELF) and DRB-sensitivity-inducing factor (DSIF) onto Pol II [69]. Release of transcriptional pausing and transition into productive elongation is mediated by p-TEFb (CDK9)-mediated phosphorylation of Pol II and DSIF [50]. In addition, BRD4 promotes SE assembly by initiating recruitment of the Mediator complex, and positively contributes to transcriptional elongation of SE-driven genes by promoting pause release [70]. Elongation is further facilitated by CDKs 12/13, which are also involved in mRNA processing [71, 72]. Thus, when considered as core regulators of the transcription cycle, the Mediator complex, BRD4, and the transcriptional CDKs offer attractive targets for inhibiting oncogenic transcription (Table 1).
Table 1.
Therapeutic targeting of SE-driven transcription in cancer
| Cancer model | Inhibitor | Effect on SE-driven transcription | Effect of SE inhibition on tumor biology | Ref. |
|---|---|---|---|---|
| DLBCL | JQ1 (BRD4) | Downregulation of SE-driven oncogenic and lineage-specific transcriptional circuits. | Decreased lymphoma infiltration in the bone marrow and improved overall survival | [29] |
| AML | JQ1 (BRD4) | Eviction of BRD4 and Mediator from select SE regions causing decreased expression of associated genes that are MYB targets and important for leukemogenesis | Impaired proliferation and triggering differentiation of leukemic blasts | [76] |
| Oncogenic Nras expression in mouse liver | iBET (BRD4) | Reduced expression of genes involved in SASP that are driven by SEs. | Decreased clearance of oncogenic senescent cells. | [81] |
| T-ALL, MYCN- amplified NB, SCLC, TNBC | THZ1 (CDK7) | Downregulation of SE-associated and tumor addictive and lineage specific gene expression, MYCN-driven transcriptional amplification | Decreased tumor volumes, growth and increased survival | [20–22, 41, 42] |
| AML | Cortistatin A (CDK8/19) | Upregulation of SE-associated genes linked to tumor suppression and lineage specification. | Reduction in disease progression, leukemic burden, and tumor volume, improved overall survival. | [85] |
| T-ALL | THZ531 (CDK12/13) | Downregulation of DNA damage response and SE-associated genes | Apoptosis | [84] |
| Ewing sarcoma | LEE011 (CDK4/6) | Downregulation of SE-associated ES dependency genes CyclinD1/CDK4 | Cytostasis and delayed growth | [87] |
SE, super-enhancer; DLBCL, diffuse large B-cell lymphoma; AML, acute myeloid leukemia; SASP, senescence-etory phenotype; T-ALL, T-cell acute lymphoblastic leukemia; NB, neuroblastoma;; SCLC, small-cell lung cancer; gative breast cancer; ES, Ewing sarcoma
In support of this idea, JQ1 selectively binds to the acetyl-lysine recognition domain of BRD4 and inhibits tumorigenesis by restricting the chromatin-dependent functions of this target at promoters and enhancers [73–75]. Consistent with the disproportionate loading of BRD4 at SEs [18, 29], and its role in positively regulating SE function [70], JQ1 treatment results in preferential depletion of BRD4 and CDK9 at these sites, resulting in Pol II stalling and impaired elongation. Such dependencies on BRD4 are also evident in SE-driven transcriptional programs required for the maintenance of pluripotency and oncogenic identity [29, 70]. For instance, in diffuse large B-cell lymphoma, JQ1 treatment abolishes the SE-driven transcription of lineage-specific TF genes and oncogenes, resulting in reduced tumor growth and improved survival [29]. Such dampening of SE-driven oncogenic transcription through BRD4 inhibition has also been demonstrated in other cancers [76–78].
The ability of BRD4 to collaborate with other chromatin regulators to mediate SE-dependent transcription opens exciting opportunities for combination therapies. In AML, for example, BRD4 inhibition leads to the eviction of the Mediator complex from a subset of SEs involved in the regulation of key leukemogenic genes, leading to tumor growth inhibition [76]. In MLL, the disruptor of telomeric silencing 1 -like (DOT1L)-mediated methylation of histone H3K79 promotes histone H4 acetylation to positively regulate BRD4 binding at SEs [79]. Dual inhibition of DOT1L and BRD4 leads to a synergistic blockade of leukemia cell growth and proliferation [79]. SE-mediated gene regulation also contributes to a senescence-associated secretory phenotype (SASP), an immune surveillance mechanism that evicts premalignant oncogenic senescent cells [80]. Activation of the SASP gene expression program is enabled by the formation of novel BRD4-enriched SEs in proximity to SASP genes and with concomitant decommissioning of SEs associated with proliferative genes [81]. In vivo, BRD4 inhibition results in the impaired clearance of oncogenic N-RAS expressing cells through disruption of SE-driven SASP transcriptional programs [81].
The extraordinary reliance of cancer cells on SE-driven transcription is further illustrated by their susceptibility to THZ1, a highly specific covalent inhibitor of CDK7 and to a lesser extent, CDK12 [20–22, 41]. THZ1 inhibits phosphorylation of the carboxyl-terminal domain (CTD) of RNA Pol II and thus hinders promoter proximal pausing [69]. As SEs are enriched in paused RNA Pol II [82, 83], THZ1-induced deficiency in pausing leads to diminished occupancy of Pol II at these enhancers, culminating in transcriptional inhibition. Indeed, THZ1 treatment leads to the collapse of SEs, resulting in massive downregulation of oncogenic transcription and inhibition of tumor growth in several cancer types (Table 1) [20–22, 41]. In all of these cancers, THZ1 specifically targets the SE-dependent expression of a subset of cell type-specific master TFs (RUNX1, MYCN and MYC) that are critical to the regulation of core transcriptional programs responsible for the maintenance of tumor cell identity, proliferation and survival.
SE-driven transcription can also be targeted by inhibiting CDK12, a positive regulator of transcriptional elongation [84], or by inhibiting the Mediator kinases CDK8/19, which negatively regulate SE function [85, 86]. In this context, selective inhibition of Mediator kinases in AML led to the upregulation of tumor suppressor genes and lineage-specific TFs, ultimately resulting in antileukemic activity [85]. Finally, guided by the principle that identification of SE-associated genes could reveal unappreciated oncogenic dependencies, Kennedy et al. have called attention to the SE-driven cyclin D1 locus in Ewing sarcoma, which may confer vulnerability to inhibition of the cyclin D/CDK4 pathway [87].
The underlying mechanism that accounts for the exquisite sensitivity of SE-associated genes to chromatin/transcriptional regulator inhibition is still uncertain. One explanation that has been put forward to account for the sensitivity of JQ1 in multiple myeloma is the disproportionate loading of BRD4 and Mediator at SE loci associated with MYC and other lineage-specific survival genes which are preferentially displaced by JQ1 [18]. This conclusion was strengthened by the demonstration of direct target engagement at SEs for BET inhibitors [73]. However, subsequent reports, albeit in AML cells, have shown that JQ1-mediated eviction of Mediator occurred in less than half of all SEs associated with cancer relevant genes, and therefore did not correlate with pre-existing levels of this transcriptional cofactor [76]. Rather, the SE-associated transcriptional programs that were most affected by JQ1 demonstrated higher levels of MYB binding.
In other studies, the heightened sensitivity of SE-associated genes to inhibition has been postulated to stem from at least two complementary mechanisms: (i) cooperativity among the constituent enhancers and (ii) the short half-lives of oncogenic TFs [20–22, 41]. Moreover, as master TFs enriched at SEs sustain their expression through autoregulatory feedforward loops, their depletion presumably results in dampened transcriptional output from the SEs that they regulate. This has been demonstrated in MYCN-amplified neuroblastoma where THZ1 treatment led to preferential downregulation of SE-associated genes, including MYCN, thus inhibiting its autoregulation, while simultaneously, suppressing MYCN-driven global transcription amplification [20]. Finally, cancer subtypes with varying transcriptional addictions have markedly different responsiveness to these inhibitors. For example, MYCN-amplified neuroblastoma and MYC-family expressing SCLC cells are much more sensitive to THZ1 than are MYCN non-amplified neuroblastoma and NSCLC respectively, due to the dependencies of the former tumors on deregulated MYC expression. [20, 21, 67, 88]. Nevertheless, the fact that these inhibitors have the potential to disrupt fundamental biological processes with eventual global consequences on gene expression cannot be discounted, especially since almost all human tissues have “normal” SEs that confer tissue specificity. It seems likely that within the established principles governing SEs, individual SE constituents may demonstrate differential sensitivities to inhibition based on the TFs and chromatin modulators that form part of the complex, the binding of each constituent, the level of dependence that such binding confers on the tumor cells, all of which would be critically context-dependent. Additional contributing factors could be oncogenic aberrations that may not be major drivers, but would contribute to the tumor phenotype and are driven, not by enhancer elements but by point mutations and post translational modifications. Clearly, further work is needed to carefully interrogate SE structure and function in each cancer type in the context of additional synthetic lethal oncogenic aberrations that are not dependent on increased enhancer activity but on factors that dictate tumor behavior, such as patient age at diagnosis (children vs. adult), tumor histology and propensity to metastasize to specific sites.
Concluding Remarks
In conclusion, the available evidence establishes a number of basic concepts regarding SEs in cancer: 1) they are present in multiple cancer types, 2) they are required for the maintenance of cancer cell identity, 3) they regulate critical oncogenes and confer tumor dependencies, and 4) they can be targeted through inhibition of chromatin and transcriptional regulators that are disproportionately bound to these regulatory elements SEs. Unresolved questions are the contributions of the individual constituents to the diverse roles of SEs as they relate to cancer phenotypes and how they selectively respond to therapeutic inhibition (see outstanding questions). Future studies focused on elucidating the three-dimensional organization of SEs into insulated neighborhoods and their perturbation by cancer-associated genetic and epigenetic changes will contribute to the understanding of their assembly and function. From a therapeutic standpoint, it will be necessary to elucidate the roles of all the components of the SE complex and how they are affected by inhibition, before additional targetable nodes can be identified. As SEs are key to the establishment and maintenance of cellular identity, elucidating the alterations that occur in SEs in drug-resistant and relapsed tumor models will be valuable in deciphering the underlying mechanisms as well as identifying new vulnerabilities. Finally, it is critical in these studies to establish that results from cell line models recapitulate the features of representative primary tumors, thus enabling assessment of the true significance of such SE-driven oncogenic dependencies.
Outstanding questions.
How do the contributions of the individual constituents of SE complexes differ in normal and cancer cells and within cancer subtypes?
How do SEs selectively respond to therapeutic inhibition?
How do oncogenic signaling pathways impinge on chromatin to influence SE assembly and function?
Does the SE landscape and organization change during the acquisition of drug resistance and relapse
Trends Box.
Super-enhancers (SEs) are large regulatory elements enabling cell type-specific gene regulation and the maintenance of cell identity.
SEs regulate cancer cell proliferation and survival as well as cell identity, through transcriptional regulation of genes that confer oncogenic traits and lineage specificity.
SEs integrate diverse oncogenic signaling pathways to effectively modulate gene expression.
Oncogenic SEs are acquired de novo during cellular transformation, often by still undefined processes.
Targeted inhibition of SE assembly and function hinders tumor viability and growth through selective abrogation of key transcriptional regulators.
Acknowledgments
We apologize to authors whose work could not be cited due to space limitations. This work was funded by National Institutes of Health/National Cancer Institute grant (NIH CA197336) and the Department of Defense (W81XWH-15-PRCRP-TTSA).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 3.Bradner JE, Hnisz D, Young RA. Transcriptional Addiction in Cancer. Cell. 2017;168(4):629–643. doi: 10.1016/j.cell.2016.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee TI, Young RA. Transcriptional regulation and its misregulation in disease. Cell. 2013;152(6):1237–51. doi: 10.1016/j.cell.2013.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nambiar M, Kari V, Raghavan SC. Chromosomal translocations in cancer. Biochim Biophys Acta. 2008;1786(2):139–52. doi: 10.1016/j.bbcan.2008.07.005. [DOI] [PubMed] [Google Scholar]
- 6.Morgan MA, Shilatifard A. Chromatin signatures of cancer. Genes Dev. 2015;29(3):238–49. doi: 10.1101/gad.255182.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Buecker C, Wysocka J. Enhancers as information integration hubs in development: lessons from genomics. Trends Genet. 2012;28(6):276–84. doi: 10.1016/j.tig.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bulger M, Groudine M. Functional and mechanistic diversity of distal transcription enhancers. Cell. 2011;144(3):327–39. doi: 10.1016/j.cell.2011.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ong CT, Corces VG. Enhancers: emerging roles in cell fate specification. EMBO Rep. 2012;13(5):423–30. doi: 10.1038/embor.2012.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Herz HM, Hu D, Shilatifard A. Enhancer malfunction in cancer. Mol Cell. 2014;53(6):859–66. doi: 10.1016/j.molcel.2014.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sur I, Taipale J. The role of enhancers in cancer. Nat Rev Cancer. 2016;16(8):483–93. doi: 10.1038/nrc.2016.62. [DOI] [PubMed] [Google Scholar]
- 12.Banerji J, Rusconi S, Schaffner W. Expression of a beta-globin gene is enhanced by remote SV40 DNA sequences. Cell. 1981;27(2 Pt 1):299–308. doi: 10.1016/0092-8674(81)90413-x. [DOI] [PubMed] [Google Scholar]
- 13.Calo E, Wysocka J. Modification of enhancer chromatin: what, how, and why? Mol Cell. 2013;49(5):825–37. doi: 10.1016/j.molcel.2013.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hnisz D, Day DS, Young RA. Insulated Neighborhoods: Structural and Functional Units of Mammalian Gene Control. Cell. 2016;167(5):1188–1200. doi: 10.1016/j.cell.2016.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shen H, et al. Suppression of Enhancer Overactivation by a RACK7-Histone Demethylase Complex. Cell. 2016;165(2):331–42. doi: 10.1016/j.cell.2016.02.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li W, Notani D, Rosenfeld MG. Enhancers as non-coding RNA transcription units: recent insights and future perspectives. Nat Rev Genet. 2016;17(4):207–23. doi: 10.1038/nrg.2016.4. [DOI] [PubMed] [Google Scholar]
- 17.Hnisz D, et al. Super-enhancers in the control of cell identity and disease. Cell. 2013;155(4):934–47. doi: 10.1016/j.cell.2013.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Loven J, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153(2):320–34. doi: 10.1016/j.cell.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Whyte WA, et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153(2):307–19. doi: 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chipumuro E, et al. CDK7 inhibition suppresses super-enhancer-linked oncogenic transcription in MYCN-driven cancer. Cell. 2014;159(5):1126–39. doi: 10.1016/j.cell.2014.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Christensen CL, et al. Targeting transcriptional addictions in small cell lung cancer with a covalent CDK7 inhibitor. Cancer Cell. 2014;26(6):909–22. doi: 10.1016/j.ccell.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kwiatkowski N, et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature. 2014;511(7511):616–20. doi: 10.1038/nature13393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parker SC, et al. Chromatin stretch enhancer states drive cell-specific gene regulation and harbor human disease risk variants. Proc Natl Acad Sci U S A. 2013;110(44):17921–6. doi: 10.1073/pnas.1317023110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li Q, et al. Locus control regions. Blood. 2002;100(9):3077–86. doi: 10.1182/blood-2002-04-1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xie W, et al. Epigenomic analysis of multilineage differentiation of human embryonic stem cells. Cell. 2013;153(5):1134–48. doi: 10.1016/j.cell.2013.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koch F, et al. Transcription initiation platforms and GTF recruitment at tissue-specific enhancers and promoters. Nat Struct Mol Biol. 2011;18(8):956–63. doi: 10.1038/nsmb.2085. [DOI] [PubMed] [Google Scholar]
- 27.Ing-Simmons E, et al. Spatial enhancer clustering and regulation of enhancer-proximal genes by cohesin. Genome Res. 2015;25(4):504–13. doi: 10.1101/gr.184986.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hnisz D, et al. Convergence of developmental and oncogenic signaling pathways at transcriptional super-enhancers. Mol Cell. 2015;58(2):362–70. doi: 10.1016/j.molcel.2015.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chapuy B, et al. Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer Cell. 2013;24(6):777–90. doi: 10.1016/j.ccr.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Drier Y, et al. An oncogenic MYB feedback loop drives alternate cell fates in adenoid cystic carcinoma. Nat Genet. 2016;48(3):265–72. doi: 10.1038/ng.3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Adam RC, Fuchs E. The Yin and Yang of Chromatin Dynamics In Stem Cell Fate Selection. Trends Genet. 2016;32(2):89–100. doi: 10.1016/j.tig.2015.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kaufman CK, et al. A zebrafish melanoma model reveals emergence of neural crest identity during melanoma initiation. Science. 2016;351(6272):aad2197. doi: 10.1126/science.aad2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pott S, Lieb JD. What are super-enhancers? Nat Genet. 2015;47(1):8–12. doi: 10.1038/ng.3167. [DOI] [PubMed] [Google Scholar]
- 34.Bojcsuk D, Nagy G, Balint BL. Inducible super-enhancers are organized based on canonical signal-specific transcription factor binding elements. Nucleic Acids Res. 2016 doi: 10.1093/nar/gkw1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brown JD, et al. NF-kappaB directs dynamic super enhancer formation in inflammation and atherogenesis. Mol Cell. 2014;56(2):219–31. doi: 10.1016/j.molcel.2014.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Proudhon C, et al. Active and Inactive Enhancers Cooperate to Exert Localized and Long-Range Control of Gene Regulation. Cell Rep. 2016;15(10):2159–69. doi: 10.1016/j.celrep.2016.04.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shin HY, et al. Hierarchy within the mammary STAT5-driven Wap super-enhancer. Nat Genet. 2016;48(8):904–11. doi: 10.1038/ng.3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mansour MR, et al. Oncogene regulation. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science. 2014;346(6215):1373–7. doi: 10.1126/science.1259037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hay D, et al. Genetic dissection of the alpha-globin super-enhancer in vivo. Nat Genet. 2016;48(8):895–903. doi: 10.1038/ng.3605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lin CY, et al. Active medulloblastoma enhancers reveal subgroup-specific cellular origins. Nature. 2016;530(7588):57–62. doi: 10.1038/nature16546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang Y, et al. CDK7-dependent transcriptional addiction in triple-negative breast cancer. Cell. 2015;163(1):174–86. doi: 10.1016/j.cell.2015.08.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jiang YY, et al. Targeting super-enhancer-associated oncogenes in oesophageal squamous cell carcinoma. Gut. 2016 doi: 10.1136/gutjnl-2016-311818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ooi WF, et al. Epigenomic profiling of primary gastric adenocarcinoma reveals super-enhancer heterogeneity. Nat Commun. 2016;7:12983. doi: 10.1038/ncomms12983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhou B, et al. INO80 governs superenhancer-mediated oncogenic transcription and tumor growth in melanoma. Genes Dev. 2016;30(12):1440–53. doi: 10.1101/gad.277178.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cejas P, et al. Chromatin immunoprecipitation from fixed clinical tissues reveals tumor-specific enhancer profiles. Nat Med. 2016;22(6):685–91. doi: 10.1038/nm.4085. [DOI] [PubMed] [Google Scholar]
- 46.Sengupta S, Henry RW. Regulation of the retinoblastoma-E2F pathway by the ubiquitin-proteasome system. Biochim Biophys Acta. 2015;1849(10):1289–97. doi: 10.1016/j.bbagrm.2015.08.008. [DOI] [PubMed] [Google Scholar]
- 47.Nabet B, et al. Deregulation of the Ras-Erk Signaling Axis Modulates the Enhancer Landscape. Cell Rep. 2015;12(8):1300–13. doi: 10.1016/j.celrep.2015.06.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Galli GG, et al. YAP Drives Growth by Controlling Transcriptional Pause Release from Dynamic Enhancers. Mol Cell. 2015;60(2):328–37. doi: 10.1016/j.molcel.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Herranz D, et al. A NOTCH1-driven MYC enhancer promotes T cell development, transformation and acute lymphoblastic leukemia. Nat Med. 2014;20(10):1130–7. doi: 10.1038/nm.3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Adelman K, Lis JT. Promoter-proximal pausing of RNA polymerase II: emerging roles in metazoans. Nat Rev Genet. 2012;13(10):720–31. doi: 10.1038/nrg3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yimlamai D, Fowl BH, Camargo FD. Emerging evidence on the role of the Hippo/YAP pathway in liver physiology and cancer. J Hepatol. 2015;63(6):1491–501. doi: 10.1016/j.jhep.2015.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang H, et al. NOTCH1-RBPJ complexes drive target gene expression through dynamic interactions with superenhancers. Proc Natl Acad Sci U S A. 2014;111(2):705–10. doi: 10.1073/pnas.1315023111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kandaswamy R, et al. Genetic Predisposition to Chronic Lymphocytic Leukemia Is Mediated by a BMF Super-Enhancer Polymorphism. Cell Rep. 2016;16(8):2061–7. doi: 10.1016/j.celrep.2016.07.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Oldridge DA, et al. Genetic predisposition to neuroblastoma mediated by a LMO1 super-enhancer polymorphism. Nature. 2015;528(7582):418–21. doi: 10.1038/nature15540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Valentijn LJ, et al. TERT rearrangements are frequent in neuroblastoma and identify aggressive tumors. Nat Genet. 2015;47(12):1411–4. doi: 10.1038/ng.3438. [DOI] [PubMed] [Google Scholar]
- 56.Groschel S, et al. A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in leukemia. Cell. 2014;157(2):369–81. doi: 10.1016/j.cell.2014.02.019. [DOI] [PubMed] [Google Scholar]
- 57.Northcott PA, et al. Enhancer hijacking activates GFI1 family oncogenes in medulloblastoma. Nature. 2014;511(7510):428–34. doi: 10.1038/nature13379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Weischenfeldt J, et al. Pan-cancer analysis of somatic copy-number alterations implicates IRS4 and IGF2 in enhancer hijacking. Nat Genet. 2017;49(1):65–74. doi: 10.1038/ng.3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang X, et al. Identification of focally amplified lineage-specific super-enhancers in human epithelial cancers. Nat Genet. 2016;48(2):176–82. doi: 10.1038/ng.3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gunnell A, et al. RUNX super-enhancer control through the Notch pathway by Epstein-Barr virus transcription factors regulates B cell growth. Nucleic Acids Res. 2016;44(10):4636–50. doi: 10.1093/nar/gkw085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhou H, et al. Epstein-Barr virus oncoprotein super-enhancers control B cell growth. Cell Host Microbe. 2015;17(2):205–16. doi: 10.1016/j.chom.2014.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dooley KE, Warburton A, McBride AA. Tandemly Integrated HPV16 Can Form a Brd4-Dependent Super-Enhancer-Like Element That Drives Transcription of Viral Oncogenes. MBio. 2016;7(5) doi: 10.1128/mBio.01446-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hahn CN, et al. Heritable GATA2 mutations associated with familial myelodysplastic syndrome and acute myeloid leukemia. Nat Genet. 2011;43(10):1012–7. doi: 10.1038/ng.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ostergaard P, et al. Mutations in GATA2 cause primary lymphedema associated with a predisposition to acute myeloid leukemia (Emberger syndrome) Nat Genet. 2011;43(10):929–31. doi: 10.1038/ng.923. [DOI] [PubMed] [Google Scholar]
- 65.Rodrigues NP, et al. Haploinsufficiency of GATA-2 perturbs adult hematopoietic stem-cell homeostasis. Blood. 2005;106(2):477–84. doi: 10.1182/blood-2004-08-2989. [DOI] [PubMed] [Google Scholar]
- 66.Yoshimoto M, et al. MYCN gene amplification. Identification of cell populations containing double minutes and homogeneously staining regions in neuroblastoma tumors. Am J Pathol. 1999;155(5):1439–43. doi: 10.1016/S0002-9440(10)65457-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lin CY, et al. Transcriptional amplification in tumor cells with elevated c-Myc. Cell. 2012;151(1):56–67. doi: 10.1016/j.cell.2012.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bhagwat AS, Vakoc CR. Targeting Transcription Factors in Cancer. Trends Cancer. 2015;1(1):53–65. doi: 10.1016/j.trecan.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nilson KA, et al. THZ1 Reveals Roles for Cdk7 in Co-transcriptional Capping and Pausing. Mol Cell. 2015;59(4):576–87. doi: 10.1016/j.molcel.2015.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Di Micco R, et al. Control of embryonic stem cell identity by BRD4-dependent transcriptional elongation of super-enhancer-associated pluripotency genes. Cell Rep. 2014;9(1):234–47. doi: 10.1016/j.celrep.2014.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bartkowiak B, et al. CDK12 is a transcription elongation-associated CTD kinase, the metazoan ortholog of yeast Ctk1. Genes Dev. 2010;24(20):2303–16. doi: 10.1101/gad.1968210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liang K, et al. Characterization of human cyclin-dependent kinase 12 (CDK12) and CDK13 complexes in C-terminal domain phosphorylation, gene transcription, and RNA processing. Mol Cell Biol. 2015;35(6):928–38. doi: 10.1128/MCB.01426-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Anders L, et al. Genome-wide localization of small molecules. Nat Biotechnol. 2014;32(1):92–6. doi: 10.1038/nbt.2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Delmore JE, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146(6):904–17. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Filippakopoulos P, et al. Selective inhibition of BET bromodomains. Nature. 2010;468(7327):1067–73. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bhagwat AS, et al. BET Bromodomain Inhibition Releases the Mediator Complex from Select cis-Regulatory Elements. Cell Rep. 2016;15(3):519–30. doi: 10.1016/j.celrep.2016.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ceribelli M, et al. A Druggable TCF4- and BRD4-Dependent Transcriptional Network Sustains Malignancy in Blastic Plasmacytoid Dendritic Cell Neoplasm. Cancer Cell. 2016;30(5):764–778. doi: 10.1016/j.ccell.2016.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yokoyama Y, et al. BET Inhibitors Suppress ALDH Activity by Targeting ALDH1A1 Super-Enhancer in Ovarian Cancer. Cancer Res. 2016;76(21):6320–6330. doi: 10.1158/0008-5472.CAN-16-0854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gilan O, et al. Functional interdependence of BRD4 and DOT1L in MLL leukemia. Nat Struct Mol Biol. 2016;23(7):673–81. doi: 10.1038/nsmb.3249. [DOI] [PubMed] [Google Scholar]
- 80.Coppe JP, et al. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010;5:99–118. doi: 10.1146/annurev-pathol-121808-102144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tasdemir N, et al. BRD4 Connects Enhancer Remodeling to Senescence Immune Surveillance. Cancer Discov. 2016;6(6):612–29. doi: 10.1158/2159-8290.CD-16-0217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Arner E, et al. Transcribed enhancers lead waves of coordinated transcription in transitioning mammalian cells. Science. 2015;347(6225):1010–4. doi: 10.1126/science.1259418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Core LJ, et al. Analysis of nascent RNA identifies a unified architecture of initiation regions at mammalian promoters and enhancers. Nat Genet. 2014;46(12):1311–20. doi: 10.1038/ng.3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhang T, et al. Covalent targeting of remote cysteine residues to develop CDK12 and CDK13 inhibitors. Nat Chem Biol. 2016;12(10):876–84. doi: 10.1038/nchembio.2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pelish HE, et al. Mediator kinase inhibition further activates super-enhancer-associated genes in AML. Nature. 2015;526(7572):273–6. doi: 10.1038/nature14904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Clarke PA, et al. Assessing the mechanism and therapeutic potential of modulators of the human Mediator complex-associated protein kinases. Elife. 2016;5 doi: 10.7554/eLife.20722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kennedy AL, et al. Functional, chemical genomic, and super-enhancer screening identify sensitivity to cyclin D1/CDK4 pathway inhibition in Ewing sarcoma. Oncotarget. 2015;6(30):30178–93. doi: 10.18632/oncotarget.4903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Nie Z, et al. c-Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell. 2012;151(1):68–79. doi: 10.1016/j.cell.2012.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]