

Abstract
The regulation of iron metabolism in biological systems centers on providing adequate iron for cellular function while limiting iron toxicity. Because mammals cannot excrete iron, mechanisms have evolved to control iron acquisition, storage, and distribution at both systemic and cellular levels. Hepcidin, the master regulator of iron homeostasis, controls iron flows into plasma through inhibition of the only known mammalian cellular iron exporter ferroportin. Hepcidin is feedback-regulated by iron status and strongly modulated by inflammation and erythropoietic demand. This review highlights recent advances that have changed our understanding of iron metabolism and its regulation.
Keywords: erythropoiesis, hepatocyte, infection, inflammation, iron metabolism
Introduction
Iron is an essential trace mineral for many biological processes, including oxygen transport and storage, oxidative phosphorylation, and the catalysis of many metabolic redox reactions. Its catalytic role depends on the ability of ferrous (Fe2+) and ferric (Fe3+) iron to respectively donate and accept electrons under conditions prevailing in biological systems. However, iron also promotes the formation of reactive oxygen species that can damage DNA, proteins, and lipids within cells (1, 2). Multiple mechanisms have evolved to chaperone iron and to regulate its concentrations through the coordinated modulation of transport, storage, and iron utilization so that adequate iron is available for physiological functions with tolerable toxicity. The other evolutionary challenge is that iron in the environment is oxidized, poorly soluble, and therefore costly to assimilate, favoring the appearance of biological mechanisms that conserve and recycle iron within the organism.
This minireview is focused on iron homeostasis in humans and other mammals where the iron economy is dominated by the production and turnover of red blood cells (erythrocytes).
The iron economy
Iron compartments and flows
Dietary iron is the sole source of iron in the body, except when erythrocyte transfusions (200–250 mg of iron/unit) or parenteral iron are given to treat anemia or iron deficiency. Because humans and other mammals lack a mechanism for controlled iron excretion, regulation of body iron content (3–4 g in adults) hinges on the control of dietary iron absorption. The two major forms of dietary are heme iron, bound within a protoporphyrin ring and abundant in animal hemoproteins such as hemoglobin or myoglobin, and non-heme iron, bound to other molecules. In humans, heme iron is absorbed more efficiently than non-heme iron. The distribution of iron to tissues relies almost entirely on the abundant plasma protein transferrin that can very tightly bind iron (K = ∼1020 m−1), but it is normally only 20–40% saturated with iron. Compared with total body iron, this is a very small compartment (∼3 mg) that turns over about eight times a day. Although all tissues require iron, most of it is in erythrocytes, as a component of heme in hemoglobin. Erythropoiesis, the process that generates erythrocytes, is the primary consumer of iron in the body, requiring in humans ∼24 mg of iron per day to generate new erythrocytes to replace those lost to senescence. In humans, nearly all (∼95%) of the plasma iron in steady-state conditions is loaded onto transferrin by macrophages that recycle iron from aged erythrocytes. Similar recycling activity on a smaller scale must operate in all tissues. Small normal losses of iron (1–2 mg per day), from sloughing of intestinal and skin cells or menstrual blood loss, are compensated for by the absorption of dietary iron, and they account for about 5% of plasma iron turnover.
Animal models
Iron metabolism in laboratory mice, the main model used to study iron metabolism, is generally similar to that of humans. However, mice consume much more food compared with their body mass, have greater iron losses relative to iron stores, and derive from the diet as much as 50% of their daily iron turnover, as estimated from ferrokinetics and the 40-day life span of murine erythrocytes. They generally do not consume meat and absorb dietary heme iron poorly (3).
Iron transport across cell membranes
Few mammalian transmembrane proteins are known to transport iron under physiological conditions. Divalent metal-ion transporter 1 (DMT1), ZRT/IRT-like protein 8 (ZIP8), and ZIP14 act as cellular importers of Fe2+ and have been functionally characterized (4–6). DMT1 transports iron optimally at an acidic pH but ZIP8 and ZIP14 at a neutral physiological pH. The lone mammalian cellular exporter of iron is ferroportin, which functions optimally at pH 7.4–8.0, near the pH of extracellular fluid. Ferroportin likely transports Fe2+ (7).
Absorption of dietary non-heme iron
Non-heme iron is absorbed in the duodenum, where acidic gastric secretions enhance iron solubility. Soluble inorganic iron is taken up in the proximal duodenum (8) by DMT1 (9, 10), which couples the transport of Fe2+ to a proton gradient. Despite the low luminal pH, the local mucosal generation of a proton gradient at the enterocyte brush-border membrane is critical for iron uptake by DMT1. The proton gradient is mainly generated by Na+/H+ exchanger-3 (NHE3), as mice lacking NHE3 have impaired iron absorption (11). Deletion of intestinal DMT1 results in very low iron absorption, severe anemia, and greatly reduced life span (12). Anemia is also seen in rare patients with loss-of-function mutations in DMT1 (13), but the interpretation of the phenotype is complicated by the parallel role of DMT1 in vacuolar export of iron within erythrocyte precursors. Intestinal DMT1 mRNA and protein are increased by iron deficiency and hypoxia (8, 14). DMT1 transports Fe2+, but luminal iron is largely Fe3+ requiring reduction prior to uptake by the enterocyte (4), in part by the apical membrane reductase duodenal cytochrome b (DCYTB). Like DMT1, DCYTB expression is increased during iron deficiency. However, the deletion of DCYTB has minor effects on iron absorption (15), suggesting that other unidentified brush-border reductases or reducing agents can compensate, including ascorbate.
Heme iron absorption
The characteristics of heme uptake suggest receptor-mediated endocytosis rather than passive diffusion. The folate transporter proton-coupled folate transporter (PCFT)2 (16) has been suggested as a candidate heme transporter (17), but its low affinity for heme and the poor absorption of heme in mice (3), despite abundant PCFT in the duodenum (18), have cast doubt on the role of PCFT in heme absorption.
Hepcidin–ferroportin axis controls major iron flows
Export of iron from enterocytes and its regulation by hepcidin
Although intestinal iron absorption is also regulated at the apical membrane of enterocytes, through increased DMT1 and DCYTB expression during iron deficiency (8), intestinal iron absorption is primarily regulated at the enterocyte basolateral membrane through control of iron release into extracellular fluid and plasma. Iron taken up at the apical membrane of enterocytes is either sequestered within the iron-storage protein ferritin or exported into the portal circulation through ferroportin. During their life span of a few days, enterocytes migrate from the crypts at the base of villi to their tips where they shed into the intestinal lumen, taking any residual ferritin-bound iron with them. In a remarkably streamlined regulatory mechanism, ferroportin also serves as the receptor for the iron-regulatory peptide hormone hepcidin, the master regulator of systemic iron homeostasis (19). Hepcidin binds to ferroportin causing its endocytosis, lysosomal degradation, and consequently reduced cellular iron export. Hepcidin is primarily produced by hepatocytes (20), and the expression of hepcidin is feedback-regulated by iron stores (Fig. 1), as well as importantly modulated by erythropoiesis and inflammation.
Figure 1.

Iron flows and their regulation by the hepcidin–ferroportin axis. Iron homeostasis depends on the balance between iron losses and absorption, and the match between iron release from stores in recycling macrophages and hepatocytes versus its utilization for biological processes, primarily erythropoiesis. Small normal losses of iron are compensated for by the absorption of dietary iron in the duodenum. The hepcidin–ferroportin interaction regulates all the major flows of iron into plasma: the release of iron from duodenal enterocytes, recycling macrophages, and hepatocyte stores.
Transport function of ferroportin
Ferroportin is essential for intestinal iron absorption, as the deletion of intestinal ferroportin results in severe anemia and iron accumulation within enterocytes (21). Ferroportin likely exports Fe2+, as judged by its ability also to transport cobalt and zinc (7), metals in which the 2+ oxidation state is more stable under laboratory conditions than for iron. The plasma iron carrier transferrin binds Fe3+, requiring the oxidation of iron exported from enterocytes. The multicopper ferroxidase hephaestin, on the enterocyte basolateral membrane, catalyzes the oxidation of iron released from enterocytes, and decreased hephaestin activity causes anemia and impaired dietary iron absorption (22). A related multicopper ferroxidase, ceruloplasmin, has both soluble and GPI-linked isoforms that convert Fe2+ to Fe3+ for loading onto transferrin in other tissues (e.g. iron-recycling macrophages, hepatocytes, retinal epithelial cells, and astrocytes). The deletion of hephaestin results in intestinal iron accumulation (22). Ferroxidases may promote iron export by maintaining low exofacial concentrations of Fe2+ and generating a gradient of Fe2+ concentration across the membrane.
Erythrophagocytosis by macrophages
Most body iron is in the hemoglobin of circulating erythrocytes (∼2–2.5 g). After ∼120 days in humans, erythrocytes display sufficient markers of senescence to be removed from the blood by macrophages through erythrophagocytosis. Most of the iron required for the production of new erythrocytes is recaptured by macrophages from phagocytosed aged or damaged erythrocytes. The major populations of macrophages responsible for steady-state erythrophagocytosis are Kupffer cells (resident tissue macrophages in the liver sinusoids) and red pulp splenic macrophages, but monocyte-derived macrophages can be temporarily recruited to the liver during increased erythrocyte clearance from circulation (23).
Recycling of erythrocytes into iron
The enzymatic breakdown of erythrocytes within the macrophage phagolysosome is followed by the degradation of hemoglobin to release the heme and finally iron, generating bilirubin and carbon monoxide as by-products. These processes may occur in different cellular compartments, with hemoglobin proteolysis in the phagolysosome and heme degradation in the cytosol (24), necessitating the export of heme from the phagolysosome prior to heme catabolism. The protein heme-responsive gene-1 (HRG-1) is expressed in macrophages and exports heme from the phagolysosome into the cytosol (25). To date, mammalian models lacking HRG-1 have not been reported, but suppression of HRG-1 in developing zebrafish results in developmental and erythropoietic defects (26). Heme degradation is catalyzed by the enzyme heme oxygenase 1 (HO-1), and the loss of HO-1 function causes macrophage death attributed to heme accumulation during erythrophagocytosis (27). An alternative mechanism for iron export from the phagolysosome may utilize DMT1 and its paralog Nramp1 transporting ferrous iron from the phagolysosome to the cytoplasm (28).
Export of iron from macrophages and its regulation by hepcidin
After release from heme, cytosolic iron within macrophages is either sequestered within ferritin or exported to plasma by ferroportin, depending on the balance between the hepcidin-induced endocytosis and lysosomal proteolysis of ferroportin versus transcriptional induction and translational derepression of ferroportin synthesis by cellular heme and iron, respectively (29, 30). Because ferroportin mediates all cellular iron export, hepcidin regulates both the acquisition of new iron from the diet and the release of iron from stores in hepatocytes and macrophages (Fig. 1) (31). Pathological iron retention in macrophages (“ferroportin disease”) (32) is associated with loss-of-function missense autosomal dominant mutations in ferroportin that do not appear to impact intestinal and placental iron transport (21). The clinical defect is selective for macrophages probably because recycling of iron-rich erythrocytes may require maximal ferroportin export capacity. By contrast, duodenal enterocytes rarely operate near their maximal transport capacity, only during recovery from hemorrhage or iron deficiency.
Regulation of hepcidin by iron-related signals
Hepcidin regulation by iron is mediated by the BMP pathway
In a classical positive homeostatic feedback loop, hepcidin production is transcriptionally regulated by iron. Sensing of iron status involves multiple pathways through which hepatocytes, the main source of hepcidin (20), sense circulating iron levels directly and also respond to iron-induced bone morphogenetic proteins (BMP) produced in neighboring hepatic sinusoidal endothelial cells (Fig. 2). BMP6 and BMP2 stimulate hepcidin mRNA production by hepatocytes, and the absence of either BMP results in iron overload caused by inadequate hepcidin production (33–36), suggesting that BMP2 and BMP6 may also function as a heterodimer. BMP signaling in hepatocytes and the control of basal and iron-stimulated hepcidin expression (37, 38) are carried out by heteromeric BMP receptors combining type 1 activin receptor-like kinase (ALK) 2 or ALK3 with type 2 BMP receptors BMPR2 or activin A receptor type 2A (ActR2a).
Figure 2.
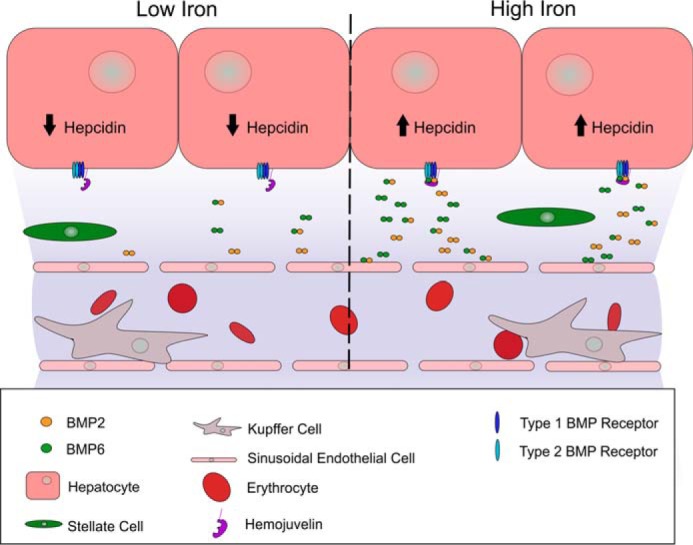
Regulation of hepcidin production by paracrine BMP signaling. Homo- and heterodimeric BMP2 and BMP6 are secreted by hepatic sinusoidal endothelial cells and bind to tetrameric complexes of type 1 and 2 BMP receptors on hepatocytes. Activation of the BMP receptor complex by BMP binding transcriptionally stimulates hepcidin production by hepatocytes. BMP secretion by sinusoidal endothelial cells increases during high-iron conditions and stimulates hepcidin production by hepatocytes. Additional iron-regulatory mechanisms in hepatocytes utilize transferrin receptors as sensors and modulate the BMP receptor signal, depending on plasma iron–transferrin concentration.
Iron sensing
Hepatocytes can directly sense plasma iron through a pathway involving the interaction between TFR1, TFR2, hemojuvelin (HJV, a GPI-linked membrane protein), and HFE, a major histocompatibility complex class 1-like protein. This pathway then modulates SMAD signaling by the BMP receptors. HFE is associated with TFR1, but this interaction is disrupted by the binding of iron-saturated transferrin (holotransferrin), resulting in HFE displacement (39, 40). HFE can then form complexes with TFR2, ALK3, and HJV to modulate hepcidin transcription. TFR2 was initially shown to interact with HFE in cell culture overexpression systems, and binding was increased by the presence of holotransferrin (41). Other studies indicate that TFR2 is stabilized by hepatocyte treatment with holotransferrin (42). In vivo, TFR2 and HFE regulate hepcidin through independent pathways as mice overexpressing HFE increase hepcidin expression even in the absence of functional TFR2. Also, the simultaneous loss of HFE and TFR2 results in a greater impairment in hepcidin regulation than either deletion individually (43). Additionally, while an interaction between TFR2 and HFE is detectable in cellular overexpression systems (41) no interaction is observed in vivo (44). HFE has also been demonstrated to interact with ALK3, stabilizing ALK3 and increasing hepcidin expression through BMP signaling (45).
Role of hemojuvelin
Mice with homozygous HJV disruption and humans with homozygous or compound heterozygous HJV mutations develop severe iron overload (46). HJV functions as a BMP co-receptor involved in hepcidin regulation by BMP signaling (47, 48). In vitro experiments have demonstrated that HJV can interact with HFE and TFR2, implicating HJV in the iron-sensing pathway. Evidence that HJV assists in iron sensing by HFE and TFR2 is provided by the lack of additive hepcidin suppression in mice lacking HJV alone compared with mice lacking both HJV and either functional HFE or TFR2 (49). Additionally, while BMP signaling and HFE/TFR2-mediated iron sensing require HJV, these pathways appear to be distinct, as the disruption of BMP6 and either HFE or TFR2 together results in additive hepcidin suppression compared with that observed in response to the loss of either BMP6 or HFE/TFR2 individually (49). Studies are needed to define the spatiotemporal molecular interactions between HFE, ALK3, HJV, and TFR2. The membrane serine protease Tmprss6 (matriptase 2) cleaves HJV and functions as a regulatory brake on this system (50). The expression of Tmprss6 is increased by iron deficiency and hypoxia (51–53), pointing to a third iron-regulated pathway controlling hepcidin.
Modulation of hepcidin and iron homeostasis by host defense and erythropoiesis
Iron and host defense
In systemic infection, iron is sequestered in macrophages, and extracellular iron concentrations decrease within hours. This response has long been thought to reduce the availability of iron to microbes, nearly all of which require iron for growth and proliferation. Supporting this concept, iron overload manifested by the saturation of transferrin with iron is associated with infections by “siderophilic” microbes.
Hepcidin, a component of innate immunity
Systemic infection increases hepcidin concentrations in plasma resulting in the sequestration of iron within macrophages and a reduction in dietary iron absorption. Mice lacking hepcidin show markedly decreased survival when infected with the siderophilic Vibrio vulnificus, and treatment with synthetic minihepcidins dramatically improves survival (54). The main mediator of increased hepcidin production during infection is IL-6 (55), which transcriptionally stimulates hepcidin synthesis via STAT3. We hypothesize that the main function of IL-6 and hepcidin-mediated iron sequestration is to reduce the availability of iron that is not bound to transferrin and so may be readily taken up by siderophilic microbes. As a side effect, the IL-6-driven increase in hepcidin and the corresponding decrease in iron availability for erythropoiesis are the major causes of the anemia of inflammation (56–58), in which iron-restricted anemia develops because of impaired mobilization of iron from stores.
Hepcidin suppression by stress erythropoiesis
Iron mobilization and increased iron absorption are early compensatory responses to anemia, such as occurs after major blood loss. Renal hypoxia stimulates the production of the hormone erythropoietin (EPO), which expands the production of erythrocytes. EPO also signals the erythrocyte progenitors (erythroblasts) to increase the production and secretion of the hormone erythroferrone (ERFE) (59). ERFE acts through an as of yet unknown receptor to suppress hepcidin production by hepatocytes, thereby increasing the release of iron from stores and its absorption from the diet, making more iron available for erythropoiesis. Mice lacking ERFE take longer to recover from anemia after blood loss or inflammation (59, 60).
Iron delivery to tissues
Transferrin and transferrin receptors
Iron released into plasma is rapidly bound by transferrin. Holotransferrin binds to transferrin receptors (TFR) 1 and 2, respectively for cellular iron acquisition (61) and iron-sensing by the liver (41). TFR1 is found in varying amounts in all cell types, but it is especially abundant in erythroblasts where it is essential for hemoglobin production. Genetic deletion of TFR1 is embryonic lethal (62). Tissue-specific knockouts showed that TFR1 is required for normal iron metabolism in skeletal muscle (63) and cardiomyocytes (64) and that TFR1 serves an unidentified iron-independent function in intestinal epithelial cells (65). Transferrin receptor 2, largely restricted to the liver and erythroid precursors (66), also interacts with holotransferrin but does not substantially contribute to iron uptake (67), acting primarily as sensor of systemic iron status (41).
Transferrin cycle
Cellular iron uptake is mediated by endocytosis of the holotransferrin–TFR1 complex, followed by acidification, which dissociates iron from transferrin, and then the release of apotransferrin from the cell. The protein six-transmembrane epithelial antigen of prostate (STEAP3) then reduces Fe3+ within the endosome. Mice lacking STEAP3 display impaired iron acquisition from transferrin in erythroblasts that manifests as anemia (68, 69). There may be other mechanisms of endocytic iron reduction, and iron sources other than transferrin may also be utilized by some cell types, as evidenced by non-lethal phenotypes (62–65, 68, 69) after STEAP3 or conditional TFR1 deletion. Erythropoiesis is impaired in rodents with loss-of-function mutations in DMT1, indicating that endosomal iron export is mediated by DMT1 in erythroblasts. However, embryos lacking DMT1 obtain iron from TFR1-mediated placental iron transport and are born with increased liver iron stores (10), indicating that other protein(s) can export iron from endosomes after iron-transferrin endocytosis.
Non-transferrin bound iron (NTBI)
Normally, practically all plasma iron is bound to transferrin. However, after ingestion of iron supplements, NTBI (iron bound to citrate, acetate, other organic anions of intermediary metabolism, or albumin) may appear in plasma (70), indicating that the binding of absorbed iron to transferrin is saturable. Hepatocytes take up NTBI and clear most NTBI from the portal circulation prior to its reaching the peripheral blood circulation. More commonly, plasma NTBI is seen in systemic iron overload, where plasma iron concentrations chronically exceed the binding capacity of transferrin. NTBI clearance does not depend on transferrin, as mice lacking transferrin readily load NTBI into tissues, including the liver, pancreas, and heart. Recent studies indicate that ZIP14 is necessary for NTBI loading by hepatocytes and pancreatic acinar cells (71) and contribute to NTBI uptake by pancreatic beta cells (72). NTBI uptake by cardiomyocytes may depend on the action of calcium channels, as calcium channel blockers prevent cardiac iron accumulation during iron overload (73, 74). The route of NTBI uptake in other tissues, such as the skeletal muscle (64), has not been determined.
Pathological tissue iron overload
Hereditary hemochromatosis is a disorder caused usually by mutations in genes (75–78) involved in regulating hepcidin expression, resulting in insufficient hepcidin production (79). Inadequate levels of hepcidin lead to excessive dietary iron absorption (80), excessive mobilization of macrophage iron stores (81), saturation of transferrin with iron, and the appearance of NTBI in plasma (82). Iron overload is also seen in β-thalassemia, a genetic disorder resulting from defective β-globin synthesis. Impaired β-globin production leads to chronic anemia with expansion of erythroblast populations that suppress hepcidin expression through the action of the hormone erythroferrone and possibly other mediators (59, 83). Similarly to hemochromatosis, reduced hepcidin levels cause increased iron absorption and tissue iron loading, further exacerbated in β-thalassemia major by a requirement for frequent blood transfusions. Tissue injury from excessive iron may cause cirrhosis, diabetes, cardiomyopathy, and endocrine failure, depending on the severity and tempo of iron accumulation.
Cellular iron homeostasis
Cellular iron regulation is distinct from its systemic regulation
Immersed in extracellular fluid whose iron concentrations are systemically regulated, each cell controls its iron uptake and storage to meet its individual iron requirement. Cell types involved in iron transport for systemic purposes (duodenal enterocytes, iron-recycling macrophages, hepatocytes, and placental trophoblast) are subject to both cellular and systemic regulation. Cellular iron is controlled post-transcriptionally by the interaction of an iron-response element (IRE) in mRNA with RNA-binding iron regulatory proteins (IRP) that bind to IREs when cellular iron levels are low. When cellular iron levels rise, IRPs undergo a conformational shift or are degraded, which ablates their interaction with IREs (84). IRPs binding to IREs have opposite effects on target protein synthesis depending on the location of the IRE in the 5′- or 3′-untranslated region (UTR) of the target mRNA. In the TFR1 mRNA, the IREs are in the 3′-UTR (85), and IRP binding stabilizes the mRNA and increases the synthesis of TFR1 protein to promote greater iron uptake during low-iron conditions. Unlike TFR1, ferritin mRNA has an IRE in the 5′-UTR (86), and when cellular iron is low, the binding of IRPs inhibits mRNA translation (87). During high-iron conditions, ferritin translation is derepressed, and cellular iron storage is favored. Other proteins involved in iron utilization and subcellular transport are also regulated by this system.
Ferritinophagy
The release of stored iron from ferritin is dependent on ferritinophagy, a regulated autophagosomal and lysosomal activity. Ferritin is first recruited to autophagosomes by the protein nuclear receptor co-activator 4 (NCOA4) (88). NCOA4 is enriched in autophagosomes and has been demonstrated to interact with ferritin prior to the fusion of autophagosomes with lysosomes in vitro (89). Mice lacking NCOA4 have increased tissue iron and an impaired ability to mobilize stored iron (90). Within lysosomes, ferritin is degraded, and its stored iron is released. How lysosomes export iron mobilized from ferritin has not been determined, but DMT1 may be responsible as DMT1 functions optimally at an acidic pH (4), and it has been reported to localize to lysosomes (91).
Iron chaperones
How iron reaches its various intracellular destinations is incompletely understood. In one model, iron taken up by a cell enters a cytosolic reservoir of weakly bound iron, the labile-iron pool (92), which can be used for metabolic functions or stored in ferritin. In a more recent model, iron is chaperoned by specific carrier proteins that limit its reactivity and target it to selected proteins. Members of the poly-r(C)-binding protein (PCBP) family, particularly the abundant PCBP1 and -2, bind iron and interact with proteins involved in iron storage and transport (93). PCBP1 has been shown to deliver iron to ferritin in vitro (94), and PCBP2 can bind iron and interact with both DMT1 and ferroportin in vitro (95, 96). Additionally, suppression of PCBP2 inhibits the cellular uptake of iron via DMT1 as well as the export of iron by ferroportin, indicating that PCBP2 may be a regulator of cellular iron transport. Studies investigating the role of PCBP1 and PCBP2 as iron chaperones in vivo have yet to be reported, but global deletion of these proteins results in embryonic lethality (97). Efficient iron transfer between endosomes and mitochondria is a major requirement for high-output synthesis of heme in erythroblasts, and it may depend on transient contact between their membranes, the “kiss and run” mechanism (98).
Local regulation of cellular iron by hepcidin
Consistent with the central role of hepatocytes in hepcidin regulation, mice with conditional deletion of hepcidin in hepatocytes recapitulate the phenotype of global hepcidin knockouts (20). However, other cell types also express hepcidin, including pancreatic beta cells, cardiomyocytes, placental syncytiotrophoblast, various kidney cell types, adipocytes, macrophages, and glial cells. The production of hepcidin in these non-hepatocyte cell populations contributes insignificantly to systemic hepcidin levels and therefore systemic iron homeostasis, but recent reports suggest a local role for extrahepatic hepcidin in the autocrine and paracrine regulation of cellular iron. Despite normal systemic iron homeostasis and circulating hepcidin levels (99), the specific deletion of hepcidin in cardiomyocytes causes cardiac iron depletion and premature death, attributed to increased ferroportin expression in cardiomyocytes and excessive cellular iron efflux. Mice with global deletion of hepcidin may be protected from cardiac iron depletion by increased levels of circulating iron, including NTBI (81). The deletion of ferroportin in cardiomyocytes yields the opposite phenotype, cardiac iron accumulation, but also results in premature death (100). These studies suggest an important role for locally produced hepcidin regulating cellular iron homeostasis in an IRP/IRE-independent manner, and they raise the possibility that similar mechanisms may operate in other cell types that express both ferroportin and hepcidin, such as macrophages, pancreatic beta cells, and adipose tissue.
Concluding remarks
In less than 2 decades, systemic iron homeostasis has been reconceptualized as a complex endocrine and paracrine system. Despite evident progress, important mechanistic details remain to be elucidated.
This work was supported by National Institutes of Health Fellowship T32 HL072752 (to R. C.) and Grant R01 DK 065029 (to T. G.). This is the first article in the Thematic Minireview series “Metals in Biology 2017: Iron transport, storage, and the ramifications.” The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- PCFT
- proton-coupled folate transporter
- GPI
- glycosylphosphatidylinositol
- ERFE
- erythroferrone
- BMP
- bone morphogenetic protein
- HJV
- hemojuvelin
- EPO
- erythropoietin
- NTBI
- non-transferrin bound iron
- IRE
- iron-response element
- IRP
- iron regulatory protein.
References
- 1. Knutson M. D., Walter P. B., Ames B. N., and Viteri F. E. (2000) Both iron deficiency and daily iron supplements increase lipid peroxidation in rats. J. Nutr. 130, 621–628 [DOI] [PubMed] [Google Scholar]
- 2. Walter P. B., Knutson M. D., Paler-Martinez A., Lee S., Xu Y., Viteri F. E., and Ames B. N. (2002) Iron deficiency and iron excess damage mitochondria and mitochondrial DNA in rats. Proc. Natl. Acad. Sci. U.S.A. 99, 2264–2269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fillebeen C., Gkouvatsos K., Fragoso G., Calvé A., Garcia-Santos D., Buffler M., Becker C., Schümann K., Ponka P., Santos M. M., and Pantopoulos K. (2015) Mice are poor heme absorbers and do not require intestinal Hmox1 for dietary heme iron assimilation. Haematologica 100, e334–e337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Illing A. C., Shawki A., Cunningham C. L., and Mackenzie B. (2012) Substrate profile and metal-ion selectivity of human divalent metal-ion transporter-1. J. Biol. Chem. 287, 30485–30496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pinilla-Tenas J. J., Sparkman B. K., Shawki A., Illing A. C., Mitchell C. J., Zhao N., Liuzzi J. P., Cousins R. J., Knutson M. D., and Mackenzie B. (2011) Zip14 is a complex broad-scope metal-ion transporter whose functional properties support roles in the cellular uptake of zinc and nontransferrin-bound iron. Am. J. Physiol. Cell Physiol. 301, C862–C871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang C. Y., Jenkitkasemwong S., Duarte S., Sparkman B. K., Shawki A., Mackenzie B., and Knutson M. D. (2012) ZIP8 is an iron and zinc transporter whose cell-surface expression is up-regulated by cellular iron loading. J. Biol. Chem. 287, 34032–34043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mitchell C. J., Shawki A., Ganz T., Nemeth E., and Mackenzie B. (2014) Functional properties of human ferroportin, a cellular iron exporter reactive also with cobalt and zinc. Am. J. Physiol. Cell Physiol. 306, C450–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gunshin H., Mackenzie B., Berger U. V., Gunshin Y., Romero M. F., Boron W. F., Nussberger S., Gollan J. L., and Hediger M. A. (1997) Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature 388, 482–488 [DOI] [PubMed] [Google Scholar]
- 9. Wang C. Y., and Knutson M. D. (2013) Hepatocyte divalent metal-ion transporter-1 is dispensable for hepatic iron accumulation and non-transferrin-bound iron uptake in mice. Hepatology 58, 788–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gunshin H., Fujiwara Y., Custodio A. O., Direnzo C., Robine S., and Andrews N. C. (2005) Slc11a2 is required for intestinal iron absorption and erythropoiesis but dispensable in placenta and liver. J. Clin. Invest. 115, 1258–1266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shawki A., Engevik M. A., Kim R. S., Knight P. B., Baik R. A., Anthony S. R., Worrell R. T., Shull G. E., and Mackenzie B. (2016) Intestinal brush-border Na+/H+ exchanger-3 drives H+-coupled iron absorption in the mouse. Am. J. Physiol. Gastrointest. Liver Physiol. 311, G423–G430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shawki A., Anthony S. R., Nose Y., Engevik M. A., Niespodzany E. J., Barrientos T., Öhrvik H., Worrell R. T., Thiele D. J., and Mackenzie B. (2015) Intestinal DMT1 is critical for iron absorption in the mouse but is not required for the absorption of copper or manganese. Am. J. Physiol. Gastrointest. Liver Physiol. 309, G635–G647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Iolascon A., and De Falco L. (2009) Mutations in the gene encoding DMT1: clinical presentation and treatment. Semin. Hematol. 46, 358–370 [DOI] [PubMed] [Google Scholar]
- 14. Shah Y. M., Matsubara T., Ito S., Yim S. H., and Gonzalez F. J. (2009) Intestinal hypoxia-inducible transcription factors are essential for iron absorption following iron deficiency. Cell Metab. 9, 152–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Choi J., Masaratana P., Latunde-Dada G. O., Arno M., Simpson R. J., and McKie A. T. (2012) Duodenal reductase activity and spleen iron stores are reduced and erythropoiesis is abnormal in Dcytb knockout mice exposed to hypoxic conditions. J. Nutr. 142, 1929–1934 [DOI] [PubMed] [Google Scholar]
- 16. Salojin K. V., Cabrera R. M., Sun W., Chang W. C., Lin C., Duncan L., Platt K. A., Read R., Vogel P., Liu Q., Finnell R. H., and Oravecz T. (2011) A mouse model of hereditary folate malabsorption: deletion of the PCFT gene leads to systemic folate deficiency. Blood 117, 4895–4904 [DOI] [PubMed] [Google Scholar]
- 17. Shayeghi M., Latunde-Dada G. O., Oakhill J. S., Laftah A. H., Takeuchi K., Halliday N., Khan Y., Warley A., McCann F. E., Hider R. C., Frazer D. M., Anderson G. J., Vulpe C. D., Simpson R. J., and McKie A. T. (2005) Identification of an intestinal heme transporter. Cell 122, 789–801 [DOI] [PubMed] [Google Scholar]
- 18. Qiu A., Jansen M., Sakaris A., Min S. H., Chattopadhyay S., Tsai E., Sandoval C., Zhao R., Akabas M. H., and Goldman I. D. (2006) Identification of an intestinal folate transporter and the molecular basis for hereditary folate malabsorption. Cell 127, 917–928 [DOI] [PubMed] [Google Scholar]
- 19. Nemeth E., Tuttle M. S., Powelson J., Vaughn M. B., Donovan A., Ward D. M., Ganz T., and Kaplan J. (2004) Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 306, 2090–2093 [DOI] [PubMed] [Google Scholar]
- 20. Zumerle S., Mathieu J. R., Delga S., Heinis M., Viatte L., Vaulont S., and Peyssonnaux C. (2014) Targeted disruption of hepcidin in the liver recapitulates the hemochromatotic phenotype. Blood 123, 3646–3650 [DOI] [PubMed] [Google Scholar]
- 21. Donovan A., Lima C. A., Pinkus J. L., Pinkus G. S., Zon L. I., Robine S., and Andrews N. C. (2005) The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 1, 191–200 [DOI] [PubMed] [Google Scholar]
- 22. Fuqua B. K., Lu Y., Darshan D., Frazer D. M., Wilkins S. J., Wolkow N., Bell A. G., Hsu J., Yu C. C., Chen H., Dunaief J. L., Anderson G. J., and Vulpe C. D. (2014) The multicopper ferroxidase hephaestin enhances intestinal iron absorption in mice. PLoS ONE 9, e98792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Theurl I., Hilgendorf I., Nairz M., Tymoszuk P., Haschka D., Asshoff M., He S., Gerhardt L. M., Holderried T. A., Seifert M., Sopper S., Fenn A. M., Anzai A., Rattik S., McAlpine C., et al. (2016) On-demand erythrocyte disposal and iron recycling requires transient macrophages in the liver. Nat. Med. 22, 945–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gottlieb Y., Truman M., Cohen L. A., Leichtmann-Bardoogo Y., and Meyron-Holtz E. G. (2012) Endoplasmic reticulum anchored heme-oxygenase 1 faces the cytosol. Haematologica 97, 1489–1493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. White C., Yuan X., Schmidt P. J., Bresciani E., Samuel T. K., Campagna D., Hall C., Bishop K., Calicchio M. L., Lapierre A., Ward D. M., Liu P., Fleming M. D., and Hamza I. (2013) HRG1 is essential for heme transport from the phagolysosome of macrophages during erythrophagocytosis. Cell Metab. 17, 261–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rajagopal A., Rao A. U., Amigo J., Tian M., Upadhyay S. K., Hall C., Uhm S., Mathew M. K., Fleming M. D., Paw B. H., Krause M., and Hamza I. (2008) Haem homeostasis is regulated by the conserved and concerted functions of HRG-1 proteins. Nature 453, 1127–1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kovtunovych G., Eckhaus M. A., Ghosh M. C., Ollivierre-Wilson H., and Rouault T. A. (2010) Dysfunction of the heme recycling system in heme oxygenase-1 deficient mice: effects on macrophage viability and tissue iron distribution. Blood 116, 6054–6062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Soe-Lin S., Apte S. S., Mikhael M. R., Kayembe L. K., Nie G., and Ponka P. (2010) Both Nramp1 and DMT1 are necessary for efficient macrophage iron recycling. Exp. Hematol. 38, 609–617 [DOI] [PubMed] [Google Scholar]
- 29. Delaby C., Pilard N., Puy H., and Canonne-Hergaux F. (2008) Sequential regulation of ferroportin expression after erythrophagocytosis in murine macrophages: early mRNA induction by haem, followed by iron-dependent protein expression. Biochem. J. 411, 123–131 [DOI] [PubMed] [Google Scholar]
- 30. Knutson M. D., Vafa M. R., Haile D. J., and Wessling-Resnick M. (2003) Iron loading and erythrophagocytosis increase ferroportin 1 (FPN1) expression in J774 macrophages. Blood 102, 4191–4197 [DOI] [PubMed] [Google Scholar]
- 31. Knutson M. D., Oukka M., Koss L. M., Aydemir F., and Wessling-Resnick M. (2005) Iron release from macrophages after erythrophagocytosis is up-regulated by ferroportin 1 overexpression and down-regulated by hepcidin. Proc. Natl. Acad. Sci. U.S.A. 102, 1324–1328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sabelli M., Montosi G., Garuti C., Caleffi A., Oliveto S., Biffo S., and Pietrangelo A. (2017) Human macrophage ferroportin biology and the basis for the ferroportin disease. Hepatology 65, 1512–1525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Meynard D., Kautz L., Darnaud V., Canonne-Hergaux F., Coppin H., and Roth M. P. (2009) Lack of the bone morphogenetic protein BMP6 induces massive iron overload. Nat. Genet. 41, 478–481 [DOI] [PubMed] [Google Scholar]
- 34. Koch P. S., Olsavszky V., Ulbrich F., Sticht C., Demory A., Leibing T., Henzler T., Meyer M., Zierow J., Schneider S., Breitkopf-Heinlein K., Gaitantzi H., Spencer-Dene B., Arnold B., Klapproth K., et al. (2017) Angiocrine Bmp2 signaling in murine liver controls normal iron homeostasis. Blood 129, 415–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Canali S., Zumbrennen-Bullough K. B., Core A. B., Wang C. Y., Nairz M., Bouley R., Swirski F. K., and Babitt J. L. (2017) Endothelial cells produce bone morphogenetic protein 6 required for iron homeostasis in mice. Blood 129, 405–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Enns C. A., Ahmed R., Wang J., Ueno A., Worthen C., Tsukamoto H., and Zhang A. S. (2013) Increased iron loading induces Bmp6 expression in the non-parenchymal cells of the liver independent of the BMP-signaling pathway. PLoS ONE 8, e60534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Steinbicker A. U., Bartnikas T. B., Lohmeyer L. K., Leyton P., Mayeur C., Kao S. M., Pappas A. E., Peterson R. T., Bloch D. B., Yu P. B., Fleming M. D., and Bloch K. D. (2011) Perturbation of hepcidin expression by BMP type I receptor deletion induces iron overload in mice. Blood 118, 4224–4230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mayeur C., Leyton P. A., Kolodziej S. A., Yu B., and Bloch K. D. (2014) BMP type II receptors have redundant roles in the regulation of hepatic hepcidin gene expression and iron metabolism. Blood 124, 2116–2123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schmidt P. J., Toran P. T., Giannetti A. M., Bjorkman P. J., and Andrews N. C. (2008) The transferrin receptor modulates Hfe-dependent regulation of hepcidin expression. Cell Metab. 7, 205–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lebrón J. A., West A. P. Jr., and Bjorkman P. J. (1999) The hemochromatosis protein HFE competes with transferrin for binding to the transferrin receptor. J. Mol. Biol. 294, 239–245 [DOI] [PubMed] [Google Scholar]
- 41. Goswami T., and Andrews N. C. (2006) Hereditary hemochromatosis protein, HFE, interaction with transferrin receptor 2 suggests a molecular mechanism for mammalian iron sensing. J. Biol. Chem. 281, 28494–28498 [DOI] [PubMed] [Google Scholar]
- 42. Johnson M. B., and Enns C. A. (2004) Diferric transferrin regulates transferrin receptor 2 protein stability. Blood 104, 4287–4293 [DOI] [PubMed] [Google Scholar]
- 43. Wallace D. F., Summerville L., Crampton E. M., Frazer D. M., Anderson G. J., and Subramaniam V. N. (2009) Combined deletion of Hfe and transferrin receptor 2 in mice leads to marked dysregulation of hepcidin and iron overload. Hepatology 50, 1992–2000 [DOI] [PubMed] [Google Scholar]
- 44. Schmidt P. J., and Fleming M. D. (2012) Transgenic HFE-dependent induction of hepcidin in mice does not require transferrin receptor-2. Am. J. Hematol. 87, 588–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wu X. G., Wang Y., Wu Q., Cheng W. H., Liu W., Zhao Y., Mayeur C., Schmidt P. J., Yu P. B., Wang F., and Xia Y. (2014) HFE interacts with the BMP type I receptor ALK3 to regulate hepcidin expression. Blood 124, 1335–1343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Huang F. W., Pinkus J. L., Pinkus G. S., Fleming M. D., and Andrews N. C. (2005) A mouse model of juvenile hemochromatosis. J. Clin. Invest. 115, 2187–2191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Babitt J. L., Huang F. W., Wrighting D. M., Xia Y., Sidis Y., Samad T. A., Campagna J. A., Chung R. T., Schneyer A. L., Woolf C. J., Andrews N. C., and Lin H. Y. (2006) Bone morphogenetic protein signaling by hemojuvelin regulates hepcidin expression. Nat. Genet. 38, 531–539 [DOI] [PubMed] [Google Scholar]
- 48. Andriopoulos B. Jr, Corradini E., Xia Y., Faasse S. A., Chen S., Grgurevic L., Knutson M. D., Pietrangelo A., Vukicevic S., Lin H. Y., and Babitt J. L. (2009) BMP6 is a key endogenous regulator of hepcidin expression and iron metabolism. Nat. Genet. 41, 482–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Latour C., Besson-Fournier C., Meynard D., Silvestri L., Gourbeyre O., Aguilar-Martinez P., Schmidt P. J., Fleming M. D., Roth M. P., and Coppin H. (2016) Differing impact of the deletion of hemochromatosis-associated molecules HFE and transferrin receptor-2 on the iron phenotype of mice lacking bone morphogenetic protein 6 or hemojuvelin. Hepatology 63, 126–137 [DOI] [PubMed] [Google Scholar]
- 50. Silvestri L., Pagani A., Nai A., De Domenico I., Kaplan J., and Camaschella C. (2008) The serine protease matriptase-2 (TMPRSS6) inhibits hepcidin activation by cleaving membrane hemojuvelin. Cell Metab. 8, 502–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Frýdlová J., Přikryl P., Truksa J., Falke L. L., Du X., Gurieva I., Vokurka M., and Krijt J. (2016) Effect of erythropoietin, iron deficiency and iron overload on liver matriptase-2 (TMPRSS6) protein content in mice and rats. PLoS ONE 11, e0148540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lakhal S., Schödel J., Townsend A. R., Pugh C. W., Ratcliffe P. J., and Mole D. R. (2011) Regulation of type II transmembrane serine proteinase TMPRSS6 by hypoxia-inducible factors: new link between hypoxia signaling and iron homeostasis. J. Biol. Chem. 286, 4090–4097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhang A. S., Anderson S. A., Wang J., Yang F., DeMaster K., Ahmed R., Nizzi C. P., Eisenstein R. S., Tsukamoto H., and Enns C. A. (2011) Suppression of hepatic hepcidin expression in response to acute iron deprivation is associated with an increase of matriptase-2 protein. Blood 117, 1687–1699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Arezes J., Jung G., Gabayan V., Valore E., Ruchala P., Gulig P. A., Ganz T., Nemeth E., and Bulut Y. (2015) Hepcidin-induced hypoferremia is a critical host defense mechanism against the siderophilic bacterium Vibrio vulnificus. Cell Host Microbe 17, 47–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Nemeth E., Rivera S., Gabayan V., Keller C., Taudorf S., Pedersen B. K., and Ganz T. (2004) IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J. Clin. Invest. 113, 1271–1276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. van Eijk L. T., John A. S., Schwoebel F., Summo L., Vauléon S., Zöllner S., Laarakkers C. M., Kox M., van der Hoeven J. G., Swinkels D. W., Riecke K., and Pickkers P. (2014) Effect of the antihepcidin Spiegelmer lexaptepid on inflammation-induced decrease in serum iron in humans. Blood 124, 2643–2646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kim A., Fung E., Parikh S. G., Valore E. V., Gabayan V., Nemeth E., and Ganz T. (2014) A mouse model of anemia of inflammation: complex pathogenesis with partial dependence on hepcidin. Blood 123, 1129–1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Gardenghi S., Renaud T. M., Meloni A., Casu C., Crielaard B. J., Bystrom L. M., Greenberg-Kushnir N., Sasu B. J., Cooke K. S., and Rivella S. (2014) Distinct roles for hepcidin and interleukin-6 in the recovery from anemia in mice injected with heat-killed Brucella abortus. Blood 123, 1137–1145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kautz L., Jung G., Valore E. V., Rivella S., Nemeth E., and Ganz T. (2014) Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat. Genet. 46, 678–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kautz L., Jung G., Nemeth E., and Ganz T. (2014) Erythroferrone contributes to recovery from anemia of inflammation. Blood 124, 2569–2574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Aisen P. (2004) Transferrin receptor 1. Int. J. Biochem. Cell Biol. 36, 2137–2143 [DOI] [PubMed] [Google Scholar]
- 62. Levy J. E., Jin O., Fujiwara Y., Kuo F., and Andrews N. C. (1999) Transferrin receptor is necessary for development of erythrocytes and the nervous system. Nat. Genet. 21, 396–399 [DOI] [PubMed] [Google Scholar]
- 63. Barrientos T., Laothamatas I., Koves T. R., Soderblom E. J., Bryan M., Moseley M. A., Muoio D. M., and Andrews N. C. (2015) Metabolic catastrophe in mice lacking transferrin receptor in muscle. EBioMedicine 2, 1705–1717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Xu W., Barrientos T., Mao L., Rockman H. A., Sauve A. A., and Andrews N. C. (2015) Lethal cardiomyopathy in mice lacking transferrin receptor in the heart. Cell Rep. 13, 533–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Chen A. C., Donovan A., Ned-Sykes R., and Andrews N. C. (2015) Noncanonical role of transferrin receptor 1 is essential for intestinal homeostasis. Proc. Natl. Acad. Sci. U.S.A. 112, 11714–11719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Fleming R. E., Migas M. C., Holden C. C., Waheed A., Britton R. S., Tomatsu S., Bacon B. R., and Sly W. S. (2000) Transferrin receptor 2: continued expression in mouse liver in the face of iron overload and in hereditary hemochromatosis. Proc. Natl. Acad. Sci. U.S.A. 97, 2214–2219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Herbison C. E., Thorstensen K., Chua A. C., Graham R. M., Leedman P., Olynyk J. K., and Trinder D. (2009) The role of transferrin receptor 1 and 2 in transferrin-bound iron uptake in human hepatoma cells. Am. J. Physiol. Cell Physiol. 297, C1567–C1575 [DOI] [PubMed] [Google Scholar]
- 68. Ohgami R. S., Campagna D. R., Greer E. L., Antiochos B., McDonald A., Chen J., Sharp J. J., Fujiwara Y., Barker J. E., and Fleming M. D. (2005) Identification of a ferrireductase required for efficient transferrin-dependent iron uptake in erythroid cells. Nat. Genet. 37, 1264–1269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Blanc L., Papoin J., Debnath G., Vidal M., Amson R., Telerman A., An X., and Mohandas N. (2015) Abnormal erythroid maturation leads to microcytic anemia in the TSAP6/Steap3 null mouse model. Am. J. Hematol. 90, 235–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Brittenham G. M., Andersson M., Egli I., Foman J. T., Zeder C., Westerman M. E., and Hurrell R. F. (2014) Circulating non-transferrin-bound iron after oral administration of supplemental and fortification doses of iron to healthy women: a randomized study. Am. J. Clin. Nutr. 100, 813–820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Jenkitkasemwong S., Wang C. Y., Coffey R., Zhang W., Chan A., Biel T., Kim J. S., Hojyo S., Fukada T., and Knutson M. D. (2015) SLC39A14 is required for the development of hepatocellular iron overload in murine models of hereditary hemochromatosis. Cell Metab. 22, 138–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Coffey R., and Knutson M. D. (2017) The plasma membrane metal-ion transporter ZIP14 contributes to non-transferrin-bound iron uptake by human beta cells. Am. J. Physiol. Cell Physiol. 312, C169–C175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Oudit G. Y., Sun H., Trivieri M. G., Koch S. E., Dawood F., Ackerley C., Yazdanpanah M., Wilson G. J., Schwartz A., Liu P. P., and Backx P. H. (2003) L-type Ca2+ channels provide a major pathway for iron entry into cardiomyocytes in iron-overload cardiomyopathy. Nat. Med. 9, 1187–1194 [DOI] [PubMed] [Google Scholar]
- 74. Kumfu S., Chattipakorn S., Chinda K., Fucharoen S., and Chattipakorn N. (2012) T-type calcium channel blockade improves survival and cardiovascular function in thalassemic mice. Eur. J. Haematol. 88, 535–548 [DOI] [PubMed] [Google Scholar]
- 75. Feder J. N., Gnirke A., Thomas W., Tsuchihashi Z., Ruddy D. A., Basava A., Dormishian F., Domingo R. Jr., Ellis M. C., Fullan A., Hinton L. M., Jones N. L., Kimmel B. E., Kronmal G. S., Lauer P., et al. (1996) A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat. Genet. 13, 399–408 [DOI] [PubMed] [Google Scholar]
- 76. Papanikolaou G., Samuels M. E., Ludwig E. H., MacDonald M. L., Franchini P. L., Dubé M. P., Andres L., MacFarlane J., Sakellaropoulos N., Politou M., Nemeth E., Thompson J., Risler J. K., Zaborowska C., Babakaiff R., et al. (2004) Mutations in HFE2 cause iron overload in chromosome 1q-linked juvenile hemochromatosis. Nat. Genet. 36, 77–82 [DOI] [PubMed] [Google Scholar]
- 77. Roetto A., Papanikolaou G., Politou M., Alberti F., Girelli D., Christakis J., Loukopoulos D., and Camaschella C. (2003) Mutant antimicrobial peptide hepcidin is associated with severe juvenile hemochromatosis. Nat. Genet. 33, 21–22 [DOI] [PubMed] [Google Scholar]
- 78. Camaschella C., Roetto A., Calì A., De Gobbi M., Garozzo G., Carella M., Majorano N., Totaro A., and Gasparini P. (2000) The gene TFR2 is mutated in a new type of haemochromatosis mapping to 7q22. Nat. Genet. 25, 14–15 [DOI] [PubMed] [Google Scholar]
- 79. Papanikolaou G., Tzilianos M., Christakis J. I., Bogdanos D., Tsimirika K., MacFarlane J., Goldberg Y. P., Sakellaropoulos N., Ganz T., and Nemeth E. (2005) Hepcidin in iron overload disorders. Blood 105, 4103–4105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Ajioka R. S., Levy J. E., Andrews N. C., and Kushner J. P. (2002) Regulation of iron absorption in Hfe mutant mice. Blood 100, 1465–1469 [DOI] [PubMed] [Google Scholar]
- 81. Lesbordes-Brion J. C., Viatte L., Bennoun M., Lou D. Q., Ramey G., Houbron C., Hamard G., Kahn A., and Vaulont S. (2006) Targeted disruption of the hepcidin 1 gene results in severe hemochromatosis. Blood 108, 1402–1405 [DOI] [PubMed] [Google Scholar]
- 82. Brissot P., Ropert M., Le Lan C., and Loréal O. (2012) Non-transferrin bound iron: a key role in iron overload and iron toxicity. Biochim. Biophys. Acta 1820, 403–410 [DOI] [PubMed] [Google Scholar]
- 83. Kautz L., Jung G., Du X., Gabayan V., Chapman J., Nasoff M., Nemeth E., and Ganz T. (2015) Erythroferrone contributes to hepcidin suppression and iron overload in a mouse model of β-thalassemia. Blood 126, 2031–2037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Guo B., Yu Y., and Leibold E. A. (1994) Iron regulates cytoplasmic levels of a novel iron-responsive element-binding protein without aconitase activity. J. Biol. Chem. 269, 24252–24260 [PubMed] [Google Scholar]
- 85. Müllner E. W., and Kühn L. C. (1988) A stem-loop in the 3′ untranslated region mediates iron-dependent regulation of transferrin receptor mRNA stability in the cytoplasm. Cell 53, 815–825 [DOI] [PubMed] [Google Scholar]
- 86. Hentze M. W., Caughman S. W., Rouault T. A., Barriocanal J. G., Dancis A., Harford J. B., and Klausner R. D. (1987) Identification of the iron-responsive element for the translational regulation of human ferritin mRNA. Science 238, 1570–1573 [DOI] [PubMed] [Google Scholar]
- 87. Gray N. K., and Hentze M. W. (1994) Iron regulatory protein prevents binding of the 43S translation pre-initiation complex to ferritin and eALAS mRNAs. EMBO J. 13, 3882–3891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Mancias J. D., Wang X., Gygi S. P., Harper J. W., and Kimmelman A. C. (2014) Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 509, 105–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Mancias J. D., Pontano Vaites L., Nissim S., Biancur D. E., Kim A. J., Wang X., Liu Y., Goessling W., Kimmelman A. C., and Harper J. W. (2015) Ferritinophagy via NCOA4 is required for erythropoiesis and is regulated by iron dependent HERC2-mediated proteolysis. Elife 4, e10308, 10.7554/eLife.10308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Bellelli R., Federico G., Matte' A., Colecchia D., Iolascon A., Chiariello M., Santoro M., De Franceschi L., and Carlomagno F. (2016) NCOA4 deficiency impairs systemic iron homeostasis. Cell Rep. 14, 411–421 [DOI] [PubMed] [Google Scholar]
- 91. Tabuchi M., Yoshimori T., Yamaguchi K., Yoshida T., and Kishi F. (2000) Human NRAMP2/DMT1, which mediates iron transport across endosomal membranes, is localized to late endosomes and lysosomes in HEp-2 cells. J. Biol. Chem. 275, 22220–22228 [DOI] [PubMed] [Google Scholar]
- 92. Kakhlon O., and Cabantchik Z. I. (2002) The labile iron pool: characterization, measurement, and participation in cellular processes. Free Radic. Biol. Med. 33, 1037–1046 [DOI] [PubMed] [Google Scholar]
- 93. Leidgens S., Bullough K. Z., Shi H., Li F., Shakoury-Elizeh M., Yabe T., Subramanian P., Hsu E., Natarajan N., Nandal A., Stemmler T. L., and Philpott C. C. (2013) Each member of the poly-r(C)-binding protein 1 (PCBP) family exhibits iron chaperone activity toward ferritin. J. Biol. Chem. 288, 17791–17802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Shi H., Bencze K. Z., Stemmler T. L., and Philpott C. C. (2008) A cytosolic iron chaperone that delivers iron to ferritin. Science 320, 1207–1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Yanatori I., Yasui Y., Tabuchi M., and Kishi F. (2014) Chaperone protein involved in transmembrane transport of iron. Biochem. J. 462, 25–37 [DOI] [PubMed] [Google Scholar]
- 96. Yanatori I., Richardson D. R., Imada K., and Kishi F. (2016) Iron export through the transporter ferroportin 1 is modulated by the iron chaperone PCBP2. J. Biol. Chem. 291, 17303–17318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Ghanem L. R., Kromer A., Silverman I. M., Chatterji P., Traxler E., Penzo-Mendez A., Weiss M. J., Stanger B. Z., and Liebhaber S. A. (2015) The poly(C) binding protein Pcbp2 and its retrotransposed derivative Pcbp1 are independently essential to mouse development. Mol. Cell Biol. 36, 304–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Hamdi A., Roshan T. M., Kahawita T. M., Mason A. B., Sheftel A. D., and Ponka P. (2016) Erythroid cell mitochondria receive endosomal iron by a “kiss-and-run” mechanism. Biochim. Biophys. Acta 1863, 2859–2867 [DOI] [PubMed] [Google Scholar]
- 99. Lakhal-Littleton S., Wolna M., Chung Y. J., Christian H. C., Heather L. C., Brescia M., Ball V., Diaz R., Santos A., Biggs D., Clarke K., Davies B., and Robbins P. A. (2016) An essential cell-autonomous role for hepcidin in cardiac iron homeostasis. Elife 5, e19804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Lakhal-Littleton S., Wolna M., Carr C. A., Miller J. J., Christian H. C., Ball V., Santos A., Diaz R., Biggs D., Stillion R., Holdship P., Larner F., Tyler D. J., Clarke K., Davies B., and Robbins P. A. (2015) Cardiac ferroportin regulates cellular iron homeostasis and is important for cardiac function. Proc. Natl. Acad. Sci. U.S.A. 112, 3164–3169 [DOI] [PMC free article] [PubMed] [Google Scholar]