

Abstract
When cells are exposed to heat shock and various other stresses, heat shock factor 1 (HSF1) is activated, and the heat shock response (HSR) is elicited. To better understand the molecular regulation of the HSR, we used 2D-PAGE–based proteome analysis to screen for heat shock–induced post-translationally modified cellular proteins. Our analysis revealed that two protein spots typically present on 2D-PAGE gels and containing heterogeneous nuclear ribonucleoprotein K (hnRNP K) with trioxidized Cys132 disappeared after the heat shock treatment and reappeared during recovery, but the total amount of hnRNP K protein remained unchanged. We next tested whether hnRNP K plays a role in HSR by regulating HSF1 and found that hnRNP K inhibits HSF1 activity, resulting in reduced expression of hsp70 and hsp27 mRNAs. hnRNP K also reduced binding affinity of HSF1 to the heat shock element by directly interacting with HSF1 but did not affect HSF1 phosphorylation–dependent activation or nuclear localization. hnRNP K lost its ability to induce these effects when its Cys132 was substituted with Ser, Asp, or Glu. These findings suggest that hnRNP K inhibits transcriptional activity of HSF1 by inhibiting its binding to heat shock element and that the oxidation status of Cys132 in hnRNP K is critical for this inhibition.
Keywords: Heat shock factor protein 1 (HSF1), heat shock protein (HSP), mass spectrometry (MS), post-translational modification (PTM), proteomics, redox regulation, hnRNP K
Introduction
When cells are exposed to heat shock stress, a cellular defense mechanism called heat shock response (HSR)2 is elicited (1). Activation of heat shock factor 1 (HSF1) followed by expression of heat shock proteins (hsps) is the most prominent feature of HSR. Upon activation by various stresses, HSF1 is phosphorylated, forms trimers cross-linked by disulfide bonds, and is translocated to the nucleus (2). In the nucleus, the activated trimeric HSF1 binds to the heat shock element (HSE) with the help of the chromatin-remodeling enzyme, BRG1 (brahma-related gene 1) (3), single-stranded DNA-binding protein RPA (replication protein A), and the histone chaperone FACT (facilitating chromatin transcription) and initiates the transcription of hsp genes. Active HSF1 trimers can be inactivated by interacting with hsp70 and hsp40, which inhibits its transactivation capacity, but not DNA-binding activity, resulting in reduced transcription of the hsp genes (4–6). Post-translational modification-dependent activation and inactivation mechanism of HSF1 has been extensively studied (2).
Heterogeneous nuclear ribonucleoprotein K (hnRNP K) is a member of RNA-binding protein complex consisting of ∼20 hnRNPs (7). hnRNP K binds preferentially to poly(C) and regulates transcription, translation, pre-mRNA splicing, RNA stability, chromatin remodeling, and signal transduction (8). Recently, hnRNP K has been reported to function in hsp105 pre-mRNA splicing in heat stressed cells (9). During its transcriptional regulation function, hnRNP K activates the transcription of μ opioid receptor (10) and eIF4E genes (11) and represses the transcription of neuronal nicotine acetylcholine receptor b4 (12), thymidine kinase (13), and CD43 genes (14). hnRNP K also binds to p53 as a co-factor and regulates the transcription of its downstream genes (15). Its affinity for p53 is increased by post-translational modifications (PTMs) such as sumoylation of Lys422 (16), methylation of Arg (17), and phosphorylation of Ser121, Thr174, Thr390, and Thr440 (18) of hnRNP K.
Various other PTMs of hnRNP K and their distinct functions have been identified. Arginine methylation of hnRNP K negatively regulates apoptosis (19). IL-1 (20), insulin (21), and oxidative stress (22) induce its phosphorylation. Src phosphorylates hnRNP K at Tyr residues and augments its function in translation (23). Phosphorylation of hnRNP K by ERK increases its cytoplasmic accumulation and its ability to inhibit translation (24), whereas phosphorylation by JNK promotes its transcriptional activation function (25). Phosphorylation by PKCδ modifies hnRNP K interaction with its binding proteins (26).
In this study, we comprehensively analyzed heat shock–induced changes in proteome profiles of mouse fibrosarcoma RIF-1 cells by combining 2D-PAGE analysis with mass spectrometry. We found that heat shock causes dramatic changes in the PTMs in hnRNP K. We also found that hnRNP K inhibits the activity of heat shock–induced HSF1 by reducing its binding to HSE and that the redox regulation of hnRNP K at Cys132 is critical for this inhibition.
Results
Cellular proteome changes in response to heat shock treatment
To investigate proteins involved in early stage of HSR, post-translationally modified proteins under heat shock stress were examined. Mouse fibrosarcoma RIF-1 cells were treated with heat shock at 45 °C for 30 min and recovered for 4, 12, or 24 h at 37 °C. The cell lysates were separated on 2D-PAGE, and the protein spots were visualized by silver staining (supplemental Fig. S1A). 149 protein spots, differentially appearing in response to heat shock during recovery, were identified by peptide sequencing (supplemental Fig. S1, B and C) and quantified using image analysis software (supplemental Fig. S1, D and E). We classified the proteins that showed more than 1.3-fold increase after heat shock treatment into four groups according to how long they took (4, 12, or 24 h) to reach their maximum levels (Fig. 1A): proteins up-regulated more than 2-fold by 4 h and remained stable up to 24 h (type I); proteins up-regulated the most at 4 h and decreased thereafter (type II); proteins up-regulated the most by 12 h and then decreased (type III); and proteins up-regulated the most by 24 h (type IV). Because heat shock at 45 °C for 30 min inhibits the synthesis of most cellular proteins except for hsp family proteins for up to 6 h (27), increased protein spots in 4 h recovery samples can be assumed to represent proteins post-translationally modified by heat shock. Therefore, the protein spots in types I and II are presumed to represent proteins with heat shock–induced PTMs that shifted on 2D gel by pI or molecular mass changes.
Figure 1.

Proteomic analysis of heat shock–induced cellular proteome changes. A, RIF-1 cells were heat shock (45 °C, 30 min) treated and recovered for 4, 12, and 24 h. Differentially appearing proteins in 2D-PAGE gels were identified by MS and quantified by image analysis. Proteins more than 1.3-fold up-regulated were analyzed and grouped based on their fold induction kinetics. B, cells were analyzed by Western blot analysis using anti-hsp25, -hsc/hsp70, -hsp60, -hsp90β, and -hnRNP K antibodies and actin antibody for loading control. Molecular mass markers (kDa) are shown alongside Western blot images. IB, immunoblot.
hnRNP K is post-translationally modified by heat shock treatment
hnRNP K was identified in five different spots on 2D gel, indicating several populations of cellular hnRNP K. Because four of these spots of the hnRNP K are of types I and II, we postulated that hnRNP K was post-translationally modified by heat shock. We selected four hsps and hnRNP K from the proteins in Fig. 1A and assessed their protein expression levels by 1D-PAGE separation and Western blot analysis (Fig. 1B). We found that hsp25, hsp70, hsp60, and hsp90β increased during recovery after heat shock. However, the level of hnRNP K protein was not changed by heat shock stress. This confirms that the changes of hnRNP K in 2D-PAGE analysis were not due to the changes in hnRNP K expression levels but are due to post-translational modifications induced by heat shock.
We examined the heat shock–induced changes in the hnRNP K protein spots in silver-stained 2D-PAGE gel as well as in 2D-PAGE Western analysis in detail (Fig. 2A). From the Western analysis results, we identified more than 10 hnRNP K populations. Among the four major spots (spots 1–4), spots 2 and 4 decreased at 4 h recovery after heat shock but were restored at 12 h after heat shock (Fig. 2, A and B). This decrease was also seen immediately after heat shock treatment (Fig. 2C). Other stress treatments such as a proteasome inhibitor MG132 and ionizing radiation did not cause this decrease in the two hnRNP K spots (Fig. 2, D and E), suggesting that these changes are specific effects of heat shock stress.
Figure 2.

Heat shock changes hnRNP K spot positions on 2D-PAGE gels. A and B, RIF-1 cells were heat shock–treated (45 °C, 30 min) and recovered for 4, 12, and 24 h. The cells were analyzed by 2D-PAGE and visualized by silver staining and Western blot analysis using anti-hnRNP K antibody (A). Western blot results of spots 1–4 in A were quantified and represented by bar graphs (B). C, RIF-1 cells were heat shock–treated (45 °C, 30 min) and harvested immediately after heat shock. The cells were analyzed by 2D-PAGE and visualized by silver staining. D, RIF-1 cells were 50 μm MG132-treated for 2 h and recovered for 2, 8.5, and 18 h. The cells were analyzed by 2D-PAGE and visualized by silver staining. E, RIF-1 cells were treated with 5-gray ionizing radiation and recovered for 15 min, 6 h, and 20 h. The cells were analyzed by 2D-PAGE and visualized by silver staining. Spots 1–4 indicate hnRNP K subpopulations. cont, control; IB, immunoblot; IR, ionizing radiation.
We attempted to comprehensively identify the PTMs representing each hnRNP K spot. We employed peptide sequencing with nanoUPLC-ESI-q-TOF MS/MS, employing selectively excluded mass screening analysis (SEMSA) for sensitive detection of low abundant PTMs (28) and MODi (29), Mascot, and Scaffold PTM algorithms for searching for unknown PTMs. Diverse PTM populations were identified in hnRNP K spots before (spots 1–4) and 4 h after heat shock (spots 1 and 3). These findings are summarized in Table 1 with representative MS/MS spectra (supplemental Fig. S2) and also presented in a schematic diagram (Fig. 3A). Phosphorylations at Ser36, Thr70, Tyr72, Ser89, Ser91, Thr107, Ser116, Ser154, Ser379, Thr438, and Thr442; acetylations at Lys52, Lys198, Lys219, and Lys405; and methylation at Arg316 were detected. Among these modifications, phosphorylation of Tyr72 was previously reported (23). Phosphorylations at Thr70, Tyr72, Ser89/Ser91, and Thr438/Thr442 and acetylation at Lys219 were decreased by heat shock treatment. In addition, various modifications of Cys including novel oxidation (30) were observed (Table 2, Fig. 3B, and supplemental Fig. S3). These included modifications of Cys132 to dehydroalanine and sulfonic acid; Cys145 to dehydroalanine and Cys-SO2-SH (Δm = +64 Da); and Cys184/185 to dehydroalanine and sulfonic acid. This is the first report on PTMs of Cys residues in hnRNP K. We concentrated on heat shock-dependent (in red in Fig. 3B) and spots 2- and 4-specific (in bold in Fig. 3B and Table 2) PTMs, because these two spots disappeared after heat shock and reappeared during recovery (Fig. 2A). Trioxidation to sulfonic acid of Cys132 was found to be heat shock–dependent and appeared only in spots 2 and 4 (Table 2).
Table 1.
hnRNP K protein spots (no. 1–4 in control gel, no. 1 and 3 in 4 h recovery gel) in silver-stained gels (Fig. 2A) were cut out of the gel and analyzed by MS and PTM analysis was performed. Identified modifications were listed with their scores.
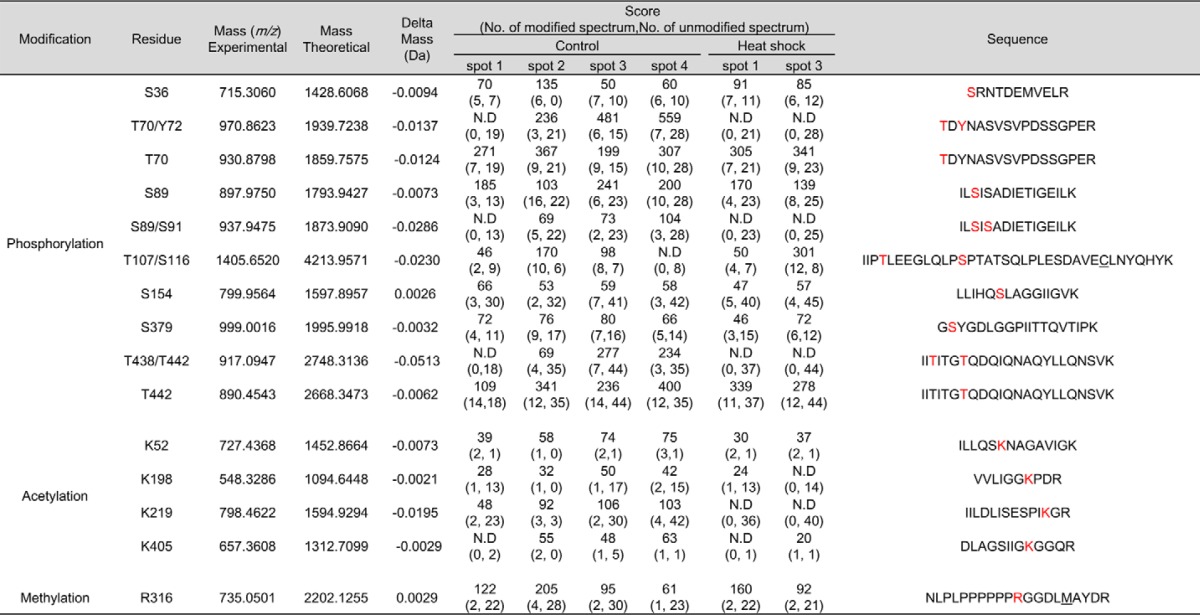
N.D, modified peptides were not detected; amino acids in red, modified residues; C, carbamidomethylation; M, oxidation.
Figure 3.

Schematic diagram of hnRNP K domains and its PTMs. hnRNP K protein spots in silver-stained gels (Fig. 1C) were analyzed for PTMs using nanoUPLC-ESI-q-TOF MS/MS. A, all of the identified sites for phosphorylation (P circles), acetylation (A circles), and methylation (M circle) are indicated. PTMs of residues in red are disappeared after heat shock treatment (45 °C, 30 min) following 4 h of recovery. B, various identified Cys modifications are indicated. PTMs in red disappeared after heat shock treatment (45 °C, 30 min) following 4 h of recovery. PTM in bold is spot 2– and 4–specific (Fig. 2A). NLS, nuclear-localization signal; KH, K homology; KI, K-protein-interactive; KNS, nuclear shuttling domain.
Table 2.
hnRNP K protein spots (no. 1–4 in control gel, no. 1 and 3 in 4 h recovery gel) in silver-stained gels (Fig. 2A) were cut out of the gel and analyzed by MS and PTM analysis was performed. Identified Cys modifications were listed with their scores.
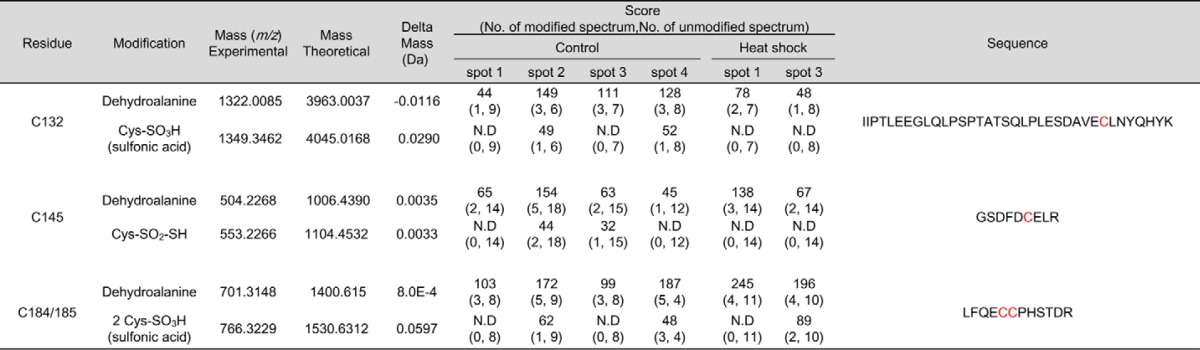
N.D, modified peptides were not detected; amino acids in red, modified residues.
hnRNP K inhibits the synthesis of hsp27 and hsp70 mRNA
Our finding that hnRNP K is the most dramatically changed protein in response to heat shock stress and recovery suggests that hnRNP K plays a key role in HSR. To investigate whether hnRNP K affects the transcriptional activity of HSF1, a major player in HSR, we examined the effect of overexpressing hnRNP K and its mutants on hsp27 mRNA levels. HEK293T cells overexpressing Flag-hnRNP K and its Cys mutants, C132S and C145S, were exposed to heat shock, and cellular levels of hsp27 mRNA were quantified and normalized to GAPDH mRNA level by RT-qPCR. Heat shock–induced hsp27 mRNA 4-fold more in cells overexpressing empty Flag vector but significantly inhibited it in cells overexpressing Flag-hnRNP K (Fig. 4A). In contrast, expression of hnRNP K C132S mutant, but not C145S, showed moderate inhibitory effect on hsp27 mRNA induction. The overexpression of each protein was confirmed by Western analysis using anti-Flag antibody (Fig. 4B). We also tested trioxidation mimic hnRNP K mutants, Flag-hnRNP K C132D and C132E mutants in HEK293T cells (Fig. 4C), and Flag-hnRNP K C132D mutant in HeLa cells (Fig. 4, E and F). These mutants showed moderate inhibitory effect on hsp70 and hsp27 mRNA expression in comparison to wild-type Flag-hnRNP K. The overexpression of each protein was confirmed by Western analysis using anti-Flag antibody (Fig. 4, D and G). Neither the oxidation-free form or the constitutive sulfonic acid mimic form of hnRNP K inhibited hsp70 and hsp27 mRNA expression as much as wild-type hnRNP K in heat shock–treated cells. This suggests that Cys132 in hnRNP K, a redox-sensitive Cys residue, is required for the inhibition of HSF1 in heat shock response; however, the oxidation status of Cys132 cannot be simply explained as a function of a fixed oxidation-free form or a sulfonic acid–mimicking form of hnRNP K.
Figure 4.

hnRNP K inhibits heat shock–induced hsp70 and hsp27 mRNA expression. A–D, HEK293T cells were transfected with Flag empty vector, Flag-hnRNP K, Flag-hnRNP K C132S, or Flag-hnRNP K C145S (A and C) and Flag empty vector, Flag-hnRNP K, Flag-hnRNP K C132D, or Flag-hnRNP K C132E (C and D), heat shock–treated at 45 °C for 15 min, and recovered at 37 °C for 4 h. hsp27 mRNAs was analyzed using quantitative real-time PCR and normalized to GAPDH mRNA. The cells were analyzed by Western blot analysis using anti-Flag antibody and anti-actin antibody as loading controls (B and D). E–G, HEK293T cells were transfected with Flag empty vector, Flag-hnRNP K, or Flag-hnRNP K C132D, heat shock–treated at 45 °C for 15 min, and recovered at 37 °C for 4 h. hsp70 (E) and hsp27 (F) mRNAs were analyzed using quantitative real-time PCR and normalized to GAPDH mRNA. The cells were analyzed by Western blot analysis using anti-Flag antibody and anti-actin antibody as loading controls (G). H–J, HeLa cells were transfected with control siRNA and hnRNP K siRNA, heat shock–treated at 45 °C for 15 min, and recovered at 37 °C for 4 h. hsp70 (H) and hsp27 (I) mRNAs were analyzed using quantitative real-time PCR and normalized to GAPDH mRNA. The cells were analyzed by Western blot analysis using anti-hnRNP K antibody and anti-actin antibody as loading controls (J). K and L, HeLa cells were transfected with control siRNA and hnRNP K siRNA, and after 48 h, the cells were transfected with Flag empty vector, Flag-hnRNP K, or Flag-hnRNP K C132S. The cells were heat shock–treated at 45 °C for 15 min and recovered at 37 °C for 4 h. hsp70 mRNA was analyzed using quantitative real-time PCR and normalized to GAPDH mRNA (K). The cells were analyzed by Western blot analysis using anti-hnRNP K antibody and anti-actin antibody as loading controls (L). M, HEK293T cells were transfected with Flag empty vector, Flag-hnRNP K, or Flag-hnRNP K C132S, heat shock–treated at 45 °C for 15 min, and recovered at 37 °C for 4 h. HSF1 mRNA was analyzed using quantitative real-time PCR and normalized to GAPDH mRNA. The data are presented as the means ± S.D. of technically triplicated experiments (t test). *, p < 0.05; **, p < 0.01; ***, p < 0.001. HS, heat shock treatment; Cont, control; IB, immunoblot.
We then tested whether depletion of hnRNP K promoted hsp70 and hsp27 mRNA expression in HeLa cells. As shown in Fig. 4, H and I, the heat shock–induced hsp70 and hsp27 mRNA levels were considerably enhanced in cells in which hnRNP K was knocked down. The depletion of hnRNP K using siRNA was confirmed by Western analysis (Fig. 4J).
We confirmed the increase of hsp mRNA in cells depleted of hnRNP K by demonstrating the converse by restoring hnRNP K. HeLa cells depleted of endogenous hnRNP K with siRNA were transfected with Flag-hnRNP K or Flag-hnRNP K mutant C132S and exposed to heat shock. Heat shock–induced hsp70 mRNA was significantly increased in cells in which hnRNP K was knocked down, and this increase was abolished by adding hnRNP K but not the hnRNP K mutant C132S (Fig. 4K). The depletion of hnRNP K and overexpression of hnRNP K was confirmed by Western analysis (Fig. 4L). These results demonstrate that hnRNP K inhibits heat shock–induced hsp70 and hsp27 mRNA expression and that Cys132 plays a critical role in this inhibitory process.
To determine whether the inhibitory effect of hnRNP K on heat shock–induced hsp70 and hsp27 mRNA expression is due to changes in HSF1 mRNA levels, we examined HSF1 mRNA levels in hnRNP K overexpressing HEK293T cells. HSF1 mRNA levels did not correlate with the amount of hnRNP K (Fig. 4M). This indicates that hnRNP K–induced inhibition of hsp70 and hsp27 mRNA expression was not caused by the reduction of HSF1 mRNA expression.
hnRNP K does not affect heat shock–induced activation nor nuclear localization of HSF1
It is well known that heat shock activation leads to the appearance of various PTMs in HSF1 (31). Active phosphorylated HSF1 can be readily detected by the appearance of a higher molecular mass band on 1D SDS-PAGE. To determine whether hnRNP K affects initial activation of HSF1, we sought to detect active form of HSF1 after heat shock in the presence and absence of Flag-hnRNP K in HEK293T cells. HSF1 band shifts to an upper position of a phosphorylated form immediately after heat shock and returns to the original position after 6 h of recovery in Flag empty vector-transfected control cells (Fig. 5A, top two panels). The HSF1 activation kinetics in cells overexpressing Flag-hnRNP K and its C132S mutant were identical to that in control cells transfected with empty vector (Fig. 5A, center and bottom sets of panels). The activation kinetics of HSF1 were also identical in both HeLa cells in which hnRNP K was knocked down and control cells (Fig. 5B). These results suggest that hnRNP K does not affect heat shock–induced HSF1 PTM-dependent initial activation.
Figure 5.

hnRNP K does not inhibit HSF1 activation nor its nuclear translocalization. A and B, HEK293T cells were transfected with Flag empty vector, Flag-hnRNP K, and Flag-hnRNP K C132S (A), and HeLa cells were transfected with control and hnRNP K siRNA (B). The cells were heat shock–treated at 45 °C for 15 min and recovered at 37 °C for the indicated times. The cells were analyzed by Western blot analysis using anti-HSF1 antibody to detect heat shock–induced band shift and anti-Flag antibody to check overexpression of Flag-tagged proteins. high, active higher molecular mass HSF1; low, inactive lower molecular mass HSF1; asterisk, nonspecific band. C and D, HEK293T cells were transfected with Flag empty vector, Flag-hnRNP K, and Flag-hnRNP K C132S (C), and HeLa cells were transfected with control and hnRNP K siRNA (D). The cells were heat shock–treated at 45 °C for 15 min. The cells were fractionated into cytosolic and nuclear fractions and analyzed by Western blot analysis using anti-HSF1 to detect its localization, anti-Flag antibody to check overexpression of Flag-tagged proteins, and anti-PRX6 and lamin B as cytosolic and nuclear markers, respectively. HS, heat shock treatment; Cont, control; IB, immunoblot; C, cytosolic fraction; N, nuclear fraction.
Because activated HSF1 forms trimers and translocates from cytosol to nucleus, we investigated whether hnRNP K regulates the translocation of activated HSF1. HEK293T cells overexpressing Flag-hnRNP K and its mutant C132S were exposed to heat shock, and cell lysates were fractionated into cytosolic and nuclear fractions (Fig. 5C). HSF1 was mainly located in cytosol prior to heat shock and translocated into nucleus after heat shock treatment. Heat shock–induced translocation into nucleus was not affected by overexpression of hnRNP K or its C132S mutant (Fig. 5C) or by depletion of hnRNP K in HeLa cells (Fig. 5D). These findings indicate that hnRNP K does not modulate translocation of the active HSF1 from the cytosol to nucleus.
hnRNP K inhibits HSF1 binding to HSE
HSF1 translocates to the nucleus, binds to HSE, and activates hsp genes. We investigated, employing the hsp70 reporter and ChIP assays, whether hnRNP K inhibits HSF1 binding to HSE. hsp70 reporter assay was performed with HA-HSF1 overexpression in the presence or absence of hnRNP K in HeLa cells. HA-HSF1 overexpression increased the luciferase activity. Depletion of hnRNP K augmented both basal and HA-HSF1-induced activities of the reporter (Fig. 6A). This suggests that hnRNP K negatively regulates HSF1 transcriptional activity.
Figure 6.

hnRNP K inhibits HSF1 binding to HSE. A, after depletion of hnRNP K with hnRNP K specific siRNA, HeLa cells were transfected with empty vector or HA-HSF1 vector both with hsp70-luciferase plasmid. After 8 h, luciferase activity was measured. B, HEK293T cells were transfected with Flag empty vector, Flag-hnRNP K, and Flag-hnRNP K C132S and heat shock–treated at 45 °C for 15 min. ChIP-qPCR was performed using control IgG and anti-HSF1 antibody. C, HEK293T cells were transfected with HA-HSF1 and Flag empty vector, Flag-hnRNP K, or Flag-hnRNP K C132S. The cells were heat shock–treated at 45 °C for 15 min and lysed, and co-immunoprecipitation using anti-Flag antibody was performed. Immunoprecipitated protein complex and input were immunoblotted with anti-HA and anti-Flag antibody. D and E, HEK293T cells were heat shock–treated at 45 °C for 15 min, and ChIP-qPCR was performed using control rabbit and mouse IgG, anti-HSF1, monoclonal anti-hnRNP K, and polyclonal anti-hnRNP K antibody. The immunoprecipitated HSE was represented as percentages of output DNA to input DNA (% input) using qPCR of ChIP samples. The PCR product size is 179 (D) and 269 base pairs (E). F, HEK293T cells were transfected with Flag empty vector or Flag-HSF1. The cells were lysed, and co-immunoprecipitation using anti-Flag antibody was performed. Immunoprecipitated protein complex were immunoblotted with anti-Flag, anti-hnRNP K, anti-hnRNP A1, and anti-hnRNP C1/C2 antibody. G, HEK293T cells were transfected with HA-HSF1 and Flag empty vector or Flag-hnRNP K. The cells were lysed in the absence or presence of 1 mg/ml RNase A and incubated at 37 °C for 15 min, and co-immunoprecipitation using anti-Flag antibody was performed. Immunoprecipitated protein complex and input were immunoblotted with anti-HA and anti-Flag antibody. H, HEK293T cells were transfected with HA-HSF1 and Flag empty vector or Flag-hnRNP K. The cells were heat shock–treated at 45 °C for 15 min and lysed, and co-immunoprecipitation using anti-Flag antibody was performed. Immunoprecipitated protein complex were immunoblotted with anti-HA, anti-Flag, and anti-hnRNP C1/C2 antibody. The data are presented as the means ± S.D. of technically triplicated experiments (t test). *, p < 0.05; **, p < 0.01; ***, p < 0.001. Cont, control; HS, heat shock treatment; IB, immunoblot; IP, immunoprecipitation.
To investigate whether hnRNP K affects the binding of HSF1 to HSE, we employed ChIP assay using anti-HSF1 antibody. HEK293T cells overexpressing Flag-hnRNP K or the C132S mutant were exposed to heat shock, and the cell lysates were immunoprecipitated with anti-HSF1 antibody or control rabbit IgG. Immunoprecipitated HSE was quantified using real-time qPCR. Heat shock treatment increased HSF1 binding to HSE as previously reported (32). Intriguingly, overexpression of hnRNP K decreased the HSF1-HSE binding in heat shock–treated cells. This inhibition was attenuated in cells overexpressing Flag-hnRNP K C132S (Fig. 6B).
Because hnRNP K inhibits HSF1 binding to HSE, we investigated whether hnRNP K interaction with HSF1 inhibits the binding of HSF1 to HSE or whether hnRNP K competes with HSF1 for binding to the HSE region. First, we overexpressed HA-HSF1 with Flag empty vector, Flag-hnRNP K, or Flag-hnRNP K C132S treated with or without heat shock, and performed co-immunoprecipitation using anti-Flag antibody. Flag-hnRNP K bound to HA-HSF1 regardless of heat shock treatment, but Flag-hnRNP K C132S mutant did not bind to HA-HSF1 in heat shock–treated cells (Fig. 6C). Second, we performed ChIP assays using anti-HSF1 antibody and monoclonal and ChIP grade polyclonal anti-hnRNP K antibodies to test HSF1 and hnRNP K binding to HSE. Unlike anti-HSF1 antibody, both anti-hnRNP K antibodies recruited negligible amounts of HSE (Fig. 6D). We also tested a wider range of DNA sequences than the 173 bp detected in Fig. 6D, including 96 bp upstream of the 173 bp (total 269 bp), but neither hnRNP K antibody recruited the HSE-containing DNA region (Fig. 6E). This means hnRNP K does not have the ability to bind to the HSE region. These results indicate that hnRNP K inhibits HSF1 binding to HSE by forming a protein complex with HSF1.
As hnRNP K is a member of protein complex hnRNP, we tested the relationship between other hnRNP members and HSF1. We transfected Flag or Flag-HSF1 and performed co-immunoprecipitation with anti-Flag antibody. When Flag-HSF1 bound to endogenous hnRNP K specifically, endogenous hnRNP A1 nor hnRNP C1/C2 did not show any specific binding to Flag-HSF1 (Fig. 6F). This suggests that HSF1 interacts with hnRNP K specifically, not hnRNP complex. To investigate whether RNA is necessary for the HSF1 and hnRNP K binding, we examined these interaction in cells overexpressing HA-HSF1 with Flag empty vector or Flag-hnRNP K with co-immunoprecipitation using anti-Flag antibody in the absence or presence of RNase A. As shown in Fig. 6G, elimination of RNA does not affect the interaction between HSF1 and hnRNP K. We also investigated whether the hnRNP complex changed during heat shock and affected the binding of HSF1 and hnRNP K. We performed same co-immunoprecipitation using anti-Flag antibody with heat shock-untreated or -treated cells. Flag-hnRNP K bound to HA-HSF1 regardless of heat shock treatment, and one of the major hnRNP complex proteins, hnRNP C1/C2, also bound stably with Flag-hnRNP K (Fig. 6H). This suggests that hnRNP K binding to HSF1 and other hnRNP proteins are not affected by heat shock treatment.
hnRNP K delays cell growth after heat shock treatment, but its C132S mutant does not
A major function of hsps is protection of cells from various stresses. Because hnRNP K inhibits heat shock–induced production of hsps mRNA, we investigated whether hnRNP K affects cell survival after heat shock treatment. HEK293T cells transfected with empty Flag vector, Flag-hnRNP K, and Flag-hnRNP K C132S mutant plasmids, respectively, were exposed to heat shock, and cell survival was monitored using WST-1 (Fig. 7A) or a real-time cell analyzer (Fig. 7B). Overexpression of Flag-hnRNP K, not its C132S mutant, discernibly inhibited the cell survival in both WST-1 and real-time cell analyzer analysis. This indicates that hnRNP K reduces cellular recovery from heat shock stress by inhibiting the HSF1 activity in Cys132 of hnRNP K–dependent manner.
Figure 7.

hnRNP K inhibits cell growth after heat shock treatment. A and B, HEK293T cells were transfected with Flag empty vector, Flag-hnRNP K, and Flag-hnRNP K C132S and heat shock–treated at 45 °C for 15 min. The cells were plated in 96-well plate and incubated at 37 °C for 2 days, and cell survival was measured using WST-1 solution (A). Cell growth was monitored under real-time cell analyzer, xCELLigence (B). The data are presented as the means ± S.D. of technically triplicated experiments (t test). *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Discussion
In the present study, we have shown that hnRNP K inhibits HSF1 binding to HSE, inhibits hsp gene activation, and reduces the survival of cells subjected to heat shock stress. Trioxidation and reduction at Cys132 is required for the inhibitory effect of hnRNP K on HSF1 transcriptional activity.
The cellular defense mechanism, HSR, was first reported in 1962 by Ritossa (33). Since then, the molecular mechanisms in HSR, including activation of HSF1 (34, 35), de novo synthesis of hsps, and induction of thermotolerance (36, 37), were defined. However, how cells sense various HSR-inducing stresses, how activation/deactivation of HSF1 and its functions are regulated, what HSF isoforms exist, and what their relationship to HSF1 are remained unknown. In the present study, we investigated, employing 2D-PAGE based proteomics, changes in cellular protein profiles caused by heat shock. One of our specific findings was that heat shock caused dramatic changes in the subpopulations of hnRNP K, without affecting its total protein level. These subpopulations of hnRNP K resulted from numerous PTMs including, among others, phosphorylation, acetylation, methylation, and oxidation or their various combinations (Tables 1 and 2). Except for the phosphorylation of Tyr72, all of the PTMs in Tables 1 and 2 were first reported PTMs of hnRNP K. Intriguingly, two of the four major hnRNP K spots from 2D-PAGE disappeared immediately after heat shock (Fig. 2C) and reappeared 12 h after heat shock during recovery (Fig. 2A). The disappearing spots contained a common PTM, trioxidation at Cys132. Because neither of hnRNP K oxidation-free mutant C132S nor constitutive oxidation mimic mutants C132D and C132E showed the HSF1 inhibitory effect as much as wild-type hnRNP K, free exchange between the two states, trioxidation and reduction at Cys132, appeared to be responsible for its inhibitory effect on HSF1.
Because the crystal structure or NMR studies of hnRNP K have not been conducted, we had no way to predict the effects of oxidation of amino acids surrounding Cys132. Therefore, we compared the PTMs of wild-type hnRNP K and C132S mutant.3 Phosphorylations at Ser36 and Ser379 increased in C132S mutant compared with wild type of hnRNP K with higher MS analysis scores. This suggests that oxidation of Cys132 affects PTMs of other residues, presumably by conformational changes, and that the inhibitory effect of HSF1 might not be solely due to modification of Cys132. Among the four reported alternative splicing variants of hnRNP K, isoforms 1 and 2 differ in their C-terminal 4-amino acid (459SGKFF463) and 5-amino acid (459ADVEGF464) sequences. Isoforms 3 and 4 differ from isoforms 1 and 2, by the absence of amino acid sequence 111–134 (exon 8) (38, 39). Because Cys132 exists in the missing region of isoforms 3 and 4, studies investigating the differences between hnRNP K isoforms might give us an insight into the functional role of Cys oxidation in the HSF1 inhibitory effect.
We also investigated whether hnRNP K regulates HSF1, one of the major HSR molecules serving as a transcription factor, because hnRNP K has been reported to function as a co-factor of a transcription factor, p53 (15). Recently its function in pre-mRNA splicing of hsp105 mRNA in heat shock–stressed cells was also reported (9). We found that in heat-shocked cells, hnRNP K inhibits HSF1, resulting in reduced hsp mRNA production. It has been reported that inhibition of HSF1 by hsp70 and hsp40 is inhibition of a transactivation capacity of HSF1 (4). In contrast, this study demonstrates that HSF1 can be inhibited by blocking its binding to HSE by hnRNP K. Our results are similar to the previously reported inhibition mechanism of HSBP1 (heat shock factor-binding protein 1) that HSBP1 binds to HSF1 and inhibits its transcriptional activity (40).
We hypothesized that hnRNP K inhibits HSF1 binding to HSE either by complexing with HSF1, which in turn prevents HSF1-HSE binding, or by competing with HSF1 for binding to HSE. To test this hypothesis, we investigated whether there is interaction between hnRNP K and HSF1 by performing co-immunoprecipitation (Fig. 6C). Because Flag-hnRNP K precipitated with HA-HSF1, but not Flag-hnRNP K C132S mutant in heat shock–treated cells, we concluded that hnRNP K inhibits HSF1 by complexing with HSF1. In regard to the second hypothesis, it has been reported that hnRNP K binds to DNA directly and affects gene expression (10, 11, 13, 14). Therefore, we also tested the alternate possibility in our alternate hypothesis, by examining whether hnRNP K binds with HSE. Employing ChIP assay with two different kinds of anti-hnRNP K antibodies, we found that hnRNP K did not interact with HSE (Fig. 6, D and E). This indicates that hnRNP K does not compete with HSF1 for binding to HSE but forms protein complex with HSF1 inhibiting HSF1 binding to HSE.
Activation and inhibition of HSF1 both are therapeutic targets for neurodegenerative diseases and various cancers, respectively. Further study of the inhibition mechanism of hnRNP K might contribute to the cancer drug discovery.
In summary, this study identifies a novel regulatory mechanism for HSF1 by demonstrating that hnRNP K inhibits HSF1 activity by preventing the binding of HSF1 to HSE and not by affecting HSF1 phosphorylation or translocation of active HSF1 to nucleus. The oxidation and reduction of Cys132 residue play critical roles in this inhibitory effect of hnRNP K presumably by modulating the binding between hnRNP K and HSF1.
Experimental procedures
Cell lines and heat shock treatment
The radiation-induced mouse fibrosarcoma cell line RIF-1 was maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum, 100 units/ml of penicillin G, and 100 μg/ml of streptomycin (all from Invitrogen) at 37 °C. HEK293T and HeLa cells were purchased from ATCC and maintained following the manufacturer's protocol. Heat shock was administered to cells grown in tissue culture dishes, by floating them in a 45 °C water bath (41).
Antibodies
Antibodies used in this study were hnRNP K (Santa Cruz), HSF1 (Enzo), Flag (Sigma), actin (Santa Cruz), PRX6 (AbClon), lamin B (Calbiochem), and Hsc/hsp70 (Stressgen).
2D-PAGE–based proteomics
2D gel electrophoresis was performed as described previously (42). Briefly, 100 μg of total protein was loaded onto the strip gels and rehydrated for 12 h (18 cm, pH 4–7) with rehydration buffer (7 m urea, 2 m thiourea, 2% (v/v) CHAPS, 2% IPG buffer, pH 4–7 (GE Healthcare)). The strip gels were then electrofocused in a manifold cup-loading system with IPGphor (GE Healthcare) and equilibrated twice. After the second equilibration, the strip gels were applied to 1.0-mm-thick 10% acrylamide gels and sealed with 0.25% agarose. SDS-PAGE was carried out at 15 mA overnight using a PROTEAN II xi 2-D Cell apparatus (Bio-Rad). Each set of gels was silver-stained simultaneously in the same tray and scanned using an Image Scanner III (GE Healthcare) in triplicate. Spot detection, matching, normalization and quantification were automatically carried out using the ProgenesisSameSpots v5.0 software (Nonlinear Dynamics, Newcastle upon Tyne, UK). All of the 2D gel images were shown with acidic part left and basic part right.
Protein identification by nanoUPLC-ESI-q-TOF tandem MS
The gel spots of differentially expressed proteins were destained and digested with trypsin, and the resulting peptides were extracted as previously described (28). The peptides were separated using trap column cartridge, injected into a C18 reversed-phase analytical column with an integrated electrospray ionization PicoTipTM using nanoAcquitTM UPLC/ESI/q-TOF MS/MS (SYNAPTTM G2Si HDMSTM; Waters Co.). Peptide solutions were injected into a column and eluted by a linear gradient of 5–40% buffer B (ACN/formic acid, 100:0.1, v/v). The mass spectrometer was programmed to record scan cycles composed of one MS scan followed by MSMS scans of the 10 most abundant ions in each MS scan. Database analysis was performed using the Scaffold PTM (version 3.0; Proteome Software Inc.), Mascot (version 2.2.06), ProteinLynx global server 2.3 (Waters Co.) and MODi (29). Only significant hits, indicated by Mascot probability analysis (probability based Mowse score p < 0.05) were considered. Phosphorylations of Ser, Thr, and Tyr; acetylation of Lys; methylation of Arg; oxidation of Met; carbamidomethylation of Cys; deamidations of Asn and Gln; oxidation, dioxidation, trioxidation of Cys; Cys-SO2-SH; dehydroalanine of Cys; oxidation of Met were used as variable modifications. MS peaks in supplemental Figs. S2 and S3 were generated by Scaffold PTM (version 3.0; Proteome Software Inc.) and MODi algorithm (29).
siRNA transfection
Control and hnRNP K siRNAs (AccuTargetTM genome-wide predesigned siRNA) were purchased from Bioneer and transfected with Lipofectamine RNAiMAX (Invitrogen) following the manufacturer's protocols.
Quantification of hsp70 and hsp27 mRNA using RT-qPCR
Total RNA was isolated using an RNeasy minikit (Qiagen), and cDNA was prepared using SuperScript II RT (Invitrogen) according to the manufacturer's protocol. Synthesized cDNA was subjected to real-time PCR analysis using SYBR green qPCR master mix (MGmed) and AB7300 real-time qPCR machine (Applied Biosystems). The following primers of human genes were used: hsp70 gene (forward primer, 5′-CCGAGAAGGACGAGTTTGAG-3′; reverse primer, 5′-CTGGTACAGTCCGCTGATGA-3′), hsp27 gene (forward primer, 5′-CATCCCAGTCACCTTCGAGT-3′; reverse primer, 5′-CTTTACTTGGCGGCAGTCTC-3′), and GAPDH gene (forward primer, 5′-AAG GTC ATC CCT GAG CTG AA-3′; reverse primer, 5′-TGC TGT AGC CAA ATT CGT TG-3′). The standard curve method was used for quantification.
ChIP assay
ChIP assay was performed using slightly modified EZChIP kit (Merck Millipore) (43). Immunoprecipitation was carried out using polyclonal anti-HSF1 (Enzo), monoclonal anti-hnRNP K (Santa Cruz), or ChIP grade polyclonal anti-hnRNP K antibody (Abcam). Immunoprecipitated DNA was analyzed by qPCR analysis using primers for heat shock element region of hsp70 gene (product size 173 bp: forward, 5′-CACTCCCCCTTCCTCTCAG-3′; reverse, 5′-TTCCCTTCTGAGCCAATCAC-3′; product size 269 bp: forward, 5′-CCGACCCTTCCTGTCAATTA-3′; reverse, 5′-TTCCCTTCTGAGCCAATCAC-3′).
Nuclear/cytosolic fractionation
The cells were lysed in hypotonic buffer (10 mm HEPES, pH 7.9, 1.5 mm MgCl2, 10 mm KCl, protease inhibitor mixture (Sigma)) by passing 31-gauge syringe 10 times and incubation on ice for 30 min. After centrifugation at 4,000 rpm for 25 min, supernatant (cytosolic fraction) was removed. The pellet (nuclear fraction) was washed with hypotonic buffer two times.
Co-immunoprecipitation
HEK293T cells were lysed in IP buffer (50 mm Tris-Cl, pH7.4, 150 mm NaCl, 1 mm EDTA, 60 mm octyl-d-β-glucopyranoside) and hypotonic buffer (10 mm HEPES, 1.5 mm MgCl2, 10 mm KCl, 60 mm octyl-d-β-glucopyranoside) 3:1 mixture supplemented with protease inhibitor mixture (Sigma). The cells were passed 31-gauge syringe 10 times and centrifuged at 12,000 rpm for 5 min. Supernatant was immunoprecipitated with anti-Flag antibody, and precipitated protein complex was analyzed by Western blot analysis.
Cell proliferation assay
The cells were plated (15,000 cells/well, 96-well plate), and after 48 h, WST-1 solution (Roche Life Science) was added. After incubation at 37 °C for 1.5 h, absorbance at 450 nm was measured. For real-time cell analyzer analysis, the same cells were plated, and cell growth was real time monitored under xCELLigence RTCA SP system (Acea Biosciences, Inc.).
Author contributions
H.-J. K. conducted most of the experiments, analyzed the results, and wrote most of the paper. J.-J. L., J. J., and W. K. conducted mass spectrometry analysis. J.-H. C. conducted 2D gel analysis. A. Y. P. conducted part of the qPCR experiment. H.-J. K. and K.-J. L. conceived the idea for the project and wrote the paper.
Supplementary Material
Acknowledgments
hsp70-GL3 and HA-HSF1 were kindly provided by Dr. Y. S. Lee at Ewha Womans University, and Flag-hnRNP K was provided by Dr. C. H. Chung at Seoul National University.
This work was supported by Global Research Lab Program Grant 2012K1A1A2045441, Basic Science Research Program Grant 2016R1A6A3A11934127, and Brain Research Program Grant 2015M3C7A1028373 of the National Research Foundation of Korea. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Figs. S1–S3.
H.-J. Kim, J.-J. Lee, J.-H. Cho, J. Jeong, A. Y. Park, W. Kang, and K.-J. Lee, unpublished data.
- HSR
- heat shock response
- HSF
- heat shock factor
- hnRNP K
- heterogeneous nuclear ribonucleoprotein K
- HSE
- heat shock element
- hsp
- heat shock protein
- PTM
- post-translational modification.
References
- 1. Lindquist S. (1986) The heat-shock response. Annu. Rev. Biochem. 55, 1151–1191 [DOI] [PubMed] [Google Scholar]
- 2. Neef D. W., Jaeger A. M., and Thiele D. J. (2011) Heat shock transcription factor 1 as a therapeutic target in neurodegenerative diseases. Nat. Rev. Drug Discov. 10, 930–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sullivan E. K., Weirich C. S., Guyon J. R., Sif S., and Kingston R. E. (2001) Transcriptional activation domains of human heat shock factor 1 recruit human SWI/SNF. Mol. Cell. Biol. 21, 5826–5837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shi Y., Mosser D. D., and Morimoto R. I. (1998) Molecular chaperones as HSF1-specific transcriptional repressors. Genes Dev. 12, 654–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Abravaya K., Myers M. P., Murphy S. P., and Morimoto R. I. (1992) The human heat shock protein hsp70 interacts with HSF, the transcription factor that regulates heat shock gene expression. Genes Dev. 6, 1153–1164 [DOI] [PubMed] [Google Scholar]
- 6. Baler R., Welch W. J., and Voellmy R. (1992) Heat shock gene regulation by nascent polypeptides and denatured proteins: hsp70 as a potential autoregulatory factor. J. Cell Biol. 117, 1151–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Piñol-Roma S., Choi Y. D., Matunis M. J., and Dreyfuss G. (1988) Immunopurification of heterogeneous nuclear ribonucleoprotein particles reveals an assortment of RNA-binding proteins. Genes Dev. 2, 215–227 [DOI] [PubMed] [Google Scholar]
- 8. Barboro P., Ferrari N., and Balbi C. (2014) Emerging roles of heterogeneous nuclear ribonucleoprotein K (hnRNP K) in cancer progression. Cancer Lett. 352, 152–159 [DOI] [PubMed] [Google Scholar]
- 9. Yamamoto K., Furukawa M. T., Fukumura K., Kawamura A., Yamada T., Suzuki H., Hirose T., Sakamoto H., and Inoue K. (2016) Control of the heat stress-induced alternative splicing of a subset of genes by hnRNP K. Genes Cells 21, 1006–1014 [DOI] [PubMed] [Google Scholar]
- 10. Choi H. S., Song K. Y., Hwang C. K., Kim C. S., Law P. Y., Wei L. N., and Loh H. H. (2008) A proteomics approach for identification of single strand DNA-binding proteins involved in transcriptional regulation of mouse mu opioid receptor gene. Mol. Cell. Proteomics 7, 1517–1529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lynch M., Chen L., Ravitz M. J., Mehtani S., Korenblat K., Pazin M. J., and Schmidt E. V. (2005) hnRNP K binds a core polypyrimidine element in the eukaryotic translation initiation factor 4E (eIF4E) promoter, and its regulation of eIF4E contributes to neoplastic transformation. Mol. Cell. Biol. 25, 6436–6453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Du Q., Melnikova I. N., and Gardner P. D. (1998) Differential effects of heterogeneous nuclear ribonucleoprotein K on Sp1- and Sp3-mediated transcriptional activation of a neuronal nicotinic acetylcholine receptor promoter. J. Biol. Chem. 273, 19877–19883 [DOI] [PubMed] [Google Scholar]
- 13. Lau J. S., Baumeister P., Kim E., Roy B., Hsieh T. Y., Lai M., and Lee A. S. (2000) Heterogeneous nuclear ribonucleoproteins as regulators of gene expression through interactions with the human thymidine kinase promoter. J. Cell. Biochem. 79, 395–406 [DOI] [PubMed] [Google Scholar]
- 14. Da Silva N., Bharti A., and Shelley C. S. (2002) hnRNP-K and Purα act together to repress the transcriptional activity of the CD43 gene promoter. Blood 100, 3536–3544 [DOI] [PubMed] [Google Scholar]
- 15. Moumen A., Masterson P., O'Connor M. J., and Jackson S. P. (2005) hnRNP K: an HDM2 target and transcriptional coactivator of p53 in response to DNA damage. Cell 123, 1065–1078 [DOI] [PubMed] [Google Scholar]
- 16. Lee S. W., Lee M. H., Park J. H., Kang S. H., Yoo H. M., Ka S. H., Oh Y. M., Jeon Y. J., and Chung C. H. (2012) SUMOylation of hnRNP-K is required for p53-mediated cell-cycle arrest in response to DNA damage. EMBO J. 31, 4441–4452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen Y., Zhou X., Liu N., Wang C., Zhang L., Mo W., and Hu G. (2008) Arginine methylation of hnRNP K enhances p53 transcriptional activity. FEBS Lett. 582, 1761–1765 [DOI] [PubMed] [Google Scholar]
- 18. Moumen A., Magill C., Dry K. L., and Jackson S. P. (2013) ATM-dependent phosphorylation of heterogeneous nuclear ribonucleoprotein K promotes p53 transcriptional activation in response to DNA damage. Cell Cycle 12, 698–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yang J. H., Chiou Y. Y., Fu S. L., Shih I. Y., Weng T. H., Lin W. J., and Lin C. H. (2014) Arginine methylation of hnRNPK negatively modulates apoptosis upon DNA damage through local regulation of phosphorylation. Nucleic Acids Res. 42, 9908–9924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Van Seuningen I., Ostrowski J., Bustelo X. R., Sleath P. R., and Bomsztyk K. (1995) The K protein domain that recruits the interleukin 1-responsive K protein kinase lies adjacent to a cluster of c-Src and Vav SH3-binding sites. Implications that K protein acts as a docking platform. J. Biol. Chem. 270, 26976–26985 [DOI] [PubMed] [Google Scholar]
- 21. Ostrowski J., Kawata Y., Schullery D. S., Denisenko O. N., Higaki Y., Abrass C. K., and Bomsztyk K. (2001) Insulin alters heterogeneous nuclear ribonucleoprotein K protein binding to DNA and RNA. Proc. Natl. Acad. Sci. U.S.A. 98, 9044–9049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ostrowski J., Schullery D. S., Denisenko O. N., Higaki Y., Watts J., Aebersold R., Stempka L., Gschwendt M., and Bomsztyk K. (2000) Role of tyrosine phosphorylation in the regulation of the interaction of heterogenous nuclear ribonucleoprotein K protein with its protein and RNA partners. J. Biol. Chem. 275, 3619–3628 [DOI] [PubMed] [Google Scholar]
- 23. Ostareck-Lederer A., Ostareck D. H., Cans C., Neubauer G., Bomsztyk K., Superti-Furga G., and Hentze M. W. (2002) c-Src-mediated phosphorylation of hnRNP K drives translational activation of specifically silenced mRNAs. Mol. Cell. Biol. 22, 4535–4543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Habelhah H., Shah K., Huang L., Ostareck-Lederer A., Burlingame A. L., Shokat K. M., Hentze M. W., and Ronai Z. (2001) ERK phosphorylation drives cytoplasmic accumulation of hnRNP-K and inhibition of mRNA translation. Nat. Cell Biol. 3, 325–330 [DOI] [PubMed] [Google Scholar]
- 25. Habelhah H., Shah K., Huang L., Burlingame A. L., Shokat K. M., and Ronai Z. (2001) Identification of new JNK substrate using ATP pocket mutant JNK and a corresponding ATP analogue. J. Biol. Chem. 276, 18090–18095 [DOI] [PubMed] [Google Scholar]
- 26. Schullery D. S., Ostrowski J., Denisenko O. N., Stempka L., Shnyreva M., Suzuki H., Gschwendt M., and Bomsztyk K. (1999) Regulated interaction of protein kinase Cdelta with the heterogeneous nuclear ribonucleoprotein K protein. J. Biol. Chem. 274, 15101–15109 [DOI] [PubMed] [Google Scholar]
- 27. Kim H. J., and Lee K. J. (2002) Heat shock and ceramide have different apoptotic pathways in radiation induced fibrosarcoma (RIF) cells. Mol. Cell. Biochem. 229, 139–151 [DOI] [PubMed] [Google Scholar]
- 28. Seo J., Jeong J., Kim Y. M., Hwang N., Paek E., and Lee K. J. (2008) Strategy for comprehensive identification of post-translational modifications in cellular proteins, including low abundant modifications: application to glyceraldehyde-3-phosphate dehydrogenase. J. Proteome Res. 7, 587–602 [DOI] [PubMed] [Google Scholar]
- 29. Kim S., Na S., Sim J. W., Park H., Jeong J., Kim H., Seo Y., Seo J., Lee K. J., and Paek E. (2006) MODi a powerful and convenient web server for identifying multiple post-translational peptide modifications from tandem mass spectra. Nucleic Acids Res. 34, W258–W263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jeong J., Jung Y., Na S., Jeong J., Lee E., Kim M. S., Choi S., Shin D. H., Paek E., Lee H. Y., and Lee K. J. (2011) Novel oxidative modifications in redox-active cysteine residues. Mol. Cell. Proteomics 10, M110 000513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Akerfelt M., Morimoto R. I., and Sistonen L. (2010) Heat shock factors: integrators of cell stress, development and lifespan. Nat. Rev. Mol. Cell Biol. 11, 545–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Trinklein N. D., Chen W. C., Kingston R. E., and Myers R. M. (2004) Transcriptional regulation and binding of heat shock factor 1 and heat shock factor 2 to 32 human heat shock genes during thermal stress and differentiation. Cell Stress Chaperones 9, 21–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ritossa F. (1962) A new puffing pattern induced by heat shock and DNP in Drosophila. Experientia 18, 571–573 [Google Scholar]
- 34. Baler R., Dahl G., and Voellmy R. (1993) Activation of human heat shock genes is accompanied by oligomerization, modification, and rapid translocation of heat shock transcription factor HSF1. Mol. Cell. Biol. 13, 2486–2496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sarge K. D., Murphy S. P., and Morimoto R. I. (1993) Activation of heat shock gene transcription by heat shock factor 1 involves oligomerization, acquisition of DNA-binding activity, and nuclear localization and can occur in the absence of stress. Mol. Cell. Biol. 13, 1392–1407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lee K. J., and Hahn G. M. (1988) Abnormal proteins as the trigger for the induction of stress responses: heat, diamide, and sodium arsenite. J. Cell. Physiol. 136, 411–420 [DOI] [PubMed] [Google Scholar]
- 37. McMillan D. R., Xiao X., Shao L., Graves K., and Benjamin I. J. (1998) Targeted disruption of heat shock transcription factor 1 abolishes thermotolerance and protection against heat-inducible apoptosis. J. Biol. Chem. 273, 7523–7528 [DOI] [PubMed] [Google Scholar]
- 38. Leopoldino A. M., Carregaro F., Silva C. H., Feitosa O., Mancini U. M., Freitas J. M., and Tajara E. H. (2007) Sequence and transcriptional study of HNRPK pseudogenes, and expression and molecular modeling analysis of hnRNP K isoforms. Genome 50, 451–462 [DOI] [PubMed] [Google Scholar]
- 39. Kimura Y., Nagata K., Suzuki N., Yokoyama R., Yamanaka Y., Kitamura H., Hirano H., and Ohara O. (2010) Characterization of multiple alternative forms of heterogeneous nuclear ribonucleoprotein K by phosphate-affinity electrophoresis. Proteomics 10, 3884–3895 [DOI] [PubMed] [Google Scholar]
- 40. Satyal S. H., Chen D., Fox S. G., Kramer J. M., and Morimoto R. I. (1998) Negative regulation of the heat shock transcriptional response by HSBP1. Genes Dev. 12, 1962–1974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kim H. J., Joo H. J., Kim Y. H., Ahn S., Chang J., Hwang K. B., Lee D. H., and Lee K. J. (2011) Systemic analysis of heat shock response induced by heat shock and a proteasome inhibitor MG132. PLoS One 6, e20252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kim M. K., Cho J. H., Lee J. J., Cheong Y. H., Son M. H., and Lee K. J. (2013) Differential protective effects of exenatide, an agonist of GLP-1 receptor and Piragliatin, a glucokinase activator in β cell response to streptozotocin-induced and endoplasmic reticulum stresses. PLoS One 8, e73340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kim J. A., Lee S., Kim D. E., Kim M., Kwon B. M., and Han D. C. (2015) Fisetin, a dietary flavonoid, induces apoptosis of cancer cells by inhibiting HSF1 activity through blocking its binding to the hsp70 promoter. Carcinogenesis 36, 696–706 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.