

Abstract
The Escherichia coli Complex II homolog quinol:fumarate reductase (QFR, FrdABCD) catalyzes the interconversion of fumarate and succinate at a covalently attached FAD within the FrdA subunit. The SdhE assembly factor enhances covalent flavinylation of Complex II homologs, but the mechanisms underlying the covalent attachment of FAD remain to be fully elucidated. Here, we explored the mechanisms of covalent flavinylation of the E. coli QFR FrdA subunit. Using a ΔsdhE E. coli strain, we show that the requirement for the assembly factor depends on the cellular redox environment. We next identified residues important for the covalent attachment and selected the FrdAE245 residue, which contributes to proton shuttling during fumarate reduction, for detailed biophysical and structural characterization. We found that QFR complexes containing FrdAE245Q have a structure similar to that of the WT flavoprotein, but lack detectable substrate binding and turnover. In the context of the isolated FrdA subunit, the anticipated assembly intermediate during covalent flavinylation, FrdAE245 variants had stability similar to that of WT FrdA, contained noncovalent FAD, and displayed a reduced capacity to interact with SdhE. However, small-angle X-ray scattering (SAXS) analysis of WT FrdA cross-linked to SdhE suggested that the FrdAE245 residue is unlikely to contribute directly to the FrdA-SdhE protein-protein interface. We also found that no auxiliary factor is absolutely required for flavinylation, indicating that the covalent flavinylation is autocatalytic. We propose that multiple factors, including the SdhE assembly factor and bound dicarboxylates, stimulate covalent flavinylation by preorganizing the active site to stabilize the quinone-methide intermediate.
Keywords: bioenergetics, flavin adenine dinucleotide (FAD), membrane enzyme, oxidation-reduction (redox), protein assembly, protein structure, respiratory chain, Complex II
Introduction
The isoalloxazine-conjugated ring system of enzyme-associated flavins can support oxidoreduction and group transfer reactions. Recent surveys of the literature identify that this flavin cofactor is covalently attached to ∼10% of flavoenzymes (1, 2). The covalent bond has been proposed to have many roles including preventing the loss of the cofactor, increasing the stability of the flavoenzyme, or raising the redox-potential of the cofactor (3–5).
One model system that has been used to investigate covalent flavin attachment is the integral-membrane heterotetramer quinol:fumarate reductase (QFR).4 QFR is a member of the Complex II superfamily and is composed of FrdABCD subunits. The FrdA and FrdB subunits are soluble and attached to the membrane through interaction with the membrane-embedded FrdC and FrdD subunits. In Escherichia coli QFR, the flavin is covalently attached to the FrdA subunit via an 8α linkage to the N-ϵ of His44. This places the flavin isoalloxazine group at the interface of two domains, termed the flavin-binding domain and the capping domain (6). These domains can move with respect to each other, which may allow the catalytic subunit to close over the substrate-bound active site and protect the reaction intermediates from solvent (7).
Physiologically, QFR catalyzes the reduction of fumarate (kcat ∼250/s) at this flavin-based active site during anaerobic respiration with fumarate as the terminal electron acceptor (8). In this process, the two electrons for fumarate reduction derive from the oxidation of menaquinol in the membrane and the two protons are likely transferred from solvent via a proton shuttle pathway consisting of the FrdAE245, FrdAR248, and FrdAR287 side chains located on the capping domain (9–12). QFR can also catalyze the reverse reaction, succinate oxidation, albeit with slower kinetics (kcat ∼30/s) and poorer catalytic efficiency (8). Other Complex II superfamily members, including bacterial succinate:quinone oxidoreductase (SQR, SdhABCD) and mammalian mitochondrial Complex II (also termed SdhABCD), kinetically favor the succinate oxidation reaction over the fumarate reduction reaction. As in QFR, the FAD cofactor is attached via a 8α-N3-histidyl linkage in the SdhA subunit (13); this linkage forms at SdhA His45 in the E. coli SQR.
An early study of covalent flavinylation performed mutagenesis of QFR-FrdAH44, which forms the histidyl linkage to FAD (3). When FrdAH44 was substituted with serine, cysteine, or tyrosine the intact QFR (FrdABCD) enzyme retained tightly associated non-covalent flavin at stoichiometric levels. Furthermore, kinetic characterization identified that QFR-FrdAH44S, QFR-FrdAH44C, and QFR-FrdAH44Y lacked detectable succinate oxidation activity but retained fumarate reduction at 25–50% of wild-type activity. This result suggested that the increase in flavin potential imparted by the covalent linkage is important for the succinate oxidation reaction catalyzed by Complex II enzymes. From this, it was proposed that one reason for the inability of the variants to oxidize succinate was because of a lowered redox potential of the non-covalent FAD (3). This was later supported by findings where similar site-directed variants of E. coli SQR result in an ∼90 mV reduction in the Em,7 of the non-covalent flavin compared with wild-type enzyme (14) and in E. coli QFR where there was a −100 mV drop in redox potential of the FAD compared with the wild-type enzyme (15).
A number of years ago, Walsh (16) proposed that the mechanism of covalent flavin bond formation was fully autocatalytic and involved a quinone-methide intermediate (Fig. 1). This mechanism only requires the FAD and a specific site of attachment; electrons and protons can be shuttled to this location to activate both the isoalloxazine ring and the amino acid where the covalent bond forms. More recently, the field has focused on the role of auxiliary proteins in covalent flavinylation. One protein of intense interest, termed SdhE (or YgfY) in bacteria, Sdh5 in yeast, and SdhAF2 in humans (17, 18), is a ∼10-kDa protein conserved throughout all kingdoms (19) that can support covalent flavinylation of human SdhA in an in vitro reconstituted system (20). Using bacterial QFR and SQR as models, the SdhE assembly factor has been shown to bind directly to the FrdA or SdhA subunits prior to assembly into the intact complex (21). The interaction involves a surface of SdhE/Sdh5/SdhAF2 containing a conserved RGXXE motif (22, 23) and a surface of FrdA/SdhA that interacts with the FrdB/SdhB subunit in the assembled heterotetramer (21). Moreover, the FrdA/SdhA subunit must be pre-associated with FAD for robust interaction with the SdhE assembly factor (20, 21).
Figure 1.
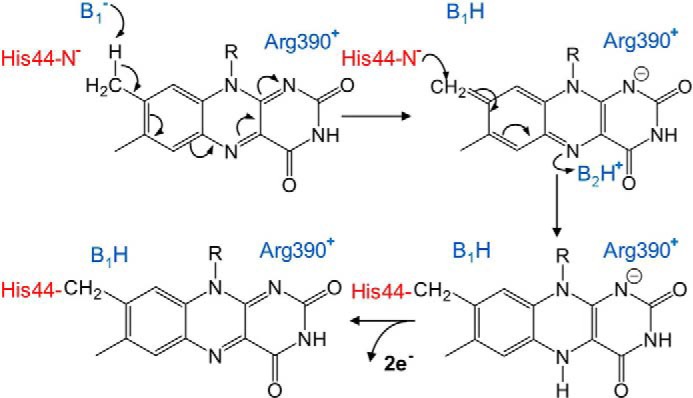
Proposed chemical mechanism of covalent flavinylation via a quinone-methide intermediate. Catalytic bases are shown in blue. The identities of the residues involved in proton abstraction and intermediate stabilization in QFR have not been unambiguously determined, but likely possibilities are shown here. The FrdAH44 side chain (red) is the nucleophile that forms a covalent bond, and may also be the base for proton abstraction.
Despite these advances, how SdhE/Sdh5/SdhAF2 promotes covalent flavinylation is not clear. We previously proposed a hypothesis where closure of the flavoprotein-capping domain over bound dicarboxylate (the term dicarboxylate is used in the general sense to refer to molecules containing two carboxylates, such as the substrate fumarate, the product succinate, the natural inhibitors oxaloacetate and malonate, etc.) at the active site promotes alignment of the FrdA/SdhA active site side chains near the bound flavin (21). This scenario could allow the FrdA/SdhA protein to adopt a conformation that stabilizes the quinone-methide tautomer at the C(8)α position on the isoalloxazine ring of the flavin. Tight closure of the flavoprotein-capping domain would be followed by nucleophilic attack by the nearby histidyl residue to form the covalent bond at the C(8) position. An alternative proposal could be that residues of SdhE/Sdh5/SdhAF2 directly participate in the chemistry of covalent bond formation. This alternative is not favored because of recent evidence that the SdhAF2 assembly factor enhances covalent flavinylation, but is not essential (24, 25). For both scenarios, the observation that SdhE and FrdB bind to the same surface of FrdA indicates that the flavoprotein-assembly factor complex must disassociate prior to assembly of FrdA/SdhA into the intact QFR/SQR heterotetramer (21).
In this study, we demonstrate that in E. coli QFR and SQR, the influence of deleting SdhE on covalent flavinylation depends upon the redox environment of the cell. To investigate SdhE-independent mechanisms of covalent flavinylation, we next identified variants of the QFR FrdA subunit with levels of covalent flavinylation below those observed in the ΔsdhE strains. We selected the FrdAE245Q variant for in-depth study; this side chain has previously been suggested as a part of the proton delivery pathway during fumarate reduction (7, 11), but has not been suggested as participating in covalent flavinylation. We identify that the QFR-FrdAE245Q variant retains a structure similar to the wild-type enzyme, but exhibits impaired dicarboxylate binding and catalytic activity. Because covalent flavinylation is normally anticipated to occur in the isolated FrdA subunit prior to assembly into the QFR (FrdABCD) complex, we additionally investigated the effects of this variant in the context of isolated FrdA subunit. We found similar stability of wild-type FrdA and FrdAE245 variants, but impaired interactions with the SdhE assembly factor. SAXS data indicate that this residue is unlikely to interact directly with SdhE. Because no single auxiliary factor is essential for the formation of the covalent flavin linkage, the most likely conclusion from these data is that covalent flavinylation is autocatalytic. One interpretation of much of the data is that factors stimulating covalent flavinylation would pre-organize the active site, suggesting that the autocatalytically formed quinone-methide tautomer originally proposed by Walsh (16) remains the most likely pathway to covalent flavinylation.
Results
SdhE influence on covalent flavin attachment
Clinical studies and yeast genetics were instrumental in the discovery of the assembly factor SdhAF2 (17, 26), and homologs have been identified in yeast (Sdh5 (17)) and bacteria (SdhE (18)). We focused on the E. coli system and used ΔsdhE strains to evaluate the impact of SdhE on covalent flavinylation of QFR and SQR complexes. We find that anaerobic expression of QFR (FrdABCD) in ΔsdhE strains results in retention of covalent flavinylation, albeit at a reduced level compared with when SdhE is present (Fig. 2, A and B). In contrast, QFR shows significant reduction of covalent FAD when expressed aerobically (Fig. 2, A and B). We then tested to see whether the SQR homolog (SdhABCD), which is kinetically optimized to function under aerobic conditions, would retain covalent FAD when expressed anaerobically (Fig. 2C). Anerobically, we observe levels of SdhA flavinylation similar to those found for FrdA when the full enzyme complexes are expressed. This suggests that the enhancement of flavinylation imparted by SdhE under aerobic conditions may be partially compensated for by anaerobiosis, when fumarate is likely to bind to the flavoprotein.
Figure 2.
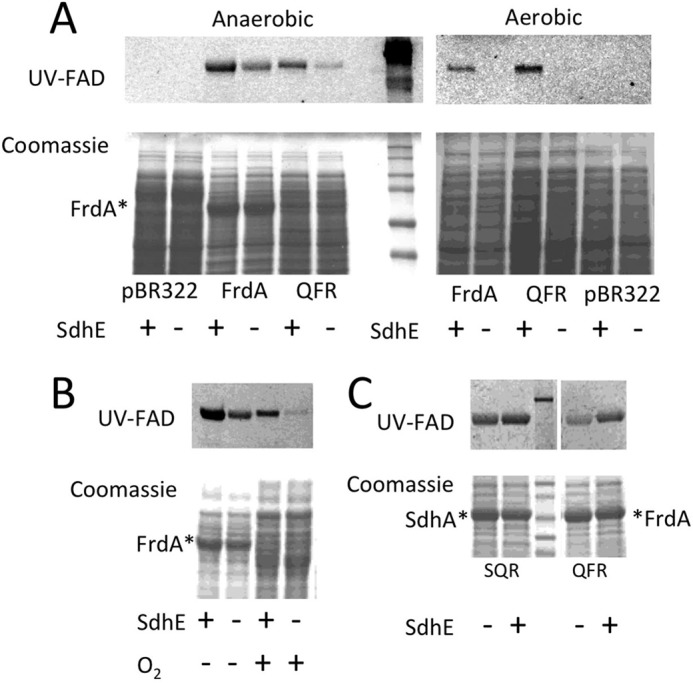
Effect of aerobic and anaerobic cell growth on flavinylation. Expression of FrdA from pFrdA and pH3 plasmids was performed in E. coli RP-2 (ΔfrdABCD, ΔsdhCDAB) and RP-3 (ΔfrdABCD, ΔsdhCDAB, ΔsdhE) under aerobic and anaerobic conditions. A, SDS-PAGE analysis of whole cell samples (20 μg of protein per lane) shows attenuated, but not abrogated covalent flavinylation under anaerobic conditions, but substantial loss of covalent flavinylation under aerobic growth conditions. The top panels indicate UV fluorescence from the covalently-bound FAD cofactor and the bottom panels are Coomassie Blue G staining of the cell extracts. Mr markers are shown on the right side of the gel for the anaerobically grown cells. Note, some of the pre-stained MW markers (Bio-Rad, number 1610374) show inherent fluorescence when exposed to a UV light. The asterisk indicates the position of FrdA in the Coomassie-stained samples. B, analysis of isolated membrane fractions from anaerobically and aerobically grown cells. Note, there is reduced expression of FrdA in the aerobically grown cells because expression is driven by the natural FRD (i.e. anaerobic) promoter. Nevertheless, there is reduced covalent flavinylation in the absence of SdhE for cells grown aerobically as compared with anaerobically. C, effect of SdhE on flavinylation in anaerobically expressed SQR and QFR. Comparison of membrane fractions isolated from RP-3 (ΔsdhE) and RP-2 (+sdhE) cells indicates that SdhE is not essential for covalent flavinylation of either SdhA or FrdA. The top panel shows UV fluorescence of the covalent FAD. The middle lane of both panels shows prestained Mr markers. The bottom panel shows Coomassie Blue G staining of proteins in the membrane fraction. The asterisk indicates the location of SdhA or FrdA, respectively. Note, both SQR and FRD are expressed anaerobically from the FRD promoter as previously described (8).
QFR Mutants deficient in covalent flavinylation
We generated a library of site-directed variants within the FrdA subunit of the E. coli QFR complex (Fig. 3A). We selected residues for mutagenesis because they exhibited one or more of the following properties: (i) they directly interact with bound substrate (FrdAR287, FrdAH355, and FrdAR390 (6)); (ii) they are either previously proposed to contribute to the shuttling of protons during catalysis (FrdAE245 and FrdAR287 (10, 11)) or could contribute to the stability of the substrate-binding site and effect the properties of other catalytic residues (FrdAD288); (iii) they may stabilize the interdomain orientations or motions important for catalysis (FrdAE245 and FrdAE250 (11)); (iv) they are in regions that may be allosterically connected to the active site (FrdAE250); or (v) they are near the proposed binding surface for the assembly factor SdhE (FrdAD129 (21)). QFR containing wild-type FrdA or FrdAR114, which is a charged residue near the FAD but without any proposed mechanistic role were used as positive controls; the previously described FrdAH44S variant of QFR, which alters the normal histidyl linkage to the FAD and retains stoichiometrically bound non-covalent FAD, was used as a negative control (3).
Figure 3.
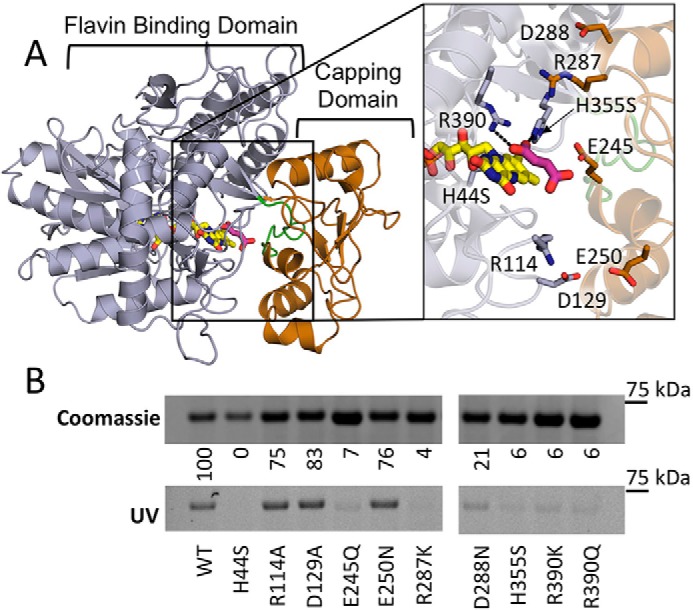
Covalent flavinylation of QFR mutants. A, location of substitutions in the FrdA subunit of QFR on the flavin-binding (gray) and -capping domains (copper) (PDB entry 3P4P (38)). Flavin (yellow) and fumarate (pink) ligands are modeled and the active site loop flanking FrdA245E is colored green (residues 234–244 in QFR). Zoomed window shows a close up of the location of the residues that were altered to evaluate their effect on covalent flavinyation. They are colored according to their location on either the flavin-binding or -capping domain. B, quantitation of covalent FAD levels in wild-type and variant QFR complexes. Coomassie-stained SDS-PAGE gels are normalized to the total amount of protein to assess differences in expression levels of variant QFR complexes. The same gel was illuminated with UV light to measure covalent flavinylation by flavin fluorescence. The ratio between the Coomassie signal and the flavin fluorescence was quantified. Wild-type covalent flavinylation was set to 100%, whereas QFR-FrdAH44 was set to 0%, and these were used as standards to quantify the percentage of covalent FAD in the remaining variants.
We evaluated the effect of these substitutions on the levels of covalent flavinylation in the intact FrdABCD enzyme produced under anaerobic conditions. To do this, we separated QFR-enriched membranes on SDS-PAGE and monitored the level of FAD fluorescence associated with the FrdA subunit (Fig. 3B). Wild-type QFR, the QFR-FrdAR114 variant, and the QFR-FrdAH44S variant were used as comparators. Five of our designed variants (FrdAE245Q, FrdAR287K, FrdAH355S, FrdAR390K, and FrdAR390Q) had almost a complete absence of covalent flavinylation, consistent with studies of the SQR protein (14). The FrdAD288N variant showed minimal residual fluorescence, suggesting covalent flavinylation was severely compromised. Each of these variants had levels of covalent flavin attachment below that observed for QFR expressed in the ΔsdhE strain that was similarly grown anaerobically (Fig. 2, A and B). This suggests that these side chains have a role in flavinylation that is independent of assembly factor function. It should be noted that with the exception of FrdAD288, each of the amino acids associated with loss of flavinylation is known to be involved in substrate binding and/or the catalytic mechanism of fumarate-succinate interconversion (6, 12, 27). Accordingly, one possibility is that these side chains also directly participate in the mechanism of flavinylation.
Of these residues, we selected QFR containing the FrdAE245Q variant for more detailed study. FrdAE245 exhibits loss of detectable flavinylation but has not previously been implicated in the covalent flavinylation process. It has been proposed as a proton shuttle during fumarate reduction within the context of both the E. coli QFR (7) and the Shewanella soluble fumarate reductase homolog (11), but is not directly involved in substrate binding. This residue is adjacent to FrdAT244, a residue implicated in the catalytic efficiency of the enzyme (7) and associated with stoichiometric covalent FAD. FrdAT244 is part of an 11-amino acid loop (FrdAT234 to FrdAT244 in E. coli QFR) that connects the flavin-binding domain and the capping domain of the flavoprotein (Fig. 3A). The location of the FrdAE245 side chain at the domain interface of the flavoprotein suggests that this side chain could have an additional role in stabilizing the orientations of the flavin-binding and capping domains of FrdA.
Catalytic activity of QFR complexes lacking covalent FAD
We began by investigating how the FrdAE245 mutation affected function within the context of the assembled QFR complex. Previous findings have shown that the QFR-FrdAH44S variant lacks covalent FAD, however, it can bind stoichiometric amounts of non-covalent FAD (3). Catalytically, the QFR-FrdAH44S variant retained ∼50% of its ability to reduce fumarate, whereas it only retains 1% of its ability to oxidize succinate (3). In the current study we found that the QFR-FrdAE245Q variant lacked detectable catalytic activity for both fumarate reduction and succinate oxidation (data not shown). This finding is consistent with the previously reported reduction of detectable fumarate reduction activity to <1% of wild-type levels upon mutation of the analogous residue in the soluble FrdA homolog from Shewanella frigidimarina, which naturally contains non-covalent FAD as its redox cofactor (22). We anticipate that the FAD cofactor remains non-covalently bound in the analogous QFR-FrdAE245Q variant. Indeed, prior studies demonstrate that in variant forms of QFR containing non-covalent FAD, the cofactor cannot be removed without KBr treatment, which is proposed to induce reversible unfolding (3).
To distinguish whether the QFR-FrdAE245Q variant lacks activity due to altered ligand binding at the bound, non-covalent FAD, we analyzed the QFR interaction with two dicarboxylates: fumarate and oxaloacetate. We evaluated the binding of fumarate because it is the natural substrate. We also evaluated oxaloacetate because oxaloacetate is a natural tight-binding inhibitor of Complex II enzymes, with nanomolar affinity for most homologs. In addition, oxaloacetate forms a charge transfer band with the isoalloxazine ring of the flavin, which results in a characteristic spectrum for Complex II proteins that is easier to follow than fumarate/succinate. For these two reasons, measuring oxaloacetate binding can be particularly useful for assessing altered dicarboxylate binding capacity in variants.
For this measurement, we used difference spectra obtained by subtraction of a ligand-free spectrum from that of a ligand-bound wild-type QFR spectrum. This experiment monitors characteristic spectral changes in the flavin spectrum that occur upon binding of dicarboxylates. These spectral changes include an increase in absorption at 500 nm indicative upon fumarate binding near the FAD cofactor and a charge-transfer band in the 600-nm region upon oxaloacetate binding near the FAD. Both of these features are observed for the wild-type QFR (Fig. 4A). The QFR-FrdAH44S variant containing noncovalent FAD shows spectral changes similar to wild-type QFR, indicating that dicarboxylate binding is similar to the wild-type enzyme (Fig. 4B). The difference spectra of the QFR-FrdAE245Q variant, however, show no distinct features (Fig. 4C). This observed lack of spectral changes from the bound FAD upon the addition of dicarboxylates is likely due to significant changes in binding capability for these ligands.
Figure 4.
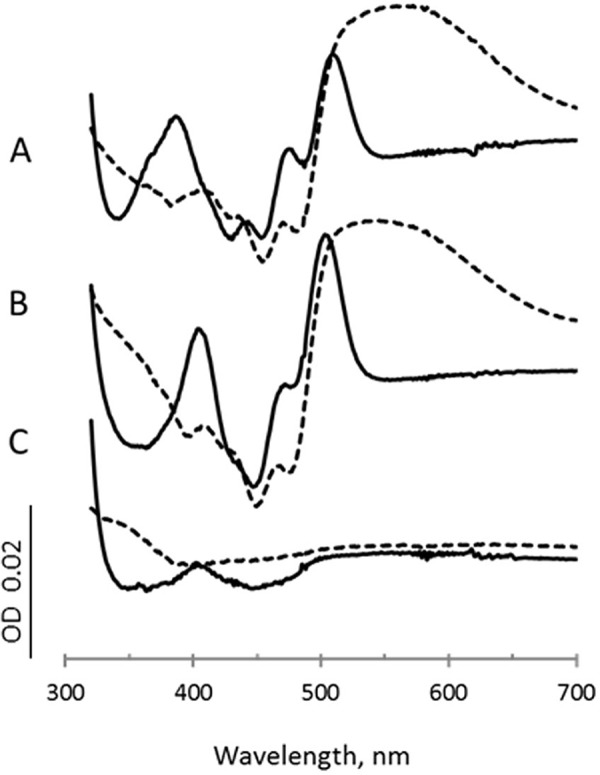
Dicarboxylate scans. Spectra are the difference between the spectrum of the enzyme after addition of each ligand minus the spectrum of the oxidized enzyme. Difference spectra for fumarate (10 mm) is shown as solid lines, and difference spectra for oxaloacetate (0.2 mm) are shown as dashed lines. A, wild-type QFR; B, QFR-FrdAH44S; C, QFR-FrdAE245Q.
Crystal structure of the QFR-FrdAE245Q mutant
To identify whether the lack of dicarboxylate binding is the result of an altered fold of the enzyme, or loss of the non-covalent FAD cofactor we determined the crystal structure. Crystallization trials of QFR-FrdAE245Q using conditions that were previously successful for wild-type and variant enzymes did not result in the appearance of crystals in this case. As a result, we screened for distinct chemical conditions to support crystallization of this variant. These crystals diffracted to low resolution, with the best data set merging to 4.25-Å resolution (Fig. 5A, Table 1). At this resolution, side chain conformations cannot be unambiguously assigned. Thus, we are not able to identify whether the FrdAE245Q substitution has allosterically altered the positions of the dicarboxylate binding residues within the active site, which would be one explanation for why dicarboxylate binding is lost. However, the capping domain is in a similar position as it is found in the wild-type enzyme. As a result, side chains from the flavin-binding and -capping domains could conceivably be presented to the active site with similar geometries as is observed in the wild-type enzyme. Indeed, the structure of the QFR-FrdAE245Q complex showed close similarity to the wild-type protein, with an root mean square deviation of 0.33 Å for all atoms of the flavoprotein. This is consistent with previous work on the Shewanella soluble fumarate reductase, where mutation of the analogous residue to aspartate (E378D) resulted in little structural perturbation (11).
Figure 5.
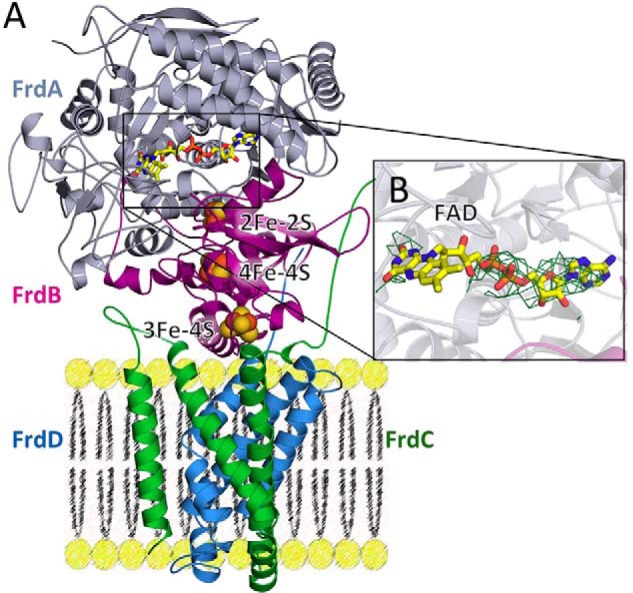
Structure of QFR containing the FrdAE245Q mutation. A, structure of QFR-FrdAE245Q. The flavoprotein (FrdA) is shown in gray, the iron-sulfur protein (FrdB) is shown in magenta, and the membrane subunits (FrdC and FrdD) are shown in green and blue, respectively. The locations of the cofactors (FAD as yellow sticks, and iron-sulfur clusters as spheres) are also shown. B, |Fo| − |Fc| electron density maps contoured at 3σ were calculated in phenix.refine after the removal of FAD from the input PDB file. The quality of the electron density is consistent with the resolution for the adenine dinucleotide, but there is no interpretable density for the isoalloxazine ring.
Table 1.
Crystallographic data collection and refinement statistics
Values in parentheses are for the highest resolution shell.
| Data statistics | |
|---|---|
| Data collection | |
| PDB entry | 5VPN |
| Beamline | APS 21-IDG |
| Wavelength | 0.97856 Å |
| Resolutiona | 4.25 Å |
| Space group | P212121 |
| Unit cell | a = 133.6 Å |
| b = 138.1 Å | |
| c = 220.1 Å | |
| Rsym | 0.166 (0.899)b |
| Rpim | 0.075 (0.448) |
| CC½ | 0.919 |
| I/σa | 10.6 (1.4) |
| Completeness | 96.8% (98.2%) |
| Redundancy | 5.5 (4.6) |
| Refinement | |
| Rcryst | 0.215 |
| Rfree | 0.270 |
| Root mean square deviation | |
| Bond lengths | 0.003 Å |
| Bond angles | 0.614° |
| Ramachandran | |
| Most favored | 93.6% |
| Allowed | 6.1% |
| Outliers | 0.3% |
a The data have an I/σ > 2 in the outer shell at 4.4-Å resolution.
b Values in parentheses are statistics for the highest resolution shell, 4.4–4.25-Å resolution.
Notably, there was a loss of strong electron density for much of the isoalloxazine ring of the FAD, despite retention of electron density for the adenine dinucleotide (Fig. 5B). This loss of electron density could arise for several reasons. One possibility is increased mobility of the isoalloxazine ring in the absence of the tethering covalent linkage. The relative structural stability of the isoalloxazine in flavoprotein homologs lacking the covalent linkage would argue against, but would not preclude, this interpretation. An alternative is that the low resolution of the data has reduced the quality of the electron density in this region. Nevertheless, it is clear from the crystal structure that the QFR complex is folded and assembled with non-covalent FAD in the presence of the FrdAE245Q mutation.
Folding and thermal stability in wild-type and variant FrdA subunits
We next assessed the function of the FrdAE245 side chain in isolated FrdA subunits. The rationale for additionally assessing the role of FrdAE245 in the isolated FrdA subunits is that FrdA containing non-covalent FAD represents the assembly intermediate, i.e. covalent flavinylation is believed to occur within the isolated subunit. We first ensured that the variant FrdA subunits were properly folded. As we were unable to identify crystallization conditions that support the growth of isolated FrdA subunits, we used CD spectroscopy and thermal denaturation to measure folding and stability. Changes in stability of the isolated FrdA subunit in the presence of mutations at the FrdAE245 could suggest whether stability affects the covalent flavinylation and maturation process, as has previously been suggested for the human complex (20). For these studies, we compared wild-type FrdA, FrdAH44S (lacking the histidine linkage to the FAD), FrdAE245Q, and two additional variants of the FrdAE245 side chain: FrdAE245R and FrdAE245D. Each of these isolated FrdAE245 variants is associated with stoichiometric non-covalent flavin (Fig. 6A), as assessed by comparing the total amount of bound FAD to the wild-type FrdA subunits and verifying that these variants lack measurable covalent FAD.
Figure 6.

Circular dichroism spectroscopy of wild-type and variant FrdA subunits. A, measurement of flavin in wild-type and variant FrdA subunits (∼67 kDa). The quantity of flavin covalently associated with FrdA was measured by separating lysates on SDS-PAGE and measuring FAD fluorescence following illumination with UV light. The relative quantity of FAD associated with wild-type and variant FrdA subunits was assessed by measuring the A280/A450 ratio of purified proteins. This value was normalized to 1.0 for wild-type protein. The ratio of A280/A450 was similar in wild-type and variant subunits, indicating that these had a similar amount of flavin associated with each subunit. As in gel UV detection indicates that flavin is not covalently associated with the subunits, the tightly associated FAD must be non-covalently bound, likely via specific binding to the active site. B, far-UV CD spectrum of wild-type FrdA compared with spectra for the FrdAH44S variant, the FrdAE245 variants, and FrdA incubated at 100 °C for 2 h. C, representative traces of thermal CD of wild-type FrdA, FrdAH44S, FrdAE245D, FrdAE245Q, and FrdAE245R. The average Tm values are indicated.
We measured the far-UV CD spectrum for wild-type FrdA, which is consistent with a predominantly helical fold (Fig. 6B). The spectra for all variants tested were similar to that of wild-type FrdA, indicating that each variant FrdA subunit is properly folded and there are no major changes in the secondary structure. We next monitored changes in the ellipticity at 220 nm as a function of temperature in thermal denaturation CD spectroscopy. There were no significant differences in the melting temperature (Tm) or midpoint of the unfolding transition for the variants as compared with wild-type FrdA (Fig. 6C). This indicates that neither the amino acid substitutions nor the loss of the covalent bond to the bound flavin significantly impacts FrdA subunit stability.
Cross-linking with the assembly factor, SdhE
To identify whether the FrdAE245 side chain influences the interaction of FrdA with the assembly factor SdhE, we monitored the ability of FrdA variants to cross-link with SdhE incorporating the artificial amino acid para-benzoyl phenylalanine (pBpF) at position 8. As previously described, this variant of SdhE, termed SdhE-R8BpF, readily cross-links with wild-type flavoprotein when illuminated with UV light (21), suggesting that this method can be used to report upon whether FrdA variants retain the ability to cross-link with SdhE.
We first measured cross-linking between wild-type FrdA and SdhE-R8BpF over the course of a 4-h illumination with UV light (Fig. 7A) and observed a time-dependent increase in FrdA-SdhE association. We selected the 3-h time point as suitable for measuring cross-linking to variants, and measured cross-linking between SdhE-R8pBpF and the FrdAE245D/Q/R and FrdAH44S variants. Using this assay, we observed robust cross-linking between SdhE and wild-type FrdA. Indeed, the cross-linking could be observed on a Coomassie-stained SDS-PAGE gel, as previously reported (21). In contrast, cross-linking to each of the variants was substantially attenuated, and we could no longer observe the formation of a stable complex when monitoring by Coomassie-stained SDS-PAGE. To assess whether a low level of cross-linking occurred, we evaluated the formation the FrdA-SdhE cross-linked complex using Western blotting analysis, probing with an antibody that recognizes that His6-affinity tag of SdhE. Even with Western blotting analysis, titration of the antibody concentrations was required before the signal was observed (Fig. 7B).
Figure 7.
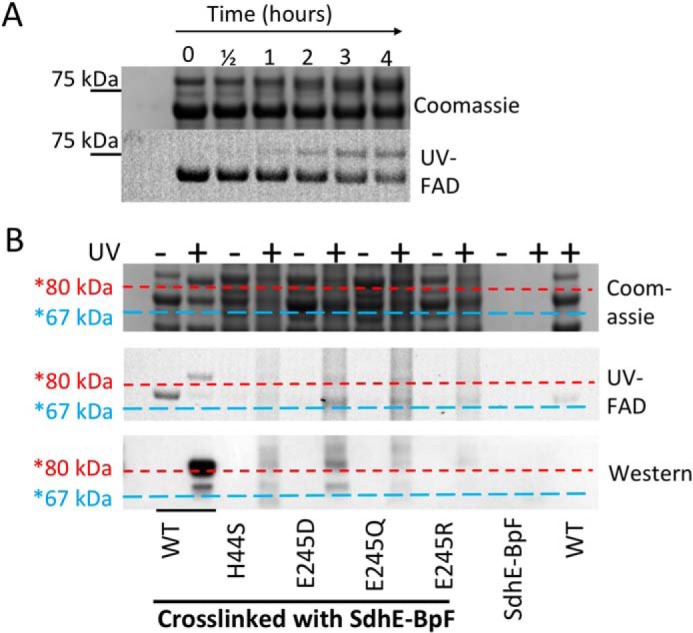
Cross-linking between wild-type and variant FrdA subunits and SdhE-R8BpF. Purified SdhE-R8BpF was mixed with lysate containing FrdA. Following UV exposure, samples were immobilized on Ni2+ resin, washed, and eluted with SDS-PAGE loading buffer prior to separation on an SDS-PAGE gel. A, time course of cross-linking of SdhE-R8BpF with wild-type flavoprotein over 4 h after exposure to UV light. The position of the 75-kDa molecular mass marker is highlighted. B, comparison of flavin fluorescence for detection of covalent FAD, Coomassie-stained SDS-PAGE and anti-His6 Western blot analysis of cross-linked samples. Horizontal lines were drawn after aligning the Coomassie, UV, and Western blot analyses to assist with identifying the molecular weights of the bands. The blue dashed lines are drawn just under the location of the FrdA subunit (∼67 kDa) and red dashed lines are drawn just under the approximate location of the FrdA-SdhE complex (∼80 kDa).
A reduction in binding between FrdA and the SdhE assembly factor is somewhat anticipated for the FrdAH44S variant, as the FrdAH44 side chain is proposed to be part of the FrdA-SdhE binding interface (21). However, the location of the FrdAE245 side chain is on the capping domain (6). In the assembled (FrdABCD) QFR heterotetramer, this side chain is buried, but mediates contacts between the FAD-binding domain and capping domain of the FrdA subunit. One possibility for the observed loss of interaction is that the cross-linking assay appears to be exquisitely sensitive to small structural changes introduced by mutations, as was shown in prior studies (21). Thus even minor structural changes accompanying these mutations could result in a loss of cross-linking. Alternatively, it is possible that the FrdA subunit undergoes conformational changes during SdhE binding that are altered in the context of FrdAE245 variants. Because FrdAE245 is positioned on the capping domain of FrdA adjacent to a loop that links the two domains, and because the capping domain of FrdA likely rotates upon the interaction with SdhE, this rotation could be altered or prohibited in the context of FrdAE245Q.
SAXS of the FrdA-SdhE-R8BpF cross-linked complex
Although there is currently not an experimental structure of the flavoprotein-SdhE complex, a computational model has been developed using restraints from site-specific cross-linking studies combined with mass spectrometry (21). This model proposed that the FrdA-SdhE interaction requires a rotation of the FrdA-capping domain, but did not suggest how the capping domain might reorient. To investigate a potential role of FrdAE245 in conformational changes in the FrdA subunit during SdhE binding, we performed SAXS analysis of the wild-type E. coli FrdA-SdhE-R8BpF cross-linked complex at protein concentrations of 1.2 and 2.4 mg/ml. Scattering intensity profiles and linear Guinier plots indicated that the protein lacks aggregation and is monodisperse (Fig. 8A). Furthermore, a Guinier approximation exhibited good linearity at both protein concentrations and yielded Rg values of 28.11 ± 1.68 and 27.79 ± 0.92 Å with an average of Rg ∼28 Å. Fourier transformation of SAXS data in the same range gave Dmax of 82–86 Å and Rg of 27–28 Å (Fig. 8B). We then developed several sets of coordinates for docking into the SAXS envelope. These coordinates began with the Rosetta-minimized computational model of the FrdA-SdhE complex that lacked the FrdA-capping domain (21). We manually assessed several plausible alternatives for capping domain positions, with the capping domain being moved as a rigid body. Each alternative that was tested reduced, but did not completely eliminate, steric clash with either SdhE or other parts of the FrdA. This procedure identified one model exhibiting a reasonable correlation between the theoretical scattering curve and the experimental scattering curves at both protein concentrations, computing χ2 values of 2.13 and 2.75, respectively (Fig. 8C). The selected computational model of the FrdA-SdhE cross-linked complex was then superimposed on the SAXS envelope by automated alignment of inertial axes (Fig. 8D). In the best model, the capping domain containing FrdAE245 is rotated so that the FrdAE245 side chain approaches the proposed SdhE-binding site, but this side chain does not appear to interact directly with SdhE. Although the resolution of SAXS data is not sufficient to place secondary structural elements or side chains, this large rotation is in contrast to small but significant structural changes observed in the flavoprotein p-cresol methyl hydroxylase upon interaction with a cytochrome subunit that promotes covalent flavinylation in that case (4). If the FrdA-SdhE model is correct, the most likely interpretation for the loss of cross-linking between FrdAE245Q and SdhE-R8pBpF is that the mutation either prevents FrdA from adopting a conformation that can bind SdhE or the mutation causes small surface changes that reduce SdhE binding without being a direct part of the binding site.
Figure 8.

SAXS of the cross-linked FrdA-SdhE-R9pBpF complex. A, intensity profiles in log scale of the cross-linked FrdA-SdhE complex acquired at protein concentrations of 1.2 (red) and 2.4 mg/ml (blue). The insets show the linear fit to the Guinier region of the respective protein concentrations. B, P(r) plot depicting the frequency distribution of interatomic vectors in the predominant scattering species after indirect Fourier transformation of the SAXS data provided an Rg and Dmax of 27.25 ± 0.32, 28.02 ± 0.13, and 82.83 and 86.81 Å, respectively, at lower and higher protein concentration. C, comparison of theoretical scattering curve (black line) of the computational model of the cross-linked protein with experimental scattering curves at different protein concentrations give χ2 values of 2.13 and 2.75, respectively. D, conceptual model of FrdA (blue) and SdhE (green) complex superimposed on wild-type FrdA (gray, PDB entry 3P4P (38)) and the SAXS envelope by automated alignment of inertial axes. Rotating views of the FrdA-SdhE-R8BpF model docked into the SAXS envelope.
Discussion
Although we know a great deal about the mechanism of succinate and quinone oxidoreduction by Complex II superfamily members, comparatively little is known about Complex II assembly. Indeed, our understanding of protein assembly in general is underdeveloped as compared with our understanding of how individual proteins function. In the case of Complex II, one critical aspect of assembly is the insertion and attachment of its cofactors. For well over 30 years, this process was considered fully autocatalytic, potentially proceeding via a quinone-methide intermediate. Only within the last 10 years were any specific assembly factors discovered, initiating a debate of the mechanisms of cofactor attachment.
SdhAF2 was identified in patients who presented clinical Complex II deficiency, but who did not have mutations in Complex II genes (17). Phylogenetic analysis of SdhAF2 (∼11 kDa) indicates that this protein evolved prior to the development of mitochondria, a proposal consistent with its strong conservation in all kingdoms of life (18). After its discovery, SdhAF2 was initially believed to be essential for covalent flavinylation of human Complex II. Indeed, its mutation recapitulates diseases associated with Complex II deficiency (17). However, recent studies of Crisper-CAS9 ΔsdhAF2 breast cancer cells suggest that covalent flavinylation can proceed in the absence of SdhAF2 (24). In addition, there are examples of thermophilic bacteria and archaea that lack SdhE genes, but still produce functional, covalently flavinylated SQR (25). Our finding that the requirement for SdhE is more important during aerobic respiration adds complexity to this debate, but further supports SdhE as a stimulating, non-essential factor for covalent flavinylation.
Intriguingly, SdhE is not the only factor that can enhance the efficiency of covalent flavinylation. It has also been shown that dicarboxylates, including citric acid cycle intermediates, stimulate covalent flavinylation of Complex II isoforms (28). Moreover, the deletion of the iron-sulfur protein (FrdB or SdhB) reduces, but does not eliminate covalent flavinylation, identifying this subunit as modestly supporting the formation of the covalent linkage (29, 30). Given that neither SdhE (24), the iron-sulfur protein (29), nor dicarboxylates (28) are absolutely required for covalent flavinylation of FrdA/SdhA, but that SdhE can enhance flavinylation of human SdhA in a purified system (20), the most logical conclusion is that the chemistry of covalent attachment is supported by the flavoprotein itself. But if covalent flavinylation does proceed autocatalytically, what are the chemical requirements, and how do SdhE, dicarboxylates, or the iron-sulfur protein enhance this?
Taken in aggregate, our findings and those from the literature support a model where autocatalytic covalent attachment via the quinone-methide intermediate requires a pre-organized active site with the capping domain closed tightly. A tightly closed structure would also be anticipated to be more stable and less sensitive to proteolysis, as has been proposed (20). This finding is consistent with observations where Sdh5 binding to yeast SdhA stabilizes Sdh5 against LON-mediated proteolysis (31). Indeed, binding of the flavoprotein to the assembly factor probably protects both from proteolysis. The tightly closed conformation may align active site residues into orientations that optimally support the chemistry of covalent flavinylation. Certainly the FrdAH44 residue must be positioned closely enough to the isoalloxazine C(8)α to allow formation of the covalent bond. In addition, other active site side chains may impact the covalent flavinylation chemistry directly, for example, FrdAR390 is positioned close to the N1-C2 of the isoalloxazine ring in the assembled complex.
Overall, the data suggest that there may be more than one way to promote active site pre-organization. The previously reported findings that bound dicarboxylates (28), the iron-sulfur protein (29, 30), and SdhE (17) can each enhance covalent flavinylation are consistent with this proposal, as each of these could function to pre-organize the active site. Bound dicarboxylates could directly interact with and orient active site residues, which would help to organize the active site and promote tight closure of the capping domain. The role of dicarboxylates in covalent flavinylation is consistent with our finding that mutation of residues that impact dicarboxylate binding directly or indirectly reduces covalent flavinylation to levels just above detection (Fig. 3), and substantially below those observed in ΔsdhE strains grown under the same conditions (Fig. 2). However, substrate turnover might not be necessary. Indeed, prior studies of other catalytically compromised QFR and SQR variants indicate that covalent flavinylation is usually retained (7, 8).
The binding of FrdA to SdhE or the iron-sulfur protein may similarly promote the organization of active site residues. A computational model developed in the context of experimental distance restraints strongly suggests that this FrdA-SdhE interaction promotes a conformational change in FrdA that closes the capping domain of the flavoprotein tightly over bound dicarboxylate (21). Similarly, the position of the iron-sulfur protein in the assembled complex may restrict the motions of the capping domain. SdhE may be particularly important during aerobic respiration with succinate. It is plausible that succinate may have lower affinity for an active site containing non-covalent FAD, the capping domain may not close as tightly prior to the formation of the covalent linkage. As a result, an auxiliary factor such as SdhE may increase the efficiency of covalent flavinylation under aerobic conditions (Fig. 2). An additional intriguing interpretation of a difference in flavinylation under aerobic versus anaerobic conditions is that SdhE may have evolved during the transition and adaption of cells to an aerobic environment. It may also be consistent with the idea that SQR formed from a QFR at the transition from anaerobic to aerobic life, with the covalent bond fulfilling the need for higher redox potential to oxidize succinate versus reduce fumarate.
Covalent flavinylation via a mechanism that uses a quinone-methide intermediate would also require a proton shuttle. Although the FrdAE245 side chain is proposed as part of a proton delivery pathway during fumarate reduction, it is not clear whether it plays the same role during covalent flavinylation. Pre-organization of the active site would have the impact of aligning the proton shuttling pathway consisting of FrdAR287, FrdAE245, and FrdAR248 (Fig. 9A). However, FrdAR248 variants retain covalent flavin (32). One interpretation of this finding is that this proton delivery pathway is used during fumarate reduction but not during covalent flavinylation. An alternative possibility could be considered in light of the rotation of the capping domain when FrdA is in complex with SdhE. The FrdA-SdhE model would suggest that this rotated capping domain could allow the FrdAE245 side chain to be directly exposed to solvent, such that the FrdAR248 side chain that normally participates in proton delivery could be bypassed during covalent flavin attachment (Fig. 9B). The amino acid residue equivalent to FrdAD288 is conserved in the Complex II family of enzymes (E. coli SdhAD287 and in the mature (i.e. 42-amino acid transit peptide processed) human SdhAD299. In the available X-ray structures of bacterial, yeast, and mammalian Complex II the aspartate residue is hydrogen bonded to a conserved Asn residue (FrdAN389, E. coli SdhAN398, and human SdhAN408), which is adjacent to FrdAR390 (E. coli SdhAR399 and human SdhAR409). FrdAR390 is known to be essential for covalent flavinylation in bacteria (Fig. 3B) and mutation of the equivalent residue in humans is associated with disease (33). FrdAR390 is thought to stabilize the negative charge at the N1-C2 position of the isoalloxazine that develops in the quinone-methide tautomer and thus is essential for development of the tautomer. It is reasonable to suggest that mutation of FrdAD288 to a neutral amino acid such as Asn impacts the architecture of the active site. This would be expected to affect dicarboxylate binding and the environment around FrdAR390 such that it is unable to sufficiently stabilize the negative charge that is required near the isoalloxazine N1-C2 position during the autocatalytic formation of the covalent flavin linkage. It is also possible, given the potential reorganization of the active site upon SdhE binding, that FrdAD288 has a role in proton shuttling in covalent flavinylation (Fig. 9B). It is likely that SdhE helps impact the active site architecture by stabilizing the environment near the flavin to help align the active site residues during the time necessary for formation of the covalent flavin bond. Further insight into the role of SdhE in covalent flavinylation requires the determination of a high-resolution structure of the FrdA-SdhE complex.
Figure 9.

Model for side chains in covalent flavinylation. A, active site organization in wild-type QFR with fumarate bound (gray, PDB entry 3P4P (38)). FAD (yellow) and fumarate (pink), and side chains or FrdA245, FrdA248, FrdA287, and FrdA288 are shown as sticks. The black arrows indicate the direction of proton transfer during fumarate reduction. B, the same view for the FrdA-SdhE-R8BpF model (blue). The black and red arrows indicate two potential alternate proton transfer pathways after reorganization of the active site as a result of domain rotation in the model. C, scheme for organization of the flavoprotein (gray) active site to promote autocatalysis of the covalent flavin bond. In this model, closure of the capping domain after binding of flavin (represented as stick model of the isoalloxazine ring) and proper orientation of the attachment site can be facilitated in several ways: (i) binding of dicarboxylate; (ii) binding of the assembly factor; (iii) enhanced stability and preorganization by sequence in thermophilic bacteria; (iv) binding of the iron-sulfur subunit. After this organization, covalent flavinylation can occur (indicated by additional bond) followed by complex assembly.
Conclusions
Together, the data argues in favor of multiple complementary mechanisms that promote the pre-organization of the Complex II active site during covalent flavinylation. One or more molecular players (bound dicarboxylate, SdhE, iron-sulfur protein, or potentially others) could facilitate the correct orientation of active site residues and promote tight domain closure over the isoalloxazine (Fig. 9C). The findings support the original proposal of Walsh (16) that covalent attachment is self-catalytic and argue against a direct role for SdhE in the chemistry of covalent flavinylation. Rather the role of SdhE or its homologous molecular chaperones (SdhAF2/Sdh5) is to stabilize the flavoprotein in a tightly closed conformation to organize the active site residues and to allow time for the covalent flavinylation to occur.
Experimental procedures
Expression and purification of the QFR complex (FrdABCD)
Wild-type and variant QFR (FrdABCD) were expressed as described (21) from plasmid pH3, which encodes the frdA+B+C+D+ operon under the control of the fumarate reductase (FRD) promoter. QFR was expressed as described in E. coli strain DW35, where both the frd and sdh operons are disrupted via the insertion of a kanamycin gene (34). Expression is induced under micro-aerophilic conditions, which is achieved in Terrific Broth medium by increasing the culture volume in the flasks (1.6 liters in a 2-liter growth flask), and reducing the shaking to 160 rpm (8). Cells were harvested by centrifugation at 9200 × g and disrupted by sonication. Membranes were isolated by ultracentrifugation. QFR comprises ∼50% of the total membrane protein when expressed using this protocol, and covalent flavinylation was assessed on this membrane-embedded QFR without further purification.
For crystallization, the QFR-FrdAE245Q variant was purified as described (35). Membranes were resuspended in 25 mm Tris, pH 7.4, 0.1 mm EDTA containing complete protease inhibitor tablets (Roche Applied Science) and solubilized in 2% thesit detergent (C12E9, Anatrace). The insoluble fraction was removed by centrifugation at 34,000 × g, and the supernatant containing membranes was filtered and applied to a Q-Sepharose column. The protein was eluted with a linear NaCl gradient from 50 mm to 1 m. The NaCl concentration was reduced via three rounds of 1:10 dilution and reconcentration using a 30-kDa molecular mass cut-off filter (Amicon). QFR was then further purified using a Poros 50HQ column, eluting with a linear NaCl gradient from 50 mm to 1 m. The protein was then concentrated as above and injected onto a Superdex 200 Increase 10/300 column. Protein concentration was determined using the Bradford assay.
Expression and purification of isolated FrdA subunits
Isolated wild-type and variant FrdA subunits were expressed in E. coli strain RP-2 (ΔfrdABCD, ΔsdhCDAB), a plasmid that inserted an additional stop codon after the frdA gene in the pH3 plasmid, as described (21). Cells were grown as described above for the intact complex. Cells were harvested by centrifugation and stored at −80 °C. The cells were then resuspended in 20 mm potassium phosphate, pH 7.4, with complete protease inhibitors (Roche Applied Science). Cells were lysed by three cycles of vortexing (3 min) with protein extraction beads (diameter <1 mm) followed by a freeze-thaw at −20 °C. Lysate was cleared by centrifugation at 34,000 × g and the supernatant was stored at −20 °C. For photocross-linking experiments, no additional purification was performed. For circular dichroism (CD) spectroscopy experiments, isolated FrdA subunits were purified using an anion-exchange Poros 50HQ column in buffer containing 25 mm Tris-HCl, pH 7.4, and a linear NaCl gradient from 50 mm to 1 m NaCl. This was followed by gel filtration in the same buffer on a Superdex 200 Increase 10/300 column. Elution was monitored by following A280. Following this procedure, the FrdA subunits were estimated to be 95% pure based on SDS-PAGE.
Expression and purification of SdhE containing the artificial amino acid pBpF
The sdhE gene with an amber codon substituting R8 was cotransformed with the pEVOL-pBpF to express pBpF-containing SdhE, as described (21). SdhE-R8BpF was grown in LB medium containing 0.5 mm pBpF. After 1 h, the temperature was adjusted to 30 °C. SdhE-R8BpF was induced by the addition of 0.1 mm isopropyl β-d-1-thiogalactopyranoside and the tRNA synthase/tRNA for pBpF was induced with 0.2 mm arabinose. Cells were harvested by centrifugation and lysed by sonication. The lysate was cleared of cellular debris by centrifugation for 45 min at 34,000 × g. SdhE-R8pBpF was purified by nickel affinity chromatography, as described (21).
Quantitation of covalent flavinylation under aerobic and anaerobic growth conditions
LB supplemented with ampicillin (100 μg/ml) (1.5 ml in a 14-ml round bottom Falcon tube) was inoculated with a single colony. The cells were grown overnight at 37 °C with 270 rpm shaking, then divided into aerobic or anaerobic cultures. For aerobic growth, 25 μl of the overnight culture was used to inoculate 1 ml of LB supplemented with ampicillin (100 μg/ml) and cells were grown for 7 h at 37 °C with shaking. For anaerobic growth, 1 ml of the overnight culture inoculated 10 ml of LB supplemented with ampicillin (100 μg/ml) in a 15-ml Falcon tube; the cells were grown at 37 °C in the Isoterm rotisserie incubator (Fisher Scientific) for 17 h with slow rotation. Prior to analysis by SDS-PAGE, the A600 was measured to normalize protein load. Cells (0.3 to 0.9 ml) were collected by centrifugation for 1 min, and the pellets resuspended in 2× SDS loading buffer (Bio-Rad). Each lane on the gel contains an equivalent amount of the cell culture (A600 = 1.0).
Quantification of covalent and non-covalent flavinylation
Covalent flavinylation of wild-type and variant QFR was detected as described (36). To analyze expression levels of the enzyme, 15 μg of isolated membranes were separated by SDS-PAGE on a 4–12% BisTris gel (NuPAGE Novex). FAD was quantified by UV fluorescence (Gel Doc EZ imager) using ImageJ software. The gel was then stained with SimplyBlue Safe Stain (ThermoFisher) and the protein was quantified by densitometry. The ratio between the UV and Coomassie was used to assess the level of covalent flavinylation, with wild-type QFR covalent flavinylation set to 100% and QFR-FrdAH44S set to 0%.
To determine the level of total FAD present in the purified FrdA variants, A280/A450 values were measured using a Nano-Drop1000 spectrophotometer. The A280/A450 absorbance ratio was calculated for wild-type FrdA containing covalent FAD, and this value was set to 100%. The A280/A450 absorbance ratio for the FrdA variants was compared with the ratio from wild-type. Because FAD was tightly associated with the variants but not covalently bound, all measured FAD absorbance was assumed to reflect non-covalent FAD specifically bound at the active site in the variants.
Measurement of enzyme activity
Enzymatic activity was determined in bacterial membranes containing wild-type and FrdAE245Q QFR variants. Succinate dehydrogenase and menaquinol-1 fumarate reductase reactions were assayed as previously described (37).
Spectroscopic evaluation of ligand binding
Ligand binding was performed using a modification of a described protocol (38). Purified wild-type and variant QFR enzymes were incubated with 3 mm malonate (to remove tightly bound oxaloacetate) in 50 mm potassium phosphate, pH 7.0, 0.01% Thesit for 20 min at room temperature and passed through a PD-10 gel filtration column in the same buffer minus malonate. The resultant enzymes were stored on ice. Optical spectra were recorded using an Agilent 8453 diode array spectrophotometer in 50 mm potassium phosphate, pH 7.0, 0.01% Thesit at 25 °C. Difference spectra were obtained by subtraction of a ligand-free spectrum from the corresponding spectrum of the enzyme in the presence of either 0.2 mm oxaloacetate or 3 mm fumarate.
Crystallization and structure determination
Crystals of the QFR-FrdAE245Q mutant were grown using the hanging-drop vapor diffusion method in droplets containing 1 μl of protein solution (15 mg/ml of QFR FrdAE245Q in 25 mm Tris-HCl, pH 7.4, 1 mm EDTA, 0.02% C12E9) and 1 μl of the reservoir solution (275 mm sodium malonate, 19% polyethylene glycol 6000, 100 mm sodium citrate, pH 4.0, 1 mm EDTA, and 0.001% dithiothreitol). Droplets were equilibrated over 1 ml of the reservoir solution at 20 °C. Crystals were cryo-protected with a solution that was 80% reservoir solution and 20% of a 1:1 mixture of glycerol and ethylene glycol prior to flash cooling with liquid nitrogen.
X-ray diffraction data were collected at the Advanced Photon Source (APS) beamline 21-ID-G at −173 °C using a wavelength of 0.9798 Å and a MarMosaic225 CCD detector. Data were processed and scaled using HKL2000 (39). The structure was determined using molecular replacement in the program Phaser (40) through the Phenix interface (41), and wild-type QFR (PDB entry 1L0V (6)) as the search model. For refinement, the capping domain was separated from the flavoprotein and the positions of each subunit plus the isolated capping domain were optimized using rigid body refinement in Phenix (41). The capping domain was then reconnected to the flavoprotein and additional refinement was performed using standard xyz refinement in Phenix (41) after constraining the secondary structural elements. As assessed by monitoring the R-factors and geometry, real space refinement decreased the quality of the model and was not used. The refined model was analyzed in Coot (42), but significant manual model building was not performed given the resolution. Data collection and refinement statistics are summarized in Table 1.
Far-UV CD spectroscopy
Spectra were collected for wild-type FrdA subunits and FrdA variants on a Jasco J-810 CD spectropolarimeter at 20 °C using a 0.1-cm path length quartz cuvette. Measurements were taken from 190 to 260 nm in 1-nm intervals. Secondary structure was analyzed using the DichroWeb analysis server (43). Data are the average of three runs and have been corrected by subtraction of the buffer spectra.
Thermal denaturation CD spectroscopy
Variable temperature CD spectra were taken by monitoring the change in the CD signal (mDeg) at 220 nm from 5 to 85 °C in 1 °C increments with a 2-min equilibration time between data points and a 4-s response. Data are the average of three runs and were corrected by subtraction of buffer spectra.
Using the lsqcurvefit function in Matlab R2015a, the CD data were fitted as a function of temperature to a modified version of the Hill equation.
| (Eq. 1) |
Where A is the maximum CD signal minus the minimum CD signal in mDeg, T is temperature in °C, B is the midpoint of the plot of CD signal versus temperature, n is the Hill exponent, and C is the minimum CD signal. To determine the melting point, the derivative of this function,
| (Eq. 2) |
was evaluated at temperature values ranging from the minimum observed temperature to the maximum observed temperature, in steps of 0.01 °C. The temperature corresponding to the maximum of the derivative was taken as the melting point.
Cross-linking of FrdA subunits to pBpF-incorporated SdhE
Purified His6-SdhE-R8BpF was mixed with E. coli strain RP-2 lysate containing overexpressed and untagged FrdA in a Corning 6-well plate on ice. Cross-linking was induced by illumination with 365-nm light (Black-Ray 100 Watt UV lamp). The SdhE-FrdA cross-linked complex was purified by Ni2+ affinity chromatography and analyzed by SDS-PAGE, where a characteristic shift in molecular weight was observed. The presence of SdhE in the shifted band was verified by Western blot analysis with a primary antibody against the His6 tag; the presence of FrdA in the shifted band was confirmed by monitoring UV fluorescence. For the time course experiment, FrdA and SdhE-R8BpF samples were mixed in a 96-well plate on ice and exposed to UV light. Samples were collected at the indicated time points, and quenched with SDS loading buffer.
SAXS data collection and analysis
SAXS data were collected at the SIBYLS beamline at the Advanced Light Source in Berkeley, CA, as described (44). Measurements of the SdhE-FrdA cross-linked complex, purified as described above and dialyzed into 50 mm MES, pH 6.0, with 5% glycerol, were carried out at 283 K. Data were collected at two different concentrations (1.2 and 2.4 mg/ml) to analyze concentration-dependent effects. Samples were placed 1.5 m from a MAR165 CCD detector arranged coaxial with the 11 keV monochromatic beam; 1012 photons/s were impingent on the sample. A series of 0.3-s exposures were collected for 10 s, resulting in 32 frames per condition. Buffer subtraction and raw image data were integrated by beamline software (45).
Guinier analysis and indirect Fourier transformation were performed in the PRIMUS (46) and GNOM (47) programs, respectively. The ab initio shape determination was carried out using DAMMIF (48). The resulting SAXS-based model was then superimposed on models of the FrdA-SdhE-R8BpF cross-linked complex by their inertial axes alignment using the SUPCOMB program (49). The experimental scattering data were further validated with the scattering profile of the computational model of cross-linked complex using CRYSOL (50).
Author contributions
C. A. S. analyzed covalent and non-covalent flavinylation in wild-type and variant QFR complexes and FrdA subunits, performed CD spectroscopy of FrdA subunits, purified, crystallized, and determined the structure of the QFR-FrdAE245Q variant with the assistance of S. Q. H., prepared samples for SAXS analysis, and analyzed the SAXS data under the direction of P. S. E. M. measured covalent flavinylation of QFR/SQR in E. coli strains grown under aerobic and anaerobic conditions, measured dicarboxylate binding to wild-type and variant QFR complexes, and performed kinetic analyses. T. M. I. and G. C. developed the study. T. M. I., G. C., and C. A. S. wrote the paper with input from all authors.
Acknowledgments
We thank B. Damon for interpreting the CD spectroscopy data in Matlab and I. Yamakawa for assistance with the FrdA-SdhE cross-linking. The Vanderbilt crystallization facility is supported by National Institutes of Health Grant S10 RR026915. This research used resources of the Advanced Photon Source, a United States Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor Grant 085P1000817. SAXS data were collected at the SIBYLS beamline at the Advanced Light Source (ALS), a national user facility operated by Lawrence Berkeley National Laboratory on behalf of the Department of Energy, Office of Basic Energy Sciences, through the Integrated Diffraction Analysis Technologies (IDAT) program, supported by DOE Office of Biological and Environmental Research. Additional support comes from National Institute of Health project MINOS Grant R01GM105404 and High-End Instrumentation Grant S10OD018483.
This work was supported in part by the U. S. Department of Veterans Affairs Biomedical Laboratory Research & Development (BLR&D) Grant BX001077 (to G. C.) and National Institutes of Health Grant GM61606 (to G. C. and T. M. I.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or Department of Veterans Affairs.
The atomic coordinates and structure factors (code 5VPN) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- QFR
- quinol:fumarate reductase
- SQR
- succinate:ubiquinone oxidoreductase
- SAXS
- small angle X-ray scattering
- FAD
- flavin adenine dinucleotide
- pBpF
- para-benzoyl phenylalanine
- PDB
- Protein Data Bank
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol.
References
- 1. Heuts D. P., Scrutton N. S., McIntire W. S., and Fraaije M. W. (2009) What's in a covalent bond?: on the role and formation of covalently bound flavin cofactors. FEBS J. 276, 3405–3427 [DOI] [PubMed] [Google Scholar]
- 2. Starbird C. A., Maklashina E., Cecchini G., and Iverson T. M. (2015) Flavoenzymes: covalent versus noncovalent. in eLS, John Wiley & Sons, Ltd., Chichester, United Kingdom [Google Scholar]
- 3. Blaut M., Whittaker K., Valdovinos A., Ackrell B. A., Gunsalus R. P., and Cecchini G. (1989) Fumarate reductase mutants of Escherichia coli that lack covalently bound flavin. J. Biol. Chem. 264, 13599–13604 [PubMed] [Google Scholar]
- 4. Efimov I., Cronin C. N., and McIntire W. S. (2001) Effects of noncovalent and covalent FAD binding on the redox and catalytic properties of p-cresol methylhydroxylase. Biochemistry 40, 2155–2166 [DOI] [PubMed] [Google Scholar]
- 5. Fraaije M. W., van den Heuvel R. H., van Berkel W. J., and Mattevi A. (1999) Covalent flavinylation is essential for efficient redox catalysis in vanillyl-alcohol oxidase. J. Biol. Chem. 274, 35514–35520 [DOI] [PubMed] [Google Scholar]
- 6. Iverson T. M., Luna-Chavez C., Cecchini G., and Rees D. C. (1999) Structure of the Escherichia coli fumarate reductase respiratory complex. Science 284, 1961–1966 [DOI] [PubMed] [Google Scholar]
- 7. Tomasiak T. M., Maklashina E., Cecchini G., and Iverson T. M. (2008) A threonine on the active site loop controls transition state formation in Escherichia coli respiratory complex II. J. Biol. Chem. 283, 15460–15468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Maklashina E., Iverson T. M., Sher Y., Kotlyar V., Andréll J., Mirza O., Hudson J. M., Armstrong F. A., Rothery R. A., Weiner J. H., and Cecchini G. (2006) Fumarate reductase and succinate oxidase activity of Escherichia coli complex II homologs are perturbed differently by mutation of the flavin binding domain. J. Biol. Chem. 281, 11357–11365 [DOI] [PubMed] [Google Scholar]
- 9. Doherty M. K., Pealing S. L., Miles C. S., Moysey R., Taylor P., Walkinshaw M. D., Reid G. A., and Chapman S. K. (2000) Identification of the active site acid/base catalyst in a bacterial fumarate reductase: a kinetic and crystallographic study. Biochemistry 39, 10695–10701 [DOI] [PubMed] [Google Scholar]
- 10. Mowat C. G., Moysey R., Miles C. S., Leys D., Doherty M. K., Taylor P., Walkinshaw M. D., Reid G. A., and Chapman S. K. (2001) Kinetic and crystallographic analysis of the key active site acid/base arginine in a soluble fumarate reductase. Biochemistry 40, 12292–12298 [DOI] [PubMed] [Google Scholar]
- 11. Pankhurst K. L., Mowat C. G., Rothery E. L., Hudson J. M., Jones A. K., Miles C. S., Walkinshaw M. D., Armstrong F. A., Reid G. A., and Chapman S. K. (2006) A proton delivery pathway in the soluble fumarate reductase from Shewanella frigidimarina. J. Biol. Chem. 281, 20589–20597 [DOI] [PubMed] [Google Scholar]
- 12. Reid G. A., Miles C. S., Moysey R. K., Pankhurst K. L., and Chapman S. K. (2000) Catalysis in fumarate reductase. Biochim. Biophys. Acta 1459, 310–315 [DOI] [PubMed] [Google Scholar]
- 13. Walker W. H., Salach J., Gutman M., Singer T. P., Hyde J. S., and Ehrenberg A. (1969) Endor studies on the covalently bound flavin at the active center of succinate dehydrogenase. FEBS Lett. 5, 237–240 [DOI] [PubMed] [Google Scholar]
- 14. Cheng V. W., Piragasam R. S., Rothery R. A., Maklashina E., Cecchini G., and Weiner J. H. (2015) Redox state of flavin adenine dinucleotide drives substrate binding and product release in Escherichia coli succinate dehydrogenase. Biochemistry 54, 1043–1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Heffron K. (2001) Studies of the redox and catalytic properties of the anaerobic respiratory enzymes of Escherichia coli. Ph.D. thesis, University of Oxford, UK [Google Scholar]
- 16. Walsh C. (1980) Flavin coenzymes: at the crossroads of biological redox chemistry. Acc. Chem. Res. 13, 148–155 [Google Scholar]
- 17. Hao H. X., Khalimonchuk O., Schraders M., Dephoure N., Bayley J. P., Kunst H., Devilee P., Cremers C. W., Schiffman J. D., Bentz B. G., Gygi S. P., Winge D. R., Kremer H., and Rutter J. (2009) SDH5, a gene required for flavination of succinate dehydrogenase, is mutated in paraganglioma. Science 325, 1139–1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. McNeil M. B., Clulow J. S., Wilf N. M., Salmond G. P., and Fineran P. C. (2012) SdhE is a conserved protein required for flavinylation of succinate dehydrogenase in bacteria. J. Biol. Chem. 287, 18418–18428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lim K., Doseeva V., Demirkan E. S., Pullalarevu S., Krajewski W., Galkin A., Howard A., and Herzberg O. (2005) Crystal structure of the YgfY from Escherichia coli, a protein that may be involved in transcriptional regulation. Proteins 58, 759–763 [DOI] [PubMed] [Google Scholar]
- 20. Zafreen L., Walker-Kopp N., Huang L. S., and Berry E. (2016) In-vitro, SDH5-dependent flavinylation of immobilized human respiratory complex II flavoprotein. Arch. Biochem. Biophys. 604, 47–56 [DOI] [PubMed] [Google Scholar]
- 21. Maklashina E., Rajagukguk S., Starbird C. A., McDonald W. H., Koganitsky A., Eisenbach M., Iverson T. M., and Cecchini G. (2016) Binding of the covalent flavin assembly factor to the flavoprotein subunit of complex II. J. Biol. Chem. 291, 2904–2916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. McNeil M. B., and Fineran P. C. (2013) The conserved RGxxE motif of the bacterial FAD assembly factor SdhE is required for succinate dehydrogenase flavinylation and activity. Biochemistry 52, 7628–7640 [DOI] [PubMed] [Google Scholar]
- 23. Eletsky A., Jeong M. Y., Kim H., Lee H. W., Xiao R., Pagliarini D. J., Prestegard J. H., Winge D. R., Montelione G. T., and Szyperski T. (2012) Solution NMR structure of yeast succinate dehydrogenase flavinylation factor Sdh5 reveals a putative Sdh1 binding site. Biochemistry 51, 8475–8477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bezawork-Geleta A., Dong L., Rohlena J., and Neuzil J. (2016) The assembly factor SDHAF2 is dispensable for flavination of the catalytic subunit of mitochondrial complex II in breast cancer cells. J. Biol. Chem. 291, 21414–21420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kounosu A. (2014) Analysis of covalent flavinylation using thermostable succinate dehydrogenase from Thermus thermophilus and Sulfolobus tokodaii lacking SdhE homologs. FEBS Lett. 588, 1058–1063 [DOI] [PubMed] [Google Scholar]
- 26. Bayley J. P., Devilee P., and Taschner P. E. (2005) The SDH mutation database: an online resource for succinate dehydrogenase sequence variants involved in pheochromocytoma, paraganglioma and mitochondrial complex II deficiency. BMC Med. Genet. 6, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cecchini G. (2003) Function and structure of complex II of the respiratory chain. Annu. Rev. Biochem. 72, 77–109 [DOI] [PubMed] [Google Scholar]
- 28. Brandsch R., and Bichler V. (1989) Covalent cofactor binding to flavoenzymes requires specific effectors. Eur. J. Biochem. 182, 125–128 [DOI] [PubMed] [Google Scholar]
- 29. Robinson K. M., and Lemire B. D. (1996) Covalent attachment of FAD to the yeast succinate dehydrogenase flavoprotein requires import into mitochondria, presequence removal, and folding. J. Biol. Chem. 271, 4055–4060 [DOI] [PubMed] [Google Scholar]
- 30. Kim H. J., Jeong M. Y., Na U., and Winge D. R. (2012) Flavinylation and assembly of succinate dehydrogenase are dependent on the C-terminal tail of the flavoprotein subunit. J. Biol. Chem. 287, 40670–40679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bezawork-Geleta A., Saiyed T., Dougan D. A., and Truscott K. N. (2014) Mitochondrial matrix proteostasis is linked to hereditary paraganglioma: LON-mediated turnover of the human flavinylation factor SDH5 is regulated by its interaction with SDHA. FASEB J. 28, 1794–1804 [DOI] [PubMed] [Google Scholar]
- 32. Schröder I., Gunsalus R. P., Ackrell B. A., Cochran B., and Cecchini G. (1991) Identification of active site residues of Escherichia coli fumarate reductase by site-directed mutagenesis. J. Biol. Chem. 266, 13572–13579 [PubMed] [Google Scholar]
- 33. Birch-Machin M. A., Taylor R. W., Cochran B., Ackrell B. A., and Turnbull D. M. (2000) Late-onset optic atrophy, ataxia, and myopathy associated with a mutation of a complex II gene. Ann. Neurol. 48, 330–335 [PubMed] [Google Scholar]
- 34. Westenberg D. J., Gunsalus R. P., Ackrell B. A., Sices H., and Cecchini G. (1993) Escherichia coli fumarate reductase frdC and frdD mutants: identification of amino acid residues involved in catalytic activity with quinones. J. Biol. Chem. 268, 815–822 [PubMed] [Google Scholar]
- 35. Luna-Chavez C., Iverson T. M., Rees D. C., and Cecchini G. (2000) Overexpression, purification, and crystallization of the membrane-bound fumarate reductase from Escherichia coli. Protein Expr. Purif. 19, 188–196 [DOI] [PubMed] [Google Scholar]
- 36. Bafunno V., Giancaspero T. A., Brizio C., Bufano D., Passarella S., Boles E., and Barile M. (2004) Riboflavin uptake and FAD synthesis in Saccharomyces cerevisiae mitochondria: involvement of the Flx1p carrier in FAD export. J. Biol. Chem. 279, 95–102 [DOI] [PubMed] [Google Scholar]
- 37. Maklashina E., and Cecchini G. (1999) Comparison of catalytic activity and inhibitors of quinone reactions of succinate dehydrogenase (succinate-ubiquinone oxidoreductase) and fumarate reductase (menaquinol-fumarate oxidoreductase) from Escherichia coli. Arch. Biochem. Biophys. 369, 223–232 [DOI] [PubMed] [Google Scholar]
- 38. Tomasiak T. M., Archuleta T. L., Andréll J., Luna-Chávez C., Davis T. A., Sarwar M., Ham A. J., McDonald W. H., Yankovskaya V., Stern H. A., Johnston J. N., Maklashina E., Cecchini G., and Iverson T. M. (2011) Geometric restraint drives on- and off-pathway catalysis by the Escherichia coli menaquinol:fumarate reductase. J. Biol. Chem. 286, 3047–3056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Otwinowski Z., and Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 40. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., and Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 43. Whitmore L., and Wallace B. A. (2008) Protein secondary structure analyses from circular dichroism spectroscopy: methods and reference databases. Biopolymers 89, 392–400 [DOI] [PubMed] [Google Scholar]
- 44. Hura G. L., Menon A. L., Hammel M., Rambo R. P., Poole F. L. 2nd, Tsutakawa S. E., Jenney F. E. Jr., Classen S., Frankel K. A., Hopkins R. C., Yang S. J., Scott J. W., Dillard B. D., Adams M. W., and Tainer J. A. (2009) Robust, high-throughput solution structural analyses by small angle X-ray scattering (SAXS). Nat. Methods 6, 606–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Classen S., Hura G. L., Holton J. M., Rambo R. P., Rodic I., McGuire P. J., Dyer K., Hammel M., Meigs G., Frankel K. A., and Tainer J. A. (2013) Implementation and performance of SIBYLS: a dual endstation small-angle X-ray scattering and macromolecular crystallography beamline at the Advanced Light Source. J. Appl. Crystallogr. 46, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Konarev P. V., Volkov V. V., Sokolova A. V., Koch M. H., and Svergun D. I. (2003) PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 36, 1277–1282 [Google Scholar]
- 47. Svergun D. I. (1992) Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 25, 495–503 [Google Scholar]
- 48. Franke D., and Svergun D. I. (2009) DAMMIF, a program for rapid ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 42, 342–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kozin M., and Svergun D. I. (2001) Automated matching of high- and low-resolution structural models. J. Appl. Crystallogr. 34, 33–41 [Google Scholar]
- 50. Svergun D. I., Barberato C., and M.J.H. K. (1995) CRYSOL: a program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 28, 768–773 [Google Scholar]