

Abstract
The type 1 ryanodine receptor (RyR1) mediates Ca2+ release from the sarcoplasmic reticulum to initiate skeletal muscle contraction and is associated with muscle diseases, malignant hyperthermia, and central core disease. To better understand RyR1 channel function, we investigated the molecular mechanisms of channel gating and ion permeation. An adequate model of channel gating requires accurate, high-resolution models of both open and closed states of the channel. To this end, we generated an open-channel RyR1 model using molecular simulations to pull Ca2+ through the pore constriction site of a closed-channel RyR1 structure determined at 3.8-Å resolution. Importantly, we find that our open-channel model is consistent with the RyR1 and cardiac RyR (RyR2) open-channel structures reported while this paper was in preparation. Both our model and the published structures show similar rotation of the upper portion of the pore-lining S6 helix away from the 4-fold channel axis and twisting of Ile-4937 at the channel constriction site out of the channel pore. These motions result in a minimum open-channel pore radius of ∼3 Å formed by Gln-4933, rather than Ile-4937 in the closed-channel structure. We also present functional support for our model by mutations around the closed- and open-channel constriction sites (Gln-4933 and Ile-4937). Our results indicate that use of ion-pulling simulations produces a RyR1 open-channel model, which can provide insights into the mechanisms of channel opening complementing those from the structural data.
Keywords: calcium channel, calcium transport, conformational change, molecular dynamics, molecular modeling, ryanodine receptor
Introduction
The skeletal muscle ryanodine receptor (RyR1)4 mediates muscle contraction by releasing Ca2+ from the sarcoplasmic reticulum (1). Provided its essential role in muscle function, RyR1 mutations are associated with muscle diseases malignant hyperthermia and central core disease (2, 3). RyR1 shares considerable homology with other Ca2+ release channels including other ryanodine receptor isoforms and isoforms of the inositol 1,4,5-triphosphate receptor. All of these receptors release Ca2+ from a Ca2+ store, are regulated by Ca2+, and form a cation conducting pore (4–9). It is likely that these receptors share an evolutionarily conserved channel gating mechanism (10). A structural understanding of RyR1 gating will provide valuable insights into mechanisms of Ca2+ signaling (11).
RyR1 forms a homotetrameric complex of 2,200 kDa. Due to the large size of the receptor structural information is limited. Early cryo-electron microscopy (cryo-EM) studies at ∼30-Å resolution revealed the global architecture of the receptor (12–14). The channel consists of a large cytoplasmic N-terminal domain that contains interaction sites for regulatory molecules, and a smaller C-terminal segment containing the pore-forming transmembrane domain. Although the N-terminal segment comprises the largest portion of RyR1, the isolated C-terminal segment (residues 3661 to 5037) can form a functional Ca2+ release channel (15), and the two C-terminal transmembrane segments, including the pore helix and connecting loops, can form a homotetrameric assembly capable of conducting K+, Cl−, and Ca2+ (16).
More detailed structures of the closed-channel RyR1 were later determined at 10-Å resolution (17–19). Open- and closed-channel structures determined at 10-Å resolution provided valuable information on the overall motions involved in channel opening, but lacked atomic detail (17). Recently, the closed and open RyR1 and RyR2 channel structures were determined at near atomic resolution of 3.6 to 4.8 Å (20–23). The pore-forming region consists of a long S6 inner helix of 50 residues, a pore helix of 15 residues, and a GGGIGDE selectivity filter. In the RyR1 closed channel, Ile-4937 lines the pore and forms the hydrophobic constriction site with a pore radius of less than 1 Å, rendering the channel impermeable to Ca2+ (20, 21). In the RyR2 open channel, Gln-4933 forms the constriction site with a minimum radius of 2.4 Å (23).
Computational methods offer a means to provide structural insights. Due to the large size of the RyR ion channels, it is not possible to reach the simulation time necessary to observe a channel opening event (microsecond to millisecond time scale). One means of addressing this limitation is to use steered molecular dynamics simulations, a technique that includes an additional external force to propel the system along a particular trajectory (24). Steered molecular dynamics simulations have previously been applied to explore channel opening for the voltage-gated potassium channel (25), the mechanosensitive channel of large conductance (26), and the nicotinic acetylcholine receptor (27).
In this work we use a novel computational method to produce an open-channel model for the RyR1 transmembrane domain. We obtain the model using a form of steered molecular dynamics in which we apply a potential to pull a Ca2+ ion through the pore constriction site. From ion pulling alone we obtain an open-channel model exhibiting a substantially larger pore and counter-clockwise twisting of the S6 helix. It was our expectation that these motions increasing the pore diameter would occur along the path of least resistance. We hypothesize that this channel opening pathway is the physiologically preferred opening pathway and that ion pulling simulations can produce an accurate open-channel model. Indeed, after refinement we find that our open-channel model is consistent with the high resolution cryo-EM structures of the open RyR1 and RyR2 channels (22, 23). We also present mutational data supporting a mechanism of channel gating in which the S6 helix rotates Ile-4937 out of the pore. In our open-channel model, Gln-4933 forms the minimum radius of the pore. Moreover, by modeling channel gating we propose a role for the conserved glycine residue Gly-4934 in channel gating. The data suggest that increasing the side chain volume of the conserved glycine residue Gly-4934 impedes movement of Ile-4937 away from the pore, which provides a plausible explanation for decreased channel gating and Ca2+ conductance.
Results
Creating the open-channel RyR1
To create the open-channel RyR1, we place a Ca2+ ion at the cytoplasmic end of the channel between residues Glu-4948 and Glu-4952 (Fig. 1, a and b). From this starting position the Ca2+ ion is pulled into the channel constriction site formed by Ile-4937, which is completely dehydrated in the closed-channel structure. Using a short 20-ns simulation, we equilibrate this system with the Ca2+ ion constrained in the pore to generate our intermediate open conformation (Fig. 1, c and d). At this point position restraints have only been applied with regard to the location of the Ca2+ ion and no additional restraints have been applied to the protein.
Figure 1.

Pore opening progression from the initial closed structure (a, b) to the intermediate structure (c, d) to the final refined open structure (e, f). Top panels (a, c, e) show the side view of the pore, whereas lower panels (b, d, f) show the pore viewed from the cytoplasmic end of the channel. Side chains for amino acids bordering Ca2+ are shown as sticks and colored according to residue type, red, acidic; green, polar; and white, aliphatic. In the lower panels (b, d, f), Gln-4933 and Ile-4937 are shown in green and white, respectively. The position of Ca2+ is shown as a magenta sphere and waters within 4 Å of Ca2+ are shown in red spheres. The initial structure represents the time point in which Ca2+ was first moved to the position between Glu-4948 and Glu-4952, before water was allowed to equilibrate around the ion.
This simulation results in an open-channel pore with a minimum pore radius of 2.8 ± 0.4 Å. The pore constriction site of this channel is formed by Gln-4933 compared with Ile-4937, which formed the constriction site in the closed-channel RyR1 (21). In this intermediate conformation the Ca2+ ion in the pore is surrounded by 7 to 8 water molecules over the 20-ns simulation, consistent with Ca2+ keeping its first hydration shell as it passes through the channel.
However, the process of pulling the Ca2+ ion into the pore resulted in distortion of the S6 helix. Given the energetic cost of breaking a number of hydrogen bonds in the S6 helix, we presumed the distortion of the S6 helix is an artifact of the ion pulling simulation. Indeed, much of the distortion occurred as the Ca2+ ion moved past the negatively charged residues lining the closed channel pore. In the final open-channel these negatively charged residues move away from the channel pore and cannot simultaneously bind Ca2+. We therefore refined the model by applying a series of restraints to the protein. We applied hydrogen bond restraints to the S6 helix to reform the distortions from Ca2+ pulling. We also applied Cα inter-helical restraints for residues Ile-4937, Asp-4945, and Glu-4948 to preserve the open channel conformation and encourage the formation of a symmetric channel. Finally, we note that the initial ion pulling caused Ile-4937 to rotate out of the pore toward the hydrophobic amino acids Ile-4931 and Leu-4935. To further encourage interaction of Ile-4937 with Ile-4931 and Leu-4935, we applied restraints between Cβ atoms of Ile-4937 and Ile-4931 or Leu-4935. After simulation with these imposed restraints we obtain a refined open-channel RyR1 model (Fig. 1, e and f).
Open pore conformation of RyR1
After our refinement simulations we obtain an open-channel model of RyR1 with a minimum channel radius of 2.8 ± 0.3 Å. This radius is moderately smaller than the experimentally derived minimum radius of 3.3 to 3.5 Å (28). However, the radius is consistent with those of the open-channel RyR1 (3.0 ± 0.2 Å) and RyR2 (2.4 Å) structures (22, 23) (Fig. 2). Consistent with the other experimental structures, Gln-4933 forms the minimum radius of the pore in our open-channel model. Although we show Gln-4933 forms the constriction site in the open-channel RyR1, we note that this constriction site was not mentioned in the original publication of the RyR1 open-channel structure (22). The original publication of the RyR1 open-channel structure presents the pore profile for only one of the three open-channel structures (22). To provide a more accurate view of the open-channel conformation, we present the average pore profile of the three open-channel structures. The structure previously presented had the side chain of Gln-4933 rotated out of the pore. However, the other two structures had conformations of the Gln-4933 side chain rotated into the pore, causing the average pore profile to reflect the constriction at Gln-4933. The results not only support our model, but also suggest a significant amount of flexibility in the Gln-4933 side chain to allow the passage of Ca2+.
Figure 2.

Pore of the RyR1 open-channel model. a, pore profiles for the open-channel model, the open-channel RyR1, and the open-channel RyR2 are shown in blue, black, and gray, respectively. For the open-channel model the pore profile is the means ± S.E. calculated over 10 frames sampled evenly over the last 20 ns of molecular dynamics simulation. For the open-channel RyR1 the error bars are the mean ± S.E. calculated for the three open-channel RyR1 structures (PDB codes 5TA3, 5TAL, and 5T9V). The pore profile is shown aligned with structures for the (b) open-channel model, (c) open-channel RyR1 (PDB code 5TAL), and (d) open-channel RyR2 (PDB code 5GOA). Pore lining residues are shown as sticks and colored according to residue type: red, acidic; blue, basic; green, polar; and white, aliphatic. Residues Gly-4934 and Gly-4941 are colored black.
We assess the accuracy of our RyR1 open-channel model by comparing the alignment of the structural model to the 3.8-Å resolution cryo-EM densities of the open-channel RyR1 (22) (Fig. 3, a and b). We note that the most significant difference between the open- and closed-channel structures occurs at the pore-lining S6 helix, particularly at the cytoplasmic end of the channel. Comparing the alignment of the S6 helix (residues 4910 to 4957) of our open-channel model to the open-channel densities we observe a correlation of 0.93 ± 0.02 (Table 1). In contrast, the alignment of the closed-channel structure (PDB code 3J8H) with the open-channel densities results in a correlation of 0.84 ± 0.02 (Fig. 3, c and d, Table 1) (21, 22).
Figure 3.

The open-channel model of RyR1 agrees with the RyR1 open-channel cryo-EM density. Side (a) and cytosolic (b) views of the RyR1 open-channel model (cyan) aligned with the 3.8-Å resolution open-channel cryo-EM density (yellow surface) (EMD 8378). For comparison, side (c) and cytoplasmic (d) views of the closed structure (PDB code 3J8H; red) (21) are shown aligned with the same open-channel cryo-EM density (EMD 8378).
Table 1.
Correlation values for fitting the S6 helix (residues 4910 to 4957) to open- and closed-channel densities
| Open densitiesa | Closed densityb | |
|---|---|---|
| Closed structure (PDB code 3J8H) | 0.84 ± 0.02 | 0.98 |
| Open model | 0.93 ± 0.02 | 0.85 |
| Open structure (PDB code 5TAL) | 0.98 | 0.81 |
a Open-channel RyR1 densities (EMD-8376, EMD-8377, and EMD-8378).
b Closed-channel RyR1 density (EMD-2807).
Conformational changes opening the channel
To analyze the mechanism of RyR1 channel gating, we compare global features of the S6 helix to the RyR1 closed-channel structure (PDB code 3J8H) (21). We perform principal component analysis of the closed-channel structure and the 10 open-channel models used to calculate the pore profile to identify the major modes of motion leading to channel opening. The first principal component explains 70% of the motion between closed and open RyR1. The major motion of the first principal component leading to channel opening is an outward rotation of the S6 helix away from the 4-fold channel axis, particularly at the cytosolic end of the channel (Fig. 4 and supplemental Video S1). We quantify this change by measuring radial and lateral tilting angles for the upper portion of the S6 helix comprised of residues 4930 to 4950. The radial tilting angle is the angle of helical tilting away from the 4-fold channel axis, whereas the lateral angle is the tilting angle between the helix and channel axis in the direction perpendicular to the plane defined by the radial angle (Fig. 4a). The outward radial tilting for the upper portion of the S6 helix is 7 ± 1°, which is in excellent agreement with values of 7 ± 1° and 7° observed for open-channel RyR1 and RyR2 structures, respectively (Fig. 4b) (22, 23). In contrast, we observe less movement for the luminal end of the S6 helix. The change in radial tilting for the lower portion of the S6 helix (residues 4910 to 4930) is 4 ± 3° compared with values of 1 ± 1° observed in the experimental structures. We also observe a relatively small amount of lateral tilting averaging 3 ± 1° tangent to the pore compared with a lateral tilting angle of 9 ± 1° observed in the experimentally determined open structures.
Figure 4.
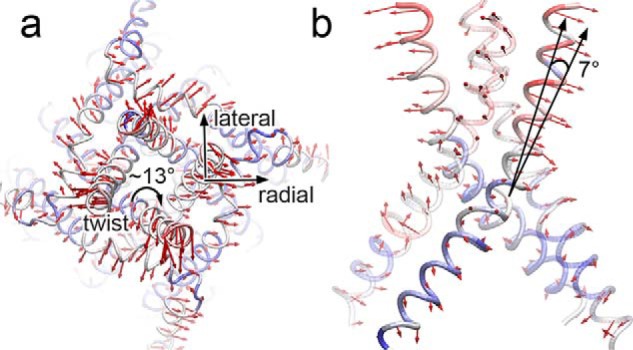
Radial tilting and lateral twisting of the S6 helix contributes to channel opening. Cytosolic (a) and side (b) views of the of the closed-channel structure (PDB code 3J8H) (21). The red arrows show the direction of the first principal component between the closed-channel structure and 10 open-channel models sampled every 2 ns over the last 20 ns of simulation. The arrow lengths are scaled according to magnitude of the motion. Likewise, the structure is colored by the magnitude of the motion from smallest (blue) to largest (red). Black arrows in a denote the planes within which radial and lateral helical tilting angles were determined for the helix the arrows are centered on. The black arrows in b highlight the region over which the helical tilting angles were computed.
We also examine the twisting motion of the S6 helix accompanying channel opening. We observe that backbone atoms for the upper portion of the S6 helix undergo a 13 ± 1° clockwise rotation viewed from the cytosolic side with respect to the closed-channel RyR1 (Fig. 4a). It is remarkable that the open-channel RyR structures are in close agreement with the degree of twisting observed in our model. The open-channel RyR1 structures show a rotation of 12 ± 2° with respect to the closed structure, whereas the open-channel RyR2 structure shows a rotation of 11 ± 2° (22, 23). The ∼13° rotation aligns Cα of Gly-4934 with the pore and brings Ile-4937 out of the pore.
Many of the motions we observe in the final structure were observed in the unrefined intermediate open model as well. Tilting angles of the open model after the initial ion pulling simulation were 5 ± 10° and 5 ± 1° for radial and lateral tilting angles, respectively. Moreover, we observe a 13 ± 4° clockwise rotation (as viewed from the cytoplasm) after the initial ion pulling simulation, before helical and inter-subunit restraints were introduced. We find that the restraints introduced during refinement effectively reduced the variance observed for rotation and radial tilting of the S6 helix without introducing much new motion.
Altering open-channel contacts in the S6 helix affects channel gating
To further evaluate our open-channel model, we compare contacts between S6 helices in our open-channel model with S6 inter-helical contacts observed in open- and closed-channel RyR structures (Fig. 5) (21–23). In the closed-channel structure Ile-4937 contacts Gly-4934 and Ile-4937 of the adjacent subunit. However, in the open-channel model Ile-4937 contacts Ala-4930, Ile-4931, and Gly-4934 in the adjacent S6 helix. We note that both Ala-4930 and Ile-4931 are aliphatic residues that are highly conserved among RyRs and these residues contact Ile-4937 in both the open-channel RyR1 and open-channel RyR2 (not shown) structures. Consistent with both open-channel RyR structures, we also observe that Gln-4933 contacts Ala-4930 and Gln-4933 of the adjacent S6 helix in the closed-channel structure. In our open-channel model Gln-4933 has rotated to contact Val-4926, rather than Gln-4933, of the adjacent subunit.
Figure 5.
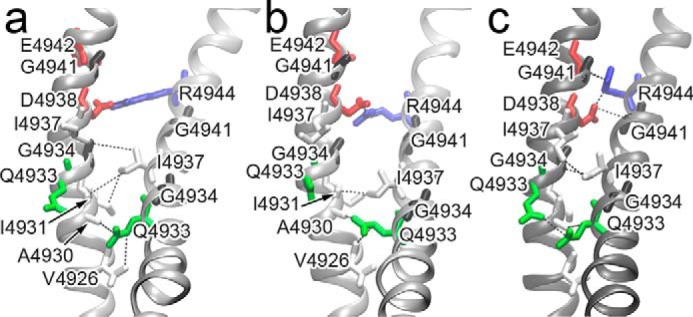
Intersubunit contacts for the S6 helix. Contacts are shown for the RyR1 open-channel model (a), the RyR1 open-channel structure (PDB code 5TAL) (b), and the RyR1 closed-channel structure (PDB code 3J8H) (c). The contacting residues are shown in stick representation and colored by residue type: red, acidic; blue, basic; green, polar; and white, aliphatic. The two S6 glycines are colored black.
To provide independent evidence for the proposed open model, we generated and determined the functional properties of 7 mutations at positions 4933, 4934, and 4937 (Table 2). The mutants were expressed in HEK293 cells, and their expression levels and retention of function were determined by immunoblot analysis, caffeine-induced cellular Ca2+ release assay, and in vitro binding assay using the highly RyR-specific ligand [3H]ryanodine. Ryanodine is known to only bind to the open-channel RyR and is therefore commonly used to assess channel function in isolated membrane fractions. Single channel recordings were performed to determine Ca2+-dependent gating and ion permeation properties of the mutants.
Table 2.
Channel properties of RyR1-WT and mutant channels
| WT-RyR1 | Q4933A | Q4933V | G4934A | G4934V | I4937A | I4937V | I4937T | |
|---|---|---|---|---|---|---|---|---|
| Immunoblot intensity (% WT) | 100 | 50.3 ± 15.5(6)a | 42.1 ± 9.6(6)a | 79.5 ± 19.5(7) | 79.6 ± 17.1(6) | 35.1 ± 17.7(6)a | 55.3 ± 14.6(6)a | 69.9 ± 16.5(6) |
| Caffeine response (% WT) | 100 | 76.4 ± 11.7(5) | 24.4 ± 10.8(5)a | 171.5 ± 16.7(4)a | 78.7 ± 23.2(6) | 103.8 ± 25.1(5) | 168.4 ± 35.7(5) | 124.2 ± 12.3(5) |
| [3H]Ryanodine binding (% WT) | 100 | 0.5 ± 0.4(7)a | 1.9 ± 1.8(5)a | 70.0 ± 10.7(4)a | 11.5 ± 1.8(5)a | 3.2 ± 2.8(7)a | 71.4 ± 8.8(5)a | 27.7 ± 2.0(6)a |
| Po (2 μm Ca2+) | 0.11 ± 0.03(13) | 0.17 ± 0.07(14) | 0.59 ± 0.22(4)a | 0.05 ± 0.01(9) | 0.10 ± 0.03(4) | 0.27 ± 0.11((7) | 0.44 ± 0.08(7)a | 0.15 ± 0.05(9) |
| Po (0.1 μm Ca2+) | 0.01 ± 0.01(9) | 0.01 ± 0.01(9) | 0.57 ± 0.23(4)a | 0.01 ± 0.01(9) | 0.10 ± 0.03(4)a | 0.34 ± 0.18(4) | 0.20 ± 0.08(7)a | 0.05 ± 0.04(3) |
| γK+ (pS) | 766 ± 11(12) | 762 ± 9(14) | 693 ± 50(4) | 635 ± 26(9)a | 539 ± 138(4)a | 597 ± 79(7) | 793 ± 21(7) | 742 ± 14(7) |
| ICa (pA) (10 mm trans Ca2+) | −2.3 ± 0.1(9) | −1.6 ± 0.1(7)a | −0.5 ± 0.2(4) | −1.5 ± 0.1(9)a | −1.1 ± 0.2(4)a | −0.1 ± 0.1(4) | −2.0 ± 0.1(7)a | −1.7 ± 0.2(5)a |
| PCa/PK | 6.6 ± 0.2(8) | 3.3 ± 0.2(7)a | 0.3 ± 0.1(4)a | 5.0 ± 0.2(9)a | 3.6 ± 0.6(4)a | NDb | 6.5 ± 0.1(7) | 4.7 ± 0.4(5)a |
| No. of events/min | 8125 ± 1086(11) | 2899 ± 937(5)a | 11,154 ± 5729(5) | 2379 ± 586(8)a | 930 ± 261(4)a | ND | 7259 ± 2236(4) | 8000 ± 2203(6) |
| To (ms) | 0.71 ± 0.14(11) | 1.57 ± 1.20(5) | 25.2 ± 23.2(5) | 0.64 ± 0.08(8) | 8.56 ± 3.74(4)a | ND | 4.26 ± 1.59(4)a | 0.70 ± 0.17(6) |
| Tc (ms) | 11.0 ± 3.5(11) | 57.9 ± 37.0(5)a | 32.8 ± 31.3(5) | 38.7 ± 9.5(8)a | 78.1 ± 22.3(4)a | ND | 5.78 ± 2.09(4) | 18.5 ± 11.0(6) |
a p < 0.05 compared to WT-RyR1.
b ND, not determined. Kinetic parameters of channel gating were obtained from recordings that contained a single channel in bilayers.
Mutation of residue Ile-4937 to alanine does not change the number of cells that show a caffeine-induced Ca2+ release response compared with wild-type RyR1 (Fig. 6). However, following isolation from HEK293 cells we observe reduced [3H]ryanodine binding, lack of Ca2+ regulation, and lack of Ca2+ conductance (Table 2, Fig. 7), which suggests that the Ile-4937 to alanine mutation results in a metastable channel in HEK293 cells. As the I4937A mutation only reduces the side chain size, but does not change the polarity, any loss of function resulting from alanine substitution should result from removing non-polar contacts. Increasing the side chain volume at position 4937 by mutating to valine restores channel function. The I4937V mutation restores [3H]ryanodine binding, Ca2+ regulation, and Ca2+ conductance in single channel experiments. The permeability ratio is also restored to a value that is not appreciably different from wild-type RyR1. Mutation of Ile-4937 to threonine also results in functional channels, however, with Ca2+ currents and permeability ratios that are significantly (p < 0.05) decreased compared with RyR1-I4937V. As the threonine side chain volume (122 Å3) is similar to the side chain volume of valine (139 Å3) (29), we expect that the reason for the decreased Ca2+ selectivity observed for RyR1-I4937T is the result of disrupting non-polar interactions that alter the open-channel structure.
Figure 6.

Caffeine-induced Ca2+ release in HEK293 cells expressing wild-type and mutant RyR1s. Ca2+ transients were determined in Ca2+-free Krebs-Ringers Henseleit bath solution as changes of Fluo-4 fluorescence (F/Fo) before and following the addition of 8 mm caffeine to the cells (arrow).
Figure 7.

Single channel recordings of WT and mutant channels. Left panels, shown are representative single channel currents at −20 mV (upper and middle traces) or 0 mV (bottom traces) as downward deflections from the closed states (c–) in symmetrical 250 mm KCl with 2 μm Ca2+ in the cis chamber (upper traces) and following the subsequent addition of EGTA to yield free Ca2+ of 0.1 μm (middle traces) or the addition of 10 mm Ca2+ to the trans chamber (bottom traces). Right panels, representative current–voltage relationships in 250 mm symmetrical KCl (●) and after the addition of 10 mm trans Ca2+ (○). Averaged Po values and ion permeation properties are summarized in Table 1.
Mutation of Gln-4933 to alanine resulted in caffeine-induced Ca2+ release in HEK293 cells (Fig. 6) but loss of ryanodine binding, increased mean close times, and decreased Ca2+ currents and permeability ratios (Table 2, Fig. 7). Comparable results were obtained with the corresponding mutation in the cardiac ryanodine receptor (RyR2-Q4863A), which showed a caffeine response in HEK293 cells and the presence of functional channels in single channel measurements but a pronounced decrease in [3H]ryanodine binding affinity (30). Mutation of Gln-4933 to valine had an even greater effect resulting in channels with significantly decreased caffeine response, Ca2+ currents, and permeability ratios compared with either wild-type RyR1 or RyR1-Q4933A. Large variability in mean open and close times suggested that channels with a different gating behavior were incorporated in lipid bilayers.
We previously investigated the single channel properties of purified RyR1-G4934A and RyR1-G4934V mutants (31). We here examine the single channel properties of these mutants in membrane fractions avoiding exposure to detergent during purification of RyR1s. Similar to our previous findings the G4934A mutation resulted in functional channels with altered gating behavior, reduced Ca2+ current, and permeability ratio (Table 2, Fig. 7). However, the G4934V mutation also resulted in functional channels with reduced Ca2+ current and permeability ratios when measured in membrane fractions. The number of channel events decreased and mean close times increased. The result suggests that the G4934V mutation possibly destabilizes the channel so it cannot retain function throughout the process of purification with detergent.
Discussion
We report an open-channel model derived by ion pulling simulations. As we did not include any EM data from an open-channel RyR (22, 23) in our modeling, the significant agreement between our model and the open-channel cryo-EM density validates the accuracy of both our model and approach. The rationale for our approach was that moving a Ca2+ ion through the channel constriction site would force structural rearrangements creating a pore capable of allowing Ca2+ to pass through. We assumed that the RyR1 structure would open via the path of least resistance and that this pathway would also be preferred physiologically. The agreement between our model and the experimental structures also supports our hypothesis that channel opening occurs via the most energetically favorable pathway.
Our model accurately reproduced the ∼12° clockwise rotation of the backbone atoms in the S6 helix viewed from the cytoplasmic end of the channel. The twisting motion rotates Cα atoms of Ile-4937 and Gln-4933 away from the pore while arranging Cα atoms of Gly-4934 to face the pore. Rotation of Ile-4937 out of the pore creates a much larger pore radius than could be achieved by widening the inter-helical distances by tilting the S6 helix. Helical rotation has been observed as a pore opening mechanism in other channels including the potassium channel from Streptomyces lividans (KcsA) (32), the NaK channel (33), and the nicotinic acetylcholine receptor (nAChR) (34) suggesting that twisting may be a common pore opening mechanism among ion channels.
Our functional measurements also support rotation of Ile-4937 out of the pore. In the closed-channel structure Ile-4937 lines the pore and forms the hydrophobic constriction site. If Ile-4937 faced the pore in the open channel conformation, we would expect that reducing the side chain volume (I4937A) or making the residue more hydrophilic (I4937T) would make the channel more open. However, we find that the I4937A and I4937T mutations abolish and reduce calcium current, respectively. Moreover, the I4937A mutation markedly reduces ryanodine binding, which suggests that this mutation removes contacts necessary for maintaining a stable channel when removed from its cellular environment. Although the side chain volumes of threonine and valine are similar, we find that the function of RyR1-I4937V is more similar to wild-type RyR1 than RyR1-I4937T. We suggest that it is the polar nature of the I4937T mutation that causes the reduced function of the RyR1-I4937T mutant. Taken together, the results suggest that in the open-channel structure Ile-4937 no longer faces the pore but rather interacts with hydrophobic residues, likely in neighboring S6 helices.
In the open-channel model, Gln-4933 faces the pore. The molecular volumes for alanine (90 Å3) and valine (139 Å3) are, respectively, smaller and similar as compared with the molecular volume of glutamine (151 Å3) (29). On the basis of molecular volume we would expect a more open channel pore, which seems to contradict the experimental finding. However, both the Q4933A and Q4933V mutations also decrease the polarity of the side chain at this site. We compute the pore profiles and apply a Poisson-Boltzmann electrostatic model (35) to determine the Ca2+ solvation energy for wild-type and mutant RyR1 channels (Fig. 8). Our model shows that the Q4933V and Q4933A mutations increase the pore radius at position 4933 from 2.8 ± 0.3 to 2.9 ± 0.1 Å and 4.0 ± 0.1 Å, respectively. Both mutations also significantly increase the Ca2+ solvation energy, suggesting that the polarity of the residue at the 4933 position is important for Ca2+ conductance. However, we cannot rule out the importance of an interaction between Gln-4933 and Val-4926 to maintain an open-channel conformation (Fig. 5).
Figure 8.
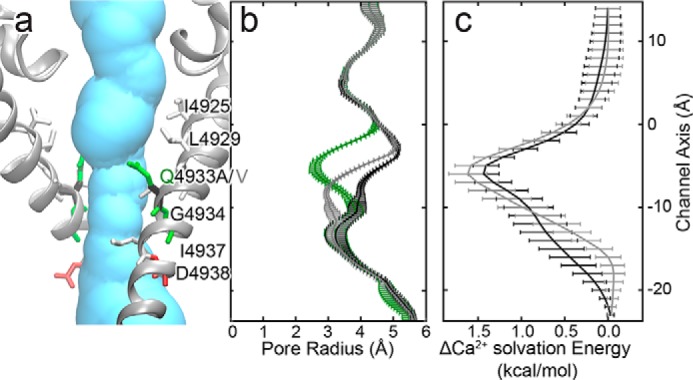
The Q4933A and Q4933V mutations increase both the minimum pore radius and Ca2+ solvation energy. a, structure of the open-channel pore surrounding position 4933. Pore lining residues are shown as sticks and colored according to residue type: red, acidic; green, polar; and white, aliphatic. Gln-4933, Q4933A, and Q4933V are colored green, black, and gray, respectively. b, RyR1 open-channel pore profiles for wild-type (green) and Q4933A (black), and Q4933V (gray) models. The pore profiles are the mean ± S.E. calculated over five structures sampled evenly over the last 20 ns of molecular dynamics simulation. c, mean change in Ca2+ solvation energies between mutant and wild-type RyR1 for Q4933A (black) and Q4933V (gray) mutations calculated over the same five structures from b. Error bars represent the 95% confidence interval for the difference between the means.
Although the radial tilting angle and the twisting angle of the S6 helix are accurately reproduced in our model, we note a difference in the lateral tilting angle for the S6 helix (3 ± 1° in our open-channel model compared with 9 ± 1° in the open-channel structures). The increased lateral tilting in the open-channel structure compared with our model has the effect of changing the contacting residues for Ile-4937. In our model Ile-4937 contacts Ile-4931 and Gly-4934. Although the contact with Ile-4931 is observed in the open-channel structure, the increased lateral tilting of the S6 helix brings Ile-4937 out of contact with Gly-4934 (Fig. 5). Our model suggests a simple rotation of Ile-4937 past Gly-4934 to open the channel. However, the open-channel structure indicates that both twisting and tilting motions of the S6 helix bring Ile-4937 past Gly-4934 during channel opening. Although the difference in lateral tilting altered contacts for Ile-4937, we note that the salt-bridge between Asp-4938 and Arg-4944 located on the cytosolic side of Ile-4937 is preserved in our open-channel model (Fig. 5). Moreover, contacts for Gln-4933 located on the luminal side of Ile-4937 are also consistent between our open-channel model and the open-channel structure.
In our open-channel model, the direction of rotation brings Ile-4937 past Gly-4934. We previously showed that the G4934A mutation reduced the Ca2+ current, whereas the G4934V mutation resulted in loss of function of purified channels (31). In the current study, we find that RyR1-G4934V can form functional channels with reduced Ca2+ current, when avoiding detergent exposure throughout the process of purification. The result affirms our previous prediction that the G4934V mutation reduces channel stability. Previously, we explained the effects of these mutations in terms of increased interaction with residues in the adjacent S6 helix (Ile-4936, Ile-4937, and Phe-4940) within the closed-channel structure. Our model and the structures of the open-channel RyR1 suggest that the G4934A mutation could also affect channel function by impeding the rotation of Ile-4937 to produce a fully open channel. This hypothesis is supported by analysis of the open- and closed-channel times. The G4934A mutation primarily lengthens mean closed times (38.7 ± 9.5 versus 11.0 ± 3.5 ms for WT, n = 8–11) rather than affect mean open times (0.64 ± 0.08 versus 0.71 ± 0.14 for WT, n = 8–11), which suggests that the mutation decreases the probability of closed to open transitions. A decrease in closed to open transitions for the G4934A mutation would be consistent with our hypothesis that rotation of Ile-4937 toward Gly-4934 in the adjacent subunit is required for channel opening.
Conclusion
Our modeling of the open-channel RyR1 reveals two primary motions responsible for creating an open-channel pore. The first is a radial tilting of the S6 helix away from the pore along the 4-fold channel axis, which results in a general widening of the pore at the cytoplasmic end of the channel. The second major motion is a twisting of the S6 helix, which rotates the hydrophobic residue Ile-4937 out of the pore. Our results indicate that this rotation brings Ile-4937 past Gly-4934 and into contact with other hydrophobic residues in the S6 helix, which may be responsible for stabilizing the open-channel conformation. Our model also indicates that the constriction site for the open-channel RyR1 is Gln-4933 and our single channel data suggest that the polarity of this residue plays an important role in Ca2+ conductance. Altogether, our modeling reveals both the major motions of the RyR1 transmembrane domain responsible for channel gating and plausible roles of RyR1–Gln-4933, RyR1–Gly-4934, and RyR1–Ile-4937 in channel gating.
Experimental procedures
Materials
[3H]Ryanodine was obtained from PerkinElmer Life Sciences, protease and phosphatase inhibitors from Sigma, and phospholipids from Avanti Polar Lipids.
Molecular dynamics simulations and pore opening of the RyR1 closed-channel
The transmembrane domain of the 3.8-Å resolution cryo-EM structure (PDB code 3J8H) (21) was inserted into a lipid bilayer with a lipid composition comparable with the one used in single channel measurements (5:3:2 molar ratio of 1-palmitoyl-2-oleoylphosphatidylethanolamine, 1-palmitoyl-2-oleoylphosphatidylserine, and 1-palmitoyl-2-oleoylphosphatidylcholine) using the GROMACS g_membed tool. The bilayer was generated using the CHARMM-GUI membrane builder (36). The RyR1 transmembrane segment modeled included residues 4545 to 5033 based on the sequence present in the cryo-EM structure. The 33-amino acid loop (residues 4588 to 4621) missing from the reported structure (21) was replaced with 3 glycine residues. Each system was solvated using the TIP3P water model and ionized with 0.02 m CaCl2 and 0.27 m KCl. All simulations were performed using GROMACS 4.6.1(37) with the CHARMM36 force field (38, 39). The simulation was performed at a constant pressure and temperature of 1 atm and 298 K. All bonds were constrained using the LINCS algorithm (40). The integration time step used for all simulations, unless otherwise noted, was 2 fs. Particle mesh Ewald was used for long-range electrostatic interactions. A 10-Å cutoff was used for non-bonded interactions.
To equilibrate the system, energy minimization was performed for 10,000 steps with harmonic position restraints of 10 kJ/(mol Å2) on the protein backbone atoms followed by 3 ns of equilibration, during which position restraints on protein backbone atoms were gradually reduced from 10 to 0 kJ/(mol Å2). Channel opening was performed using the molecular pulling code implemented in GROMACS. A Ca2+ ion was inserted at the cytosolic side of the channel and pulled through the channel gate at a rate of 0.1 Å/ns over 100 ns of simulation using a force constant of 10 kJ/(mol Å2). The Ca2+ ion was fixed to the channel constriction site for an additional 20 ns of simulation with a harmonic potential of 10,000 kJ/(mol Å2) along the channel axis. The ion was free to move within the membrane plane.
To further refine the open-channel inter-subunit distance, restraints were added between Cβ atoms of Ile-4937 and Ile-4931 in the previous subunit and between Cβ atoms Ile-4937 and Leu-4935 in the previous subunit. The inter-subunit distance restraints used were 7.8 and 7.0 Å for residues 4931 and 4935, respectively. The restraints were designed to further rotate Ile-4937 out of the pore in the direction observed after the initial pulling. As the additional torque of twisting was likely to distort the helices, helical distance restraints were added between backbone nitrogen and oxygen atoms for residues 4930 to 4953. The upper boundaries used for the distance restraints were obtained from distances observed in the cryo-EM structure plus 0.5 Å. Lower boundaries were all 2.7 Å. Inter-subunit distance restraints of 12, 15, and 16 Å were added between Cα atoms of adjacent subunits for residues Ile-4937, Asp-4945, and Glu-4948, respectively. The listed distances were obtained from the mean distance among four chains observed after the initial ion pulling simulation. The restraints were added gradually over the course of 14 ns of simulation using a time step of 1 fs. Restraint potentials were all 1000 kJ/(mol Å2). A final production run of 20 ns was performed that was used for data analyses.
Alignment with EM density
Initial alignment of structures with cryo-EM maps was performed using the colors function in Situs (41). The last structural snapshot from the pore opening calculations was aligned to the cryo-EM map using a Gaussian density map generated at 4-Å resolution. Rigid body fitting was performed sampling the rotational search space with a 5° step size. The resulting fit was refined using the fit-in-map function implemented in the Chimera software package. The Chimera fit-in-map function was also used to evaluate the correlation between the structural models and the experimental EM data. For all structures we fit the map of the backbone and Cβ atoms in the structure generated at 4-Å resolution for residue ranges 4820 to 4957 and 4910 to 4957. Residues in loops (residues 4861 to 4879 and residues 4891 to 4909) were excluded from the fitting. The atoms included and the resolution for fitting were chosen to maximize the correlation of the experimentally derived structure with the experimental data. The correlations of the open structure (PDB code 5TAL) to the open densities (EMD-8376, EMD-8377, and EMD-8378) and the closed structure (PDB code 3J8H) to the closed density (EMD-2807)(23) were 0.98 and 0.97, respectively.
Structural analyses
For all structural analyses 10 structures sampled every 2 ns from the last 20 ns of simulation were used. Principal component analysis was performed on the 10 open-channel structures from simulation combined with the closed-channel cryo-EM structure (21) using ProDy (42). The eigenvectors of the first principal component were visualized using the NMWiz plugin in VMD (43). Radial and lateral tilting angles were calculated for both upper (4930 to 4950) and lower (4910 to 4930) portions of the S6 helix according to the previous method (44). The mean ± S.E. are reported in the results. The twist angle was measured as the average angle between the respective backbone atoms of the open-channel model and closed-channel cryo-EM structure within the membrane plane. The angle was calculated after centering both over the residue range 4930 to 4950. The reported value is the mean ± S.E. from 10 structures and four chains.
Pore profile and Poisson-Boltzmann calculations
All pore profiles were calculated using the HOLE program (45). To generate pore profiles for RyR1-WT, RyR1-Q4934A, and RyR1-Q4934V, we extracted five structures sampled evenly over the last 20 ns of the pore opening simulation. Amino acid substitution and rotamer optimization were performed using Eris (46, 47). For each structure the Eris program was used to perform 200 independent Monte Carlo optimizations allowing side chain repacking within 10 Å of the mutation site for both wild-type and mutant proteins. From each set of calculations we selected the minimum energy structure for both the mutant and wild-type protein (five structures each) and calculated the pore profile for each of these structures.
The same structures were used in the Poisson-Boltzmann calculations performed using APBSmem (48). The charge model used for calculations was the Swanson charge model (49), which was applied using the PDB2PQR software (50). Two focusing layers were used. The finest grid included 97 grid points in the x, y, and z dimension and had a grid length of 90 Å. Ion concentrations were set to 0.1 m. Non-linear Poisson-Boltzmann method was implemented in the Adaptive Poisson-Boltzmann Solver (35). Upper and lower exclusions were set at 16 Å to exclude the membrane from the pore region. Membrane thickness was 42.5 Å with a head group thickness of 7 Å similar to values previously used to calculate the Ca2+ solvation energy profile of the TRPV1 receptor (51). Dielectrics for membrane and protein were set to 2.0 and dielectrics for solvent and head groups were set to 80.0. The ion step size was 1.0 Å along the channel axis and the ionic radius used for Ca2+ was 1.03. The temperature was set at 298 K.
Expression and preparation of wild-type and mutant channels
pCMV5-RyR1 mutants were prepared as described previously (52) or by a gene synthesis method using a proprietary protocol (Genewiz Inc., South Plainfield, NJ). Wild-type and mutant expression vectors were transiently expressed in HEK293 cells with jetPRIME (Polyplus, New York), according to the manufacturer's instructions. Cells were maintained in Dulbecco's minimum essential medium (DMEM/F-12 (1:1)) containing 10% fetal bovine serum and 1× Antibiotic Antimycotic Solution at 37 °C and 5% CO2 and plated the day before transfection. For each 10-cm tissue culture dish, 10 μg of DNA was used at a DNA/jetPRIME ratio of 1:2. Following transfection, cells were kept at 35 °C and harvested 66–72 h after transfection. Keeping transfected cells at 35 °C instead at 37 °C previously used (53) increased the number of functional RyR1s, as determined using [3H]ryanodine ligand binding method. Membrane fractions were prepared in the presence of protease inhibitors and 1 mm oxidized glutathione (GSSG) as described (53).
Cellular Ca2+ release
Retention of the function in HEK293 cells was studied by cellular Ca2+ release assay using the Ca2+ releasing drug caffeine described in the previous publication (52). Briefly, HEK293 cells grown on glass coverslips were incubated with 5 μm Fluo 4-AM. Cellular Ca2+ release was induced by the addition of ∼8 mm caffeine and measured in individual cells using EasyRatioPro (Photon Technology International, Lawrenceville, NJ).
SDS-PAGE and immunoblot analysis
Proteins in membrane preparations were separated using 3–12% gradient SDS-PAGE, transferred to nitrocellulose membranes, probed with primary rabbit anti-RyR1 polyclonal antibody 6425, and quantified using Image Lab 4.1 Analysis Software (Bio-Rad) (31).
[3H]Ryanodine binding
Expression of functional RyR1 mutants in membrane preparations was assessed using [3H]ryanodine-binding assay. Ryanodine binds with high specificity to RyRs and is widely used to probe for RyR activity and content (54). Expression levels were determined by measuring maximum binding capacity (Bmax) of [3H]ryanodine binding of membrane preparations for 4–5 h at 24 °C with a near-saturating concentration of 20 nm [3H]ryanodine in 20 mm imidazole, pH 7.0, 0.6 m KCl, protease inhibitors, 1 mm GSSG, and 0.1 mm Ca2+. Nonspecific binding was determined with a 1000-fold excess of unlabeled ryanodine. Bound [3H]ryanodine binding was determined using a filter assay (52).
Single channel recordings
Single channel measurements were performed by the fusion of membrane preparations with Mueller-Rudin type planar lipid bilayers containing a 5:3:2 mixture of bovine brain phosphatidylethanolamine, phosphatidylserine, and phosphatidylcholine (25 mg of total phospholipid/ml of n-decane) (52). Single channels were recorded in symmetric 0.25 m KCl, 20 mm K-HEPES, pH 7.4, buffers with additions as indicated in the text. The trans side of the bilayer was defined as ground. Electrical signals were filtered at 2 kHz (0.5 kHz for Ca2+ currents at 0 mV), digitized at 10 kHz, and analyzed at 50% threshold setting (52). Data acquisition and analysis were performed with commercially available software package (pClamp, Axon Instruments, CA), using 2-min recordings for analysis. To determine permeability ratios, single channel activities were recorded in symmetrical 250 mm KCl solution with 10 mm Ca2+ on the trans side and the reversal potential (Erev) was measured. The permeability ratio of Ca2+ versus K+ (PCa/PK) was calculated using a modified form of the Goldman-Hodgkin-Katz equation.
| (Eq. 1) |
Biochemical assays and data analysis
Free Ca2+ concentrations were established by adding varying amounts of Ca2+ to 1 mm EGTA solutions and determined with the use of a Ca2+ selective electrode. Differences between samples were analyzed using Student's t test. p < 0.05 was considered significant.
Author contributions
D. D. M., G. M., and N. V. D. designed the study and wrote the paper. L. X. performed and analyzed the single channel experiments. Y. M. constructed vectors for expression of mutant proteins and Y. M. and D. A. P. analyzed the mutant phenotypes expressed in HEK293 cells. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grant AR018687. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Video S1.
- RyR1
- type 1 ryanodine receptor
- PDB
- Protein Data Bank.
References
- 1. Franzini-Armstrong C., and Protasi F. (1997) Ryanodine receptors of striated muscles: a complex channel capable of multiple interactions. Physiol. Rev. 77, 699–729 [DOI] [PubMed] [Google Scholar]
- 2. McCarthy T. V., Quane K. A., and Lynch P. J. (2000) Ryanodine receptor mutations in malignant hyperthermia and central core disease. Hum. Mutat. 15, 410–417 [DOI] [PubMed] [Google Scholar]
- 3. Betzenhauser M. J., and Marks A. R. (2010) Ryanodine receptor channelopathies. Pflugers Arch. 460, 467–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Smith J. S., Coronado R., and Meissner G. (1986) Single channel measurements of the calcium release channel from skeletal muscle sarcoplasmic reticulum: activation by Ca2+ and ATP and modulation by Mg2+. J. Gen. Physiol. 88, 573–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lai F. A., Erickson H. P., Rousseau E., Liu Q. Y., and Meissner G. (1988) Purification and reconstitution of the calcium release channel from skeletal muscle. Nature 331, 315–319 [DOI] [PubMed] [Google Scholar]
- 6. Boehning D., Mak D. O., Foskett J. K., and Joseph S. K. (2001) Molecular determinants of ion permeation and selectivity in inositol 1,4,5-trisphosphate receptor Ca2+ channels. J. Biol. Chem. 276, 13509–13512 [DOI] [PubMed] [Google Scholar]
- 7. Clapham D. E. (1995) Calcium signaling. Cell 80, 259–268 [DOI] [PubMed] [Google Scholar]
- 8. Bezprozvanny I., Watras J., and Ehrlich B. E. (1991) Bell-shaped calcium-response curves of Ins(1,4,5)P3- and calcium-gated channels from endoplasmic reticulum of cerebellum. Nature 351, 751–754 [DOI] [PubMed] [Google Scholar]
- 9. Meissner G., Darling E., and Eveleth J. (1986) Kinetics of rapid Ca2+ release by sarcoplasmic reticulum: effects of Ca2+, Mg2+, and adenine nucleotides. Biochemistry 25, 236–244 [DOI] [PubMed] [Google Scholar]
- 10. Seo M.-D., Velamakanni S., Ishiyama N., Stathopulos P. B., Rossi A. M., Khan S. A., Dale P., Li C., Ames J. B., Ikura M., and Taylor C. W. (2012) Structural and functional conservation of key domains in InsP3 and ryanodine receptors. Nature 483, 108–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Seo M.-D., Enomoto M., Ishiyama N., Stathopulos P. B., and Ikura M. (2015) Structural insights into endoplasmic reticulum stored calcium regulation by inositol 1,4,5-trisphosphate and ryanodine receptors. Biochim. Biophys. Acta 1853, 1980–1991 [DOI] [PubMed] [Google Scholar]
- 12. Radermacher M., Wagenknecht T., Grassucci R., Frank J., Inui M., Chadwick C., and Fleischer S. (1992) Cryo-EM of the native structure of the calcium release channel/ryanodine receptor from sarcoplasmic reticulum. Biophys. J. 61, 936–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Radermacher M., Rao V., Grassucci R., Frank J., Timerman A. P., Fleischer S., and Wagenknecht T. (1994) Cryo-electron microscopy and three-dimensional reconstruction of the calcium release channel/ryanodine receptor from skeletal muscle. J. Cell Biol. 127, 411–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Serysheva I. I., Orlova E. V., Chiu W., Sherman M. B., Hamilton S. L., and van Heel M. (1995) Electron cryomicroscopy and angular reconstitution used to visualize the skeletal muscle calcium release channel. Nat. Struct. Biol. 2, 18–24 [DOI] [PubMed] [Google Scholar]
- 15. Bhat M. B., Zhao J., Takeshima H., and Ma J. (1997) Functional calcium release channel formed by the carboxyl-terminal portion of ryanodine receptor. Biophys. J. 73, 1329–1336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Euden J., Mason S. A., and Williams A. J. (2013) Functional characterization of the cardiac ryanodine receptor pore-forming region. PLoS ONE 8, e66542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Samsó M., Feng W., Pessah I. N., and Allen P. D. (2009) Coordinated movement of cytoplasmic and transmembrane domains of RyR1 upon gating. PLos Biol. 7, e85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ludtke S. J., Serysheva I. I., Hamilton S. L., and Chiu W. (2005) The pore structure of the closed RyR1 channel. Structure 13, 1203–1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Samsó M., Wagenknecht T., and Allen P. D. (2005) Internal structure and visualization of transmembrane domains of the RyR1 calcium release channel by cryo-EM. Nat. Struct. Mol. Biol. 12, 539–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zalk R., Clarke O. B., des Georges A., Grassucci R. A., Reiken S., Mancia F., Hendrickson W. A., Frank J., and Marks A. R. (2015) Structure of a mammalian ryanodine receptor. Nature 517, 44–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yan Z., Bai X. C., Yan C., Wu J., Li Z., Xie T., Peng W., Yin C., Li X., Scheres S. H., Shi Y., and Yan N. (2015) Structure of the rabbit ryanodine receptor RyR1 at near-atomic resolution. Nature 517, 50–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. des Georges A., Clarke O. B., Zalk R., Yuan Q., Condon K. J., Grassucci R. A., Hendrickson W. A., Marks A. R., and Frank J. (2016) Structural basis for gating and activation of RyR1. Cell 167, 145.e17–157.e17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Peng W., Shen H., Wu J., Guo W., Pan X., Wang R., Chen S. R., and Yan N. (2016) Structural basis for the gating mechanism of the type 2 ryanodine receptor RyR2. Science 354, aah5324-1–aah5324-10 [DOI] [PubMed] [Google Scholar]
- 24. Isralewitz B., Baudry J., Gullingsrud J., Kosztin D., and Schulten K. (2001) Steered molecular dynamics investigations of protein function. J. Mol. Graph. Model 19, 13–25 [DOI] [PubMed] [Google Scholar]
- 25. Monticelli L., Robertson K. M., MacCallum J. L., and Tieleman D. P. (2004) Computer simulation of the KvAP voltage-gated potassium channel: steered molecular dynamics of the voltage sensor. FEBS Lett. 564, 325–332 [DOI] [PubMed] [Google Scholar]
- 26. Gullingsrud J., and Schulten K. (2003) Gating of MscL studied by steered molecular dynamics. Biophys. J. 85, 2087–2099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liu X., Xu Y., Li H., Wang X., Jiang H., and Barrantes F. J. (2008) Mechanics of channel gating of the nicotinic acetylcholine receptor. PLoS Comput. Biol. 4, e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tinker A., and Williams A. J. (1993) Probing the structure of the conduction pathway of the sheep cardiac sarcoplasmic reticulum calcium-release channel with permeant and impermeant organic cations. J. Gen. Physiol. 102, 1107–1129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tsai J., Taylor R., Chothia C., and Gerstein M. (1999) The packing density in proteins: standard radii and volumes. J. Mol. Biol. 290, 253–266 [DOI] [PubMed] [Google Scholar]
- 30. Wang R., Zhang L., Bolstad J., Diao N., Brown C., Ruest L., Welch W., Williams A. J., and Chen S. R. (2003) Residue Gln4863 within a predicted transmembrane sequence of the Ca2+ release channel (ryanodine receptor) is critical for ryanodine interaction. J. Biol. Chem. 278, 51557–51565 [DOI] [PubMed] [Google Scholar]
- 31. Mei Y., Xu L., Mowrey D. D., Mendez Giraldez R., Wang Y., Pasek D. A., Dokholyan N. V., and Meissner G. (2015) Channel gating dependence on pore lining helix glycine residues in skeletal muscle ryanodine receptor. J. Biol. Chem. 290, 17535–17545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shimizu H., Iwamoto M., Konno T., Nihei A., Sasaki Y. C., and Oiki S. (2008) Global twisting motion of single molecular KcsA potassium channel upon gating. Cell 132, 67–78 [DOI] [PubMed] [Google Scholar]
- 33. Alam A., and Jiang Y. (2009) High-resolution structure of the open NaK channel. Nat. Struct. Mol. Biol. 16, 30–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Miyazawa A., Fujiyoshi Y., and Unwin N. (2003) Structure and gating mechanism of the acetylcholine receptor pore. Nature 423, 949–955 [DOI] [PubMed] [Google Scholar]
- 35. Baker N. A., Sept D., Joseph S., Holst M. J., and McCammon J. A. (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wu E. L., Cheng X., Jo S., Rui H., Song K. C., Dávila-Contreras E. M., Qi Y., Lee J., Monje-Galvan V., Venable R. M., Klauda J. B., and Im W. (2014) CHARMM-GUI Membrane Builder toward realistic biological membrane simulations. J. Comput. Chem. 35, 1997–2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hess B., Kutzner C., van der Spoel D., and Lindahl E. (2008) GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 4, 435–447 [DOI] [PubMed] [Google Scholar]
- 38. Best R. B., Zhu X., Shim J., Lopes P. E., Mittal J., Feig M., and Mackerell A. D. Jr. (2012) Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone ϕ, ψ and side-chain χ(1) and χ(2) dihedral angles. J. Chem. Theory Comput. 8, 3257–3273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Klauda J. B., Monje V., Kim T., and Im W. (2012) Improving the CHARMM force field for polyunsaturated fatty acid chains. J. Phys. Chem. B 116, 9424–9431 [DOI] [PubMed] [Google Scholar]
- 40. Hess B. (2008) P-LINCS: a parallel linear constraint solver for molecular simulation. J. Chem. Theory Comput. 4, 116–122 [DOI] [PubMed] [Google Scholar]
- 41. Chacón P., and Wriggers W. (2002) Multi-resolution contour-based fitting of macromolecular structures. J. Mol. Biol. 317, 375–384 [DOI] [PubMed] [Google Scholar]
- 42. Bakan A., Meireles L. M., and Bahar I. (2011) ProDy: protein dynamics inferred from theory and experiments. Bioinformatics 27, 1575–1577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Humphrey W., Dalke A., and Schulten K. (1996) VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38, 27–28 [DOI] [PubMed] [Google Scholar]
- 44. Cheng X., Ivanov I., Wang H., Sine S. M., and McCammon J. A. (2007) Nanosecond-timescale conformational dynamics of the human alpha7 nicotinic acetylcholine receptor. Biophys. J. 93, 2622–2634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Smart O. S., Neduvelil J. G., Wang X., Wallace B. A., and Sansom M. S. (1996) HOLE: a program for the analysis of the pore dimensions of ion channel structural models. J. Mol. Graph. 14, 354–360, 376 [DOI] [PubMed] [Google Scholar]
- 46. Yin S., Ding F., and Dokholyan N. V. (2007) Eris: an automated estimator of protein stability. Nat. Methods 4, 466–467 [DOI] [PubMed] [Google Scholar]
- 47. Yin S., Ding F., and Dokholyan N. V. (2007) Modeling backbone flexibility improves protein stability estimation. Structure 15, 1567–1576 [DOI] [PubMed] [Google Scholar]
- 48. Callenberg K. M., Choudhary O. P., de Forest G. L., Gohara D. W., Baker N. A., and Grabe M. (2010) APBSmem: a graphical interface for electrostatic calculations at the membrane. PLoS ONE 5, e12722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Swanson J. M., Wagoner J. A., Baker N. A., and McCammon J. A. (2007) Optimizing the poisson dielectric boundary with explicit solvent forces and energies: lessons learned with atom-centered dielectric functions. J. Chem. Theory Comput. 3, 170–183 [DOI] [PubMed] [Google Scholar]
- 50. Dolinsky T. J., Nielsen J. E., McCammon J. A., and Baker N. A. (2004) PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 32, W665–W667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Marcoline F. V., Bethel N., Guerriero C. J., Brodsky J. L., and Grabe M. (2015) Membrane protein properties revealed through data-rich electrostatics calculations. Structure 23, 1526–1537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wang Y., Xu L., Pasek D. A., Gillespie D., and Meissner G. (2005) Probing the role of negatively charged amino acid residues in ion permeation of skeletal muscle ryanodine receptor. Biophys. J. 89, 256–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gao L., Tripathy A., Lu X., and Meissner G. (1997) Evidence for a role of C-terminal amino acid residues in skeletal muscle Ca2+ release channel (ryanodine receptor) function. FEBS Lett. 412, 223–226 [DOI] [PubMed] [Google Scholar]
- 54. Sutko J. L., Airey J. A., Welch W., and Ruest L. (1997) The pharmacology of ryanodine and related compounds. Pharmacol. Rev. 49, 53–98 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.