

Abstract
The protein mini-chromosome maintenance 10 (Mcm10) was originally identified as an essential yeast protein in the maintenance of mini-chromosome plasmids. Subsequently, Mcm10 has been shown to be required for both initiation and elongation during chromosomal DNA replication. However, it is not fully understood how the multiple functions of Mcm10 are coordinated or how Mcm10 interacts with other factors at replication forks. Here, we identified and characterized the Mcm2–7-interacting domain in human Mcm10. The interaction with Mcm2–7 required the Mcm10 domain that contained amino acids 530–655, which overlapped with the domain required for the stable retention of Mcm10 on chromatin. Expression of truncated Mcm10 in HeLa cells depleted of endogenous Mcm10 via siRNA revealed that the Mcm10 conserved domain (amino acids 200–482) is essential for DNA replication, whereas both the conserved and the Mcm2–7-binding domains were required for its full activity. Mcm10 depletion reduced the initiation frequency of DNA replication and interfered with chromatin loading of replication protein A, DNA polymerase (Pol) α, and proliferating cell nuclear antigen, whereas the chromatin loading of Cdc45 and Pol ϵ was unaffected. These results suggest that human Mcm10 is bound to chromatin through the interaction with Mcm2–7 and is primarily involved in the initiation of DNA replication after loading of Cdc45 and Pol ϵ.
Keywords: cell cycle, DNA binding protein, DNA helicase, DNA replication, DNA-protein interaction, Mcm10, Mcm2–7
Introduction
Eukaryotic DNA replication is tightly coupled with cell cycle progression to ensure that the entire genome is duplicated only once per cell cycle. Recent analyses using different systems have shown that the process that initiates eukaryotic DNA replication from a large number of origins is highly conserved (1). The initiation of DNA replication consists of two major processes: the formation of a pre-replication complex (pre-RC)2 in G1 phase and the activation of pre-RCs in S phase. During G1 phase, the origin recognition complex (ORC) recruits Cdc6 and Cdt1, which facilitate the loading of the double-hexameric Mcm2–7 complex to form a pre-RC (2–6). At the G1/S transition, the pre-RC is activated by Cdc7/Dbf4 kinase (DDK) and S phase cyclin-dependent kinases (S-Cdks) (7, 8). DDK phosphorylates Mcm4 and Mcm6 to promote its interaction with Cdc45 on chromatin and the recruitment of Sld3/7 onto a pre-RC (9–11). Cdk phosphorylates Sld2 and Sld3 and facilitates their interaction with Dpb11 (12, 13), which triggers the recruitment of Sld2, Dpb11, Pol ϵ, and GINS (7, 14). Finally, Mcm2–7 interacts with Cdc45 and GINS to form the active replicative helicase complex called the CMG complex (15–17).
After the DNA is unwound at the replication origin, the single-stranded DNA-binding protein RPA (replication protein A), Pol α, and other elongation factors including proliferating cell nuclear antigen (PCNA) are recruited (18, 19). Additionally, the components of the checkpoint pathway as well as chromatin remodeling factors are recruited to the replication fork to monitor the progression of S phase (20–22).
Mcm10 was originally identified in Saccharomyces cerevisiae as an essential protein in screens for genes that are required for maintenance of mini-chromosome plasmids and subsequently shown to be required for chromosomal DNA replication (23). The phenotype of the mcm10 mutant is similar to that of several known initiation mutants (23). Budding yeast Mcm10 has multiple functions: interacting with Mcm2–7 and DNA (23–25), interacting with Pol α as a molecular chaperone (26), and interacting with PCNA as diubiquitinated Mcm10, which is essential for cell growth (27). These results suggest that budding yeast Mcm10 is involved in both the initiation and the elongation of DNA replication. Mcm10 is also involved in checkpoint control (28) as well as transcriptional silencing (29, 30), which is independent from its roles in DNA replication. Mcm10 homologs have been identified in metazoans and reported to interact with DNA and various proteins including Mcm2–7, Pol α, And-1, RECQL4, Cdc45, and Sirt1 (31–36). However, it is not fully understood how Mcm10 interacts with multiple replication factors at replication forks and how the interaction is regulated throughout the cell cycle.
Additionally, its exact contribution to chromosomal DNA replication is still controversial, because there are multiple discrepancies among studies on Mcm10. Studies in budding yeast, fission yeast, Xenopus, Drosophila, and humans have shown that Mcm10 is required for the recruitment of Cdc45 to origins and formation of the CMG complex (37–41). The chromatin loading of Mcm10 is independent of Cdk2 and Cdc7 in a Xenopus egg extract system (37). Conversely, recent studies in budding and fission yeast using degron mutants have shown that a stable CMG complex forms in the absence of Mcm10 (42–44). The results from an in vitro reconstitution system using budding yeast extract also suggest that the CMG complex associates with the origin DNA in the absence of Mcm10 (7, 8). Furthermore, recruitment of Mcm10 is dependent on Cdc45, GINS, DDK, and S-CDK (7, 8). Recent results obtained in a Xenopus egg extract system also support that chromatin loading of Mcm10 requires DDK and S-CDK (45).
In budding yeast and mammalian cells, Mcm10 is reported to stabilize Pol α (26, 32). Alternatively, another study has shown that depletion of Mcm10 does not affect the stability of Pol α (33). As for its role in the elongation step, chromatin immunoprecipitation analysis revealed that Mcm10 migrates with the replisome in budding and fission yeasts (26, 46). In contrast, other studies suggest that Mcm10 is not a stable component of the replisome (42, 43). Thus far, Mcm10 has been found to exist as a monomer, dimer, trimer, and hexamer in vitro (25, 47, 48), whereas the oligomeric state of Mcm10 in vivo remains unknown.
Limited proteolysis and mass spectrometry analyses revealed that Mcm10 in higher eukaryotes contains at least three functional domains (49): the N-terminal domain that is involved in multimer formation (49, 50), the conserved internal domain that interacts with DNA as well as Pol α (49), and the C-terminal domain that is specific to metazoans and interacts with DNA, Pol α, Cdc45, and Sirt1 (35, 36, 49). X-ray crystallography and NMR spectroscopy have shown that the oligonucleotide/oligosaccharide binding-fold (OB-fold) of the internal domain competitively interacts with DNA and the catalytic subunit of Pol α (51, 52). The zinc finger motifs in the internal domain and C-terminal domain are also involved in DNA binding (48, 49, 53).
Recently, budding yeast Mcm10 was reported to interact with double hexameric Mcm2–7 through the C-terminal region and be involved in double hexamer splitting (54, 55). However, the C-terminal region is not conserved between yeast and human; the domain required for the association with Mcm2–7 has not yet been identified in higher eukaryotes. In this report, we determined the domain of human Mcm10 required for the association with Mcm2–7. This domain was important for supporting the efficient firing of replication origins and the stable chromatin association of Mcm10. Depletion of Mcm10 resulted in the inhibition of initiation and perturbed chromatin loading of RPA, Pol α, and PCNA without affecting the chromatin loading of Cdc45 and Pol ϵ, suggesting that Mcm10 is involved in origin unwinding downstream of the recruitment of Cdc45.
Results
Chromatin-associated Mcm10 interacts with Mcm2–7
We previously reported that Mcm10 associated with DNase I–resistant nuclear structures (31). However, we found that Mcm10 was extracted with benzonase, a potent nuclease that degrades all forms of DNA and RNA and is active even at low temperature (<16 °C). The extraction of Mcm10 by benzonase was temperature dependent, and Mcm10 was not extracted efficiently at 30 °C although chromosomes were digested to the mononucleosomal level by an unknown mechanism (data not shown). The nuclease treatment at 30 °C may denature Mcm10 proteins and enhance the hydrophobic interactions with nuclease-resistant structures. To confirm that fractionation was performed properly, the localization of several replication factors as well as lamin B1 was investigated (Fig. 1A). As demonstrated previously, more than half of the Mcm2, Mcm6, and PCNA were free in soluble fractions, whereas the remaining proteins were bound to chromatin and extracted by benzonase treatment (56, 57). On the other hand, lamin B1 was localized in nuclease-resistant structures.
Figure 1.
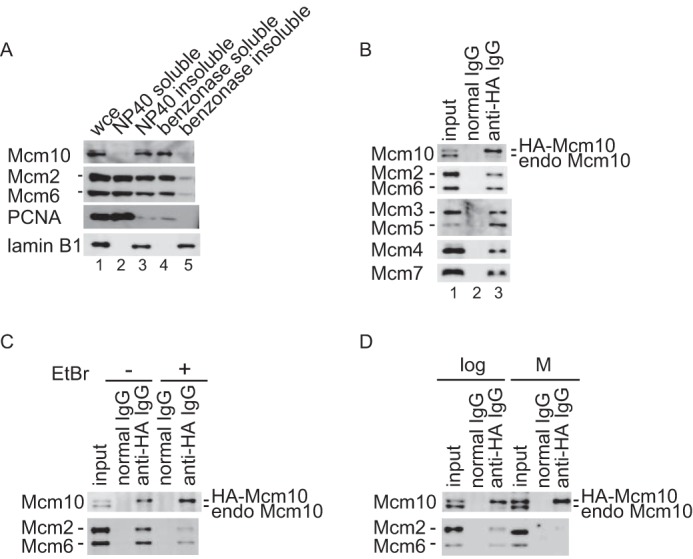
Subcellular distribution of Mcm10 and association with Mcm2–7 were studied. A, HeLa cells were first fractionated into Nonidet P-40-soluble (lane 2) and insoluble (lane 3) fractions, and then chromatin-bound fractions solubilized with benzonase digestion (lane 4) and benzonase-unextractable fraction (lane 5) were obtained from the Nonidet P-40-insoluble fraction. Whole cell extract was loaded onto lane 1. Subcellular localization of Mcm10, as well as several replication proteins, was investigated by immunoblotting. Mcm10 was mainly localized in the benzonase-soluble fraction. B, the benzonase-soluble fraction was prepared from a HeLa cell line that stably expressed HA-tagged Mcm10 and immunoprecipitated with control antibody (lane 2) or anti-HA antibody (lane 3). Precipitates were subjected to immunoblot analysis using anti-Mcm10 or Mcm2–7 antibodies. Lane 1 contained 12.5 or 3% of the input material for the detection of Mcm10 or Mcm2–7, respectively. Chromatin-associated human HA-Mcm10 was co-purified with the Mcm2–7 complex as well as a small amount of endogenous Mcm10. C, the benzonase-soluble fraction was prepared and immunoprecipitated with anti-HA antibody in the presence of ethidium bromide. Precipitates were subjected to immunoblot analysis using anti-Mcm10, Mcm2, and Mcm6 antibodies. The amount of co-purified Mcm2–7 was decreased substantially in the presence of ethidium bromide, suggesting that the DNA binding of either Mcm10 or Mcm2–7 facilitated the complex formation. D, the benzonase-soluble fractions from an asynchronous culture and the Nonidet P-40–soluble fractions from metaphase cells were prepared and immunoprecipitated with anti-HA antibody. Precipitates were subjected to immunoblot analysis. Mcm10 did not interact with Mcm2–7 at metaphase.
Next, we obtained a benzonase-soluble fraction from HeLa cells stably expressing HA-tagged Mcm10 and then purified chromatin-associated HA-Mcm10. Chromatin-associated HA-Mcm10 was co-purified with Mcm2–7 as described previously (33) (Fig. 1B). Additionally, a small amount of endogenous Mcm10 was co-purified with HA-Mcm10, suggesting that HA-Mcm10 forms a complex with endogenous Mcm10. We observed the reproducible co-immunoprecipitation with a substantial amount of Mcm2–7, but no co-immunoprecipitation was observed with components of the replisome such as And-1, PCNA, and Pol α (data not shown), which may reflect the lability of these interactions in solution.
To confirm that Mcm10 interacts with Mcm2–7 independently of DNA, the immunoprecipitation was performed in the presence of ethidium bromide to inhibit the interaction of proteins with double-stranded DNA (58). The amount of co-purified Mcm2–7 was decreased by 75% in the presence of ethidium bromide, suggesting that the DNA binding of either Mcm10 or Mcm2–7 cooperates for the interaction (Fig. 1C). To determine whether the interaction is regulated through the cell cycle, HA-Mcm10 was immunoprecipitated from either a benzonase-soluble fraction of cells that were growing asynchronously or a Nonidet P-40 soluble fraction of cells that had been arrested in metaphase as both Mcm10 and Mcm2–7 are released from chromatin at metaphase (Fig. 1D). The interaction between Mcm10 and Mcm2–7 was dependent on the cell cycle and was not observed at metaphase.
Identification of the Mcm10 domain that interacts with Mcm2–7
To determine whether a discrete part of Mcm10 is required for the interaction with the Mcm2–7 complex, a set of truncation mutants of HA-tagged Mcm10 was transiently expressed in HeLa cells and a co-immunoprecipitation assay was performed using Nonidet P-40-soluble fractions (Fig. 2). Mcm10 contains several domains: an N-terminal domain required for oligomerization, a conserved domain in the middle that interacts with DNA and Pol α, and a C-terminal domain that interacts with DNA, Pol α, Cdc45, and Sirt1 (Fig. 2A). We found that Mcm10 interacted with Mcm2–7 through the domain that contained amino acids 530–655, to which no functions have been attributed so far (Fig. 2, A and B). To examine whether Mcm10 interacts with Mcm2–7 directly, the GST-tagged mouse Mcm10(447–684) and the mouse Mcm4/6/7 complex were overexpressed in Escherichia coli and mixed in vitro. Mcm4/6/7 was co-immunoprecipitated with GST-tagged Mcm10(447–684), suggesting that Mcm10 and Mcm2–7 interact directly (Fig. 2C).
Figure 2.

The domain of Mcm10 required for the interaction with Mcm2–7 was mapped using a transient expression system. A, the domain structures and the summary of deletion derivatives of human Mcm10. The striped box indicates the N-terminal domain, which is involved in oligomerization. The gray box indicates the conserved domain among species. The hatched box indicates the C-terminal domain, which is involved in the interaction with DNA, Pol α, Cdc45, and Sirt1. Black lines indicate the mutants that interacted with Mcm2–7. Gray lines indicate the mutants that did not interact with Mcm2–7. The light gray box indicates the region required for the interaction with Mcm2–7. B, the soluble fractions were prepared from HeLa cells where HA-tagged truncated mutants were transiently overexpressed and immunoprecipitated with anti-HA antibody. Precipitates were subjected to immunoblot analysis using anti-HA, Mcm2, and Mcm6 antibodies. The asterisk indicates a nonspecific protein. Open circles indicate the immunoglobulin. C, Mcm10 interacts with the Mcm4/6/7 complex directly. Purified mouse Mcm4/6/7 complex and GST-tagged mouse Mcm10(447–684) were mixed in vitro and incubated with glutathione-Sepharose to precipitate the complex containing the mouse Mcm10 mutant. Precipitates were subjected to immunoblot analysis using anti-Mcm4 and GST antibodies.
To confirm that the interaction between overexpressed, truncated Mcm10 and Mcm2–7 in Nonidet P-40-soluble fractions is not an artifact, because a major fraction of Mcm10 associates with chromatin in vivo, we next investigated whether Mcm10 interacts with Mcm2–7 through the domain that contains amino acids 530–655 in vivo. We established HeLa cell lines expressing the HA-tagged truncated Mcm10 mutants (Fig. 3, A and B). The stable expression of truncated mutants did not have dominant negative effects on cell cycle progression (data not shown), indicating that the expression levels were insufficient to replace endogenous Mcm10. The cellular proteins were fractionated as described in Fig. 1, and the subcellular localization of proteins was examined (Fig. 3C). The truncated mutants Mcm10(200–482) and Mcm10(200–543) were not efficiently extracted from chromatin with benzonase, whereas the mutants Mcm10(200–655), Mcm10(200–693), and Mcm10(473–874) showed similar localization to the endogenous Mcm10. Alternatively, Mcm10(530–874) was primarily localized in the soluble fraction and a small fraction was extracted from chromatin with benzonase.
Figure 3.

The domain of Mcm10 required for the interaction with Mcm2–7 was mapped using a stable expression system. A, the domain structures and the summary of deletion derivatives of human Mcm10. Closed boxes indicate the mutants that interacted with Mcm2–7. Open boxes indicate the mutants that did not interact with Mcm2–7. The light gray box indicates the region required for the interaction with Mcm2–7. B, whole cell extracts were prepared from HeLa cell lines expressing HA-tagged truncated Mcm10, and HA-tagged mutants were detected by immunoblotting with anti-Mcm10 or HA antibody. Because the epitope for anti-Mcm10 antibody was amino acids 166–436, Mcm10(473–874) and Mcm10(530–874) were detected by anti-HA antibody. The open circle indicates the endogenous Mcm10 protein. Asterisks indicate the nonspecific proteins. C, the stable cell lines were fractionated as described in Fig. 1A, and subjected to immunoblotting. Mcm10(200–482) and Mcm10(200–543), which did not interact with Mcm2–7, were not efficiently extracted from chromatin with benzonase treatment. D, the benzonase-soluble fractions were prepared from stable cell lines and immunoprecipitated with anti-HA antibody. Precipitates were subjected to immunoblot analysis using anti-HA, Mcm2, and Mcm6 antibodies. The interaction with Mcm2–7 required the domain that contained amino acids 530–655. Lane 1 contained 12.5 or 3% of the input material for the detection of Mcm10 or Mcm2/6, respectively.
Next, we examined whether the truncated mutants were co-immunoprecipitated with Mcm2–7 (Fig. 3D). The truncated mutants Mcm10(200–482) and Mcm10(200–543) were not co-purified with Mcm2–7. In contrast, Mcm10(200–655), Mcm10(200–693), and Mcm10(473–874) were co-purified with Mcm2–7. Mcm10(530–874) was also co-purified with Mcm2–7 but with lower affinity. Taken together, the interaction with Mcm2–7 in vivo required the domain that contained amino acids 530–655. Additionally, the efficient extraction of Mcm10 by benzonase seems to require the Mcm2–7-interacting domain (Fig. 3C), suggesting that the interaction with Mcm2–7 is important for proper nuclear localization of Mcm10.
To confirm that the interaction with Mcm2–7 is important for the localization of Mcm10, we examined whether the depletion of Mcm2–7 affects the extraction efficiency of Mcm10 by benzonase digestion (supplemental Fig. S1A). Mcm6-depleted cells proliferated normally, whereas Mcm2-depleted cells proliferated at a slightly slower pace at 72 h after siRNA transfection (data not shown), suggesting that the knockdown of Mcm2 or Mcm6 still permitted the formation of sufficient amount of Mcm2–7 to support DNA replication. We synchronized cells at the G1/S boundary because Mcm2-depleted cells show a slightly increased G1 and G2 population (59) and the protein level and the subcellular localization of Mcm10 are dependent on the cell cycle (60) (supplemental Fig. S1B). Then the cellular proteins were fractionated as described in Fig. 1A, and the subcellular localization of proteins was examined by immunoblotting (supplemental Fig. S1C). Mcm10 was efficiently extracted from Nonidet P-40-insoluble fraction with benzonase treatment in mock- or control siRNA-transfected cells. Conversely, more than half of Mcm10 was localized in benzonase-unextractable fraction in Mcm2–7-depleted cells. These results also suggest that the interaction with Mcm2–7 is necessary for proper nuclear localization of Mcm10.
Depletion of Mcm10 perturbs new origin firing
To explore the functions of Mcm10, we depleted Mcm10 using siRNAs in HeLa cells. We used three independent siRNAs designed for different sequences of the open reading frame (Fig. 4A), and a significant reduction in the amount of Mcm10 was observed 24 h after transfection (down to less than 10% of the original level, Fig. 4B). We first examined whether down-regulation of Mcm10 inhibited S phase progression (Fig. 4C). The population of bromodeoxyuridine (BrdU)-positive cells increased slightly in the first 24 h after transfection, which was caused by prolonged S phase as described below, and then decreased at a later time point.
Figure 4.

Knockdown of Mcm10 results in the delay of S phase progression. A, domain structures of human Mcm10 and the positions of the target sequence of siRNAs (arrowheads #1–#3). B, nonspecific control or Mcm10 siRNAs were transfected into HeLa cells (HeLa, upper panel) or HeLa cells expressing the HA-tagged siRNA-resistant form of Mcm10 (siRNA-resistant HeLa, lower panel) for 24 h. Whole cell extracts were prepared and subjected to immunoblotting for the expression of Mcm10 and lamin B1 as a loading control. C, control or Mcm10 siRNAs were transfected into an asynchronous population of HeLa cells. To monitor S phase progression, cells were pulse-labeled with BrdU at indicated time points. D, HeLa cells transfected with either control siRNA or Mcm10 siRNA were released from the G1/S boundary, and DNA synthesis was estimated by BrdU incorporation. E, HeLa cells were synchronized at the G1/S boundary after siRNA transfection and then released into S phase. Whole cell extracts (wce) and Triton-insoluble fractions (ppt) were prepared 1 h from the G1/S boundary and subjected to immunoblotting. Depletion of Mcm10 by siRNA slowed S phase progression and interfered with chromatin loading of RPA, Pol α, and PCNA (indicated by asterisks), whereas loading of Cdc45 and Pol ϵ were not affected.
Next, we depleted Mcm10 in synchronous HeLa cells. HeLa cells were transfected with siRNAs and synchronized at the G1/S boundary by a sequential excess thymidine-aphidicolin block. Cells were then released into S phase, and DNA replication was analyzed by immunostaining of the incorporated BrdU (Fig. 4D, upper panel). Fifty percent of the Mcm10-depleted cells completed DNA replication 13 h after entering S phase, whereas 50% of control cells completed DNA replication within 8 h. To confirm that the prolonged S phase was because of the depletion of endogenous Mcm10, we examined whether expression of the siRNA-resistant Mcm10 mutant protein could restore S phase progression. We introduced silent mutations into the target sequences of Mcm10 and established cell lines that stably expressed siRNA-resistant Mcm10. As expected, the expression of Mcm10 siRNA-resistant mutant protein was unaffected in Mcm10 siRNA-transfected cells (Fig. 4B, lower panel). The transfection of Mcm10 siRNAs failed to slow S phase progression in the cell line expressing siRNA-resistant Mcm10 (Fig. 4D, lower panel), suggesting that the delay in S phase progression was caused by the depletion of Mcm10, not by the off-target effects of Mcm10-specific siRNA or the cytotoxic effects of transfection.
To determine whether the depletion of Mcm10 affected other replication factors, several replication proteins in whole cell extracts and Triton-insoluble chromatin fractions were analyzed by immunoblotting (Fig. 4E). Whole cell extracts and Triton-insoluble fractions were prepared from synchronized cells 1 h after release from the aphidicolin block, the time point at which the replication factors for elongation (PCNA, RPA, Cdc45, and Pols) should be sufficiently loaded onto chromatin. We did not detect a down-regulation of Pol α as previously reported (32). When Triton-insoluble fractions were examined, the chromatin loading of pre-RC proteins (Orc2 and Mcm7) was unaffected. Additionally, the chromatin loading of Dbf4, Cdc45, and Pol ϵ, which are assembled at origins before unwinding, was unaffected. On the other hand, RPA level in chromatin-bound fraction was reduced by 60% in Mcm10-depleted cells, suggesting that origin unwinding was inhibited. Moreover, the levels of PCNA and Pol α in chromatin-bound fractions, which are recruited during elongation of DNA replication, were reduced by 70 and 60% in Mcm10-depleted cells, respectively.
To address whether the inhibition of S phase progression was a consequence of a defect in DNA initiation and/or slowed elongation, a DNA fiber assay was performed to estimate the rate of replication fork migration (Fig. 5). Cells were pulse-labeled sequentially with iododeoxyuridine (IdU) and chlorodeoxyuridine (CldU), and the labeled DNA fiber spread on a glass slide was detected with specific antibodies as described in the “Experimental Procedures” section. As shown in Fig. 5A, the distribution of labeled track length as well as the average length did not change after Mcm10 knockdown. Based on a conversion factor of 1 μm = 2.59 kb (61), replication forks grew at an average rate of 2.1 kb/min, which is in agreement with measurements reported by others (61).
Figure 5.

Depletion of Mcm10 in S phase results in prolonged DNA synthesis and inhibits new origin firing. HeLa cells were synchronized at the G1/S boundary 24 h after siRNA transfection and then released into S phase. Cells were then labeled sequentially with IdU and CldU. DNA fibers were spread onto glass slides and immunostained with anti-IdU and CldU antibodies. The frequency histograms show the distribution of the track length (A) or inter-origin distance (B) in cells transfected with each siRNA. Mean values ± S.D. are indicated.
We next performed a DNA combing assay to examine whether Mcm10 depletion affects origin firing. The inter-origin distance increased in Mcm10-depleted cells, suggesting that Mcm10 depletion perturbs origin firing (Fig. 5B). Although the length of S phase was nearly double in Mcm10-depleted cells, the average inter-origin distance was increased by only 30% in those cells. This may be because of the limit of this method; the distance spanning more than a few hundred kilobases of DNA is difficult to detect, which leads to an underestimation. These results suggest that prolonged S phase in Mcm10-depleted cells was caused by the reduction of initiation frequency, not by the slowed fork rate.
Knockdown of endogenous Mcm10 was rescued by the truncated mutants of Mcm10
Next, we established additional cell lines expressing HA-tagged truncated Mcm10 mutants and performed knockdown and replacement studies to analyze the contribution of the Mcm2–7-interacting domain to S phase progression (Fig. 6, A and B). The knockdown of endogenous Mcm10 was confirmed by immunoblotting (Fig. 6C), and the S phase progression was monitored by BrdU incorporation (Fig. 6D). Leptomycin B was added to the cell line expressing Mcm10(1–482) and Mcm10(1–693) in S phase to induce the nuclear transport of the mutant protein because these mutants were mainly localized in cytoplasm because of the leucine-rich nuclear export signal in the N-terminal region (amino acids 126–135, MKALQEQLKV) (supplemental Fig. S2). The typical nuclear export signal consists of four spaced hydrophobic residues (Φ1-Φ4) and follows the consensus sequence Φ1-X2-3-Φ2-X2-3-Φ3-X-Φ4, where X2-3 represents any two or three amino acids. Four critical hydrophobic (Φ) residues are underlined. The Mcm10 knockdown phenotype was rescued by exogenous expression of Mcm10(1–693), Mcm10(200–874), Mcm10(200–655), and Mcm10(200–693), indicating that the N-terminal (1–200) and C-terminal (655–874) domains were not essential for DNA replication. In contrast, Mcm10(Δ165–437) and Mcm10(473–874), which lacked the conserved domain (200–473), could not rescue the knockdown of endogenous Mcm10 at all. Mcm10(1–482), Mcm10(200–482), and Mcm10(200–543), which lack the Mcm2–7 binding domain, could partially rescue the knockdown phenotype. Therefore, both the conserved domain and the Mcm2–7 binding domain were necessary for full activity of Mcm10, and the Mcm2–7 binding domain seems to function as an auxiliary domain for DNA replication.
Figure 6.

Knockdown and replacement studies of Mcm10 are shown. A, the domain structures and summary of deletion derivatives of human Mcm10. Closed boxes or gray boxes indicate the mutants that complemented the knockdown of endogenous Mcm10 completely (++) or partially (+), respectively. Open boxes indicate the mutants that did not complement Mcm10 function. B, whole cell extracts from stably transfected HeLa cell lines were prepared and subjected to immunoblotting. Mutant Mcm10 proteins were detected by anti-HA or anti-Mcm10 antibody. The open circle indicates the endogenous Mcm10. Asterisks indicate nonspecific proteins. C, HeLa cells expressing the HA-tagged truncated Mcm10 were transfected with nonspecific control or Mcm10 siRNAs and synchronized at the G1/S boundary 24 h after siRNA treatment. Whole cell extracts were prepared and subjected to immunoblotting for the expression of endogenous Mcm10, HA-tagged Mcm10 mutants, and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as a loading control. D, HeLa cells transfected with either control siRNA or Mcm10 siRNA were released from the G1/S boundary, and DNA synthesis was monitored by BrdU incorporation.
Localization of GFP-Mcm10 mutants in S phase
Fractionation analysis showed that the Mcm2–7-interacting domain is correlated with efficient extraction from chromatin using benzonase (Fig. 3C and supplemental Fig. S1C). Next, we tried to detect the subcellular localization of HA-Mcm10 mutants in HeLa cells. However, several mutants were difficult to detect with immunofluorescence staining, which may be because of epitope masking. Therefore, we established cell lines expressing GFP-tagged Mcm10 mutants (Fig. 7A). We first examined the localization of mutant Mcm10 proteins in an asynchronous cell population without Triton extraction (Fig. 7B, left panels). The mutants Mcm10(1–482) and Mcm10(1–693) were localized in the cytoplasm because of the nuclear export signal. The mutants Mcm10(200–874), Mcm10(438–874), and Mcm10(Δ166–436) were localized in the nucleus because of the authentic nuclear import signal (amino acids 687–760), which seemed to dominate the nuclear export signal. The mutants Mcm10(200–482) and Mcm10(200–655) were also localized in the nucleus, which may be because of another cryptic nuclear import signal(s) or interaction with component(s) in the nucleus. The distribution of mutant Mcm10 proteins in the nucleus was relatively homogeneous.
Figure 7.

The segments of Mcm10 required for the stable association with chromatin were mapped. A, the whole cell extracts were prepared from HeLa cells expressing GFP-tagged truncated Mcm10 mutants and subjected to immunoblot analysis using anti-Mcm10 or anti-GFP antibody. The open circle indicates the endogenous Mcm10. Asterisks indicate nonspecific proteins. B, HeLa cells that stably expressed GFP-Mcm10 were synchronized at the G1/S boundary and released into S phase. Cells were extracted with 0.1% Triton X-100 for 10 min and fixed with 4% paraformaldehyde at 0 (early S phase) or 4.5 h (mid S phase) from G1/S boundary. DNA was stained with DAPI, and the cells were observed by fluorescence microscopy.
A different distribution was observed when the cells were extracted with 0.1% Triton X-100 before fixation (Fig. 7B, right panels). The mutant Mcm10 constructs capable of interacting with Mcm2–7, Mcm10(200–874), Mcm10(438–874), Mcm10(200–655), and Mcm10(Δ166–436) were retained in the nucleus after extraction and observed in discrete replication foci. The distribution patterns of mutants were similar to those of the full-length Mcm10. In addition, these mutants showed a similar transition in distribution pattern with the progression of S phase. Unexpectedly, a small fraction of Mcm10(1–693) was also observed in similar nuclear foci after extraction, whereas a major fraction was found in the cytoplasm. It may be that a fraction of Mcm10(1–693) was retained in insoluble nuclear foci after being transported into the nucleus through a piggyback mechanism or shuttling between the nucleus and cytoplasm, because Mcm10 possesses both nuclear localization and export signals. In contrast, the fluorescence signals of Mcm10(1–482) disappeared after extraction. On the other hand, Mcm10(200–482) was mainly localized in the nucleoli. Therefore, the Mcm2–7-interacting domain is necessary for stable nuclear retention of Mcm10 at replication foci.
Discussion
Although the interaction between Mcm10 and Mcm2–7 has been observed in various species (23, 31, 62, 63), the physiological meaning of the interaction and the exact roles of Mcm10 in replication remain controversial. Recent studies showed that budding and fission yeast Mcm10 interacts with Mcm2–7 via the C-terminal domain, which in turn is essential for DNA unwinding at the origins (54, 55). However, the Mcm2–7-interacting domain in yeast is not conserved in higher eukaryotes. In the present study, we identified the Mcm2–7-interacting domain of human Mcm10 and found that it was important for Mcm10 function.
The chromatin-associated Mcm10 preferentially bound to Mcm2–7 in HeLa cells. Mcm10 interacted with Mcm2–7 in the absence of DNA; however, its interaction was stimulated by DNA. It is suggested that monomeric budding yeast Mcm10 binds to dsDNA cooperatively (25), which may be induced by protein-protein interactions or a conformational change in proximal DNA for which Mcm10 may have an enhanced affinity. Additionally, the interaction of budding yeast Mcm10 with ssDNA induces the trimer formation (25). Therefore, the oligomer formation or conformational change of DNA-bound Mcm10 may facilitate the interaction with Mcm2–7.
A small fraction of endogenous Mcm10 was co-immunoprecipitated with HA-tagged Mcm10 as well, indicating that Mcm10 oligomerizes in vivo. Whereas HA-Mcm10 and endogenous Mcm10 were detected in the cell extract with similar amounts, the amount of co-purified endogenous Mcm10 was much lower than that of HA-Mcm10, suggesting that only a small fraction of Mcm10 forms oligomer in vivo. It is possible that Mcm10 exists mainly as a monomer and forms multimeric configurations in response to its environment. For example, upon pre-RC activation, Mcm10 may oligomerize to stabilize the interaction with other replication factors or facilitate strand unwinding. The interaction with various replication factors and the lack of enzymatic activity suggest that Mcm10 serves as a scaffold to orchestrate protein and DNA interactions within the replisome. Oligomerization would provide multiple platforms to associate with various replication factors and DNA.
Human Mcm10 interacted with Mcm2–7 through the amino acids 530–655, which are not present in budding yeast and fission yeast. The conservation from yeast to human is limited to the central domain of Mcm10, although the internal domain and the Mcm2–7-interacting domain are separated by over 100 amino acids in both yeasts and human as a common feature. Unexpectedly, no structural motifs or little secondary structure was predicted for the Mcm2–7-interacting domain of human Mcm10. This region has an amino acid compositional bias, with a low content of hydrophobic amino acids and a high content of particular polar and charged amino acids, which is typical for intrinsically disordered proteins (IDPs) (64). Contrary to the traditional view that protein function requires a stable three-dimensional structure, recent studies reveal that disordered regions are often functional and fold upon binding to their biological target. Furthermore, some IDPs can bind several different targets, and it has been suggested that IDPs adopt different structures (65). Although further spectrometric methods such as NMR or CD spectrum are necessary to determine whether the Mcm2–7-interacting domain has a disordered structure, it is intriguing to examine whether this domain can interact with other replication factors or whether post-translation modification affects the structure because the interaction between Mcm10 and Mcm2–7 was dependent on the cell cycle. In general, the disordered region evolves significantly more rapidly than the ordered region (66), which may explain why this region is not highly conserved even among higher eukaryotes.
The Mcm2–7-interacting domain overlapped with the domain required for the stable retention of chromatin in S phase progression and the efficient extraction from chromatin with nuclease digestion. Furthermore, depletion of Mcm2–7 with siRNA reduced the extraction efficiency of Mcm10 with nuclease digestion. Taken together, Mcm10 is likely bound onto chromatin through the interaction with Mcm2–7, whereas the central conserved domain (amino acids 236–435) and the C-terminal conserved domain (amino acids 700–860), which have DNA binding activity (48, 49), appear to be insufficient for chromatin binding.
To define the function of Mcm10, we performed knockdown analysis using siRNA. Mcm10 depletion slowed S phase, but did not inhibit DNA replication completely, probably because Mcm10 was highly abundant (67) and the residual proteins that escaped knockdown by siRNA supported DNA replication. The depletion of Mcm10 reduced the frequency of replication initiation, whereas the fork velocity of each replication fork remained unchanged. The chromatin loading of Dbf4, Cdc45, Pol ϵ, and pre-RC components was not affected in Mcm10-depleted cells, whereas the chromatin loading of RPA, Pol α, and PCNA, which are loaded after origin unwinding, was inhibited in Mcm10-depleted cells. Similar results were obtained in budding and fission yeast using degron mutants (42–44). Our results are also consistent with the recent in vitro observation that chromatin loading of Cdc45 is independent of Mcm10 (7, 8). Therefore, our results favor the hypothesis that Mcm10 primarily interacts with Mcm2–7 and is involved in a step during origin unwinding after formation of the CMG complex. However, we cannot rule out the possibility that Mcm10 is also involved in elongation. For example, Mcm10 may recruit Pol α or PCNA to the replication fork because Mcm10 physically interacts with these proteins (26, 27). Alternatively, Mcm10 may affect the stability of CMG complex and stimulate elongation as reported recently (68).
Contrary to our DNA fiber assay results, Fatoba et al. reported that the inter-origin distances slightly decreased when Mcm10 was knocked down in HCT116 cells, whereas fork velocities did not change in those cells (36). In their report, DNA replication was analyzed 48 h after siRNA transfection without synchronization. They also found that Mcm10 knockdown produced a significant decrease (>2-fold) in the number of S phase cells and accumulation of the phosphorylated form of histone H2AX. Therefore, the reduction of inter-origin distance may result from the activation of dormant origins, which is not a direct consequence of Mcm10 depletion but rather a secondary event caused by activation of the DNA-damage checkpoint pathway.
We also investigated the functions of domains using replacement analysis and found that either the N-terminal (1–200) or C-terminal (693–874) domain was dispensable for S phase progression. Alternatively, the residual amount of Mcm10 in our system could support the function of the N-terminal or C-terminal domain. On the other hand, the internal conserved domain (200–498) was essential for S phase progression, whereas the Mcm2–7-interacting domain was important for supporting the efficient firing of replication origins. Because the internal conserved domain could only partially rescue the knockdown of endogenous Mcm10, a stable interaction with chromatin did not appear critical for S phase progression.
Mcm10 is known to interact with several subunits of Mcm2–7 proteins in budding yeast (23, 41, 55, 68). We have also found that human Mcm10 interacts with Mcm2 and Mcm6 with higher affinity compared with other subunits of the Mcm2–7 complex in a yeast two-hybrid screen (31). Further investigation is necessary to determine the molecular architecture of the interaction between Mcm10 and Mcm2–7. Budding yeast Mcm10 specifically associates with double hexameric Mcm2–7, but not soluble single hexamers (54), whereas Mcm10 interacts with single hexamers in vitro (41). Our results also showed that human Mcm10 interacted with soluble Mcm2–7 when overexpressed (Fig. 3). Other replication factors, which interact with Mcm10, might ensure that Mcm10 associates specifically with the double hexameric Mcm2–7 in vivo. In our previous report, Mcm10 is recruited to the replication sites 30–60 min before they replicate in mammalian cells (67). It would be interesting to elucidate how the interaction with Mcm2–7 is temporally and spatially regulated throughout S phase and whether DDK or S-CDK is involved in the interaction.
Experimental procedures
Plasmid construction
To construct pHA-IREShyg and pGFP-IREShyg, the HA epitope tag and GFP tag from either ptetHA11 (67) or ptetGFP (69) was amplified by PCR using the primers 5′-ATT AGG ATC CAG CCT CCG CCT AGC-3′ and 5′-CTA CAA ATG TGG TAT GGC TG-3′, digested with BamHI, and subcloned into the compatible site of pIREShyg (Clontech). To construct a series of deletion constructs, the appropriate region of human Mcm10 cDNA was amplified by PCR using primers containing the SmaI site and a stop codon, digested with SmaI, and then subcloned into the compatible site of pHA-IREShyg or pGFP-IREShyg. Sequence analysis confirmed that no mutations had been introduced into the amplified sequence during PCR. An siRNA-resistant mutant cDNA was generated by introducing silent mutations at codons Ile-217 (ATT→ATC), Ser-218 (TCT→TCA), Arg-219 (CGG→AGA), Leu-378 (TTA→CTG), Ile-379 (ATT→ATC), Gly-381 (GGT→GGA), Ser-487 (AGT→AGC), Asn-488 (AAT→AAC), and Leu-489 (CTG→TTA) using the QuikChange Site-Directed Mutagenesis Kit (Agilent Technologies), and specific mutations were confirmed by sequencing. To construct the mouse expression vector, Mcm10 was amplified by PCR using a mouse embryonic cDNA library (Clontech) with the forward primer 5′-GGT GAT CAT GGA TGT GGA GGA AGA CGA CT-3′ and reverse primer 5′-CCG TCG ACT TGA GGC TGT TCA GAA ACT TG-3′. The PCR fragment was subcloned into the vector pSKB4 as described (70).
Cell culture and siRNA transfection
HeLa cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% calf serum. To establish stable cell lines expressing HA- or GFP-tagged Mcm10 mutants, the expression vectors were linearized with SspI and transfected into HeLa cells using Lipofectamine 2000 reagent (Thermo Fisher) according to the manufacturer's protocol. Stably transformed cell lines were selected with 400 μg/ml hygromycin B and maintained as described previously (67).
Stealth siRNAs (Thermo Fisher) were synthesized against the following target sequences: #1, 5′-GGT CTT AAT TAT GGG TGA AGC TCT T-3′; #2, 5′-CCA CTC TGA GTA ATC TGG TTG TTA A-3′; and #3, 5′-GAC GAT TTC TCG GAA CAA A-3′. For siRNA transfection using an asynchronous cell population, cells plated at 104 cells per well of 24-well dishes were grown for 24 h. Then, cells were transfected with 100 nm Stealth RNAi with RNAiMax (Thermo Fisher) according to the manufacture's protocol for 24 h. For siRNA transfection using a synchronous cell population, cells plated at 2 × 104 cells per well of 6-well dishes or at 5 × 103 cells per well of 24-well dishes were grown for 24 h. Then, the cells were treated with 2.5 mm thymidine for 14 h, washed twice with fresh medium, and transfected with 100 nm Stealth RNAi with RNAiMax (Thermo Fisher) according to the manufacture's protocol. Ten h later, aphidicolin was added at 15 μm and incubated for another 14 h to synchronize cells at the G1/S boundary. Stealth RNAi Negative Control Low GC Duplexes (Thermo Fisher) were used as a control. To monitor DNA synthesis, cells were pulse-labeled with BrdU for 10 min, and incorporated BrdU was detected by immunofluorescence as described (67). In knockdown studies using HA-Mcm10(1–482) or HA-Mcm10(1–693), leptomycin B (10 nm) was added to cells after release from the G1/S boundary because these mutants were primarily localized in the cytoplasm due to the nuclear export signal.
Fractionation of cellular proteins and co-immunoprecipitation analysis
Whole cell extracts were prepared as described previously (31). To obtain nuclease-soluble fraction from stable cell lines expressing HA-tagged Mcm10 mutant proteins, 2 × 106 cells were lysed in 200 μl of lysis buffer containing 25 mm Hepes, pH 7, 0.1 m NaCl, 2.5 mm EGTA, 3 mm MgCl2, 1 mm DTT, 0.1% Nonidet P-40, 10 mm NaF, 0.25 mm Na3VO4, 10% glycerol, and 2× cOmpleteTM (Roche) at 4 °C for 10 min and subjected to low-speed centrifugation (700 × g for 2 min). After Nonidet P-40–extracted nuclei were washed once with lysis buffer, DNA and RNA were digested with 0.25 unit/μl benzonase (Millipore) in lysis buffer at 10 °C for 1 h. The sample was cleared by centrifugation at 700 × g for 5 min, and the supernatant containing the solubilized chromatin proteins was obtained. The chromatin proteins were then incubated with 1 μg of anti-HA rabbit antibody (Medical & Biological Laboratories Co., Ltd.) or anti-HA rat monoclonal antibody (Roche, 3F10) at 4 °C for 1 h, after which 10 μl of Protein G Sepharose 4 Fast Flow (GE Healthcare) was added and incubated for an additional 1 h. As a control, normal rabbit antibody or normal rat antibody was used (Jackson ImmunoResearch Laboratories). The beads were washed five times with lysis buffer, and the precipitates were dissolved in 40 μl of 2× SDS-PAGE sample buffer and subjected to immunoblotting.
For immunoprecipitation from HeLa cells where mutant proteins were transiently expressed, HeLa cells were plated at 106 cells in 6-cm dishes and transfected with pIREShyg expressing HA-tagged mutant Mcm10 using Lipofectamine 2000 Reagent (Thermo Fisher) according to the manufacturer's protocol for 24 h. The cells were then lysed in 200 μl of lysis buffer at 4 °C for 10 min and centrifuged at 3000 rpm for 5 min. The lysates were subjected to immunoprecipitation as described above. For chromatin fractionation, shown in Fig. 4E, chromatin-unbound and -bound proteins were obtained as described previously (60). GST-tagged mouse Mcm10 and mouse Mcm4/6/7 were overproduced in the E. coli strain BL21 (DE3) and purified as described (70). The GST-pulldown assay was carried out as described (70).
Protein bands were detected using the chemiluminescent reagent SuperSignal (Thermo Fisher) and analyzed by a luminoimage LAS-3000 analyzer (GE Healthcare). The anti-human Mcm10 and Mcm3 antibodies used were described previously (31, 71). Anti-Orc2 (no. M055–3) and anti-HA rabbit (no. 561–5) antibodies were purchased from Medical & Biological Laboratories. Anti-replication protein A (no. NA19L) antibody was purchased from Oncogene Research Products. Anti-Mcm2 (no. 9839), anti-Mcm4 (no. 48407), anti-Mcm5 (no. 22780), anti-Mcm6 (no. 9843), anti-Mcm7 (no. 9966), anti-Cdc45 (no. 20685), anti-PCNA (no. 56), anti-Pol α (no. 5921), anti-RFC (no. 20993), anti-lamin B1 (no. 6216), anti-glyceraldehyde 3-phosphate dehydrogenase (no. 20357), and anti-GFP (no. 8334) antibodies were purchased from Santa Cruz Biotechnology. Anti-Dbf4 (no. ab70134) and anti-Pol ϵ (no. ab104) were purchased from Abcam.
DNA fiber assay
Cells were pulse-labeled with 20 μm IdU for 20 min, washed twice with PBS, and then labeled with 20 μm CldU for 15 min. DNA was spread onto glass slides as described previously (61). After DNA was depurinated with 2 n HCl for 30 min, IdU-labeled tracks were detected with anti-BrdU antibody (no. 347580; BD Biosciences) and FITC-conjugated anti-mouse IgG (no. 715–096-151; Jackson ImmunoResearch Laboratories). CldU-labeled tracks were detected with anti-BrdU antibody (no. OBT0030; Oxford Biotechnology Ltd.) and Texas Red-conjugated anti-rat IgG (no. 712–076-153; Jackson ImmunoResearch Laboratories). Images of labeled DNA fibers were acquired using an Olympus AX70 fluorescent microscope equipped with a 100× UplanApo (1.35 NA) oil-immersion objective. The track length was measured using IP Lab software (BD Biosciences) and converted to kilobase pairs using a conversion factor of 1 μm = 2.59 kb (61).
DNA combing assay
Cells were pulse-labeled with 100 μm IdU for 20 min, washed twice with PBS, and then labeled with 100 μm CldU for 20 min. Genomic DNA was prepared and combed onto the silanated coverslips (Matsunami Glass) as described previously (72). After DNA was denatured with formamide, IdU-labeled DNA was detected with mouse anti-BrdU monoclonal antibody (BD Biosciences) and Alexa Fluor 555-conjugated goat anti-mouse IgG (Thermo Fisher). CldU-labeled DNA was detected with rat anti-BrdU monoclonal antibody (Oxford Biotechnology) and Alexa Fluor 488-conjugated rabbit anti-rat IgG (Thermo Fisher). Images of labeled DNA fibers were acquired using a Zeiss Axioplan 2 MOT equipped with a 63× Plan Apochromat (NA 1.4) objective. Fluorescent signals were measured by using MetaMorph version 6.1 software (MDS Analytical Technologies) and converted to kilobase pairs using a conversion factor of 1 μm = 2.32 ± 0.11 kbp (72).
Fluorescence microscopy
Cells grown on coverslips were washed three times with ice-cold PBS and fixed with 4% paraformaldehyde at 4 °C for 15 min. In experiments in which cells were first extracted, cells were washed three times with ice-cold PBS and then incubated in the buffer containing 25 mm Hepes, pH 7.5, 0.1 m NaCl, 2.5 mm EGTA, 3 mm MgCl2, 1 mm DTT, 0.1% Nonidet P-40, 10 mm NaF, 0.25 mm Na3VO4, 10% glycerol, and 2× cOmplete (Roche) at 4 °C for 10 min before fixation. GFP-tagged Mcm10 mutants were visualized using a Zeiss microscope equipped with a 60× oil-immersion objective and ApoTome system. Images were taken with a CCD camera and assembled using ZEN software.
Author contributions
M. I., T. M., and F. H. conceived and designed the study. M. I. conducted most of the experiments and wrote the paper. T. M. and K. Y. performed GST-pulldown assays. K. S. and K. O. performed DNA combing assays. N. I. and T. A. provided the helpful advice. F. H. wrote the paper and coordinated the study. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Dr. Bruce Stillman for the kind gift of the anti-Mcm3 antibody, the Support Unit for Bio-Material Analysis, and RIKEN BSI Research Resources Center for DNA sequencing.
This work was supported by grants from the Ministry of Education, Culture, Sports, Science, and Technology of Japan and the Hayashi Memorial Foundation for Female Natural Scientists. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental “Experimental Procedures” and reference and supplemental Figs. S1 and S2.
- pre-RC
- pre-replication complex
- Mcm
- mini-chromosome maintenance
- Pol
- DNA polymerase
- DDK
- Cdc7/Dbf4 kinase
- S-Cdks
- S phase cyclin-dependent kinases
- RPA
- replication protein A
- PCNA
- proliferating cell nuclear antigen
- BrdU
- bromodeoxyuridine
- CldU
- chlorodeoxyuridine
- IdU
- iododeoxyuridine.
References
- 1. Masai H., Matsumoto S., You Z., Yoshizawa-Sugata N., and Oda M. (2010) Eukaryotic chromosome DNA replication: Where, when, and how? Annu. Rev. Biochem. 79, 89–130 [DOI] [PubMed] [Google Scholar]
- 2. Cocker J. H., Piatti S., Santocanale C., Nasmyth K., and Diffley J. F. (1996) An essential role for the Cdc6 protein in forming the pre-replicative complexes of budding yeast. Nature 379, 180–182 [DOI] [PubMed] [Google Scholar]
- 3. Coleman T. R., Carpenter P. B., and Dunphy W. G. (1996) The Xenopus Cdc6 protein is essential for the initiation of a single round of DNA replication in cell-free extracts. Cell 87, 53–63 [DOI] [PubMed] [Google Scholar]
- 4. Nishitani H., Lygerou Z., Nishimoto T., and Nurse P. (2000) The Cdt1 protein is required to license DNA for replication in fission yeast. Nature 404, 625–628 [DOI] [PubMed] [Google Scholar]
- 5. Evrin C., Clarke P., Zech J., Lurz R., Sun J., Uhle S., Li H., Stillman B., and Speck C. (2009) A double-hexameric MCM2–7 complex is loaded onto origin DNA during licensing of eukaryotic DNA replication. Proc. Natl. Acad. Sci. U.S.A. 106, 20240–20245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Remus D., Beuron F., Tolun G., Griffith J. D., Morris E. P., and Diffley J. F. (2009) Concerted loading of Mcm2–7 double hexamers around DNA during DNA replication origin licensing. Cell 139, 719–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Heller R. C., Kang S., Lam W. M., Chen S., Chan C. S., and Bell S. P. (2011) Eukaryotic origin-dependent DNA replication in vitro reveals sequential action of DDK and S-CDK kinases. Cell 146, 80–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yeeles J. T., Deegan T. D., Janska A., Early A., and Diffley J. F. (2015) Regulated eukaryotic DNA replication origin firing with purified proteins. Nature 519, 431–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Masai H., Taniyama C., Ogino K., Matsui E., Kakusho N., Matsumoto S., Kim J. M., Ishii A., Tanaka T., Kobayashi T., Tamai K., Ohtani K., and Arai K. (2006) Phosphorylation of MCM4 by Cdc7 kinase facilitates its interaction with Cdc45 on the chromatin. J. Biol. Chem. 281, 39249–39261 [DOI] [PubMed] [Google Scholar]
- 10. Sheu Y. J., and Stillman B. (2006) Cdc7-Dbf4 phosphorylates MCM proteins via a docking site-mediated mechanism to promote S phase progression. Mol. Cell 24, 101–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Deegan T. D., Yeeles J. T., and Diffley J. F. (2016) Phosphopeptide binding by Sld3 links Dbf4-dependent kinase to MCM replicative helicase activation. EMBO J. 35, 961–973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tanaka S., Umemori T., Hirai K., Muramatsu S., Kamimura Y., and Araki H. (2007) CDK-dependent phosphorylation of Sld2 and Sld3 initiates DNA replication in budding yeast. Nature 445, 328–332 [DOI] [PubMed] [Google Scholar]
- 13. Zegerman P., and Diffley J. F. (2007) Phosphorylation of Sld2 and Sld3 by cyclin-dependent kinases promotes DNA replication in budding yeast. Nature 445, 281–285 [DOI] [PubMed] [Google Scholar]
- 14. Muramatsu S., Hirai K., Tak Y. S., Kamimura Y., and Araki H. (2010) CDK-dependent complex formation between replication proteins Dpb11, Sld2, Pol ϵ, and GINS in budding yeast. Genes Dev. 24, 602–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Moyer S. E., Lewis P. W., and Botchan M. R. (2006) Isolation of the Cdc45/Mcm2–7/GINS (CMG) complex, a candidate for the eukaryotic DNA replication fork helicase. Proc. Natl. Acad. Sci. U.S.A. 103, 10236–10241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pacek M., Tutter A. V., Kubota Y., Takisawa H., and Walter J. C. (2006) Localization of MCM2–7, Cdc45, and GINS to the site of DNA unwinding during eukaryotic DNA replication. Mol. Cell 21, 581–587 [DOI] [PubMed] [Google Scholar]
- 17. Ilves I., Petojevic T., Pesavento J. J., and Botchan M. R. (2010) Activation of the MCM2–7 helicase by association with Cdc45 and GINS proteins. Mol. Cell 37, 247–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mimura S., and Takisawa H. (1998) Xenopus Cdc45-dependent loading of DNA polymerase α onto chromatin under the control of S-phase Cdk. EMBO J. 17, 5699–5707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Walter J., and Newport J. (2000) Initiation of eukaryotic DNA replication: Origin unwinding and sequential chromatin association of Cdc45, RPA, and DNA polymerase α. Mol. Cell 5, 617–627 [DOI] [PubMed] [Google Scholar]
- 20. Van Hatten R. A., Tutter A. V., Holway A. H., Khederian A. M., Walter J. C., and Michael W. M. (2002) The Xenopus Xmus101 protein is required for the recruitment of Cdc45 to origins of DNA replication. J. Cell Biol. 159, 541–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lee J., Kumagai A., and Dunphy W. G. (2003) Claspin, a Chk1-regulatory protein, monitors DNA replication on chromatin independently of RPA, ATR, and Rad17. Mol. Cell 11, 329–340 [DOI] [PubMed] [Google Scholar]
- 22. Gambus A., Jones R. C., Sanchez-Diaz A., Kanemaki M., van Deursen F., Edmondson R. D., and Labib K. (2006) GINS maintains association of Cdc45 with MCM in replisome progression complexes at eukaryotic DNA replication forks. Nat. Cell Biol. 8, 358–366 [DOI] [PubMed] [Google Scholar]
- 23. Merchant A. M., Kawasaki Y., Chen Y., Lei M., and Tye B. K. (1997) A lesion in the DNA replication initiation factor Mcm10 induces pausing of elongation forks through chromosomal replication origins in Saccharomyces cerevisiae. Mol. Cell. Biol. 17, 3261–3271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Homesley L., Lei M., Kawasaki Y., Sawyer S., Christensen T., and Tye B. K. (2000) Mcm10 and the MCM2–7 complex interact to initiate DNA synthesis and to release replication factors from origins. Genes Dev. 14, 913–926 [PMC free article] [PubMed] [Google Scholar]
- 25. Eisenberg S., Korza G., Carson J., Liachko I., and Tye B. K. (2009) Novel DNA binding properties of the Mcm10 protein from Saccharomyces cerevisiae. J. Biol. Chem. 284, 25412–25420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ricke R. M., and Bielinsky A. K. (2004) Mcm10 regulates the stability and chromatin association of DNA polymerase-α. Mol. Cell 16, 173–185 [DOI] [PubMed] [Google Scholar]
- 27. Das-Bradoo S., Ricke R. M., and Bielinsky A. K. (2006) Interaction between PCNA and diubiquitinated Mcm10 is essential for cell growth in budding yeast. Mol. Cell. Biol. 26, 4806–4817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Alver R. C., Zhang T., Josephrajan A., Fultz B. L., Hendrix C. J., Das-Bradoo S., and Bielinsky A. K. (2014) The N-terminus of Mcm10 is important for interaction with the 9–1-1 clamp and in resistance to DNA damage. Nucleic Acids Res. 42, 8389–8404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Douglas N. L., Dozier S. K., and Donato J. J. (2005) Dual roles for Mcm10 in DNA replication initiation and silencing at the mating-type loci. Mol. Biol. Rep. 32, 197–204 [DOI] [PubMed] [Google Scholar]
- 30. Liachko I., and Tye B. K. (2005) Mcm10 is required for the maintenance of transcriptional silencing in Saccharomyces cerevisiae. Genetics 171, 503–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Izumi M., Yanagi K., Mizuno T., Yokoi M., Kawasaki Y., Moon K.-Y., Hurwitz J., Yatagai F., and Hanaoka F. (2000) The human homolog of Saccharomyces cerevisiae Mcm10 interacts with replication factors and dissociates from nuclease-resistant nuclear structures in G2 phase. Nucleic Acids Res. 28, 4769–4777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chattopadhyay S., and Bielinsky A. K. (2007) Human Mcm10 regulates the catalytic subunit of DNA polymerase-α and prevents DNA damage during replication. Mol. Biol. Cell 18, 4085–4095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhu W., Ukomadu C., Jha S., Senga T., Dhar S. K., Wohlschlegel J. A., Nutt L. K., Kornbluth S., and Dutta A. (2007) Mcm10 and And-1/CTF4 recruit DNA polymerase α to chromatin for initiation of DNA replication. Genes Dev. 21, 2288–2299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xu X., Rochette P. J., Feyissa E. A., Su T. V., and Liu Y. (2009) MCM10 mediates RECQ4 association with MCM2–7 helicase complex during DNA replication. EMBO J. 28, 3005–3014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Di Perna R., Aria V., De Falco M., Sannino V., Okorokov A. L., Pisani F. M., and De Felice M. (2013) The physical interaction of Mcm10 with Cdc45 modulates their DNA-binding properties. Biochem. J. 454, 333–343 [DOI] [PubMed] [Google Scholar]
- 36. Fatoba S. T., Tognetti S., Berto M., Leo E., Mulvey C. M., Godovac-Zimmermann J., Pommier Y., and Okorokov A. L. (2013) Human SIRT1 regulates DNA binding and stability of the Mcm10 DNA replication factor via deacetylation. Nucleic Acids Res. 41, 4065–4079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wohlschlegel J. A., Dhar S. K., Prokhorova T. A., Dutta A., and Walter J. C. (2002) Xenopus Mcm10 binds to origins of DNA replication after Mcm2–7 and stimulates origin binding of Cdc45. Mol. Cell 9, 233–240 [DOI] [PubMed] [Google Scholar]
- 38. Gregan J., Lindner K., Brimage L., Franklin R., Namdar M., Hart E. A., Aves S. J., and Kearsey S. E. (2003) Fission yeast Cdc23/Mcm10 functions after prereplicative complex formation to promote Cdc45 chromatin binding. Mol. Biol. Cell 14, 3876–3887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sawyer S. L., Cheng I. H., Chai W., and Tye B. K. (2004) Mcm10 and Cdc45 cooperate in origin activation in Saccharomyces cerevisiae. J. Mol. Biol. 340, 195–202 [DOI] [PubMed] [Google Scholar]
- 40. Im J. S., Ki S. H., Farina A., Jung D. S., Hurwitz J., and Lee J. K. (2009) Assembly of the Cdc45-Mcm2–7-GINS complex in human cells requires the Ctf4/And-1, RecQL4, and Mcm10 proteins. Proc. Natl. Acad. Sci. U.S.A. 106, 15628–15632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Perez-Arnaiz P., Bruck I., and Kaplan D. L. (2016) Mcm10 coordinates the timely assembly and activation of the replication fork helicase. Nucleic Acids Res. 44, 315–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Watase G., Takisawa H., and Kanemaki M. T. (2012) Mcm10 plays a role in functioning of the eukaryotic replicative DNA helicase, Cdc45-Mcm-GINS. Curr. Biol. 22, 343–349 [DOI] [PubMed] [Google Scholar]
- 43. van Deursen F., Sengupta S., De Piccoli G., Sanchez-Diaz A., and Labib K. (2012) Mcm10 associates with the loaded DNA helicase at replication origins and defines a novel step in its activation. EMBO J. 31, 2195–2206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kanke M., Kodama Y., Takahashi T. S., Nakagawa T., and Masukata H. (2012) Mcm10 plays an essential role in origin DNA unwinding after loading of the CMG components. EMBO J. 31, 2182–2194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chadha G. S., Gambus A., Gillespie P. J., and Blow J. J. (2016) Xenopus Mcm10 is a CDK-substrate required for replication fork stability. Cell Cycle, 15, 2183–2195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Taylor M., Moore K., Murray J., Aves S. J., and Price C. (2011) Mcm10 interacts with Rad4/Cut5(TopBP1) and its association with origins of DNA replication is dependent on Rad4/Cut5(TopBP1). DNA Repair 10, 1154–1163 [DOI] [PubMed] [Google Scholar]
- 47. Cook C. R., Kung G., Peterson F. C., Volkman B. F., and Lei M. (2003) A novel zinc finger is required for Mcm10 homocomplex assembly. J. Biol. Chem. 278, 36051–36058 [DOI] [PubMed] [Google Scholar]
- 48. Okorokov A. L., Waugh A., Hodgkinson J., Murthy A., Hong H. K., Leo E., Sherman M. B., Stoeber K., Orlova E. V., and Williams G. H. (2007) Hexameric ring structure of human MCM10 DNA replication factor. EMBO Rep. 8, 925–930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Robertson P. D., Warren E. M., Zhang H., Friedman D. B., Lary J. W., Cole J. L., Tutter A. V., Walter J. C., Fanning E., and Eichman B. F. (2008) Domain architecture and biochemical characterization of vertebrate Mcm10. J. Biol. Chem. 283, 3338–3348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Du W., Josephrajan A., Adhikary S., Bowles T., Bielinsky A. K., and Eichman B. F. (2013) Mcm10 self-association is mediated by an N-terminal coiled-coil domain. PLoS One 8, e70518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Warren E. M., Vaithiyalingam S., Haworth J., Greer B., Bielinsky A. K., Chazin W. J., and Eichman B. F. (2008) Structural basis for DNA binding by replication initiator Mcm10. Structure 16, 1892–1901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Warren E. M., Huang H., Fanning E., Chazin W. J., and Eichman B. F. (2009) Physical interactions between Mcm10, DNA, and DNA polymerase α. J. Biol. Chem. 284, 24662–24672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Robertson P. D., Chagot B., Chazin W. J., and Eichman B. F. (2010) Solution NMR structure of the C-terminal DNA binding domain of Mcm10 reveals a conserved MCM motif. J. Biol. Chem. 285, 22942–22949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Quan Y., Xia Y., Liu L., Cui J., Li Z., Cao Q., Chen X. S., Campbell J. L., and Lou H. (2015) Cell-cycle-regulated interaction between Mcm10 and double hexameric Mcm2–7 is required for helicase splitting and activation during S phase. Cell Rep. 13, 2576–2586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Douglas M. E., and Diffley J. F. (2016) Recruitment of Mcm10 to sites of replication initiation requires direct binding to the minichromosome maintenance (MCM) complex. J. Biol. Chem. 291, 5879–5888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Fujita M., Yamada C., Goto H., Yokoyama N., Kuzushima K., Inagaki M., and Tsurumi T. (1999) Cell cycle regulation of human CDC6 protein. Intracellular localization, interaction with the human mcm complex, and CDC2 kinase-mediated hyperphosphorylation. J. Biol. Chem. 274, 25927–25932 [DOI] [PubMed] [Google Scholar]
- 57. Méndez J., and Stillman B. (2000) Chromatin association of human origin recognition complex, Cdc6, and minichromosome maintenance proteins during the cell cycle: Assembly of prereplication complexes in late mitosis. Mol. Cell. Biol. 20, 8602–8612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lai J. S., and Herr W. (1992) Ethidium bromide provides a simple tool for identifying genuine DNA-independent protein associations. Proc. Natl. Acad. Sci. U.S.A. 89, 6958–6962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ibarra A., Schwob E., and Méndez J. (2008) Excess MCM proteins protect human cells from replicative stress by licensing backup origins of replication. Proc. Natl. Acad. Sci. U.S.A. 105, 8956–8961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Izumi M., Yatagai F., and Hanaoka F. (2001) Cell cycle-dependent proteolysis and phosphorylation of human Mcm10. J. Biol. Chem. 276, 48526–48531 [DOI] [PubMed] [Google Scholar]
- 61. Jackson D. A., and Pombo A. (1998) Replicon clusters are stable units of chromosome structure: Evidence that nuclear organization contributes to the efficient activation and propagation of S phase in human cells. J. Cell Biol. 140, 1285–1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lee J. K., Seo Y. S., and Hurwitz J. (2003) The Cdc23 (Mcm10) protein is required for the phosphorylation of minichromosome maintenance complex by the Dfp1-Hsk1 kinase. Proc. Natl. Acad. Sci. U.S.A. 100, 2334–2339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Apger J., Reubens M., Henderson L., Gouge C. A., Ilic N., Zhou H. H., and Christensen T. W. (2010) Multiple functions for Drosophila Mcm10 suggested through analysis of two Mcm10 mutant alleles. Genetics 185, 1151–1165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wright P. E., and Dyson H. J. (1999) Intrinsically unstructured proteins: Re-assessing the protein structure-function paradigm. J. Mol. Biol. 293, 321–331 [DOI] [PubMed] [Google Scholar]
- 65. Tompa P. (2005) The interplay between structure and function in intrinsically unstructured proteins. FEBS Lett. 579, 3346–3354 [DOI] [PubMed] [Google Scholar]
- 66. Brown C. J., Takayama S., Campen A. M., Vise P., Marshall T. W., Oldfield C. J., Williams C. J., and Dunker A. K. (2002) Evolutionary rate heterogeneity in proteins with long disordered regions. J. Mol. Evol. 55, 104–110 [DOI] [PubMed] [Google Scholar]
- 67. Izumi M., Yatagai F., and Hanaoka F. (2004) Localization of human Mcm10 is spatially and temporally regulated during the S phase. J. Biol. Chem. 279, 32569–32577 [DOI] [PubMed] [Google Scholar]
- 68. Lõoke M., Maloney M. F., and Bell S. P. (2017) Mcm10 regulates DNA replication elongation by stimulating the CMG replicative helicase. Genes Dev. 31, 291–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Izumi M., and Gilbert D. M. (1999) Homogeneous tetracycline-regulatable gene expression in mammalian fibroblasts. J. Cell. Biochem. 76, 280–289 [DOI] [PubMed] [Google Scholar]
- 70. Yanagi K., Mizuno T., You Z., and Hanaoka F. (2002) Mouse geminin inhibits not only Cdt1-MCM6 interactions but also a novel intrinsic Cdt1 DNA binding activity. J. Biol. Chem. 277, 40871–40880 [DOI] [PubMed] [Google Scholar]
- 71. Prasanth S. G., Méndez J., Prasanth K. V., and Stillman B. (2004) Dynamics of pre-replication complex proteins during the cell division cycle. Philos. Trans. R. Soc. Lond. B Biol. Sci. 359, 7–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Sugimura K., Takebayashi S., Taguchi H., Takeda S., and Okumura K. (2008) PARP-1 ensures regulation of replication fork progression by homologous recombination on damaged DNA. J. Cell Biol. 183, 1203–1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.