

Abstract
Hypothalamic agouti-related peptide (AgRP) neurons potently stimulate food intake, whereas proopiomelanocortin (POMC) neurons inhibit feeding. Whether AgRP neurons exert their orexigenic actions, at least in part, by inhibiting anorexigenic POMC neurons remains unclear. Here, the connectivity between GABA-releasing AgRP neurons and POMC neurons was examined in brain slices from male and female mice. GABA-mediated spontaneous IPSCs (sIPSCs) in POMC neurons were unaffected by disturbing GABA release from AgRP neurons either by cell type-specific deletion of the vesicular GABA transporter or by expression of botulinum toxin in AgRP neurons to prevent vesicle-associated membrane protein 2-dependent vesicle fusion. Additionally, there was no difference in the ability of μ-opioid receptor (MOR) agonists to inhibit sIPSCs in POMC neurons when MORs were deleted from AgRP neurons, and activation of the inhibitory designer receptor hM4Di on AgRP neurons did not affect sIPSCs recorded from POMC neurons. These approaches collectively indicate that AgRP neurons do not significantly contribute to the strong spontaneous GABA input to POMC neurons. Despite these observations, optogenetic stimulation of AgRP neurons reliably produced evoked IPSCs in POMC neurons, leading to the inhibition of POMC neuron firing. Thus, AgRP neurons can potently affect POMC neuron function without contributing a significant source of spontaneous GABA input to POMC neurons. Together, these results indicate that the relevance of GABAergic inputs from AgRP to POMC neurons is state dependent and highlight the need to consider different types of transmitter release in circuit mapping and physiologic regulation.
SIGNIFICANCE STATEMENT Agouti-related peptide (AgRP) neurons play an important role in driving food intake, while proopiomelanocortin (POMC) neurons inhibit feeding. Despite the importance of these two well characterized neuron types in maintaining metabolic homeostasis, communication between these cells remains poorly understood. To provide clarity to this circuit, we made electrophysiological recordings from mouse brain slices and found that AgRP neurons do not contribute spontaneously released GABA onto POMC neurons, although when activated with channelrhodopsin AgRP neurons inhibit POMC neurons through GABA-mediated transmission. These findings indicate that the relevance of AgRP to POMC neuron GABA connectivity depends on the state of AgRP neuron activity and suggest that different types of transmitter release should be considered when circuit mapping.
Keywords: arcuate nucleus, circuit mapping, electrophysiology, energy balance, IPSC, optogenetics
Introduction
Agouti-related peptide (AgRP) and proopiomelanocortin (POMC) neurons are two well characterized neuron types located in the arcuate nucleus (ARC) of the hypothalamus that play important roles in the modulation of energy homeostasis (Cone, 2005; Mercer et al., 2013). POMC neurons can inhibit food intake and are critical for the maintenance of normal body weight. Deletion of POMC neurons (Xu et al., 2005), their peptide product α-MSH (Yaswen et al., 1999; Smart et al., 2006), or receptors of α-MSH (Butler et al., 2000; Balthasar et al., 2005) all result in obesity. AgRP neurons act in opposition to POMC neurons with their activation stimulating food intake through the release of inhibitory transmitters and neuromodulators (Cone, 2005; Tong et al., 2008; Krashes et al., 2013). AgRP neurons are activated by energy deficit (Takahashi and Cone, 2005; Liu et al., 2012; Betley et al., 2013), and recent in vivo photometric studies demonstrated an increase in AgRP activity during food restriction, an effect that was relieved following the presentation of food (Betley et al., 2015; Chen et al., 2015). Further, optogenetic (Aponte et al., 2011) or chemogenetic (Nakajima et al., 2016) stimulation of AgRP neurons triggers feeding behavior. These findings, together with earlier anatomic studies (Horvath et al., 1997; Cowley et al., 2001; Pinto et al., 2004) have led many to propose that AgRP neurons stimulate feeding, at least partially, by direct inhibition of the anorexigenic POMC neurons (Cone, 2005; Tong et al., 2008; Zeltser et al., 2012; Nuzzaci et al., 2015).
In addition to anatomic evidence, the idea that GABA-releasing AgRP terminals directly inhibit POMC neurons is supported by a study showing a dramatic reduction in spontaneous IPSCs (sIPSCs) in POMC neurons following toxin-induced ablation of AgRP neurons in adult mice (Wu et al., 2008). However, basal sIPSC frequency in POMC neurons was not affected by deletion of the vesicular GABA transporter (VGAT) to disrupt GABA release from AgRP neurons (Tong et al., 2008) questioning the presence of the suggested GABAergic connection from AgRP neurons to POMC neurons. More recent studies have used an optogenetic approach to demonstrate that photostimulation of AgRP neurons results in evoked IPSCs in POMC neurons (Atasoy et al., 2012; Dicken et al., 2015), indicating that AgRP neurons can release GABA onto POMC neurons when stimulated.
Given the importance of AgRP and POMC neuron activity in the regulation of food intake and energy balance and their often reciprocal roles, we set out to determine whether types of transmitter release examined could account for the disparate connectivity results found to date. Collectively, the present results provide additional evidence that light-evoked depolarization of AgRP neurons triggers GABA release onto POMC cells and that coordinated activity of AGRP neurons is sufficient to reduce POMC firing. However, using a variety of transgenic, pharmacologic, and electrophysiological tools we provide evidence that AgRP neurons are not a primary contributor of spontaneously released GABA onto POMC neurons, even in the fasted state. The ability to detect evoked AgRP, but not spontaneous AgRP, in POMC neuron inputs may be accounted for by differential molecular mechanisms for evoked versus spontaneous fusion of neurotransmitter-filled vesicles (Ramirez and Kavalali, 2011; Schneggenburger and Rosenmund, 2015), separate presynaptic vesicle populations (Fredj and Burrone, 2009), differential targeting of postsynaptic receptors (Otis and Mody, 1992), or divergent target locations on the postsynaptic cell (Atasoy et al., 2008; Zenisek, 2008). Collectively, our results suggest that physiological dissection of neuronal circuits and function should consider both evoked and spontaneous release of neurotransmitter release.
Materials and Methods
Animals.
All experiments were performed in accordance with the Colorado State University Animal Care and Use Committee and the Guide for the Care and Use of Laboratory Animals set forth by the National Institutes of Health. The following mice were procured from The Jackson Laboratory: POMC enhanced green fluorescent protein (eGFP) [C57BL/6J-Tg(Pomc-EGFP)1Low/J; stock #009593]; AgRPcre [AgRPtm1(cre)Lowl/J, stock #012899]; Cre-dependent channelrhodopsin (ChR2) mice [B6.Cg-Gt(ROSA)26Sortm32(CAG-COP4*H134R/EYFP)Hze/J; stock #024109]; VGATflox/flox [slc32a1tm1lowl/J; stock #012897]; MORflox/flox [B6.129S2-Oprm1tm1Kff/J; stock #007559]; and Cre-dependent hM4Di mice [B6N.129-Gt(ROSA)26Sortm1(CAG-CHRM4*,-mCitrine)ute/J; stock #026219]. The Cre-dependent botulinum toxin-expressing mice (iBot mice) were obtained from Dr. Frank Pfrieger (University of Strasbourg, Strasbourg, France; Slezak et al., 2012). POMC DsRed mice (Hentges et al., 2009) were originally a gift from Dr. Malcolm Low (University of Michigan, Ann Arbor, MI). To detect transgenes and floxed alleles, standard PCR techniques were used.
Animals were housed on a 12 h light/dark schedule, and had ad libitum access to standard rodent chow and tap water. Where noted, animals were fasted overnight with food removed 1 h before lights out. Brain slices were prepared for electrophysiology 17 h later.
Stereotaxic surgery for in vivo gene delivery.
Six–eight-week-old mice were induced into a deep anesthetic plane with isoflurane and were placed into a stereotaxic apparatus (David Kopf Instruments) fitted with a nose cone for the delivery of isoflurane for the entirety of the operation. Viral vectors allowing for Cre recombinase-dependent expression of ChR2 [AAV9.EF1.dflox.hChR2(H134R)-mCherry.WPRE.hGH, 200 nl, 7.24e13 genome copies (GC)/ml; Penn Vector Core, University of Pennsylvania, Philadelphia, PA] or Cre recombinase-dependent expression of the inhibitory designer receptor hM4Di [rAVV8/hSyn-DIO-hm4Di (Gi)-mcherry, 200 nl, 7e12 GC/ml; Virus Vector Core, University of North Carolina at Chapel Hill) were delivered over the course of 60 s into each side of the arcuate nucleus. For each injection, the needle was left in place for 150 s following the end of solution delivery to allow diffusion and minimize leak. Brain slices were prepared 10–24 d after virus injection.
Electrophysiology.
Following the induction of a deep anesthetic plane with isoflurane, mice were decapitated and their brains were removed and placed into ice-cold artificial CSF (aCSF) consisting of the following (in mm): 126 NaCl, 2.5 KCl, 1.2 MgCl2∧6H2O, 2.4 CaCl2∧2H2O, 1.2 NaH2PO4, 11.1 glucose, and 21.4 NaHCO3, bubbled with 95% O2 and 5% CO2. Sagittal slices containing the ARC were cut at a thickness of 240 μm using a model VT1200S vibratome (Leica Microsystems). After resting [≥1 h at 37°C in aCSF containing the NMDA receptor blocker MK-801 (15 μm)], slices were transferred to the recording chamber and perfused with oxygenated 37°C aCSF at a flow rate of ∼2 ml/min. For whole-cell recordings, the internal recording solution contained the following (in mm): KCL 57.5, K-methyl sulfate 57.5, NaCl 20, MgCl2 1.5, HEPES 5; EGTA 0.1; ATP 2; GTP 0.5, and phosphocreatine 10. The pH was adjusted to 7.3. Recording electrodes had a resistance of 1.6–2.2 MΩ when filled with this solution. Loose patch recordings were made with aCSF in the recording pipette. POMC cells were visually identified for recording based on the transgenic expression of DsRed or eGFP. Whole-cell patch-clamp recordings were acquired in voltage-clamp mode at a holding potential of −60 mV using an Axopatch 200B Amplifier (Molecular Devices). Electrophysiological data were collected and analyzed using Axograph X software running on a Mac OS X operating system (Apple).
Light activation of AgRP neurons expressing ChR2 was achieved via a 2 ms 470 nm light pulse with an interstimulus interval of 20 s through the use of a 470 nm LED (Thorlabs) driven by an LEDD1B driver (Thorlabs) triggered through the TTL output on an ITC-18 computer interface board (HEKA Instruments). In some cases, three consecutive 2 ms light pulses were delivered with an interstimulus interval of 800 ms to better visualize light-evoked IPSCs against the background of sIPSCs. Electrically evoked IPSCs were elicited by passing current through a bipolar stimulating electrode that was inserted into the mid-dorsal region of the ARC. For both light-evoked and electrically evoked IPSCs, the stimulus intensity was reduced at the beginning of the experiment to produce events with amplitudes ∼50% of the maximal amplitude to allow detection of both increased or decreased amplitudes. sIPSCs were continuously collected in 60 s epochs, and three to five epochs were combined for frequency and amplitude analysis. sIPSC events were detected by sliding an event template over the raw data trace, and data were then visually inspected to exclude spurious events. All electrophysiological data were collected at 10 kHz and filtered at 5 kHz. Recordings were excluded from analysis if the series resistance exceeded 20 MΩ or changed significantly over the course of the experiment. Evoked and spontaneous IPSCs were pharmacologically isolated with the addition of 10 μm 6,7-dinitroquinoxaline-2,3-dione (DNQX) into the recording solution. IPSCs were confirmed to be GABAA mediated by the perfusion of bicuculline (BIC; 10 μm) before the cessation of the experiment. Previous studies have shown that the majority of spontaneous inputs to POMC neurons are TTX insensitive (Cowley et al., 2001; Pinto et al., 2004; Vong et al., 2011; Pennock and Hentges, 2016), indicating that within this preparation sIPCS represent action potential-independent events.
Immunohistochemistry and confocal imaging.
To confirm the presence of the botulinum toxin in the same cells that contain ChR2, AgRPcre; iBoteGFP mice were injected with an adeno-associated virus (AAV) encoding a cre-dependent ChR2 tagged with mCherry. Fifteen to 21 d after injection, brain slices from these mice were used for electrophysiological experiments. After recording, slices were transferred to a multiwell plate and stored overnight in potassium PBS (KPBS) containing 4% paraformaldehyde (PFA). The following day, slices were washed in KPBS and incubated in 2% normal donkey serum and 0.6% Triton X-100. eGFP was detected using the chicken anti-GFP primary antibody (Abcam; RRID: AB_300798; overnight at 4°C; 1:2000). Slices were washed in KPBS and incubated with an Alexa Fluor 488-conjugated donkey anti-chicken secondary antibody (Jackson ImmunoResearch; RRID: AB_2340375; 1:400). Tissue was washed once more, and slices were mounted on slides. To confirm the presence of hM4Di receptors following electrophysiological experiments, slices were postfixed and washed as described above. Slices were then incubated in 2% normal goat serum and 0.6% Triton X-100 prepared in KPBS. Human influenza hemagglutinin (HA)-tagged hM4Di receptors were detected using the rabbit anti-HA antibody (Cell Signaling Technology; RRID: AB_1549585; overnight at 4°C; 1:1000). The following day, slices were washed in KPBS and incubated with an Alexa Fluor 488-conjugated goat anti-rabbit secondary antibody for 1 h (Thermo Fisher; RRID: AB_143165; 1:250). Slices were washed again in KPBS and mounted for imaging. In separate experiments in which fluorescent images were obtained but immunohistochemistry was not, the necessary slices were transferred to a multiwell plate following electrophysiology and postfixed overnight in KPBS containing 4% PFA. Slices were then washed three times in KPBS and mounted on slides. All images were acquired on a Zeiss 880 Confocal Microscope.
Drugs.
BIC was purchased from Tocris Bioscience, and MK-801, DNQX, and [d-Ala(2),N-Me-Phe(4),Gly(5)-ol]-enkephalin (DAMGO) were purchased from Sigma-Aldrich. Clozopine-N-oxide (CNO) was purchased from Enzo Life Sciences. Drugs were prepared in distilled water (BIC, DAMGO, and CNO) or DMSO (MK-801 and DNQX) as 1000 or 10,000× concentrates and diluted in aCSF to achieve the desired concentration immediately before use.
Experimental design and statistical analysis.
Data were compared using paired and unpaired Student's t tests, as indicated. Individual points on each figure represent a recording from a single cell. Only one recording was made per slice and a maximum of three recordings were made from each animal. Male and female mice were used for all experiments, and results were initially analyzed for sex differences. No sex differences were detected, and, therefore, data from both sexes were pooled for final analysis and data presentation. All data were analyzed using GraphPad Prism version 6. Data are presented as the mean ± SEM, and differences were considered to be significant at p < 0.05.
Results
Deletion of VGAT from AgRP cells blocks light-evoked GABA release onto POMC cells without affecting the frequency of spontaneous GABA release onto POMC cells
A previous study (Wu et al., 2008) found evidence for a strong input of spontaneous GABA from AgRP neurons to POMC neurons as the ablation of AgRP neurons resulted in a profound decrease in GABA-mediated sIPSCs in POMC neurons. However, other investigators deleted the VGAT, which is used to package GABA into vesicles for subsequent release (Wojcik et al., 2006), from AgRP cells and found no difference in sIPSCs recorded from POMC neurons (Tong et al., 2008). To replicate the VGAT deletion study within our experimental recoding conditions, cre/lox technology was used to generate mice that lack VGAT on AgRP neurons (i.e., AgRPcre; VGATflox/flox; referred to here as AgRP-VGATKO mice). sIPSCs were collected from visually identified POMC cells in hypothalamic slices from these mice to determine whether the deletion of GABA release from AgRP cells attenuated spontaneous GABAergic inputs onto POMC cells. The frequency of sIPSC inputs onto POMC cells in slices from AgRP-VGATWT mice was not significantly altered relative to AGRP-VGATKO mice [6.05 ± 0.8 Hz (n = 17) vs 6.22 ± 1.3 Hz (n = 14), respectively; t(29) = 0.113, p = 0.91, unpaired t test; Fig. 1A,B]. The deletion of VGAT from AgRP neurons also did not affect the amplitude of sIPSCs recorded from POMC cells (AgRP-VGATWT: n = 17; 36.28 ± 2.6 pA; AgRP-VGATKO: n = 14; 32.74 ± 2.2 pA; t(29) = 0.998, p = 0.326, unpaired t test).
Figure 1.

Constitutive deletion of the VGAT from AgRP cells does not affect the frequency of spontaneous IPSCs recorded from POMC cells despite abolishing evoked IPSCs between these cells. A, sIPSCs recorded from POMC cells with intact VGAT in AgRP neurons (top) and after deletion of VGAT from AgRP cells (bottom). All voltage-clamp experiments were conducted at a holding potential of −60 mV. B, Summary figure comparing the frequency of sIPSCs recorded from POMC cells in mice with intact GABA release from AgRP neurons and mice where GABA release has been removed from AgRP neurons by deleting VGAT in these cells (AgRP-VGATWT, n = 17; AgRP-VGATKO, n = 14; p = 0.917, unpaired t test). Data are presented as the mean ± SEM. C, Twenty consecutive traces demonstrating the reliability of light-evoked GABA release from AgRP neurons onto POMC neurons (top trace) and the ablation of this connection following deletion of VGAT from AgRP cells (bottom trace). A 2 ms flash of 470 nm light is indicated by a blue dash above traces.
To confirm that AgRP-VGATKO disrupted GABA release from AgRP neurons, whole-cell voltage-clamp recordings were made from POMC cells, while AgRP cells expressing ChR2 were photostimulated with 2 ms pulses of 470 nm light. In contrast to AgRP-VGATWT mice (41 of 48 cells connected; n = 14 mice; Fig. 1C, top trace), light-evoked IPSCs recorded from POMC cells were not observed in AgRP-VGATKO mice (0 of 14 cells connected; n = 3 mice; Fig. 1C, bottom trace), indicating the loss of GABA release from AgRP neurons following VGAT deletion. Together, these results provide support for previous reports suggesting that AgRP neurons are not a significant source of GABA-mediated sIPSCs recorded from POMC neurons (Tong et al., 2008; Vong et al., 2011). However, the ability to reliably evoke IPSCs in POMC neurons upon AgRP neuron stimulation when VGAT is intact (Atasoy et al., 2012; Dicken et al., 2015; Fig. 1C) indicates that a connection does exist between AgRP and POMC neurons, leaving open the possibility that increased AgRP activity may reveal a significant contribution of sIPSCs to POMC cells.
Fasting-induced increase of spontaneous IPSCs recorded from POMC cells are not attenuated by deletion of VGAT from AgRP cells
Consistent with their orexigenic nature, caloric deficit or fasting elevates AgRP neuron activity (Liu et al., 2012; Betley et al., 2015; Chen et al., 2015) and increases the expression of glutamate decarboxylase mRNA in AgRP neurons (Dicken et al., 2015). Fasting also increases spontaneous release of GABA onto POMC cells (Vong et al., 2011), raising the possibility that fasting-induced increases in AgRP neuron activity may be responsible for increased GABA release onto POMC cells. To test this hypothesis, recordings of sIPSCs were made from POMC cells in slices from both AgRP-VGATWT and AgRP-VGATKO mice fed ad libitum or following an overnight (17 h) fast. Fasting resulted in a significant increase in sIPSC frequency in POMC cells in slices from both AgRP-VGATWT mice (fed: n = 17; 6.05 ± 0.8 Hz; fasted: n = 15; 9.12 ± 1.3; t(30) = 2.063, p = 0.04, unpaired t test; Fig. 2A,B) and AgRP-VGATKO mice (fed: n = 14; 6.22 ± 1.3 Hz; fasted: n = 10; 11.24 ± 1.5; t(22) = 2.549, p = 0.02, unpaired t test; Fig. 2A,C). This result provides further support to the theory that AgRP neurons do not contribute significant spontaneous GABA to POMC neurons even during fasting, which increases AgRP neuron activity, while also indicating that the neurons responsible for sIPSCs released onto POMC cells are regulated by energy state.
Figure 2.

Increased frequency of sIPSCs in POMC cells following overnight fast is not attenuated by deletion of VGAT in AgRP cells. A, Representative traces showing that an overnight fast increases sIPSC frequency in POMC cells in recordings from both mice with intact VGAT in AgRP neurons (AgRP-VGATWT) and in mice where VGAT was deleted from AgRP neurons (AgRP-VGATKO). B, Overnight fast increases spontaneous GABA release onto POMC cells in hypothalamic slices obtained from animals with intact VGAT in AgRP cells (fed, n = 17; fasted, n = 15; *p = 0.04, unpaired t test). C, In hypothalamic slices obtained from mice with deleted VGAT in AgRP cells, the fasting-induced increase in spontaneous GABA release onto POMC cells persists (fed, n = 14; fasted, n = 10; *p = 0.02, unpaired t test). All data are presented as the mean ± SEM.
Pharmacological investigation of spontaneous GABA release from AgRP to POMC neurons
The preceding experiments indicate that AgRP cells do not contribute spontaneous GABA onto POMC cells. However, constitutive deletion of GABA release from AgRP neurons could result in compensatory changes wherein adaptive mechanisms from other cell types contribute greater GABAergic inhibition to maintain homeostatic inhibition of POMC cells. This compensation could mask a normal contribution from AgRP cells when approaches rely on constitutive disruption of GABA release. To address this important caveat, we designed a strategy wherein the inhibition of evoked and spontaneous IPSCs onto POMC cells was assessed following bath perfusion of the μ-opioid receptor (MOR) agonist DAMGO (10 μm) in animals following the deletion of MORs from AgRP neurons. This alternative approach allows for experimentation where GABA release from AgRP neurons is not grossly disturbed and is based on the observation that MOR agonists potently inhibit both spontaneous and evoked GABA-mediated currents in POMC neurons (Pennock and Hentges, 2016) and that AgRP neurons express MORs (Barnes et al., 2010). If AgRP neurons are a significant source of spontaneous GABA inputs to POMC neurons, then the deletion of MORs only from AgRP cells should blunt the ability of MOR agonist to inhibit sIPSCs to POMC neurons. To validate this approach, we generated mice lacking MORs on AgRP neurons (i.e., AgRPcre; MORflox/flox; referred to here as AgRP-MORKO). A portion of the offspring received virus containing the Cre-dependent version of ChR2 to allow for light-evoked GABA release from AgRP neurons. Light-evoked IPSCs from AgRP-MORWT cells were significantly inhibited by the addition of 10 μm DAMGO to the superfusate (29.12 ± 6.1% of baseline; n = 12; t(11) = 5.356, p = 0.0002, paired t test; Fig. 3A,B), confirming that AgRP cells express MORs in presynaptic terminals contacting POMC cells. Conversely, in recordings made from POMC cells in AgRP-MORKO mice 10 μm DAMGO was ineffective at inhibiting light-evoked IPSCs (106.3 ± 3.1% of baseline; n = 13; t(12) = 1.117, p = 0.286, paired t test; Fig. 3A,B), demonstrating that we effectively deleted MORs from AgRP cells.
Figure 3.
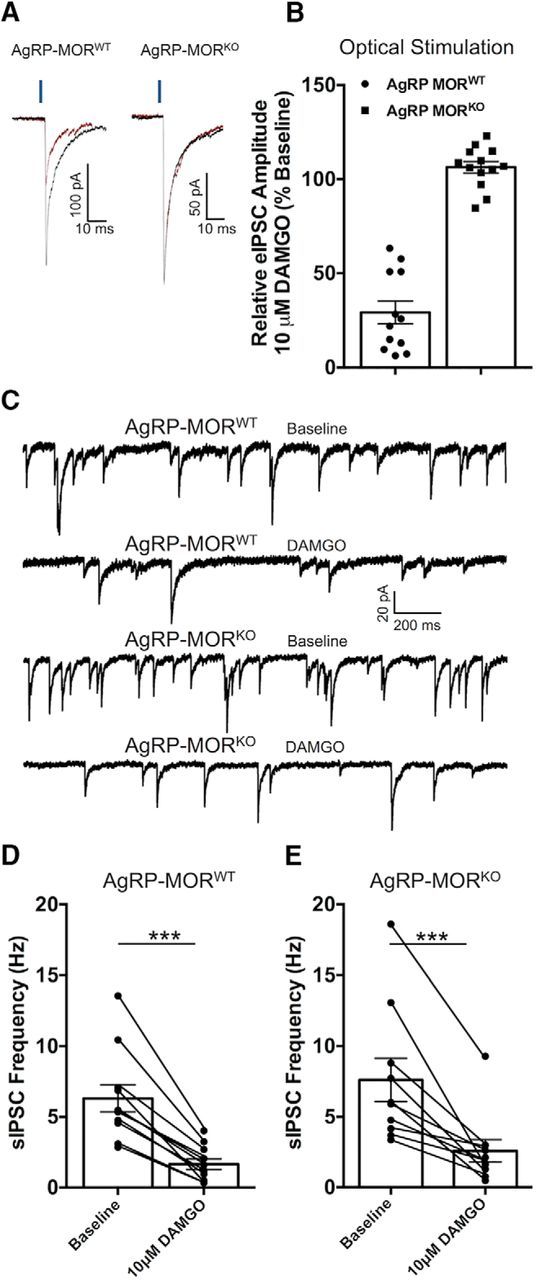
Deletion of MORs from AgRP neurons does not attenuate the inhibition of sIPSCs in POMC neurons produced by MOR activation. A, Ten to 20 consecutive traces averaged to demonstrate the inhibition of light-evoked IPSCs caused by the MOR agonist DAMGO (10 μm) in recordings from POMC neurons in mice with intact MORs in AgRP cells (AgRP-MORWT; left traces) and the block of this effect in mice where MORs were deleted form AgRP cells (AgRP-MORKO; right traces). Blue bar above traces indicates a 2 ms pulse of 470 nm light. B, Comparison of the effect of 10 μm DAMGO on light-evoked IPSCs onto POMC cells from AgRP-MORWT and AgRP-MORKO mice. C, Traces representative of the effect of DAMGO (10 μm) on the frequency of sIPSCs recorded from POMC cells in AgRP-MORWT and AgRP-MORKO mice. D, Compiled data showing the frequency of sIPSCs recorded from POMC cells at baseline and after bath perfusion of DAMGO (10 μm) from mice with intact MORs on AgRP neurons (***p = 0.00003, paired t test), E, Compiled data showing the frequency of sIPSCs recorded from POMC neurons at baseline and following bath perfusion of DAMGO (10 μm) from mice where MORs were deleted from AgRP neurons (***p = 0.001; paired t test). All data are presented as the mean ± SEM.
We next investigated whether sIPSCs onto POMC cells are differentially inhibited by the addition of 10 μm DAMGO in recordings from AgRP-MORWT and AgRP-MORKO mice. DAMGO (10 μm) significantly and dramatically reduced the frequency of sIPSCs in recordings from POMC cells in both AgRP-MORWT mice (n = 11; from 6.31 ± 0.9 to 1.65 ± 0.4 Hz; t(10) = 7.031, p = 0.00003; paired t test; Fig. 3C–E) and AgRP-MORKO mice (n = 10; from 7.61 ± 1.5 to 2.57 ± 0.7 Hz; t(9) = 4.717, p = 0.001; paired t test; Fig. 3C–E). Additionally, the magnitude of inhibition was not different between the two genotypes (AgRP-MORWT mice: n = 11; 24.57 ± 3.1% baseline; AgRP-MORKO mice: n = 10; 31.10 ± 5.2% at baseline; t(19) = 1.104, p = 0.283, unpaired t test), further suggesting that the source of GABA released spontaneously onto POMC cells is not AgRP neurons.
Chemogenetic inhibition of AgRP neurons does not affect sIPSCs recorded from POMC neurons
As an additional approach with low potential for compensatory mechanisms, we expressed the Cre-dependent, pharmacologically selective designer Gi-protein-coupled designer receptor hM4Di in AgRPcre neurons using a transgenic approach (i.e., AgRPcre; hM4Di; referred to as AgRPhM4Di). Activation of the hM4Di receptors with its ligand CNO inhibits evoked release probability (Stachniak et al., 2014) and spontaneous release frequency (Mahler et al., 2014) without affecting action potential propagation (Stachniak et al., 2014). Thus, the expression of hM4Di receptors on AgRP neurons allows for the normal function of these neurons in the absence of agonist. The presence of hM4Di receptors was confirmed immunohistochemically using an antibody against the HA-tagged hM4Di receptors (Fig. 4A). In all slices, robust penetrance of hM4Di receptors was observed in AgRP neurons within close proximity to POMC cells. Activation of hM4Di receptors on AgRP neurons by bath application of 10 μm CNO did not affect the frequency of sIPSCs recorded from POMC neurons (n = 11; from 7.53 ± 1.8 to 7.16 ± 1.6 Hz; t(10) = 0.5609, p = 0.56; paired t test; Fig. 4B,C). We also injected an AAV encoding Cre-dependent hM4Di receptors into the ARC of AgRPcre mice to allow expression of the hM4Di receptor in adulthood. Bath application of 10 μm CNO again did not affect the frequency of sIPSCs in recordings from POMC cells (n = 6; from 6.69 ± 2.0 to 6.91 ± 2.1 Hz; t(5) = 0.7446, p = 0.49; paired t test). Collectively, these two experiments support our previous experiments by again demonstrating that AgRP neurons are not a significant source of GABA released spontaneously onto POMC cells. Importantly, these two strategies allow for normal developmental function of AgRP neurons, which should minimize any compensation from other neuron types.
Figure 4.
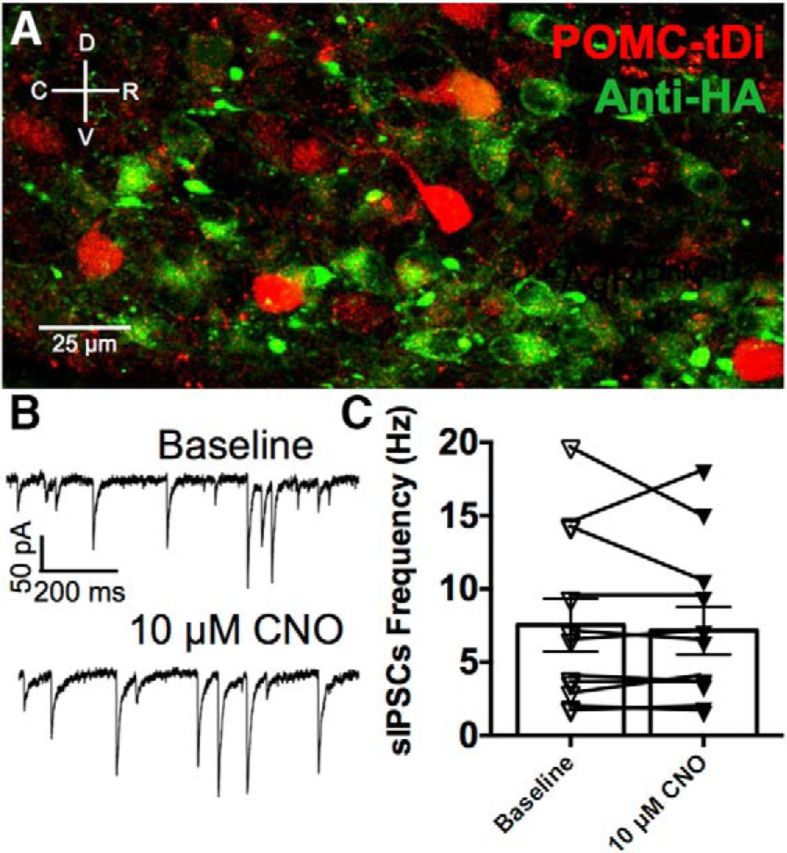
Activation of the Gi-protein-coupled designer receptor hM4Di on AgRP neurons does not affect the frequency of sIPSCs recorded from POMC cells. A, Immunodetection of HA-tagged hM4Di receptors (green) on AgRP neurons in close proximity to POMC cells (red) in the ARC. Verification of hM4Di expression was conducted on all slices following electrophysiological experiments. B, Representative trace demonstrating that bath application of 10 μm CNO does not alter the frequency of sIPSCs recorded from POMC neurons in mice expressing hM4Di receptors on AgRP neurons. C, Compiled data showing the frequency of sIPSCs recorded from POMC neurons at baseline and following bath perfusion of 10 μm CNO (p = 0.56, paired t test).
Disrupting vesicle fusion in AgRP neurons does not alter release of sIPSCs onto POMC neurons
As an additional strategy to investigate the contribution of GABA released from AgRP neurons on the frequency of sIPSCs in POMC neurons, we used cell type-specific expression of the clostridial botulinum neurotoxin serotype B light chain (BoNT/B) in AgRP cells. Botulinum toxin inhibits both evoked and spontaneous vesicle exocytosis (Dreyer and Schmitt, 1983; Dreyer et al., 1987; Molgó et al., 1989) by recognizing and cleaving proteins critically involved in exocytotic machinery (Humeau et al., 2000), including the vesicle-associated membrane protein 2 (VAMP2; also termed synaptobrevin), a portion of the soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complex. A transgenic mouse carrying a Cre recombinase-dependent, eGFP-tagged BoNT/B construct (iBot mouse), allows for cell type-specific expression and visualization of this toxin (Slezak et al., 2012). We used these animals to generate mice with BoNT/B expressed exclusively in AgRP cells (i.e., AgRPcre; iBot; referred to here as AgRPiBOT). A subset of these mice were injected with an AAV encoding Cre-dependent ChR2 tagged with mCherry to both validate the expression pattern of BoNT/B within AgRP cells and to determine whether light-evoked GABAergic connectivity from AgRP cells to POMC cells was indeed blocked by the introduction of BoNT/B. In AgRPiBOT mice injected with ChR2-mCherry, BoNT/B expression was confined to the ARC and was coexpressed with ChR2-positive cells (Fig. 5A,B). Further, we did not detect light-evoked IPSCs in any of our whole-cell recordings from POMC cells (0 of 7 connected; Fig. 5C), indicating that BoNT/B-induced cleaving of VAMP2 in AgRP neurons blocks evoked release of GABA from these cells. In subsequent recordings from AgRPiBOT mice, we found that the frequency of sIPSCs in POMC cells (n = 11; 7.60 ± 1.1 Hz; Fig. 5D,F) were not significantly inhibited relative to recordings from mice not expressing BoNT/B (t(26) = 1.159, p = 0.257, unpaired t test), providing further support to our findings that AgRP neurons do not contribute spontaneously released GABA to POMC cells.
Figure 5.

Disruption of exocytotic machinery in AgRP neurons blocks evoked GABA release from these cells but does not alter the frequency of sIPSCs recorded from POMC cells. A, Confocal image of a midsagittal brain slice from an AgRPcre transgenic mouse showing the expression pattern of a Cre-dependent ChR2 tagged with an mCherry protein in the arcuate nucleus of the hypothalamus as well as eGFP immunoreactivity, indicating that the Cre-dependent BoNT/B is localized to the same region. Scale bar, 100 μm. B, Zoomed image from the same brain slice demonstrating a high degree of colocalization in the merged image. Scale bar, 10 μm. C, Twenty consecutive traces recorded from a POMC cell showing that light-evoked GABAergic IPSCs are absent in recordings from mice where BoNT/B was transgenically expressed in AgRP cells (AgRPiBot). D, Single representative trace depicting the frequency of sIPSCs recorded from a POMC cell in a slice from an AgRPiBot mouse. E, The frequency of sIPSCs recorded from POMC cells in slices taken from AgRPiBot mice. Data are presented as the mean ± SEM. For comparison, the dashed line indicates the mean frequency of sIPSCs recorded from POMC neurons where BoNT/B was not introduced into AgRP neurons (from Fig. 1B).
Activation of AgRP neurons reliably produces GABAergic IPSCs in POMC cells and inhibits POMC cell firing
Despite the apparent lack of contribution of AgRP neurons to spontaneous GABA release onto POMC neurons, synchronous activation of AgRP neurons results in evoked IPSCs in POMC neurons (Dicken et al., 2015, Atasoy et al., 2012). This synchronous release of GABA from AgRP neurons has been previously shown to reduce POMC neuron excitability (Dicken et al., 2015; Atasoy et al., 2012), and we replicated that finding here. ChR2 was virally or transgenically expressed in AgRPcre cells, and electrophysiological recordings were made from POMC cells visually identified with a fluorescent marker (Fig. 6A,B). Light activation of AgRP cells reliably produced GABAergic IPSCs in POMC cells (41 of 48 cells connected; n = 14 mice; Fig. 3C). To determine the effect of GABA released from AgRP neurons on POMC cell firing, cell-attached recordings were made from POMC cells. Photoexcitation of AgRP cells resulted in a cessation of action potential firing in POMC cells (Fig. 6D), and this effect was blocked by the addition of the GABAA receptor antagonist BIC (Fig. 1E; see also Dicken et al., 2015). The earlier studies (Atasoy et al., 2012; Dicken et al., 2015), together with the current results, provide strong evidence that coordinated activation of AgRP neurons is sufficient to silence the firing of POMC neurons.
Figure 6.

Viral expression of ChR2 in AgRP neurons allows for the dissection of an AgRP neuron-to-POMC neuron GABAergic connection. A, Confocal image of a midsagittal hypothalamic section demonstrating the expression of virally delivered ChR2-mCherry in AgRPcre neurons and transgenically expressed eGFP in POMC neurons in the arcuate nucleus. B, Schematic representation of stimulation and recording parameters. C, Light stimulation of AgRP neurons containing ChR2 with 470 nm light (indicated by blue dash above traces) results in an IPSC in POMC cells that was confirmed to be GABA mediated by the addition of the GABAA receptor antagonist BIC to the bath. D, E, Cell-attached recordings demonstrating that light activation of ChR2-containing AgRP cells (indicated by blue dash above trace) terminates firing in POMC cells (D) and that this effect is blocked when GABAA receptors are antagonized with BIC (E).
Non-AgRP neurons likely provide a significant portion of evoked GABA to POMC neurons
Synchronous GABA release from AgRP neurons can clearly inhibit POMC neuron activity, as indicated above. However, the relative contribution of AgRP terminals to evoked GABA inputs to POMC neurons has not been determined. To address this, we again used the MOR pharmacologic approach and compared MOR agonist inhibition of evoked IPSCs in POMC neurons. First, IPSCs were electrically evoked onto POMC cells in slices from both AgRP-MORWT and AgRP-MORKO mice, and inhibition of the amplitude of IPSCs following bath perfusion of 10 μm DAMGO was compared between genotypes. The deletion of MORs from AgRP neurons had no effect on the magnitude of inhibition seen following DAMGO application (AgRP-MORWT: n = 18; 47.96 ± 5.3% baseline; AgRP-MORKO: n = 10; 44.01 ± 7.7% baseline; t(26) = 0.432, p = 0.67, unpaired t test; Fig. 7A,B). The fact that deleting MORs from AgRP terminals caused a loss of MOR agonist-induced inhibition of light-evoked IPSCs from AgRP to POMC neurons (Fig. 3A,B) without significantly affecting electrically evoked IPSCs (Fig. 7) suggests that AgRP neurons supply only a modest portion of the total inputs that can supply electrically evoked GABA to POMC neurons. Next, we compared the inhibition of light-evoked and electrically evoked IPSCs in POMC cells where MORs were intact on AgRP cells (i.e., AgRP-MORWT). We found that MOR-mediated inhibition of light-evoked IPSCs from AgRP neurons (Fig. 3A,B) resulted in a significantly larger inhibition relative to that for electrically evoked IPSCs from nonselective inputs (Fig. 7A,B; t(28) = 2.299, p = 0.03, unpaired t test), indicating that a higher portion of AgRP terminals may express MORs compared with the overall inputs that contribute to the electrically evoked IPSCs.
Figure 7.

Deletion of MORs from AgRP neurons does not affect the inhibition of electrically evoked IPSCs in POMC neurons. A, Average of 15–20 consecutive electrically evoked IPSCs recorded from POMC cells at baseline (black trace) and following bath application of 10 μm DAMGO (red trace) in which AgRP cells contained MORs (AgRP-MORWT, left traces) and where MORs were constitutively deleted from AgRP neurons (AgRP-MORKO, right traces). B, The inhibition of electrically evoked IPSC release onto POMC cells seen following bath application of 10 μm DAMGO is not affected by the presence or absence of MORs on AgRP neurons (AgRP-MORWT: n = 18; 47.96 ± 5.3% of baseline; AgRP-MORKO: n = 10; 44.01 ± 7.7% of baseline; p = 0.67, unpaired t test). Data are presented as the mean ± SEM.
Discussion
Non-AgRP afferents deliver spontaneous inhibition onto POMC cells
Despite the importance of both AgRP and POMC neurons in the regulation and modulation of energy homeostasis, the relevance of communication between these cell types has not been clearly defined. For example, the deletion of GABA release from AgRP neurons, achieved by constitutive knockout of VGAT, does not affect sIPSC frequency in POMC cells (Tong et al., 2008). This observation contrasts with results obtained when diphtheria toxin was targeted to AgRP neurons to ablate them, which caused a robust inhibition of sIPSCs recorded from POMC cells (Wu et al., 2008). This result argues that AgRP neurons provide significant GABA spontaneously to POMC neurons. However, the ablation of AgRP neurons using diphtheria toxin completely removes AgRP neuron contribution from the circuit, including the actions of peptides from these cells. Therefore, it could be that indirect modulation of POMC cells through the release of peptides was also removed. This contrasts the VGAT deletion approach where peptidergic transmission would presumably remain, which could explain why sIPSC frequency was not affected in these studies. Regardless, the physiological connectivity between these cell types warrants further investigation and clarity.
To address the disparate findings, we used transgenic manipulations and subsequent ex vivo electrophysiological experiments to provide clarity regarding the relevance of GABA-mediated transmission from AgRP neurons onto POMC neurons. The results indicate that AgRP neurons do not contribute significantly to spontaneous release of GABA to POMC cells. Our evidence for this is fivefold, as follows: (1) constitutive deletion of GABA release from AgRP neurons achieved via deletion of VGAT does not alter the frequency of sIPSCs in POMC cells; (2) the increase in sIPSC frequency in POMC cells following fasting is not blocked by the deletion of VGAT from AgRP cells; (3) cell type-specific expression of BoNT/B to disrupt neurotransmitter release in AgRP neurons does not affect the frequency of sIPSCs in POMC cells; (4) the inhibition of sIPSC frequency onto POMC cells following MOR activation is not significantly affected by constitutive deletion of MORs from AgRP neurons; and (5) chemogenetic inhibition of AgRP neurons does not inhibit sIPSC release onto POMC cells. Although developmental compensation may contribute to some of these observations, the convergence of evidence from the use of multiple approaches strongly suggests that AgRP neurons are not a primary contributor of the spontaneous release of GABA onto POMC neurons. These findings are particularly interesting considering that we (Figs. 1, 3, 5; Dicken et al., 2015) and others (Atasoy et al., 2012) have observed reliable evoked GABA release from AgRP neurons onto POMC cells.
The finding that AgRP neurons do not provide direct spontaneous GABA release onto POMC cells raises the important question as to where these inhibitory afferents originate. Current data indicate that a diverse array of neuron types and brain regions provides synaptic input to POMC neurons (van den Pol, 2003; Wang et al., 2015), and a combination of these inputs likely contributes to spontaneous GABA release onto POMC cells. Additionally, AgRP neurons represent a small fraction of GABAergic cells within the arcuate nucleus (Dennison et al., 2016), suggesting that other local GABAergic cells could play an important role in spontaneous inhibitory modulation of POMC neurons. Interestingly, the frequency of sIPSCs recorded from POMC cells increased after fasting even when GABA release was blocked in AgRP neurons, indicating that the non-AgRP neurons contributing sIPSCs to POMC cells are metabolically sensitive. It may be that these neurons are responding to the fasting-induced decline in leptin (Vong et al., 2011). Despite the apparent lack of spontaneous GABAergic communication from AgRP neurons to POMC cells, we provide further support for the observation that synchronized activity of AgRP neurons can exert robust inhibitory influence over POMC cell activity.
Coordinated activity of AgRP neurons plays an important role in modulating POMC neuron activity
Using cell type-specific optical stimulation, we replicated the findings of others (Atasoy et al., 2012; Dicken et al., 2015) indicating that the majority of POMC neurons receive direct GABAergic inputs from AgRP cells. The synchronized activity of AgRP neurons is sufficient to silence firing in POMC neurons (Atasoy et al., 2012; Dicken et al., 2015). This finding, along with others demonstrating a critical role in AgRP neuron activity in the regulation of feeding behaviors (Gropp et al., 2005; Tong et al., 2008; Aponte et al., 2011; Atasoy et al., 2012; Betley et al., 2015; Nakajima et al., 2016; Padilla et al., 2016), indicates that the modulation of AgRP neuron activity plays an important role in energy homeostasis. Efferent modulators of AgRP neurons may coordinate the activity of AgRP cells during episodes of caloric deficit, facilitating a silencing of POMC neurons, and a number of recent studies have identified neuronal populations that modulate AgRP activity. For example, thyrotropin-releasing hormone and pituitary adenylate cyclase-activating polypeptide expressing neurons in the paraventricular nucleus of the hypothalamus release glutamate in AgRP neurons to drive feeding (Krashes et al., 2011); while leptin receptor-expressing neurons in the ventral aspect of the dorsal medial hypothalamus inhibit AgRP neurons through GABAergic connections to signal the availability of food (Garfield et al., 2016). Together, it appears as though a number of brain regions are capable of modulating the activity of AgRP neurons during depleted energy states. These convergent signals may help to synchronize the activity of AgRP neurons, which in turn can silence POMC activity. Indeed, in vivo calcium imaging reveals that AgRP neurons increase their activity during caloric deficit (Betley et al., 2015; Chen et al., 2015), and although synchronicity was not directly measured in these studies, an overall increase in activity across a population of cells could result in coordinated activity of a subset of that population.
Despite the reliability and functional consequences of evoked GABAergic transmission from AgRP to POMC neurons that we and others (Atasoy et al., 2012) have shown, the results presented here provide compelling evidence that GABA spontaneously released onto POMC cells originates from non-AgRP sources. Considering the high frequency of sIPSCs conferred onto POMC cells and the energy state dependence of these events, resolving the functional role and afferent origin of sIPSCs in POMC neurons may provide important insight into the neuronal circuits governing feeding behaviors but also other behaviors modulated by POMC neurons and their peptide products.
Spontaneous release of neurotransmitter may play a fundamentally different role than evoked release
Recent debate has emerged regarding the differential release of neurotransmitters through evoked versus spontaneous vesicle fusion (Kavalali, 2015). Central to this debate is whether spontaneous release imparts functionally relevant postsynaptic signals. Indeed, stimulus-independent release events can target different postsynaptic receptor populations (Atasoy et al., 2008; Sara et al., 2011) that can be located at separate postsynaptic sites (Zenisek, 2008; Melom et al., 2013).
The presynaptic mechanisms governing evoked versus spontaneous release also appear to be different (Peled et al., 2014; Schneggenburger and Rosenmund, 2015). For example, the fusion interactions between SNARE proteins appear to differentially regulate these two types of release events (Deák et al., 2006), and a growing consensus suggests that different vesicle populations exist for evoked and spontaneous release of neurotransmitters (Sara et al., 2005; Fredj and Burrone, 2009; Hablitz et al., 2009). This convergent evidence argues that spontaneous vesicle release is not stochastic and likely plays a fundamental role in communication between neurons. Indeed, the relevance of spontaneous neurotransmission may be particularly important when the frequency of sIPSCs is as high as we observe in POMC cells.
Postsynaptically, the reception of spontaneously released neurotransmitters can modulate homeostatic plasticity (Jin et al., 2012) and dendritic protein expression (Sutton et al., 2007; Autry et al., 2011) and can alter spike timing (Otmakhov et al., 1993; Carter and Regehr, 2002). As the spike timing of other neuron types plays an important role in motivated behaviors (Grace, 1991; Schultz, 2007; Mikhailova et al., 2016), it seems plausible that changes in the frequency of sIPSC release onto POMC cells may impart changes in feeding or other behaviors regulated by POMC cells by setting the frequency at which POMC neurons fire action potentials into downstream targets. Further, POMC cells are heterogeneous. They express transcriptional markers for both GABA and glutamate (Hentges et al., 2009; Jarvie and Hentges, 2012; Wittmann et al., 2013) and can release these neurotransmitters in addition to their peptide products (Hentges et al., 2009). The corelease of neurotransmitters from a single cell can play a critical role in modulating target brain regions (Kavalali, 2015), and the excitability of the coreleasing cell appears to modulate how and when multiple neurotransmitters are released (Lee et al., 2010; Borisovska et al., 2013). Therefore, regulating the excitability of POMC neurons through changes in sIPSC frequency may contribute to the manner and circumstances in which POMC cells release individual or multiple neurotransmitters into downstream targets.
Collectively, our data provide evidence that while AgRP neurons can provide robust inhibition of POMC neuron firing through coordinated GABAergic activity, they provide a negligible frequency of spontaneous GABA onto POMC cells. Studies investigating the afferent origin of spontaneous GABAergic inputs onto POMC neurons will shed valuable insight into how a multitude of behavioral processes linked to POMC activity are modulated and may provide critical points of therapeutic intervention to combat overeating and obesity.
Footnotes
This work was supported by National Institutes of Health Grants DK-078749 and DA-032562 (to S.T.H.) and by an award from the Monfort Family Foundation (to S.T.H.). We thank Connie King for her assistance with animal colony management and genotyping.
The authors declare no competing financial interests.
References
- Aponte Y, Atasoy D, Sternson SM (2011) AGRP neurons are sufficient to orchestrate feeding behavior rapidly and without training. Nat Neurosci 14:351–355. 10.1038/nn.2739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atasoy D, Ertunc M, Moulder KL, Blackwell J, Chung C, Su J, Kavalali ET (2008) Spontaneous and evoked glutamate release activates two populations of NMDA receptors with limited overlap. J Neurosci 28:10151–10166. 10.1523/JNEUROSCI.2432-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atasoy D, Betley JN, Su HH, Sternson SM (2012) Deconstruction of a neural circuit for hunger. Nature 488:172–177. 10.1038/nature11270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Autry AE, Adachi M, Nosyreva E, Na ES, Los MF, Cheng PF, Kavalali ET, Monteggia LM (2011) NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature 475:91–95. 10.1038/nature10130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balthasar N, Dalgaard LT, Lee CE, Yu J, Funahashi H, Williams T, Ferreira M, Tang V, McGovern RA, Kenny CD, Christiansen LM, Edelstein E, Choi B, Boss O, Aschkenasi C, Zhang CY, Mountjoy K, Kishi T, Elmquist JK, Lowell BB (2005) Divergence of melanocortin pathways in the control of food intake and energy expenditure. Cell 123:493–505. 10.1016/j.cell.2005.08.035 [DOI] [PubMed] [Google Scholar]
- Barnes MJ, Argyropoulos G, Bray GA (2010) Preference for a high fat diet, but not hyperphagia following activation of mu opioid receptors is blocked in AgRP knockout mice. Brain Res 1317:100–107. 10.1016/j.brainres.2009.12.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betley JN, Cao ZF, Ritola KD, Sternson SM (2013) Parallel, redundant circuit organization for homeostatic control of feeding behavior. Cell 155:1337–1350. 10.1016/j.cell.2013.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betley JN, Xu S, Cao ZF, Gong R, Magnus CJ, Yu Y, Sternson SM (2015) Neurons for hunger and thirst transmit a negative-valence teaching signal. Nature 521:180–185. 10.1038/nature14416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borisovska M, Bensen AL, Chong G, Westbrook GL (2013) Distinct modes of dopamine and GABA release in a dual transmitter neuron. J Neurosci 33:1790–1796. 10.1523/JNEUROSCI.4342-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler AA, Kesterson RA, Khong K, Cullen MJ, Pelleymounter MA, Dekoning J, Baetscher M, Cone RD (2000) A unique metabolic syndrome causes obesity in the melanocortin-3 receptor-deficient mouse. Endocrinology 141:3518–3521. 10.1210/endo.141.9.7791 [DOI] [PubMed] [Google Scholar]
- Carter AG, Regehr WG (2002) Quantal events shape cerebellar interneuron firing. Nat Neurosci 5:1309–1318. 10.1038/nn970 [DOI] [PubMed] [Google Scholar]
- Chen Y, Lin YC, Kuo TW, Knight ZA (2015) Sensory detection of food rapidly modulates arcuate feeding circuits. Cell 160:829–841. 10.1016/j.cell.2015.01.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cone RD. (2005) Anatomy and regulation of the central melanocortin system. Nat Neurosci 8:571–578. 10.1038/nn1455 [DOI] [PubMed] [Google Scholar]
- Cowley MA, Smart JL, Rubinstein M, Cerdán MG, Diano S, Horvath TL, Cone RD, Low MJ (2001) Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature 411:480–484. 10.1038/35078085 [DOI] [PubMed] [Google Scholar]
- Deák F, Shin OH, Kavalali ET, Südhof TC (2006) Structural determinants of synaptobrevin 2 function in synaptic vesicle fusion. J Neurosci 26:6668–6676. 10.1523/JNEUROSCI.5272-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennison CS, King CM, Dicken MS, Hentges ST (2016) Age-dependent changes in amino acid phenotype and the role of glutamate release from hypothalamic proopiomelanocortin neurons. J Comp Neurol 524:1222–1235. 10.1002/cne.23900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dicken MS, Hughes AR, Hentges ST (2015) Gad1 mRNA as a reliable indicator of altered GABA release from orexigenic neurons in the hypothalamus. Eur J Neurosci 42:2644–2653. 10.1111/ejn.13076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreyer F, Schmitt A (1983) Transmitter release in tetanus and botulinum A toxin-poisoned mammalian motor endplates and its dependence on nerve stimulation and temperature. Pflugers Archiv 399:228–234. 10.1007/BF00656720 [DOI] [PubMed] [Google Scholar]
- Dreyer F, Rosenberg F, Becker C, Bigalke H, Penner R (1987) Differential effects of various secretagogues on quantal transmitter release from mouse motor nerve terminals treated with botulinum A and tetanus toxin. Naunyn-Schmiedebergs Arch Pharmacol 335:1–7. 10.1007/BF00165027 [DOI] [PubMed] [Google Scholar]
- Fredj NB, Burrone J (2009) A resting pool of vesicles is responsible for spontaneous vesicle fusion at the synapse. Nat Neurosci 12:751–758. 10.1038/nn.2317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garfield AS, Shah BP, Burgess CR, Li MM, Li C, Steger JS, Madara JC, Campbell JN, Kroeger D, Scammell TE, Tannous BA, Myers MG Jr, Andermann ML, Krashes MJ, Lowell BB (2016) Dynamic GABAergic afferent modulation of AgRP neurons. Nat Neurosci 19:1628–1635. 10.1038/nn.4392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace AA. (1991) Phasic versus tonic dopamine release and the modulation of dopamine system responsivity: a hypothesis for the etiology of schizophrenia. Neuroscience 41:1–24. 10.1016/0306-4522(91)90196-U [DOI] [PubMed] [Google Scholar]
- Gropp E, Shanabrough M, Borok E, Xu AW, Janoschek R, Buch T, Plum L, Balthasar N, Hampel B, Waisman A, Barsh GS, Horvath TL, Brüning JC (2005) Agouti-related peptide-expressing neurons are mandatory for feeding. Nat Neurosci 8:1289–1291. 10.1038/nn1548 [DOI] [PubMed] [Google Scholar]
- Hablitz JJ, Mathew SS, Pozzo-Miller L (2009) GABA vesicles at synapses: are there 2 distinct pools? Neuroscientist 15:218–224. 10.1177/1073858408326431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hentges ST, Otero-Corchon V, Pennock RL, King CM, Low MJ (2009) Proopiomelanocortin expression in both GABA and glutamate neurons. J Neurosci 29:13684–13690. 10.1523/JNEUROSCI.3770-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath TL, Bechmann I, Naftolin F, Kalra SP, Leranth C (1997) Heterogeneity in the neuropeptide Y-containing neurons of the rat arcuate nucleus: GABAergic and non-GABAergic subpopulations. Brain Res 756:283–286. 10.1016/S0006-8993(97)00184-4 [DOI] [PubMed] [Google Scholar]
- Humeau Y, Doussau F, Grant NJ, Poulain B (2000) How botulinum and tetanus neurotoxins block neurotransmitter release. Biochimie 82:427–446. 10.1016/S0300-9084(00)00216-9 [DOI] [PubMed] [Google Scholar]
- Jarvie BC, Hentges ST (2012) Expression of GABAergic and glutamatergic phenotypic markers in hypothalamic proopiomelanocortin neurons. J Comp Neurol 520:3863–3876. 10.1002/cne.23127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin I, Udo H, Rayman JB, Puthanveettil S, Kandel ER, Hawkins RD (2012) Spontaneous transmitter release recruits postsynaptic mechanisms of long-term and intermediate-term facilitation in Aplysia. Proc Natl Acad Sci U S A 109:9137–9142. 10.1073/pnas.1206846109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavalali ET. (2015) The mechanisms and functions of spontaneous neurotransmitter release. Nat Rev Neurosci 16:5–16. 10.1038/nrn3875 [DOI] [PubMed] [Google Scholar]
- Krashes MJ, Koda S, Ye C, Rogan SC, Adams AC, Cusher DS, Maratos-Flier E, Roth BL, Lowell BB (2011) Rapid, reversible activation of AgRP neurons drives feeding behavior in mice. J Clin Invest 121:1424–1428. 10.1172/JCI46229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krashes MJ, Shah BP, Koda S, Lowell BB (2013) Rapid versus delayed stimulation of feeding by the endogenously released AgRP neuron mediators GABA, NPY, and AgRP. Cell Metab 18:588–595. 10.1016/j.cmet.2013.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Kim K, Zhou ZJ (2010) Role of ACh-GABA cotransmission in detecting image motion and motion direction. Neuron 68:1159–1172. 10.1016/j.neuron.2010.11.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Kong D, Shah BP, Ye C, Koda S, Saunders A, Ding JB, Yang Z, Sabatini BL, Lowell BB (2012) Fasting activation of AgRP neurons requires NMDA receptors and involves spinogenesis and increased excitatory tone. Neuron 73:511–522. 10.1016/j.neuron.2011.11.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahler SV, Vazey EM, Beckley JT, Keistler CR, McGlinchey EM, Kaufling J, Wilson SP, Deisseroth K, Woodward JJ, Aston-Jones G (2014) Designer receptors show role for ventral pallidum input to ventral tegmental area in cocaine seeking. Nat Neurosci 17:577–585. 10.1038/nn.3664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melom JE, Akbergenova Y, Gavornik JP, Littleton JT (2013) Spontaneous and evoked release are independently regulated at individual active zones. J Neurosci 33:17253–17263. 10.1523/JNEUROSCI.3334-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer AJ, Hentges ST, Meshul CK, Low MJ (2013) Unraveling the central proopiomelanocortin neural circuits. Front Neurosci 7:19. 10.3389/fnins.2013.00019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikhailova MA, Bass CE, Grinevich VP, Chappell AM, Deal AL, Bonin KD, Weiner JL, Gainetdinov RR, Budygin EA (2016) Optogenetically-induced tonic dopamine release from VTA-nucleus accumbens projections inhibits reward consummatory behaviors. Neuroscience 333:54–64. 10.1016/j.neuroscience.2016.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molgó J, Siegel LS, Tabti N, Thesleff S (1989) A study of synchronization of quantal transmitter release from mammalian motor endings by the use of botulinal toxins type A and D. J Physiol 411:195–205. 10.1113/jphysiol.1989.sp017568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima K, Cui Z, Li C, Meister J, Cui Y, Fu O, Smith AS, Jain S, Lowell BB, Krashes MJ, Wess J (2016) Gs-coupled GPCR signalling in AgRP neurons triggers sustained increase in food intake. Nat Commun 7:10268. 10.1038/ncomms10268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuzzaci D, Laderrière A, Lemoine A, Nédélec E, Pénicaud L, Rigault C, Benani A (2015) Plasticity of the melanocortin system: determinants and possible consequences on food intake. Front Endocrinol (Lausanne) 6:143. 10.3389/fendo.2015.00143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otis TS, Mody I (1992) Differential activation of GABAA and GABAB receptors by spontaneously released transmitter. J Neurophysiol 67:227–235. [DOI] [PubMed] [Google Scholar]
- Otmakhov N, Shirke AM, Malinow R (1993) Measuring the impact of probabilistic transmission on neuronal output. Neuron 10:1101–1111. 10.1016/0896-6273(93)90058-Y [DOI] [PubMed] [Google Scholar]
- Padilla SL, Qiu J, Soden ME, Sanz E, Nestor CC, Barker FD, Quintana A, Zweifel LS, Rønnekleiv OK, Kelly MJ, Palmiter RD (2016) Agouti-related peptide neural circuits mediate adaptive behaviors in the starved state. Nat Neurosci 19:734–741. 10.1038/nn.4274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peled ES, Newman ZL, Isacoff EY (2014) Evoked and spontaneous transmission favored by distinct sets of synapses. Curr Biol CB 24:484–493. 10.1016/j.cub.2014.01.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennock RL, Hentges ST (2016) Desensitization-resistant and -sensitive GPCR-mediated inhibition of GABA release occurs by Ca2+-dependent and -independent mechanisms at a hypothalamic synapse. J Neurophysiol 115:2376–2388. 10.1152/jn.00535.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto S, Roseberry AG, Liu H, Diano S, Shanabrough M, Cai X, Friedman JM, Horvath TL (2004) Rapid rewiring of arcuate nucleus feeding circuits by leptin. Science 304:110–115. 10.1126/science.1089459 [DOI] [PubMed] [Google Scholar]
- Ramirez DM, Kavalali ET (2011) Differential regulation of spontaneous and evoked neurotransmitter release at central synapses. Curr Opin Neurobiol 21:275–282. 10.1016/j.conb.2011.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sara Y, Virmani T, Deák F, Liu X, Kavalali ET (2005) An isolated pool of vesicles recycles at rest and drives spontaneous neurotransmission. Neuron 45:563–573. 10.1016/j.neuron.2004.12.056 [DOI] [PubMed] [Google Scholar]
- Sara Y, Bal M, Adachi M, Monteggia LM, Kavalali ET (2011) Use-dependent AMPA receptor block reveals segregation of spontaneous and evoked glutamatergic neurotransmission. J Neurosci 31:5378–5382. 10.1523/JNEUROSCI.5234-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneggenburger R, Rosenmund C (2015) Molecular mechanisms governing Ca2+ regulation of evoked and spontaneous release. Nat Neurosci 18:935–941. 10.1038/nn.4044 [DOI] [PubMed] [Google Scholar]
- Schultz W. (2007) Multiple dopamine functions at different time courses. Annu Rev Neurosci 30:259–288. 10.1146/annurev.neuro.28.061604.135722 [DOI] [PubMed] [Google Scholar]
- Slezak M, Grosche A, Niemiec A, Tanimoto N, Pannicke T, Münch TA, Crocker B, Isope P, Härtig W, Beck SC, Huber G, Ferracci G, Perraut M, Reber M, Miehe M, Demais V, Lévêque C, Metzger D, Szklarczyk K, Przewlocki R, et al. (2012) Relevance of exocytotic glutamate release from retinal glia. Neuron 74:504–516. 10.1016/j.neuron.2012.03.027 [DOI] [PubMed] [Google Scholar]
- Smart JL, Tolle V, Low MJ (2006) Glucocorticoids exacerbate obesity and insulin resistance in neuron-specific proopiomelanocortin-deficient mice. J Clin Invest 116:495–505. 10.1172/JCI25243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stachniak TJ, Ghosh A, Sternson SM (2014) Chemogenetic synaptic silencing of neural circuits localizes a hypothalamus→midbrain pathway for feeding behavior. Neuron 82:797–808. 10.1016/j.neuron.2014.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton MA, Taylor AM, Ito HT, Pham A, Schuman EM (2007) Postsynaptic decoding of neural activity: eEF2 as a biochemical sensor coupling miniature synaptic transmission to local protein synthesis. Neuron 55:648–661. 10.1016/j.neuron.2007.07.030 [DOI] [PubMed] [Google Scholar]
- Takahashi KA, Cone RD (2005) Fasting induces a large, leptin-dependent increase in the intrinsic action potential frequency of orexigenic arcuate nucleus neuropeptide Y/Agouti-related protein neurons. Endocrinology 146:1043–1047. 10.1210/en.2004-1397 [DOI] [PubMed] [Google Scholar]
- Tong Q, Ye CP, Jones JE, Elmquist JK, Lowell BB (2008) Synaptic release of GABA by AgRP neurons is required for normal regulation of energy balance. Nat Neurosci 11:998–1000. 10.1038/nn.2167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Pol AN. (2003) Weighing the role of hypothalamic feeding neurotransmitters. Neuron 40:1059–1061. 10.1016/S0896-6273(03)00809-2 [DOI] [PubMed] [Google Scholar]
- Vong L, Ye C, Yang Z, Choi B, Chua S Jr, Lowell Bradford BB (2011) Leptin action on GABAergic neurons prevents obesity and reduces inhibitory tone to POMC neurons. Neuron 71:142–154. 10.1016/j.neuron.2011.05.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, He X, Zhao Z, Feng Q, Lin R, Sun Y, Ding T, Xu F, Luo M, Zhan C (2015) Whole-brain mapping of the direct inputs and axonal projections of POMC and AgRP neurons. Front Neuroanat 9:40. 10.3389/fnana.2015.00040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittmann G, Hrabovszky E, Lechan RM (2013) Distinct glutamatergic and GABAergic subsets of hypothalamic pro-opiomelanocortin neurons revealed by in situ hybridization in male rats and mice. J Comp Neurol 521:3287–3302. 10.1002/cne.23350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojcik SM, Katsurabayashi S, Guillemin I, Friauf E, Rosenmund C, Brose N, Rhee JS (2006) A shared vesicular carrier allows synaptic corelease of GABA and glycine. Neuron 50:575–587. 10.1016/j.neuron.2006.04.016 [DOI] [PubMed] [Google Scholar]
- Wu Q, Howell MP, Cowley MA, Palmiter RD (2008) Starvation after AgRP neuron ablation is independent of melanocortin signaling. Proc Natl Acad Sci U S A 105:2687–2692. 10.1073/pnas.0712062105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu AW, Kaelin CB, Morton GJ, Ogimoto K, Stanhope K, Graham J, Baskin DG, Havel P, Schwartz MW, Barsh GS (2005) Effects of hypothalamic neurodegeneration on energy balance. PLoS Biol 3:e415. 10.1371/journal.pbio.0030415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaswen L, Diehl N, Brennan MB, Hochgeschwender U (1999) Obesity in the mouse model of pro-opiomelanocortin deficiency responds to peripheral melanocortin. Nat Med 5:1066–1070. 10.1038/12506 [DOI] [PubMed] [Google Scholar]
- Zeltser LM, Seeley RJ, Tschöp MH (2012) Synaptic plasticity in neuronal circuits regulating energy balance. Nat Neurosci 15:1336–1342. 10.1038/nn.3219 [DOI] [PubMed] [Google Scholar]
- Zenisek D. (2008) Vesicle association and exocytosis at ribbon and extraribbon sites in retinal bipolar cell presynaptic terminals. Proc Natl Acad Sci U S A 105:4922–4927. 10.1073/pnas.0709067105 [DOI] [PMC free article] [PubMed] [Google Scholar]