

Abstract
Calcium-dependent nuclear export of histone deacetylase 1 (HDAC1) was shown previously to precede axonal damage in culture, but the in vivo relevance of these findings and the potential posttranslational modifications of HDAC1 remained elusive. Using acute hippocampal slices from mice of either sex with genetic conditional ablation of Hdac1 in CA1 hippocampal neurons (i.e., Camk2a-cre;Hdac1fl/fl), we show significantly diminished axonal damage in response to neurotoxic stimuli. The protective effect of Hdac1 ablation was detected also in CA3 neurons in Grik4-cre;Hdac1fl/f mice, which were more resistant to the excitotoxic damage induced by intraventricular injection of kainic acid. The amino acid residues modulating HDAC1 subcellular localization were identified by site-directed mutagenesis, which identified serine residues 421 and 423 as critical for its nuclear localization. The physiological phosphorylation of HDAC1 was decreased by neurotoxic stimuli, which stimulated the phosphatase enzymatic activity of calcineurin. Treatment of neurons with the calcineurin inhibitors FK506 or cyclosporin A resulted in nuclear accumulation of phospho-HDAC1 and was neuroprotective. Together, our data identify HDAC1 and the phosphorylation of specific serine residues in the molecule as potential targets for neuroprotection.
SIGNIFICANCE STATEMENT The importance of histone deacetylation in normal brain functions and pathological conditions is unquestionable, yet the molecular mechanisms responsible for the neurotoxic potential of histone deacetylase 1 (HDAC1) and its subcellular localization are not fully understood. Here, we use transgenic lines to define the in vivo relevance of HDAC1 and identify calcineurin-dependent serine dephosphorylation as the signal modulating the neurotoxic role of HDAC1 in response to neurotoxic stimuli.
Keywords: axon, neurodegeneration, neuroprotection
Introduction
Axonal damage is a common pathological feature of several neurodegenerative disorders, including multiple sclerosis, Parkinson's disease, and Alzheimer's disease (Crews and Masliah, 2010; Burke and O'Malley, 2013; Friese et al., 2014). Key morphological changes occurring during axonal degeneration include localized swelling (also known as axonal beading) and axonal fragmentation, followed by irreversible retrograde breakdown of the proximal axon and ultimately death of the affected neuron (Kim et al., 2010; Wang et al., 2012; Friese et al., 2014). These neuropathological changes are shared among many distinct neurodegenerative disorders, thereby suggesting the existence of common molecular mechanisms of axonal damage.
Histone deacetylases (HDACs) are a large family of enzymes that are capable of removing acetyl groups from lysine residues in nucleosomal histones and nonhistone proteins, regulating a diverse range of biological processes (Haberland et al., 2009a). Based on sequence homology, HDACs can be categorized into four classes, including class I (HDAC 1, 2, 3, and 8), class II (HDAC 4, 5, 6, 7, 9, and 10), class III (Sirt1–7), and class IV (HDAC 11). Class II HDACs exhibit tissue specificity and are known to shuttle between the cytoplasm and the nucleus in a stimuli-dependent manner (Cho and Cavalli, 2014). Class III HDACs, sirtuins, are structurally and functionally distinct from other classes (Fiorino et al., 2014). HDAC11 is the sole member of the class IV with a function that is largely unknown. Class I HDACs are more ubiquitously expressed and their levels are higher in the brain (Fischer et al., 2010). Of the class I members, HDAC1 and HDAC2 share a high level of sequence homology and are almost exclusively expressed in the nucleus in normal physiological conditions (Gregoretti et al., 2004). Accordingly, HDAC1 and HDAC2 show functional redundancy in a variety of tissue types and biological processes, such as neuronal development (Montgomery et al., 2009), oligodendrocyte differentiation (Ye et al., 2009), tumorigenesis and survival (Haberland et al., 2009b; Jurkin et al., 2011), and myocardial development (Montgomery et al., 2007). In the adult brain, distinct functions for HDAC1 and HDAC2 have been reported. Although overexpression of HDAC2 in forebrain neurons dramatically impaired synaptic plasticity and memory formation, overexpression of HDAC1 did not lead to any cognitive change (Guan et al., 2009). Similarly, only HDAC1 was shown to exhibit nucleo–cytoplasmic translocation in inflammatory demyelinating lesions. In addition, only shRNA-mediated knock-down of Hdac1, but not Hdac2 or other HDAC members, prevented axonal damage induced by excitotoxic amino acid and cytokine exposure (Kim et al., 2010). However, the in vivo role of HDAC1 in axonal damage is not fully understood. The underlying molecular mechanism that regulates its aberrant subcellular localization is also not known.
HDACs are known to be posttranslationally modified and to regulate a wide range of biological functions such as protein stability, enzymatic activity, complex formation, and subcellular localization (Brandl et al., 2009; Segré and Chiocca, 2011; Mathias et al., 2015). In the nervous system, previous studies showed that injury to sensory axons resulted in HDAC5 nuclear export and phosphorylation by PKC, which promoted axon regeneration (Cho et al., 2013). Because multiple posttranslational modification (PTM) sites are present in the sequence of HDAC1, including phosphorylation, acetylation, carbonylation, and SUMOylation (Brandl et al., 2009; Segré and Chiocca, 2011; Mathias et al., 2015), it is conceivable that HDAC1 nuclear export might also be regulated through PTM mechanisms.
In the current study, we provide ex vivo and in vivo evidence that decreasing HDAC1 levels by genetic ablation is neuroprotective against acute neurotoxicity. In addition, we identify phosphorylation of serine 421 and 423 residues as a critical signal responsible for HDAC1 subcellular localization.
Materials and Methods
Animals.
The following lines were obtained from The Jackson Laboratory: Camk2a-cre (#005359), Grik4-cre (#006474), and Rosa-loxSTOPlox-tdTomato (#007909). Hdac1fl/fl mice (Montgomery et al., 2007) were kindly donated by Dr. Eric Olson. All mice were backcrossed to C57BL/6 for at least six generations. Timed pregnant (embryonic day 15, E15) Sprague Dawley rats were purchased from Charles River Laboratories. Mice of both sexes were used in this study.
Primary culture of hippocampal neurons.
Primary neurons were prepared from embryos of E18 rat as described previously (Haines et al., 2015). Briefly, hippocampi were dissociated into a single cell suspension and were grown in Neurobasal medium supplemented with B-27 (Invitrogen), GlutaMax (Invitrogen), and penicillin/streptomycin (Invitrogen) and maintained at 37°C and 5% CO2. At the third day of culture, neurons were treated with 5 μm AraC in Neurobasal medium for 18 h and replenished with fresh medium. Half of the medium was subsequently changed every 3 d. Cells were used for experiments at 12 d in vitro (DIV). For glutamate/TNFα treatment, cells were treated with 50 μm glutamate (Sigma-Aldrich) and 200 ng/ml recombinant TNFα (Millipore Bioscience Research Reagents, catalog #GF027) for 1 h or longer as specified in the text. For calcineurin inhibition experiments, neurons were pretreated with 1 μm FK506 (Enzo Biochem, catalog #ALX-380-008-M005) or 2 μm cyclosporin A (Enzo Biochem, catalog #BML-A195-0100) dissolved in DMSO for 1 h, followed by treatment with 50 μm glutamate and 200 ng/ml TNFα in the presence of the previously added drugs. In the control group, DMSO was added to the cells in the same amount as the drug treatment groups, followed by glutamate and TNFα treatment.
Hippocampal slice preparation.
Acute dorsal hippocampal slices were prepared from 3- to 4-week-old C57BL/6 mice as described previously (Bozdagi et al., 2000). Briefly, mice were deeply anesthetized with isofluorane and decapitated. The brain was rapidly removed and cooled in chilled (∼4°C) oxygenated (95% O2-5% CO2) sucrose–aCSF consisting of the following (in mm): 233.7 sucrose, 26 NaHCO3, 3 KCl, 8 MgCl2, 0.5 CaCl2, 20 glucose, and 0.4 ascorbic acid. Acute sagittal hippocampal slices (350 μm) were prepared using a Leica VT1000S. Slices were allowed to equilibrate in recording aCSF at room temperature for ∼1 h before transferring to the recording chamber. The recording aCSF was composed of the following (in mm): 117 NaCl, 4.7 KCl, 1.2 MgSO4, 2.5 CaCl2, 1.2 NaH2PO4, 24.9 NaHCO3, and 11.5 glucose. Slices were maintained at 31°C and perfused (1.5 ml/min) with oxygenated aCSF in an immersion chamber during both whole-cell patch-clamp and field recordings.
Electrophysiological recording.
Whole-cell recordings were performed with glass micropipettes pulled from borosilicate glass capillaries. The pipette resistance was 2–4 MΩ. Electrodes were filled with an intracellular solution containing the following (in mm): 124 K+-gluconate, 10 HEPES, 10 phosphocreatine di(tris), 0.2 EGTA, 4 Mg2ATP, and 0.3 Na2GTP, pH 7.3, and 280–290 mOsm. All recordings were made in current-clamp mode using a Multiclamp 200B amplifier (Molecular Devices). Analog signals were low-pass filtered at 2 kHz and digitized at 5 kHZ using a Digidata 1440A interface and pClamp10 software (Molecular Devices). All whole-cell recordings were conducted on CA1 pyramidal neurons. Neurons were visualized using an upright microscope (BX50WI; Olympus) with an X40 water-immersion lens and an IR-1000 infrared CCD monochrome video camera (Dage-MTI). Gigaseal and further access to the intracellular neuronal compartment was achieved in voltage-clamp mode with the holding potential set at −60 mV. Soon after rupturing the membrane, the intracellular solution reached equilibrium with the pipette solution without significant changes in either series resistance or membrane capacitance values. The following membrane properties were characterized: action potential (AP) frequency (Hz), amplitude (mV), and AP half-width (msec). Maximal AP firing of CA1 pyramidal neurons was determined in current-clamp mode as described previously (Simon et al., 2010) using 500 ms depolarizing pulses that increased in intensity by 20 pA per step until AP firing plateaued or until APs failed.
Field EPSPs (fEPSPs) were recorded from the CA1 region of hippocampus evoked by stimulation of the Schaffer collaterals every 30 s with 300 μs pulses through a concentric bipolar tungsten electrode (FHC) (Bozdagi et al., 2000). The input/output (I/O) relationships were measured for amplitudes of fEPSPs elicited by stimulating currents with increasing intensity (0.2–2 mA). Four stimuli were induced and response amplitude averaged for every current intensity step. Slices were allowed to equilibrate for ∼1 h before recording; all field recordings were conducted at 31°C. Recording pipettes had a resistance of 2–4 MΩ and were filled with aCSF.
Offline analysis was performed using Clampfit software (Molecular Devices). AP data were analyzed using two-tailed Student's t tests and fEPSP data using one-way ANOVA with Bonferroni post hoc test. p < 0.05 was considered statistically significant.
Stereotaxic injection of kanic acid.
Male 8- to 12-week-old Hdac1fl/fl or Grik4-cre;Hdac1fl/fl mice were anesthetized (ketamine/xylazine, 100 mg/15 mg/kg, i.p.) and placed into a stereotaxic apparatus (Stoelting). Mice were injected with kainic acid (0.2 μg in 1 μl PBS) at the speed of 250 nl/min into the right ventricle using a pulled borosilicate glass needle (WPI, 1.0 outer diameter/0.75 inner diameter). The tip of the glass needle was trimmed to a diameter of 40–50 μm. Stereotaxic coordinates are in reference to bregma (mm): AP = −0.94, ML = 2.25, DV = −2.1. After each injection, the needle was kept in the brain for 2 min before being slowly retracted. Animals were sacrificed 20 h after injection to assess axonal damage in the hippocampus.
Immunohistochemistry and immunocytochemistry.
For immunohistochemistry, mice were perfused transcardially with 4% paraformaldehyde (PFA). Mouse brains were extracted and postfixed for 24 h in 4% PFA, followed by soaking in 30% sucrose and embedding in optimal cutting temperature medium. Coronal brain sections were cut at 16 μm using a cryostat and permeabilized with blocking buffer (0.1 m PB, 0.1% gelatin, 1% BSA, 0.002% azide, 0.1% Triton X-100, and 10% normal goat serum) for 1 h at room temperature. Sections were then incubated with primary antibodies at 4°C overnight, followed by incubation with corresponding secondary antibodies and DAPI counterstaining. For immunocytochemistry, cells were fixed with 4% PFA for 20 min at room temperature, permeabilized with the same blocking buffer, and incubated with primary and secondary antibodies similarly as described above. The following primary antibodies were used: anti-SMI32 (Calbiochem, catalog #NE1023, 1:500), anti-neurofilament medium chain (NFM; Millipore, catalog #ab1987, 1:500), anti-neurofilament heavy chain (anti-NFH; Millipore, catalog #MAB5448, 1:1000), anti-HDAC1 (Invitrogen, catalog #PA1-860, 1:1000), anti-NeuN (Millipore, catalog #MAB377B, 1:500), anti-FLAG (Sigma-Aldrich, catalog #F1804, 1:2000), anti-phosphoSer421, 423-HDAC1 (Thermo Fisher, catalog #PA5-36911, 1:1000). All images were taken on a Zeiss LSM-780 confocal microscope with Zen software. Relative SMI32 immunofluorescence was quantified using the JACoP plugin V2.1.1 (Bolte and Cordelieres, 2006) in ImageJ by measuring the percentage of NFH+ (or NFM+) area, which colocalized with SMI32+ pixels. Quantification of relative SMI32 immunofluorescence in the CA3 was limited to regions 100 μm adjacent to the CA3 pyramidal cell layer (100 μm toward the stratum lucidum and another 100 μm toward the stratum oriens).
Overexpression.
At 6 DIV, 3 × 104 hippocampal neurons plated in 8-well chamber slides were transfected with C-terminal FLAG-tagged HDAC1 constructs in the pBJ5 mammalian expression vector (a gift from Dr. Stuart Schreiber at Broad Institute) using Invitrogen Lipofectamine 2000 according to the manufacturer's instructions. Point mutations in WT HDAC1 were generated using the QuikChange Site-Directed Mutagenesis Kit (Agilent Technologies) following the manufacturer's instructions at positions specified in the text to disrupt HDAC1 acetylation, carbonylation, and phosphorylation. For lentivirus-mediated overexpression, the same C-terminal FLAG-tagged HDAC1 constructs [WT and the serine421, 423 mutated (SA) mutant] were cloned into pLVX-IRES-ZsGreen1 Vector (Clontech) at C&M Biolabs. Fresh medium mixed with lentivirus at a multiplicity of infection of 5 were mixed with neurons at 6 DIV. After 18 h of incubation, the virus-containing medium was replaced with fresh medium and neurons were maintained at 37°C and 5% CO2 until use at 12 DIV.
Coimmunoprecipitation and Western blot.
Total proteins from cultured neurons were extracted using a lysis buffer containing 50 mm HEPES, pH 7.4, 150 mm NaCl, 1% NP-40, 1 mm dithiothreitol, 1 mm EDTA, 0.01% phenylmethylsulfonyl fluoride, 1× protease inhibitor mixture (Thermo Scientific, catalog #88265), and 1× phosphatase inhibitor (Sigma-Aldrich, catalog #p0044). Collected cells were incubated in lysis buffer for 20 min on ice followed by cell lysis using a TissueLyser LT (Qiagen) (50 oscillations/s for 2 min). Protein concentrations were determined using the Lowry protein assay (Bio-Rad, catalog #5000112). For immunoprecipitation of HDAC1 variants, 2 mg of total protein was incubated overnight at 4°C with 2 μg of polyclonal rabbit anti-FLAG antibody (Sigma-Aldrich, catalog #F7425), followed by incubation with protein A agarose (Millipore, catalog #16-156) at room temperature for 1 h. Immunoprecipitated samples and total protein (20 μg per lane) was resolved by SDS-PAGE on 4–12% precast gels (Sigma-Aldrich TruPAGE, catalog #PCG2011) and transferred to PVDF membranes. Membranes were blocked in 5% (w/v) BSA in TBS-Tween (TBST) and incubated overnight with the following primary antibodies diluted in the blocking buffer: anti-HDAC1 (Invitrogen, catalog #PA1-860, 1:1000), anti-phosphoSer421, 423-HDAC1(Thermo Fisher, catalog #PA5-36911, 1:1000; Abnova, catalog #PAB16973, 1:1000), anti-FLAG (Sigma-Aldrich, catalog #F7425, 1:2000), anti-β-actin (Sigma-Aldrich, catalog #A5441, 1:1000), anti-KPNB1 (Abcam, catalog #ab45938, 1:1000), anti-CRM1 (BD Biosciences, catalog #611832, 1:1000), anti-KPNA1 (Proteintech, catalog #18137-1-AP, 1:1000), and anti-KPNA4 (Proteintech, catalog #12463-1-AP, 1:1000). Immunoreactive bands were visualized using horseradish peroxidase-conjugated IgG light-chain-specific secondary antibodies for 1 h followed by chemiluminescence with ECL-Plus Western Blotting Detection System (GE Healthcare). Images were taken on a Bio-Rad molecular imager and quantified by ImageJ.
Calcineurin activity assay.
Calcineurin activity was determined using a Calcineurin Cellular Activity Assay Kit (Enzo Biochem, catalog #BML-AK816-0001) following the manufacturer's instructions. Briefly, treated neurons were collected using the lysis buffer provided in the kit with addition of proteinase inhibitors. The extracts were then passed through desalting resins to remove free phosphate and then incubated with the calcineurin substrate RII phosphopeptide and 5 μm okadaic acid (OA) to inhibit the activity of the phosphatases PP1 and PP2A in the assay buffer containing calcium and calmodulin. Background phosphatase activity (OA + EGTA) was determined by incubating the desalted lysates with the RII substrate and 5 μm okadaic acid in the EGTA buffer in the absence of calcium and calmodulin. Human recombinant calcineurin (40 U) was used as a positive control. After incubation at 30°C for 30 min, 100 μl of BIOMOL Green were added to terminate the reaction and plates were kept at room temperature for 20 min to allow color development immediately before absorbance detection at 620 nm using a microplate reader. The absorbance contributed by calcineurin enzymatic activity was calculated using the following equation: A620 [OA – (OA + EGTA)], which was further converted to absolute phosphate release (nmol) using a standard curve prepared for each assay. Finally, absolute phosphate release values were normalized to protein concentrations determined by a modified Lowry assay (Bio-rad, catalog #5000112).
Experimental design and statistical analysis.
Primary neurons prepared from embryos of E18 rats were challenged by glutamate/TNFα (see Figs. 1; 6), overexpressed with various Hdac1 constructs (see Fig. 5), or rescued by calcineurin inhibitors after glutamate/TNFα treatment (see Fig. 7), followed by assessment of neurotoxicity. A minimum of three individual neuronal preparations from three distinct rats were used for each experiment and each experiment was performed in triplicate. Acute dorsal hippocampal slices were prepared from 3- to 4-week-old WT or mutant mice and incubated in aCSF in the presence or absence of glutamate/TNFα, followed by electrophysiological recordings (see Fig. 2). After verification of Grik4-cre recombination efficiency and specificity (see Fig. 3), kainic acid was injected into the right ventricle of WT or mutant mice (8–12 weeks old), followed by perfusion, cryopreservation, sectioning, and immunohistochemical assessment of axonal damage (see Fig. 4). All transgenic mice used in the study were backcrossed to C57BL/6 for at least six generations. Detailed experimental procedures can be found in the corresponding Materials and Methods subsections.
Figure 1.

Glutamate and TNFα induce cytoplasmic localization of HDAC1 in hippocampal neurons in culture. A, Primary cultures of rat hippocampal neurons (12 DIV) either untreated (control) or treated with 50 μm glutamate and 200 ng/ml TNFα (Glut/TNFα) for 1 h and stained with DAPI and antibodies against HDAC1 (green) or NFM (red). B, Primary cultures of rat hippocampal neurons treated the same way as described in A and stained with DAPI and antibodies against HDAC1 (green) or nonphosphorylated NFH (SMI32, red), a marker of axonal damage. Scale bars, 25 μm.
Figure 6.

Glutamate- and TNFα-induced HDAC1 translocation coincides with HDAC1 dephosphorylation. A, HEK293 cells naive (uninfected, NA) or after overexpression of FLAG-tagged WT or SA HDAC1. Total proteins were extracted, immunoprecipitated with anti-FLAG antibody, and probed with indicated antibodies. B, Western blot analysis of phosphorylated S421, 423-HDAC1 (P-HDAC1) and total HDAC1 from rat hippocampal neurons (12 DIV) treated with Glut/TNFα for 30 min to 3 h. Quantification from three independent experiments and P-HDAC1 levels referred to total HDAC1. Data represent average ± SEM. Data were analyzed with one-way ANOVA with Bonferroni post hoc test, *p < 0.05, **p < 0.01 compared with the untreated control (the first bar), p > 0.05 (NS). C, Rat hippocampal neurons treated with Glut/TNFα for 1 h were immunostained for HDAC1 or P-HDAC1 (green), SMI32 (red), NFH (gray), and DAPI. Scale bar, 20 μm.
Figure 5.
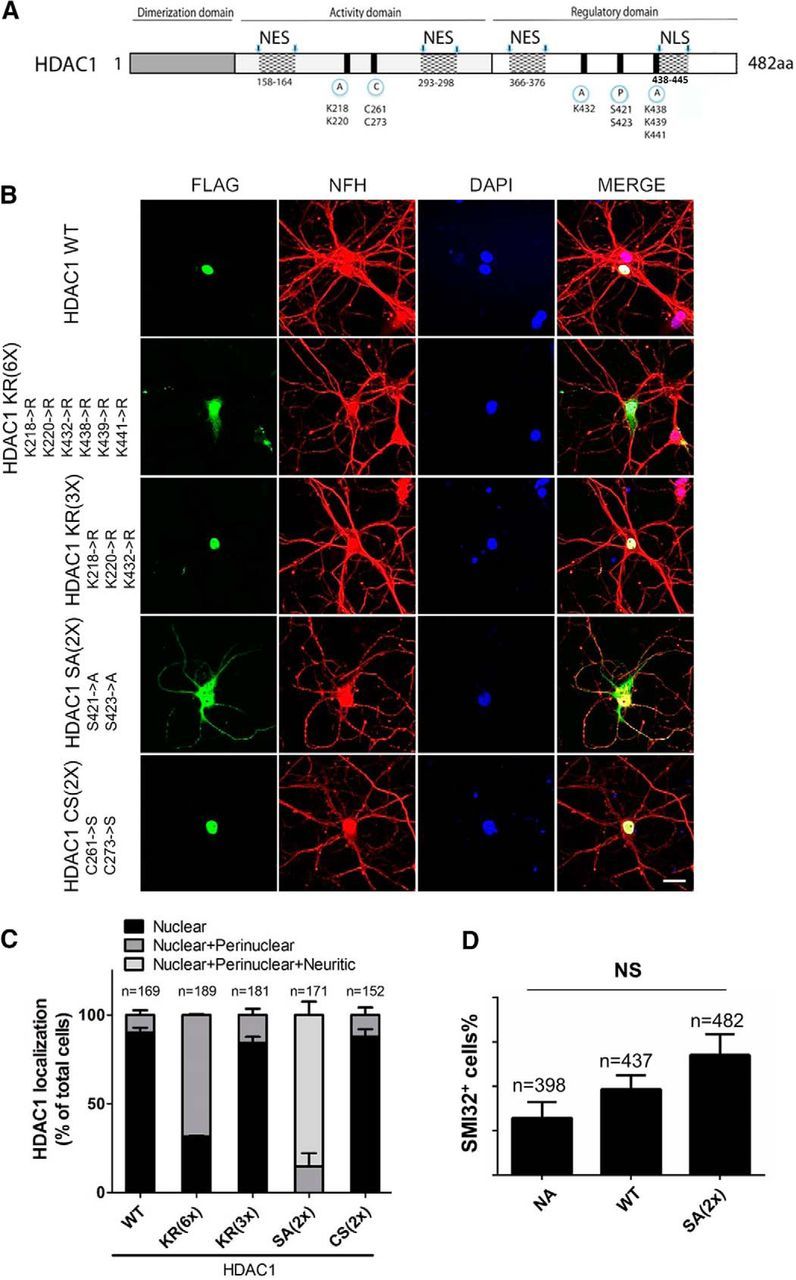
Mutation of phosphorylation sites induce HDAC1 nuclear export. A, Schematic representation of HDAC1 protein sequence displaying its domains and PTM sites. A, Acetylation; C, carbonylation; P, phosphorylation; NES, nuclear export signal. B, Primary cultures of rat hippocampal neurons transfected with FLAG-tagged HDAC1 constructs containing different mutations on PTM sites were immunostained against FLAG (green) and NFH (red) and counterstained with DAPI (blue). Scale bar, 20 μm. C, Quantification of subcellular localization of FLAG signal from B showing that only mutation of phosphorylation sites (S421, 423→A) induced localization of recombinant HDAC1 to the neuronal cytosolic processes. Error bars indicate mean ± SEM. D, Quantification of neurons with SMI32+ processes over total DAPI+ cells after lentivirus-mediated overexpression of either WT or the nonphosphorylated mutant HDAC1 HDAC1 SA(2×). NA, Uninfected naive neurons. Error bars indicate mean ± SEM. Data were analyzed with one-way ANOVA with Bonferroni post hoc test, p > 0.05 (NS).
Figure 7.
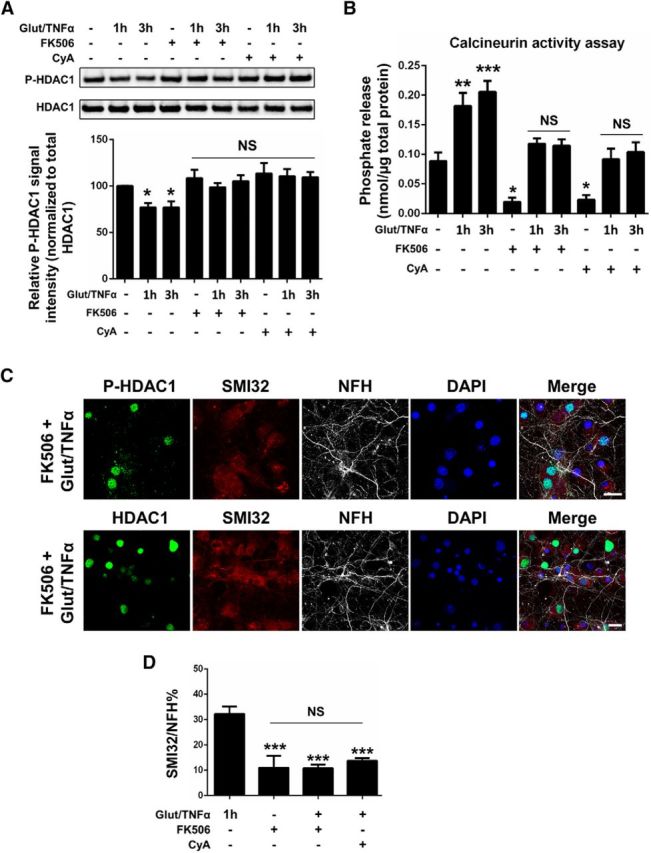
HDAC1 dephosphorylation is blocked by the calcineurin-specific inhibitors FK506 and cyclosporin A. A, Western blot analysis of phosphorylated S421, 423-HDAC1 (P-HDAC1) and total HDAC1 from primary hippocampal neurons treated with Glut/TNFα in the presence or absence of calcineurin-specific inhibitors FK506 (1 μm) or CyA (2 μm). Quantification from three independent experiments and P-HDAC1 levels referred to total HDAC1. Data represent average ± SEM. Data were analyzed with one-way ANOVA with Bonferroni post hoc test, *p = 0.02 compared with the untreated control (the first bar). B, Calcineurin activity of neurons treated the same way as in A. Data are expressed as phosphate release (nmol) per micrograms total protein. Data represent average ± SEM. Data were analyzed with one-way ANOVA with Bonferroni post hoc test, ***p = 0.0002, **p = 0.0025, *p = 0.03 (FK506+), *p = 0.04 (CyA+) compared with the untreated control (the first bar), p > 0.05 (NS). C, Rat hippocampal neurons pretreated with FK506, followed by Glut/TNFα treatment for 1 h, were immunostained for HDAC1 or P-HDAC1 (green), SMI32 (red), NFH (gray), and DAPI. Scale bar, 20 μm. D, Percentage of NFH+ neurites colocalized with SMI32+ neurites was quantified in six to eight independent images (0.27 mm2 per image) containing at least 400 DAPI+ cells. Data represent average ± SEM. Data were analyzed with one-way ANOVA with Bonferroni post hoc test, ***p < 0.0001 compared with the Glut/TNFα only group (the first bar), p > 0.05 (NS).
Figure 2.
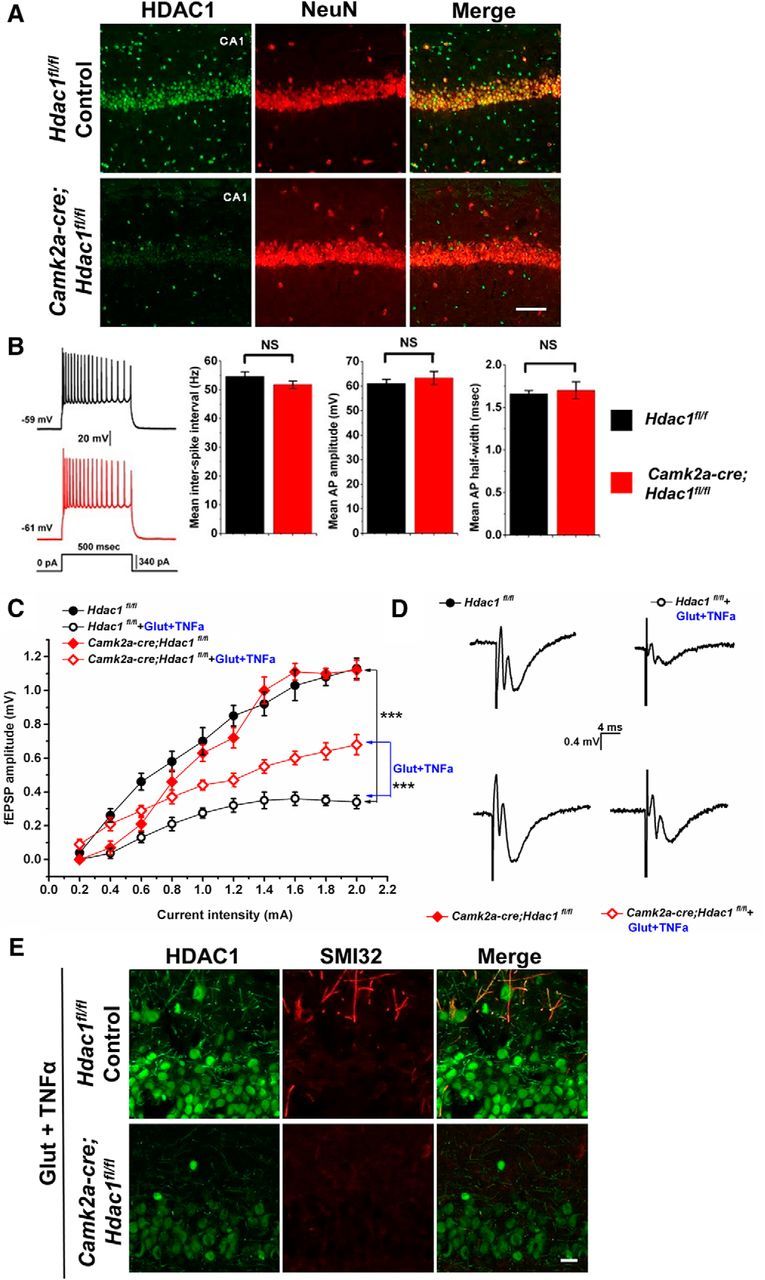
Conditional ablation of Hdac1 in Camk2a-cre;Hdac1fl/fl CA1 pyramidal neurons is protective from axonal damage caused by exposure to glutamate and TNFα. A, Micrographs through area CA1 from adult Hdac1fl/fl control (top row) or Camk2a-cre;Hdac1fl/fl conditional knock-out mice (bottom row) immunostained for HDAC1 (green) and NeuN (red). HDAC1 immunoreactivity is undetectable in CA1 pyramidal cells, but is still present in interneurons, as expected because such neurons do not express Camk2a. Scale bar, 100 μm. B, Representative examples of APs in Hdac1fl/fl control (black record; 9 cells from 4 animals) or Camk2a-cre;Hdac1fl/fl (red record; 7 cells from 4 animals) CA1 pyramidal neurons in response to depolarizing current injection. There were no significant differences in the mean interspike interval (p = 0.47), mean amplitude (p = 0.17), or mean half-width (p = 0.48) of APs in the floxed control CA1 neurons relative to the mutant CA1 neurons. Error bar indicates mean ± SEM. Data were analyzed with two-tailed Student's t tests. C, Comparison of I/O relationships across genotypes and experimental conditions. There were no differences in I/O relationships between untreated Hdac1fl/fl controls (black filled circles, n = 7 from 4 animals) or Camk2a-cre;Hdac1fl/fl mice (red filled diamonds, n = 7 from 4 animals). Floxed control slices challenged with 100 mm glutamate + 200 ng/ml TNFα for 1 h (black open circles, n = 5 from 4 animals) show a larger reduction in fEPSP amplitude compared with HDAC1 knock-out slices receiving the same glutamate/TNFα treatment (red filled diamonds, n = 7 from 4 animals) relative to baseline, suggesting that ablation of Hdac1 was partially protective of the damaging effects of Glut and TNFα on axonal and synaptic function. Data were analyzed with one-way ANOVA with Bonferroni post hoc test, **p = 0.0099, ***p = 7.9 × 10−11. D, Representative fEPSP recordings from the four different conditions as indicated in C. E, Acute hippocampal slices treated with 100 μm glutamate and 200 ng/ml TNFα for 1 h were immunostained for HDAC1 (green) and nonphosphorylated neurofilament-H (SMI32, red) to assess axonal damage. Scale bar, 20 μm.
Figure 3.
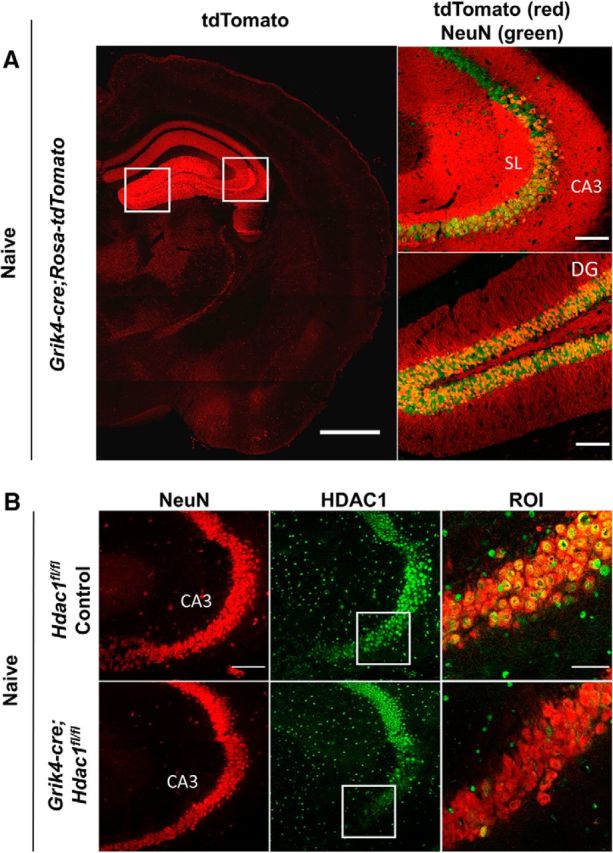
Immunohistochemical analysis of Grik4-cre;Hdac1fl/fl mice validates the selective knock-down of HDAC1 in CA3. A, Confocal image of cryosections from the reporter Grik4-cre;Rosa-tdTomato mice reveals the expression of the recombinase Cre mostly limited to CA3 pyramidal neurons and, to a lower degree, also in dentate gyrus (DG) granule neurons. Recombined cells are marked by tdTomato expression (red) and neurons are labeled by NeuN (green) staining (n = 3). Scale bar, 1000 μm in the left panel. Boxed regions are enlarged in the right panel. Scale bar, 100 μm. SL, Stratum lucidum. B, Representative confocal images of hippocampal CA3 neurons in cryosections from Hdac1fl/fl controls or Grik4-cre;Hdac1fl/fl mice (8–12 weeks old, n = 3 in each group) immunostained with NeuN (red) and HDAC1 (green) (Scale bar, 100 μm). Boxed regions are enlarged in ROI. Scale bar, 50 μm.
Figure 4.

CA3 pyramidal neurons in Grik4-cre;Hdac1fl/fl mice display protection from acute axonal damage induced by kainic acid. A, Hippocampal CA3 from 8- to 12-week-old Hdac1fl/fl controls (n = 7) or from Grik4-cre;Hdac1fl/fl mice (n = 8) received unilateral intracerebroventricular injection of 0. 2 μg (in 1 μl PBS) kainic acid and were perfused 20 h later. Cryosections were immunostained for NFM (red) and nonphosphorylated NFH (SMI32, green) and counterstained with DAPI (blue). Scale bar, 100 μm. Boxed regions are enlarged in ROI. Scale bar, 50 μm. B, Quantification of SMI32 intensity relative to NFM in a region spanning 100 μm radius from the CA3 pyramidal cell layer (marked by dense DAPI staining). Regions used for quantification are depicted by dashed yellow lines in A. Error bars indicate mean ± SEM. Data were analyzed by Student's t test, *p = 0.001.
Detailed sample size, statistical comparisons, and the precise results for all experiments can be found in the corresponding text describing the experiment and in the figure legends.
Two-tailed Student's t test was used when only two experimental variables were compared, one-way ANOVA with Bonferroni post hoc test was used when more than two experimental variables were compared. Statistical analysis for the electrophysiological data was performed using Clampfit software. All other statistical analyses were performed using GraphPad Prism 6.
Results
Ablation of neuronal Hdac1 protects neurons from acute exposure to excitotoxicity and inflammatory cytokines
Previous work from our laboratory focused on the importance of nuclear–cytoplasmic transport of various proteins in neurotoxicity (Kim et al., 2010; Haines et al., 2015). One of these molecules was HDAC1, which showed a typical nuclear localization in untreated rat hippocampal neurons (Fig. 1A, left), but became aberrantly colocalized with NFM, after treatment with 50 μm glutamate and 200 ng/ml TNFα (Fig. 1A, right) for 1 h. Neuronal processes characterized by the presence of cytosolic HDAC1, also showed immunoreactivity for the antibody SMI32 (Fig. 1B), recognizing the nonphosphorylated form of NFH and previously described as marker of axonal damage and neurotoxicity (Trapp et al., 1998).
To further define the functional relevance of HDAC1 in vivo, we generated two mouse lines with conditional ablation of Hdac1 in neurons. The first line (Camk2a-cre;Hdac1fl/fl) was characterized by the neuronal expression of the Cre recombinase driven by the Camk2a (calcium/calmodulin-dependent protein kinase II α) promoter, which is widely expressed in the forebrain and hippocampal area CA1 (Tsien et al., 1996). The efficiency and specificity of Hdac1 deletion was validated by immunohistochemistry of hippocampal sections from 8- to 12-week-old adult mice. In Hdac1fl/fl control mice, a very strong immunoreactive signal for HDAC1 was detected in NeuN+ pyramidal cells and other cells in the CA1 region, whereas in the Camk2a-cre;Hdac1fl/fl mutant mice, the signal was barely detectable in pyramidal cells of CA1, although it was retained in other cells (Fig. 2A). The dentate gyrus and the CA3 regions did not show clear differences between controls and mutants (data not shown).
We next used acute hippocampal slices from floxed controls or from Camk2a-cre;Hdac1fl/fl mutants to investigate whether conditional ablation of Hdac1 affected the baseline properties of intrinsic excitability using whole-cell patch-clamp recordings from CA1 pyramidal cells. In response to depolarizing current steps, we found no significant differences between genotypes in mean interspike interval (p = 0.47), mean amplitude (p = 0.17), or half-width (p = 0.48) of APs at their maximal activation state (i.e., steps during plateau or immediately before AP failure) (Fig. 2B). These data suggest that ablation of Hdac1 in CA1 neurons did not affect intrinsic membrane excitability.
Next, we assessed the effect of Hdac1 ablation on evoked functional properties of CA1 neurons in acute hippocampal slices exposed to bath-application of excitotoxins (i.e., 100 μm glutamate) and inflammatory cytokines (200 ng/ml TNFα). Baseline I/O curves were generated by monitoring postsynaptic responses in CA1 neurons after Schaffer collateral stimulation in Hdac1fl/fl control and Camk2a-cre;Hdac1fl/fl mutant mice. No difference was detected in I/O curves between untreated Hdac1fl/fl controls and Camk2a-cre;Hdac1fl/fl mutant mice (Fig. 2C), demonstrating that Hdac1 ablation did not by itself affect evoked baseline responses of CA1 neurons. The experiment was then repeated after bath application of glutamate/TNFα for 1 h. Under these conditions, we found significantly altered I/O responses in the Hdac1fl/fl control slices, with diminished responses at each intensity level of stimulation (p = 7.9 × 10−11; Fig. 2C,D). In contrast, conditional ablation of Hdac1 partially protected CA1 knock-out neurons from the excitotoxic effects of glutamate/TNFα. In Camk2a-cre;Hdac1fl/fl mutant slices challenged with glutamate/TNFα, neurons displayed I/O responses that were significantly greater at each intensity level compared with treated Hdac1fl/fl control slices (p = 0.0099; Fig. 2C,D). After recordings, slices were fixed and processed for immunohistochemistry, which revealed less prominent axonal damage in CA1 neurons from the Camk2a-cre;Hdac1fl/fl mutants compared with controls (Fig. 2E). Together, these data suggest that ablation of Hdac1 partially protected hippocampal neurons from the damaging effect of exposure to excitotoxic amino acids and inflammatory cytokines.
Genetic ablation of Hdac1 protects neurons from kainic-acid-induced damage in vivo
To further define whether the protective effect of Hdac1 ablation could also be observed in live mice, we tested the response of control and mutant mice to kainic acid injection. Kainic acid is a nondegradable glutamate analog that induces excitotoxic neurodegeneration most effectively in neurons expressing high levels of its receptor (e.g., CA3 hippocampal neurons compared with those in CA1 or dentate gyrus; Wang et al., 2005). To address specifically the potential neuroprotective effect of Hdac1 ablation on CA3 neurons, we generated a second line of mutant mice expressing Cre recombinase under the control of the Grik4 (glutamate receptor, ionotropic, kainate 4) promoter (Grik4-cre;Hdac1fl/fl). The specificity of expression of the recombinase was first validated by crossing the Grik4-cre mice with the reporter Rosa-tdTomato line, which confirmed the high levels of expression in the CA3 region and in the dentate gyrus granule neurons (Fig. 3A) in a pattern that was consistent with previous reports (Filosa et al., 2009). In agreement with the distribution of the recombinase, HDAC1 immunoreactivity was lost in several CA3 neurons of Grik4-cre;Hdac1fl/fl mice, whereas adjacent CA1 region and dentate gyrus retained the expression detected in controls (Fig. 3B).
Kainic acid was injected into the right ventricle and axonal damage was assessed in the ipsilateral hippocampus. The relative immunoreactivity of SMI32 was then quantified within the area immediately adjacent to the CA3 pyramidal cell layer (Fig. 4B), in mice of either genotype and normalized to the intensity of neurofilament. Consistent with the neuroprotective effect of Hdac1 ablation, we detected higher SMI32 immunoreactivity in the ipsilateral CA3 region of control mice than in that of Grik4-cre;Hdac1fl/fl mice (p = 0.001; Fig. 4B). Together, these data present strong evidence that HDAC1 plays an important role in neurotoxicity after exposure to pathological stimuli.
Subcellular localization of HDAC1 is modulated by serine phosphorylation
To begin unraveling the molecular mechanisms underlying the subcellular localization of HDAC1, we adopted a site-directed mutagenesis approach and targeted amino acids that are sites of PTM unique to HDAC1. The specific residues included: lysine (K218, 220, 432, 438, 439, 441) to target acetylation (Luo et al., 2009; Yang et al., 2012), cysteine (C261, 273) to target carbonylation (Doyle and Fitzpatrick, 2010), and serine (S421, 423) to target phosphorylation (Cai et al., 2001; Pflum et al., 2001). A series of FLAG-tagged (C terminus) HDAC1 constructs were generated and the nomenclature indicated the number and type of residues that were replaced. For instance, HDAC1 KR(3×) had 3 lysine residues replaced with arginines (K218, 220, 432→R), whereas HDAC1 KR(6×) had all 6 lysine residues replaced (K218, 220, 432, 438, 439, 441→R). Similarly, HDAC1 CS(2×) indicated that two cysteine residues were replaced with serines (C261, 273→S), whereas HDAC1 SA(2×) described that two serine residues were substituted with alanines (S421, 423→A). Transfection of the naive WT HDAC1 molecule or the mutant constructs into rat hippocampal neurons was followed by immunocytochemistry using an antibody against the FLAG peptide to define the subcellular localization of HDAC1. Neurofilament antibodies were used to highlight the neuronal processes and DAPI was used to delineate the nuclei. Nuclear localization of FLAG in neurons transfected with the WT HDAC1 was used as a reference and validation for the experimental approach (HDAC1 WT; Fig. 5B). Nuclear localization was also detected for HDAC1 CS(2×) and for HDAC1 KR(3×), whereas HDAC1 KR(6×) became perinuclear (Fig. 5B), possibly because the mutation partially interfered with the nuclear localization signal (NLS) (Fig. 5A). The most dramatic difference in localization was detected for HDAC1 SA(2×), which was mostly localized to neuronal processes (Fig. 5B) and therefore resembled the distribution of endogenous HDAC1 in neurons undergoing neuritic damage (Fig. 1A). Because HDAC1 serine421, 423 have been reported previously to be constitutively phosphorylated in other cell types (Karwowska-Desaulniers et al., 2007), we reasoned that their substitution with alanines would mimic the dephosphorylated form of the molecule. Therefore, our results suggest that serine phosphorylation is an important determinant of the subcellular localization of HDAC1.
To further investigate whether overexpression of the HDAC1 serine mutant HDAC1 SA(2×) was sufficient to induce neurotoxicity even in the absence of external noxious stimuli, we infected rat hippocampal neurons with lentiviruses encoding either FLAG-tagged WT HDAC1 or the SA(2×) mutant. Uninfected naïve neurons were used as controls. Immunostaining of FLAG revealed 90% transduction efficiency (n = 1152 cells counted) and a clear nuclear localization of WT HDAC1, whereas the SA(2×) mutant was mostly localized to cell body and cytoplasmic processes (data not shown), thereby replicating the results obtained by plasmid-mediated overexpression (Fig. 5B). Quantification of SMI32+ immunoreactivity as marker of damage revealed a nonsignificant trend toward increased neurotoxicity in the SA(2×)-expressing cells compared with neurons overexpressing full-length HDAC1 and compared with untransduced controls (Fig. 5D). Together, these data suggest that nuclear retention of HDAC1 requires phosphorylation of serine residues 421 and 423 and reveal that cytoplasmic localization of the nonphosphorylated HDAC1 per se is not sufficient to induce neurotoxicity in cultured hippocampal neurons.
HDAC1 dephosphorylation is induced by neurotoxic stimuli that favor its distribution to cytoplasmic processes
Since we identified serine421, 423 as important for HDAC1 subcellular localization, we used two antibodies raised against the phosphorylated form of the enzyme in order to define whether experimental conditions favoring cytosolic localization of HDAC1 also modulated the phosphorylation state of these residues. We first assessed antibody specificity by Western blot analysis of protein extracts from HEK293 cells in three conditions: WT overexpressors (expressing both endogenous HDAC1 as well as the transfected full-length FLAG-tagged HDAC1), SA-HDAC1 overexpressors (expressing both endogenous HDAC1 as well as the FLAG-tagged serine421, 423 to alanine mutant HDAC1) and naive cells expressing only the endogenous HDAC1. Whole-cell protein extracts were probed with antibodies for HDAC1, which detected similar levels of the enzyme in all three conditions. They were also probed for phospho-HDAC1, which recognized a band (P-HDAC1) in all three conditions, consistent with the nuclear localization of the endogenous enzyme (Fig. 6A). The same extracts were then immunoprecipitated with anti-FLAG antibodies to capture only the FLAG-tagged overexpressed HDAC1 (either WT or SA mutant). As expected, no FLAG immunoprecipitation could be detected in untransfected cells. Probing the immunoprecipitated proteins with phosphospecific antibodies from two independent sources (Thermo Fisher and Abnova) revealed an immunoreactive band only in the FLAG-immunoprecipitated extracts from the WT overexpressing cells, not in those overexpressing the serine SA-HDAC1 mutant or in the naive cells (Fig. 6A), thereby confirming the specificity of the phosphospecific HDAC1 antibodies.
Next, we investigated whether treatment of cultured rat hippocampal neurons with glutamate/TNFα, previously shown to favor localization of HDAC1 to cytoplasmic processes, would also decrease phosphorylation of serine residues. Western blot analysis revealed high levels of P-HDAC1 in untreated neurons, consistent with the nuclear distribution of HDAC1 in healthy cells and a progressive decline of immunoreactivity after 1–3 h of glutamate/TNFα treatment (Fig. 6B). Immunocytochemical analysis of P-HDAC1 using phosphospecific antibodies confirmed the nuclear localization of the phosphorylated form (Fig. 6C), whereas the form that colocalized with NFH and SMI32 could only be detected by HDAC1 and not by the phospho-HDAC1 antibodies. These results indicate a correspondence between the nucleo-cytosolic distribution of HDAC1 and phosphorylation of serine residues 421 and 423.
To define the direct effect of HDAC1 serine phosphorylation on its binding to nuclear transport molecules, we used transfected HEK293 cells and assessed binding of FLAG-tagged WT or SA HDAC1 (Fig. 6A) to several karyopherins, including major exportins such as CRM1 (also known as Exportin 1), and importins, including KPNB1 (also known as Importin β-1), KPNA1 (also known as Importin α-5), and KPNA4 (also known as Importin α-3). In these conditions, we were unable to detect direct binding of the WT or SA mutant HDAC1 to either CRM1 or KPNA1. Even though we detected the presence of faint bands of KPNA1 and KPNA4 after immunoprecipitation (Fig. 6A), the levels of these karyopherins were too low to allow reliable quantification and statistical comparison. Since the experiment was conducted in the HEK293 cell line at baseline conditions, whereas we previously reported transient binding of endogenous HDAC1 to CRM1 in neurons treated with neurotoxic stimuli (Kim et al., 2010), we hypothesize that HDAC1 nucleo-cytosolic shuttling is a complex process requiring more than simple interaction between two proteins, possibly involving additional components activated by neurotoxic stimuli.
HDAC1 dephosphorylation is modulated by the calcium-dependent phosphatase calcineurin
The data identify dephosphorylation of critical serine residues in HDAC1 as being important for its cytosolic localization. Because our previous data reported nuclear export of HDAC1 as being dependent on calcium entry (Kim et al., 2010), we hypothesized that the calcium-dependent serine/threonine phosphatase calcineurin (also known as protein phosphatase 2B, PP2B), which is highly expressed in neurons (Asai et al., 1999), might be involved in HDAC1 dephosphorylation. To test this hypothesis, we pretreated primary neurons with two independent calcineurin-specific pharmacological inhibitors, FK506 and cyclosporin A (CyA) (Liu et al., 1991; Azzi et al., 2013), and then exposed them to treatment with glutamate/TNFα. Western blot analysis of P-HDAC1 and total HDAC1 revealed that addition of FK506 or CyA prevented the glutamate/TNFα-induced dephosphorylation of HDAC1 in primary neurons even after prolonged neuronal exposure to glutamate/TNFα (Fig. 7A). Next, we measured calcineurin enzymatic activity in cultured rat neurons using a highly sensitive and well established colorimetric assay. The inclusion of a highly specific substrate and the presence of okadaic acid to inhibit potentially interfering nonspecific phosphatases such as PP1 and PP2A (Fruman et al., 1996) enhanced specificity. Consistent with the progressive decrease of P-HDAC1 in response to glutamate/TNFα treatment, we detected a concomitant increase of calcineurin activity in treated neurons compared with untreated controls (p = 0.0025 and 0.0002, respectively; Fig. 7B). We also showed that pretreatment of cultured neurons with the inhibitors FK506 or CyA before glutamate/TNFα treatment prevented the upregulation of calcineurin activity, which remained at control levels (Fig. 7B). The same inhibitors also prevented glutamate/TNFα-induced HDAC1 localization to cytosolic processes (Figs. 7C, 6C) and reduced the percentage of neurites showing signs of damage, as revealed by SMI32 immunoreactivity (p = 0.0001; Fig. 7D). Inhibition of calcineurin activity favored the persistence of nuclear immunoreactivity of P-HDAC1 in neuronal cultures treated with FK506 and challenged with glutamate/TNFα (Fig. 7C). Pretreatment with a distinct calcineurin inhibitor (i.e., CyA) elicited similar results and significantly reduced the neurotoxicity of glutamate/TNFα treatment, as revealed by SMI32 immunoreactivity (p < 0.0001; Fig. 7D). Together, these experimental results suggest that calcium-dependent dephosphorylation of serine residues in HDAC1 is important for its subcellular distribution and could be explored as a potential neuroprotective target.
Discussion
HDACs include a family of proteins implicated in neuronal survival and in neurodegeneration (Kazantsev and Thompson, 2008). Class II HDACs, such as HDAC4 and HDAC6, have the ability to shuttle between nucleus and cytosol and are reported to facilitate neuroprotection. In contrast, inhibition of class I HDACs has been proposed as being potentially neuroprotective (Sleiman et al., 2009; Kim et al., 2010). HDAC1 was reported as being upregulated in mouse models of Huntington's disease (Bardai et al., 2012) and tauopathy (Kim et al., 2008) and was shown to enhance apoptosis of cerebellar granule cells (Bardai et al., 2012). The first scope of this study was defining role of HDAC1 in axonal damage in vivo by characterizing two new lines of Hdac1 conditional mutants generated in our laboratory. The Grik4-cre;Hdac1fl/fl lines, targeting the CA3 regions of the hippocampus, unveiled the protective effect of Hdac1 ablation against damage induced by acute kainic acid injection. The Camk2a-cre;Hdac1fl/fl line, targeting the CA1 region of the hippocampus, revealed a neuroprotective role after exposure to excitotoxic stimuli and inflammatory cytokines. Importantly, loss of HDAC1 function did not alter membrane excitability or baseline electrophysiological properties in acute hippocampal slices and decreased the effect of excitotoxicity and inflammatory burden on neurons.
Different models have been proposed to explain the neurotoxic effect of HDAC1, including its interaction with HDAC3 (Bardai et al., 2012), the RNA/DNA-binding protein FUS (Wang et al., 2013), and the axonal transport protein KIF2 (Kim et al., 2010). Intriguingly, both HDAC3 and FUS are actively shuttled in and out of the nucleus and we previously characterized the interaction between HDAC1 and KIF2 in cytoplasmic processes in response to excitotoxic stimuli and inflammatory cytokines (Kim et al., 2010).
Nucleocytoplasmic transport of macromolecules through the nuclear pore in eukaryotic cells is a complex process mediated by specialized proteins. At least seven human exportins have been identified (Kimura and Imamoto, 2014). They bind to nuclear export signals and favor nuclear export. Among them, we previously identified CRM1 as being critical for nuclear export of HDACs (McKinsey et al., 2001; Kim et al., 2010). However, the presence of NLS on HDAC1 also suggested a potential interaction with importins, which include the 7α and 10β proteins (Xu et al., 2010; Pumroy and Cingolani, 2015). Of these, KPNB1 (i.e., Importin β-1), KPNA1 (i.e., Importin α-5), and KPNA4 (i.e., Importin α-3) are highly expressed in hippocampal neurons (see the Allen Brain Atlas). To understand whether the process of nucleo-cytoplasmic shuttling of HDAC1 was purely dependent on interaction between cargo (e.g., HDAC1) and specialized proteins, we overexpressed WT or mutant HDAC1 in HEK293 cells and conducted coimmunoprecipitation experiments with selected exportins (i.e., CRM1) and importins (i.e., KPNB1, KPNA1, and KPNA4). Remarkably, in these conditions, we did not detect any significant interaction beween HDAC1 and either importins or exportins. This suggested that the subcellular localization of HDAC1 is a complex event possibly requiring neuron-specific adaptor molecules modified in response to neurotoxic stimuli. Future studies using unbiased proteomic approaches from neurons lacking endogenous HDAC1 and overexpressing only the mutant cytosolic HDAC1 will be required to identify adequately and comprehensively the neuronal binding partners induced by pathological stimuli.
This study was designed to define the precise amino acids responsible for the subcellular localization of HDAC1 using a site-directed mutagenesis approach, followed by transfection in primary neurons. This approach enabled us to identify two specific serine residues in HDAC1 as being critical for translocation. Because the substitution of serine to alanine residues resulted in a nonphosphorylatable form of HDAC1 that was distributed to neuronal cytoplasmic processes, we further investigated the relationship between HDAC1 phosphorylation and its subcellular distribution using antibodies recognizing the phosphorylated serine residues in the molecule. Interestingly, the phosphorylated form of HDAC1 was only detected in the nucleus and stimuli that promoted cytoplasmic localization of the molecule also favored its dephosphorylation. We previously reported that a calcium-dependent signal could be involved in the cytoplasmic localization of HDAC1 (Kim et al., 2010), therefore we reasoned that a calcium-dependent phosphatase must be important for this effect. Calcineurin is such a phosphatase, having been shown to catalyze dephosphorylation of several molecules (Hogan et al., 2003; Faure et al., 2007; Cereghetti et al., 2008; Medina et al., 2015), and can be inhibited by specific pharmacological inhibitors such as FK506 and cyclosporin A. The use of calcineurin-specific inhibitors in the experimental conditions defined by exposure to excitatory amino acids and proinflammatory cytokines prevented nucleo-cytosolic shuttling and diminished overall neurotoxicity.
Therefore, we propose a model of HDAC1 nuclear export initiated by a calcium-dependent signal activated by pathological stimuli such as glutamate and TNFα and involving calcineurin for the induction of the very early and still reversible events of axonal damage. Repeated exposure to these stimuli and increased calcium influx in the cells, however, might trigger the activation of calcium-dependent proteases that start digesting neurofilaments, thereby rendering the damage irreversible.
These findings might shed some light on the mechanism of the early stages of axonal damage and perhaps provide insights for the selection of suitable targets promoting neuroprotection and repair in neurological disorders.
Footnotes
This study was supported by the National Institutes of Health (Grants R01-NS069835-11 and R37NS042925), the National Multiple Sclerosis Society, and the Department of the Army. We thank Dr. Eric Olson at University of Texas Southwestern Medical Center for generously donating the Hdac1fl/fl mice and Dr. Stuart Schreiber at Broad Institute for his kind gift of the pBJ5 expression vector.
The authors declare no competing financial interests.
References
- Asai A, Qiu Jh, Narita Y, Chi S, Saito N, Shinoura N, Hamada H, Kuchino Y, Kirino T (1999) High level calcineurin activity predisposes neuronal cells to apoptosis. J Biol Chem 274:34450–34458. 10.1074/jbc.274.48.34450 [DOI] [PubMed] [Google Scholar]
- Azzi JR, Sayegh MH, Mallat SG (2013) Calcineurin inhibitors: 40 years later, can't live without. J Immunol 191:5785–5791. 10.4049/jimmunol.1390055 [DOI] [PubMed] [Google Scholar]
- Bardai FH, Price V, Zaayman M, Wang L, D'Mello SR (2012) Histone deacetylase-1 (HDAC1) is a molecular switch between neuronal survival and death. J Biol Chem 287:35444–35453. 10.1074/jbc.M112.394544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolte S, Cordelières FP (2006) A guided tour into subcellular colocalization analysis in light microscopy. J Microsc 224:213–232. 10.1111/j.1365-2818.2006.01706.x [DOI] [PubMed] [Google Scholar]
- Bozdagi O, Shan W, Tanaka H, Benson DL, Huntley GW (2000) Increasing numbers of synaptic puncta during late-phase LTP: N-cadherin is synthesized, recruited to synaptic sites, and required for potentiation. Neuron 28:245–259. 10.1016/S0896-6273(00)00100-8 [DOI] [PubMed] [Google Scholar]
- Brandl A, Heinzel T, Krämer OH (2009) Histone deacetylases: salesmen and customers in the post-translational modification market. Biol Cell 101:193–205. 10.1042/BC20080158 [DOI] [PubMed] [Google Scholar]
- Burke RE, O'Malley K (2013) Axon degeneration in Parkinson's disease. Exp Neurol 246:72–83. 10.1016/j.expneurol.2012.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai R, Kwon P, Yan-Neale Y, Sambuccetti L, Fischer D, Cohen D (2001) Mammalian histone deacetylase 1 protein is posttranslationally modified by phosphorylation. Biochem Biophys Res Commun 283:445–453. 10.1006/bbrc.2001.4786 [DOI] [PubMed] [Google Scholar]
- Cereghetti GM, Stangherlin A, Martins de Brito O, Chang CR, Blackstone C, Bernardi P, Scorrano L (2008) Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc Natl Acad Sci U S A 105:15803–15808. 10.1073/pnas.0808249105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho Y, Cavalli V (2014) HDAC signaling in neuronal development and axon regeneration. Curr Opin Neurobiol 27:118–126. 10.1016/j.conb.2014.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho Y, Sloutsky R, Naegle KM, Cavalli V (2013) Injury-induced HDAC5 nuclear export is essential for axon regeneration. Cell 155:894–908. 10.1016/j.cell.2013.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews L, Masliah E (2010) Molecular mechanisms of neurodegeneration in Alzheimer's disease. Hum Mol Genet 19:R12–R20. 10.1093/hmg/ddq160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle K, Fitzpatrick FA (2010) Redox signaling, alkylation (carbonylation) of conserved cysteines inactivates class I histone deacetylases 1, 2, and 3 and antagonizes their transcriptional repressor function. J Biol Chem 285:17417–17424. 10.1074/jbc.M109.089250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faure C, Corvol JC, Toutant M, Valjent E, Hvalby O, Jensen V, El Messari S, Corsi JM, Kadaré G, Girault JA (2007) Calcineurin is essential for depolarization-induced nuclear translocation and tyrosine phosphorylation of PYK2 in neurons. J Cell Sci 120:3034–3044. 10.1242/jcs.009613 [DOI] [PubMed] [Google Scholar]
- Filosa A, Paixão S, Honsek SD, Carmona MA, Becker L, Feddersen B, Gaitanos L, Rudhard Y, Schoepfer R, Klopstock T, Kullander K, Rose CR, Pasquale EB, Klein R (2009) Neuron-glia communication via EphA4/ephrin-A3 modulates LTP through glial glutamate transport. Nat Neurosci 12:1285–1292. 10.1038/nn.2394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorino E, Giudici M, Ferrari A, Mitro N, Caruso D, De Fabiani E, Crestani M (2014) The sirtuin class of histone deacetylases: regulation and roles in lipid metabolism. IUBMB Life 66:89–99. 10.1002/iub.1246 [DOI] [PubMed] [Google Scholar]
- Fischer A, Sananbenesi F, Mungenast A, Tsai LH (2010) Targeting the correct HDAC(s) to treat cognitive disorders. Trends Pharmacol Sci 31:605–617. 10.1016/j.tips.2010.09.003 [DOI] [PubMed] [Google Scholar]
- Friese MA, Schattling B, Fugger L (2014) Mechanisms of neurodegeneration and axonal dysfunction in multiple sclerosis. Nat Rev Neurol 10:225–238. 10.1038/nrneurol.2014.37 [DOI] [PubMed] [Google Scholar]
- Fruman DA, Pai SY, Klee CB, Burakoff SJ, Bierer BE (1996) Measurement of calcineurin phosphatase activity in cell extracts. Methods 9:146–154. 10.1006/meth.1996.0020 [DOI] [PubMed] [Google Scholar]
- Gregoretti IV, Lee YM, Goodson HV (2004) Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J Mol Biol 338:17–31. 10.1016/j.jmb.2004.02.006 [DOI] [PubMed] [Google Scholar]
- Guan JS, Haggarty SJ, Giacometti E, Dannenberg JH, Joseph N, Gao J, Nieland TJ, Zhou Y, Wang X, Mazitschek R, Bradner JE, DePinho RA, Jaenisch R, Tsai LH (2009) HDAC2 negatively regulates memory formation and synaptic plasticity. Nature 459:55–60. 10.1038/nature07925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberland M, Montgomery RL, Olson EN (2009a) The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet 10:32–42. 10.1038/nrg2485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberland M, Johnson A, Mokalled MH, Montgomery RL, Olson EN (2009b) Genetic dissection of histone deacetylase requirement in tumor cells. Proc Natl Acad Sci U S A 106:7751–7755. 10.1073/pnas.0903139106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haines JD, Herbin O, de la Hera B, Vidaurre OG, Moy GA, Sun Q, Fung HY, Albrecht S, Alexandropoulos K, McCauley D, Chook YM, Kuhlmann T, Kidd GJ, Shacham S, Casaccia P (2015) Nuclear export inhibitors avert progression in preclinical models of inflammatory demyelination. Nat Neurosci 18:511–520. 10.1038/nn.3953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan PG, Chen L, Nardone J, Rao A (2003) Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev 17:2205–2232. 10.1101/gad.1102703 [DOI] [PubMed] [Google Scholar]
- Jurkin J, Zupkovitz G, Lagger S, Grausenburger R, Hagelkruys A, Kenner L, Seiser C (2011) Distinct and redundant functions of histone deacetylases HDAC1 and HDAC2 in proliferation and tumorigenesis. Cell Cycle 10:406–412. 10.4161/cc.10.3.14712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karwowska-Desaulniers P, Ketko A, Kamath N, Pflum MK (2007) Histone deacetylase 1 phosphorylation at S421 and S423 is constitutive in vivo, but dispensable in vitro. Biochem Biophys Res Commun 361:349–355. 10.1016/j.bbrc.2007.06.167 [DOI] [PubMed] [Google Scholar]
- Kazantsev AG, Thompson LM (2008) Therapeutic application of histone deacetylase inhibitors for central nervous system disorders. Nat Rev Drug Discov 7:854–868. 10.1038/nrd2681 [DOI] [PubMed] [Google Scholar]
- Kim D, Frank CL, Dobbin MM, Tsunemoto RK, Tu W, Peng PL, Guan JS, Lee BH, Moy LY, Giusti P, Broodie N, Mazitschek R, Delalle I, Haggarty SJ, Neve RL, Lu Y, Tsai LH (2008) Deregulation of HDAC1 by p25/Cdk5 in neurotoxicity. Neuron 60:803–817. 10.1016/j.neuron.2008.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Shen S, Dietz K, He Y, Howell O, Reynolds R, Casaccia P (2010) HDAC1 nuclear export induced by pathological conditions is essential for the onset of axonal damage. Nat Neurosci 13:180–189. 10.1038/nn.2471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M, Imamoto N (2014) Biological significance of the importin-beta family-dependent nucleocytoplasmic transport pathways. Traffic 15:727–748. 10.1111/tra.12174 [DOI] [PubMed] [Google Scholar]
- Liu J, Farmer JD Jr, Lane WS, Friedman J, Weissman I, Schreiber SL (1991) Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell 66:807–815. 10.1016/0092-8674(91)90124-H [DOI] [PubMed] [Google Scholar]
- Luo Y, Jian W, Stavreva D, Fu X, Hager G, Bungert J, Huang S, Qiu Y (2009) Trans-regulation of histone deacetylase activities through acetylation. J Biol Chem 284:34901–34910. 10.1074/jbc.M109.038356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathias RA, Guise AJ, Cristea IM (2015) Post-translational modifications regulate class IIa histone deacetylase (HDAC) function in health and disease. Mol Cell Proteomics 14:456–470. 10.1074/mcp.O114.046565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinsey TA, Zhang CL, Olson EN (2001) Identification of a signal-responsive nuclear export sequence in class II histone deacetylases. Mol Cell Biol 21:6312–6321. 10.1128/MCB.21.18.6312-6321.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina DL, Di Paola S, Peluso I, Armani A, De Stefani D, Venditti R, Montefusco S, Scotto-Rosato A, Prezioso C, Forrester A, Settembre C, Wang W, Gao Q, Xu H, Sandri M, Rizzuto R, De Matteis MA, Ballabio A (2015) Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat Cell Biol 17:288–299. 10.1038/ncb3114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery RL, Davis CA, Potthoff MJ, Haberland M, Fielitz J, Qi X, Hill JA, Richardson JA, Olson EN (2007) Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes Dev 21:1790–1802. 10.1101/gad.1563807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery RL, Hsieh J, Barbosa AC, Richardson JA, Olson EN (2009) Histone deacetylases 1 and 2 control the progression of neural precursors to neurons during brain development. Proc Natl Acad Sci U S A 106:7876–7881. 10.1073/pnas.0902750106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pflum MK, Tong JK, Lane WS, Schreiber SL (2001) Histone deacetylase 1 phosphorylation promotes enzymatic activity and complex formation. J Biol Chem 276:47733–47741. 10.1074/jbc.M105590200 [DOI] [PubMed] [Google Scholar]
- Pumroy RA, Cingolani G (2015) Diversification of importin-alpha isoforms in cellular trafficking and disease states. Biochem J 466:13–28. 10.1042/BJ20141186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segré CV, Chiocca S (2011) Regulating the regulators: the post-translational code of class I HDAC1 and HDAC2. J Biomed Biotechnol 2011:690848. 10.1155/2011/690848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon C, Kezunovic N, Ye M, Hyde J, Hayar A, Williams DK, Garcia-Rill E (2010) Gamma band unit activity and population responses in the pedunculopontine nucleus. J Neurophysiol 104:463–474. 10.1152/jn.00242.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleiman SF, Basso M, Mahishi L, Kozikowski AP, Donohoe ME, Langley B, Ratan RR (2009) Putting the ‘HAT’ back on survival signalling: the promises and challenges of HDAC inhibition in the treatment of neurological conditions. Expert Opin Investig Drugs 18:573–584. 10.1517/13543780902810345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mörk S, Bö L (1998) Axonal transection in the lesions of multiple sclerosis. N Engl J Med 338:278–285. 10.1056/NEJM199801293380502 [DOI] [PubMed] [Google Scholar]
- Tsien JZ, Chen DF, Gerber D, Tom C, Mercer EH, Anderson DJ, Mayford M, Kandel ER, Tonegawa S (1996) Subregion- and cell type-restricted gene knockout in mouse brain. Cell 87:1317–1326. 10.1016/S0092-8674(00)81826-7 [DOI] [PubMed] [Google Scholar]
- Wang JT, Medress ZA, Barres BA (2012) Axon degeneration: molecular mechanisms of a self-destruction pathway. J Cell Biol 196:7–18. 10.1083/jcb.201108111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Yu S, Simonyi A, Sun GY, Sun AY (2005) Kainic acid-mediated excitotoxicity as a model for neurodegeneration. Mol Neurobiol 31:3–16. 10.1385/MN:31:1-3:003 [DOI] [PubMed] [Google Scholar]
- Wang WY, Pan L, Su SC, Quinn EJ, Sasaki M, Jimenez JC, Mackenzie IR, Huang EJ, Tsai LH (2013) Interaction of FUS and HDAC1 regulates DNA damage response and repair in neurons. Nat Neurosci 16:1383–1391. 10.1038/nn.3514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D, Farmer A, Chook YM (2010) Recognition of nuclear targeting signals by Karyopherin-beta proteins. Curr Opin Struct Biol 20:782–790. 10.1016/j.sbi.2010.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Jian W, Luo Y, Fu X, Noguchi C, Bungert J, Huang S, Qiu Y (2012) Acetylation of histone deacetylase 1 regulates NuRD corepressor complex activity. J Biol Chem 287:40279–40291. 10.1074/jbc.M112.349704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye F, Chen Y, Hoang T, Montgomery RL, Zhao XH, Bu H, Hu T, Taketo MM, van Es JH, Clevers H, Hsieh J, Bassel-Duby R, Olson EN, Lu QR (2009) HDAC1 and HDAC2 regulate oligodendrocyte differentiation by disrupting the beta-catenin-TCF interaction. Nat Neurosci 12:829–838. 10.1038/nn.2333 [DOI] [PMC free article] [PubMed] [Google Scholar]