


Abstract
Cells respond to DNA damage by activating both cellular growth arrest and DNA repair processes. In Saccharomyces cerevesiae the RAD9 gene controls DNA damage-mediated cell cycle arrest that is known to allow efficient repair. To ascertain whether RAD9 plays a role in DNA repair per se, the removal of UV-induced photolesions was assessed in synchronized isogenic normal and rad9Δ cells using the high resolution primer extension technique. The results show that RAD9 is indeed involved in the removal of photolesions from both the transcribed and the non-transcribed strands of the reporter GAL10 gene, in G1- as well as G2/M-arrested cells. Interestingly, these data also reveal that in both normal and rad9 mutant, the repair strand bias towards the transcribed stand is more pronounced in G2/M- than in G1-arrested cells. These data indicate that RAD9 coordinate the cellular response to DNA damage by activating both cell cycle checkpoint and excision repair.
INTRODUCTION
DNA, the keeper of genetic information, is vulnerable to structural damage introduced by both endogenous and environmental agents (1).
Cells possess various DNA repair pathways that operate on different types of DNA damage (2,3). Nucleotide excision repair (NER) is a ubiquitous DNA repair process that deals with a broad range of DNA lesions, including the mutagenic and carcinogenic UV light-induced pyrimidine dimers (PDs). There are two main forms of UV lesions, 6-4 photoproducts and cyclobutane pyrimidine dimers (CPDs) (4). The efficiency of the NER process is modulated in vivo by several factors such as chromatin structure and transcription (5). Indeed, several connections between NER and the RNA polymerase II (RNAP II) transcription machinery contribute to fast and efficient excision of transcription-blocking lesions, leading to preferential repair of RNAP II-transcribed DNA. Impairment of the strand-specific repair pathway leads, in humans, to Cockayne syndrome, a rare autosomal disease. Beside this crucial NER subpathway, there is the global genome repair subpathway that deals with the non-transcribed parts of the genome. Most of the NER components are common to both subpathways but some are specific (6,7).
In proliferating cells, an adequate response to the introduction of DNA damage requires, in addition to the participation of many DNA repair processes, a delay of the cell cycle machinery at specific points in the cell cycle called checkpoints. These represent locations at which DNA repair and cell cycle events are coordinated to ensure efficient repair of DNA (4).
In Saccharomyces cerevisiae, the response to DNA damage is tightly regulated by a battery of genes responsible for sensing DNA damage and delaying the cell cycle at different checkpoints (8,9). A key member of this family of genes is RAD9, which is required for activation of both G1 and G2 checkpoints (10–14). DNA damage results in the generation of hyperphosphorylated forms of the Rad9 protein (15,16).
rad9 mutants are sensitive to the killing effects of UV light and ionizing radiation and are defective in DNA damage-induced cell cycle delays in G1 and G2/M phases (10–14). They are also defective in UV-dependent transactivation of a battery of DNA repair and replication genes including all known DNA damage-inducible NER genes (RAD2, RAD7, RAD16 and RAD23) (17,18).
One important unanswered question is whether the radiosensitivity of rad9 mutants is attributable to their cell cycle checkpoint defect only, or whether they are additionally defective in DNA repair per se. In other words, does RAD9 modulate NER independently of cell cycle control? There are indications that cell cycle checkpoint genes may also participate in DNA repair of UV-induced DNA damage, but so far this hypothesis is still awaiting a direct demonstration.
Using a high resolution primer extension technique, we demonstrate in this report that the absence of the RAD9 gene product reduces the repair efficiency of PDs in both G1- and G2/M-arrested cells, affecting both transcribed strands (TS) and non-transcribed strands (NTS). This shows that in response to UV-induced DNA damage, RAD9 has dual functions, namely mediating cellular growth arrest and inducing DNA repair. In addition, our data indicate that differential repair at the GAL10 gene is more pronounced in G2/M-arrested cells than in late G1-held cells, in both normal and rad9 mutant.
MATERIALS AND METHODS
Strains and media
The yeast strains used were FF181268 (MATa; bar1::LEU2; leu2; ura3; trp1; his7; lys1) and FF181270 (MATa; bar1::LEU2; leu2; ura3; trp1; his7; lys1; rad9::URA3) (17). Cells were grown at 30°C in complete medium containing 2% galactose (YEPG) (19) to a density of ∼107 cells/ml. α-Factor (Sigma) and methyl 2-benzimidazil carbamate (MBC) (Aldrich) were prepared and used as described previously (17).
Cell cycle blocks and UV-irradiation
Exponentially growing cells were arrested in late G1 or in G2/M with the appropriate agent, i.e., α-factor and MBC, respectively. Cells were then monitored microscopically for unbudded (G1 arrest) or large budded cells (G2/M arrest). When a significant number of cells were in the desired phase (essentially 100% with α-factor and 70% with MBC), cells were collected by centrifugation, resuspended in water containing the synchronization agent (α-factor or MBC) and irradiated with a dose of 200 Jm–2. UV-irradiation was performed using a germicidal UV lamp (predominantly 254 nm), with a UV-fluence rate of 1 Jm–2/s. The UV dosimetry was performed using a UV meter (Spectronics Corporation, NY).
DNA repair
Once irradiated, cells were reincubated in the dark in the same conditions either in the presence of the cell cycle blocking agent (to maintain them in the desired phase), or in the absence of the agent in the case of asynchronous cells. Cells were then collected at various repair times (0, 1, 2 and 3 h) and chilled immediately on ice to stop DNA repair. The percentage of cells in G1 or in G2/M phases was monitored microscopically during the entire repair period. A non-irradiated sample served as a sham control.
DNA preparation and enzyme digestion
Genomic DNA was prepared from samples corresponding to different repair times using Qiagen columns and protocols (Qiagen Genomic DNA Handbook). The DNA was incised with the EcoRI restriction enzyme.
Primer extension analysis
Primer labeling and extension were carried out as previously described (20), using the GAL10 gene as a template and the following primers:
Bottom strand: 5′-TCTTCTGCTACTGCTTATGGTGATG-3′
Top strand: 5′-CTAGATCAACTACGTGGATATAATC-3′
Primer extension was achieved by 30 cycles of repeated denaturation (95°C for 1 min), annealing (60°C for 4 min) and extension (72°C for 3 min) reaction, with 0.2 U of Taq polymerase (Qiagen). The reaction products were ethanol precipitated and analyzed on a 5% polyacrylamide, urea (w/v, 40%) gel. DNA sequencing reactions were performed in parallel, using the Sanger chain termination technique with the same primer. The gels were subsequently dried on 3MM paper and then either exposed to X-ray film (Kodak) or analyzed with a phosphorimager (Bio-Rad).
Quantification
The sequencing gels were used for the quantification of the relative repair of UV-induced DNA damage, as detailed previously (20,21). Briefly, a volume box was made around each band obtained from a UV-induced DNA lesion and the corresponding gel background was subtracted using a volume box of the same size outside of the loaded lanes. The value obtained was then divided by the value obtained for a volume box that covered the whole lane to correct for inter-lane loading differences. The values obtained for the non-irradiated DNA were then subtracted in order to correct for non-specific background signal due to DNA nicking or non-specific Taq DNA polymerase blockage. For standardization, the corrected values obtained at time 0 (no repair) were defined as 100% damage.
RESULTS
RAD9 is required for efficient repair of the GAL10 TS in G1-arrested cells
To study the role of the RAD9 gene in the repair of UV-induced DNA damage, use was made of the very sensitive primer extension technique that allows the assessment of NER at nucleotide resolution in single-copy genes (20,21). By this method, PDs were revealed as blocks to DNA synthesis by Taq DNA polymerase and the corresponding signal was amplified by linear PCR. Since cells compromised for Rad9 protein are deficient in cell cycle transient arrest, DNA repair experiments were performed on synchronized cell populations, arrested in a particular phase of the cell cycle throughout the entire DNA repair period. These studies were performed using the RAD9-deleted strain FF181270 (rad9Δ) and its normal isogenic counterpart FF181268 (wild-type). Log-phase cells grown in YPEG were synchronized at late G1 with α-factor, then either sham-treated (control) or UV-irradiated. Next, the irradiated and non-irradiated cells were reincubated to permit repair in the presence of α-factor. PDs were then mapped by primer extension along the GAL10 gene, which was used as a template. Figure 1A shows the initial distribution pattern of dimers formed almost exclusively at adjacent pyrimidines (Fig. 1A, lanes 1), and the repair kinetics that occurred along the TS of the portion of the GAL10 gene chosen for NER analysis, in both rad9Δ and wild-type cells. A close visual inspection of the representative autoradiograms shows a faster decrease in the intensities of the bands corresponding to the PDs formed in the wild-type cells, compared with those formed in the rad9Δ cells, suggesting that PD removal is less efficient in the mutant. Quantification of the autoradiograms, expressed as the percentage of repaired PDs formed at different pyrimidine clusters, is presented in Figure 1B. On average, within the 3 h of repair, the wild-type cells removed >70% of PDs, while only ∼35% were excised in the rad9Δ cells (Fig. 1B). The repair assessment at the pyrimidine cluster (550)-CTTTTTT, where the frequency of PD formation is similar in both wild-type and rad9Δ cells (0.046 and 0.04, respectively), indicated that within 3 h of repair the wild-type cells removed >76% of the photolesions, whereas the rad9Δ cells repaired only 35%. Intriguingly, at 1 h post-irradiation, the difference in repair rate between normal and mutant cells was very small (42 and 35%, respectively). The autoradiograms as well as the corresponding quantitative results show that in the rad9Δ cells PD removal took place during the first hour only, then no repair was detected during the following 2 h, while in the wild-type cells PD removal increased during the entire 3 h of repair (Fig. 1). These results show that the rad9Δ cells are deficient in the removal of UV-induced photolesions from the TS of the GAL10 gene.
Figure 1.
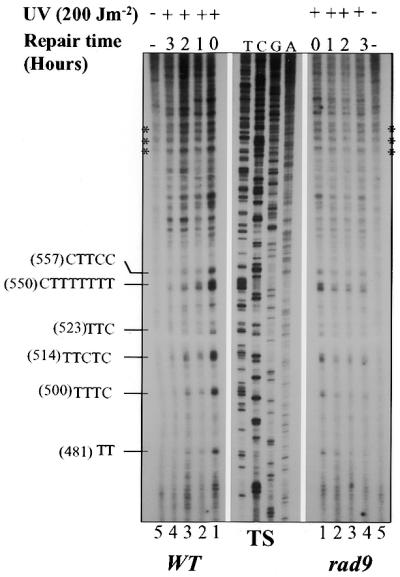

Repair of UVC-induced PDs in the GAL10 TS in G1-arrested wild-type and rad9 mutant cells. α-Factor-synchronized S.cerevisae cells were UV-irradiated and allowed to repair in the presence of α-factor for the indicated periods. NER was analyzed by primer extension. (A) Autoradiograms showing the primer extension products. Asterisks, non-specific Taq polymerase arrests; T, C, G and A, sequencing reactions. Each pyrimidine track on the left represent a PD cluster in the GAL10 gene with the accompanying number in parentheses referring to the 5′ nucleotide of the cluster. (B) Quantitative analysis of PD removal, illustrating the fraction (%) of PDs removed at each repair time. Each data point corresponds to an average value for the repair of several PD clusters. Inter-lane loading differences were corrected as described in the Materials and Methods. Each error bar represents the standard deviation of three experiments.
RAD9 is also required for efficient repair of the GAL10 NTS in G1-arrested cells
To test whether the role of RAD9 in PD removal is confined to the TS or also includes the NTS, the effect of RAD9 deletion on the removal rate of PDs from the GAL10 NTS was measured by primer extension, employing the appropriate primer (Fig. 2). The autoradiograms show that, in both WT and mutant cells, the removal of the photolesions formed along the NTS is, in general, slower than that seen in the TS, which is in accord with the TS-preferential repair phenomenon. A detailed analysis of the band intensities over repair times shows that PDs are slowly removed in the rad9Δ cells as compared to the wild-type cells (Fig. 2A). Consistent with previous reports (21,22), the autoradiograms and their quantification show a heterogeneity in the rate of PD removal along the NTS. Therefore, it was decided to compare the efficiency of DNA repair in wild-type versus rad9Δ cells based on the percentage of PD removal occurring in only one pyrimidine cluster, (575)-TTTT, where PD yield is one of the highest (Fig. 2B). Figure 2B shows that in this pyrimidine cluster, PD removal is more efficient in the wild-type than in the rad9Δ cells. As in the case of the TS, the repair rate of the NTS in rad9Δ cells reached its maximum (15%) during the first hour, and then plateaued during the following 2 h, indicating an arrest in DNA repair (Fig. 2B). Wild-type cells, however, exhibited a further linear in repair rate, reaching a level of ∼45% (Fig. 2B). These results reveal a deficiency of the rad9Δ cells in the removal of DNA photodimers from the NTS of the GAL10 gene. Taken together, these results indicate that in G1 phase, the product of the RAD9 cell cycle checkpoint gene is also involved in DNA repair.
Figure 2.
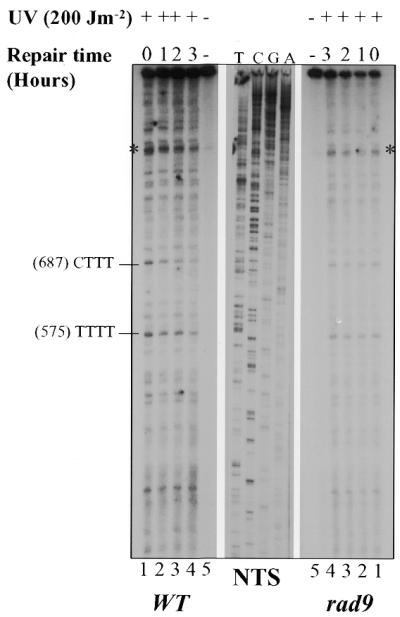

Repair of UVC-induced PDs in the GAL10 NTS in G1-arrested wild-type and rad9 mutant cells. (A) Autoradiograms legend is as in Figure 1A. (B) Quantitative analysis showing the percentage of PD removal occurring in the pyrimidine cluster (575)-TTTT. Each error bar represents the standard deviation of three experiments. Error bars not shown are obscured by the datum points.
G2/M-arrested RAD9-deleted cells are compromised for DNA repair of photodimers
DNA damage-mediated transient arrest in G2/M phase represents the most prominent cell cycle checkpoint in S.cerevisiae. To test whether the RAD9 gene is also involved in the repair of UV-induced DNA damage in the G2/M phase of the cell cycle, normal and rad9Δ cells were synchronized in this phase and challenged with UV light. Synchronization was achieved with the aid of the microtubule inhibitory drug MBC, which was also present after UV treatment. As seen for G1-arrested cells, it is obvious from the autoradiograms that the photoproducts were repaired more rapidly in wild-type than in rad9Δ cells (Figs 3 and 4A). While wild-type cells demonstrated efficient removal of PDs from the TS, with 60% removed after 1 h and 90% after 3 h, RAD9-deficient cells exhibited a lower repair rate, removing only 35% of the PDs during the first hour and 55% by 2 h post-irradiation (Fig. 4B). Interestingly, in G2/M-arrested rad9Δ cells, the plateauing phenomenon was observed only during the third hour of repair while only 5% of the remaining PDs were removed (Fig. 4B).
Figure 3.
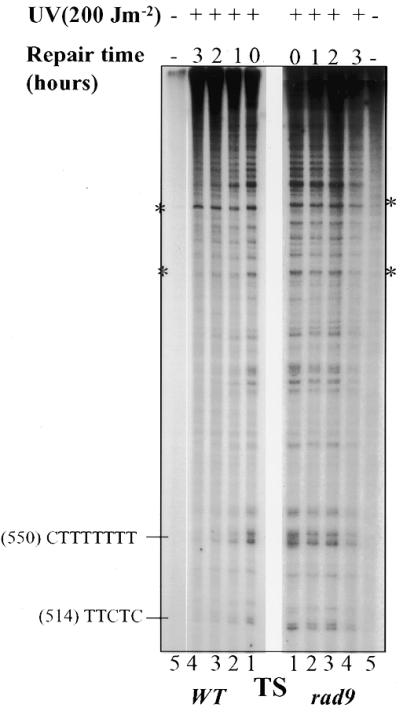
Repair of UVC-induced PDs in the GAL10 TS in G2/M-arrested wild-type and rad9 mutant. MBC-arrested cells were UV-irradiated and incubated for repair in the presence of the drug for the times indicated. Autoradiograms legend is as in Figure 1A. The corresponding quantification is in Figure 4B.
Figure 4.
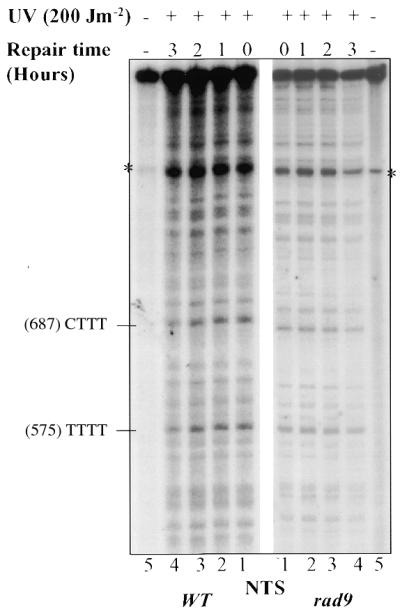

Repair of UVC-induced PDs in the GAL10 NTS in G2/M-arrested wild-type and rad9 mutant cells. (A) Autoradiograms legend is as in Figure 2A. (B) Quantitative analysis of PD removal from TS (unbroken lines) and NTS (broken lines). The quantification was performed as described in Figures 1B and 2B, respectively. Each error bar represents the standard deviation of three experiments. Error bars not shown are obscured by the datum points.
Intriguingly, repair of the NTS was very low in both wild-type and rad9Δ cells. After 2 h of repair, only 20% of the photolesions were removed in wild-type cells, while PD removal was not detectable in rad9 mutant cells (Fig. 4B). These results show that the RAD9 gene is also required for the repair of PDs from both the TS and the NTS at G2/M phase.
Preferential repair of the GAL10 TS is more pronounced in G2/M cells
In agreement with the transcription-coupled repair phenomenon, the data presented above show a clear preferential repair of the TS of the GAL10 gene in both G1- and G2/M-synchronized cells. Interestingly, however, the repair bias towards the TS in both rad9Δ and wild-type cells was stronger in G2/M phase. Indeed, the repair efficiency of the GAL10 TS was higher in G2/M-arrested cells than in G1-blocked cells (Fig. 5). On the other hand, the NTS showed a lower repair rate in the G2/M-arrested cells compared to G1 cells (Fig. 5). For the site (575)-TTTT (NTS) for instance, where the frequency of PD formation is similar in both G1- and G2/M-arrested cells (0.023), the removal of PDs after 3 h of repair reached 45% in α-factor-blocked cells, however, it did not exceed 25% in the MBC-arrested cells (Fig. 5). This difference in strand-specific repair may be due to a stronger focus of the repair apparatus on the template strand at the expense of the NTS in the G2/M cells as compared with the G1-arrested cells. A similar phenomenon is observed in rad9 mutant (compare Figs 1B and 2B with 4B). Since in both situations DNA repair was analyzed in arrested cells, it was decided to measure PD removal in both the TS and NTS of the GAL10 gene in exponentially growing cells, and compare the level of repair strand bias obtained in these conditions with those realized in G1- and G2/M-arrested cells. Exponentially growing cells were challenged with UV light, and allowed to repair in growing conditions. Figure 5 shows that within the 3 h of incubation, photodimers were removed from the TS and the NTS of the GAL10 gene with a repair rate of ∼80 and 40%, respectively. These values and consequently the repair strand bias appear intermediate between those obtained in G1- and G2/M-arrested cells (Fig. 5). This is most likely due to the fact that asynchronized cells are in different phases of the cell cycle (40, 20 and 40% for G1, S and G2/M, respectively). Hence, these results suggest that the level of repair strand bias could be modulated throughout the cell cycle.
Figure 5.

Repair of UVC-induced PDs in the TS and the NTS of the GAL10 gene in asynchronous wild-type cells. Exponentially growing cells were UV-irradiated and incubated to allow repair. Quantitative analysis of PD removal, illustrating the fraction (%) of PDs removed at each repair time. Each datum point corresponds to an average value for the repair of several PD clusters. The fraction of PD removed from the TS (filled triangles) and the NTS (open triangles). The quantification was performed as described in Figures 1B and 2B, respectively. Quantification results from Figures 1B, 2B and 4B are also shown for comparison.
DISCUSSION
Upon sustaining DNA damage, cells trigger an SOS stress response, which concomitantly arrests cell cycle progression and activates DNA repair processes. These two responses are linked through one or more signal transduction pathways, the components of which have been extensively characterized in S.cerevisiae. In this lower eukaryotic organism, the cell cycle checkpoint gene RAD9 controls both arrest in G1 and G2 phases of the cell cycle and DNA damage-mediated induction of several genes involved in NER (8). We report here that RAD9 is also required for fully proficient removal of UV-induced photolesions in G1- and G2/M-arrested cells, from both TS and NTS of the GAL10 gene. Interestingly, in α-factor-held rad9Δ cells, the repair kinetics of the TS as well as the NTS were biphasic with an increasing DNA repair rate during the first phase, and a constant DNA repair rate during the second one. After 3 h of repair, while 75 (TS) and 45% (NTS) of the induced photolesions were excised from the wild-type cells, only 35 (TS) and 15% (NTS) were removed in rad9Δ cells. These repair rates obtained in the rad9 mutant strain were achieved during the first hour of repair, followed by a complete cessation of PD removal (Figs 1B and 2B). These results indicate that the G1-arrested RAD9-deleted cells have a limited ability to remove PDs, and that this effect appears to be more severe for the NTS. Accordingly, RAD9 clearly plays a role in DNA repair independently of its cell cycle checkpoint function.
The premature termination of PD repair observed in the rad9Δ cells may have several explanations, but the more plausible one could be that these cells are defective in DNA damage-dependent de novo repair protein synthesis, associated with a rapid turnover of these proteins. Since the RAD9 gene product is known to be essential for DNA damage-dependent transactivation of several NER proteins (17,18), without itself being inducible (11,15), it is reasonable to assume that the DNA repair defect observed in the rad9Δ cells may be due to a failure in the up-regulation of the RAD9-controlled NER proteins. It was indeed previously suggested that NER is inducible in S.cerevisiae (23). It is hence possible that the role of RAD9 in NER is to up-regulate the expression of the UV-inducible NER genes (RAD2, RAD7, RAD16, RAD23), without itself being part of the core NER reaction.
The reduced repair capability of the rad9Δ cells was not confined to G1-arrested cells but was also observed in the G2/M-held cells, although the repair kinetics were different. PD removal was impaired in both the TS and the NTS, however, as in G1 phase, the effect was more pronounced on the NTS (Fig. 4B). It is noteworthy that the difference in repair rate between wild-type and rad9Δ cells is more pronounced in G2/M-arrested cells. This difference is greater in the first hour of repair, during which ∼60% of PDs were excised from the TS in the wild-type cells (35% in G1) but only 30% were removed from the rad9Δ cells (30% in G1) (Figs 1B and 4B). This showed that in wild-type cells the repair rate of the TS during the first hour of incubation is almost 2-fold higher in G2/M-arrested cells compared to G1-held cells. Likewise, in rad9Δ cells the repair rate of the TS is higher in G2/M phase than in G1 phase. The cessation of PD removal was not observed in G2/M phase until the third hour of repair (Fig. 4B).
Interestingly, the repair of the NTS in both wild-type and rad9Δ cells is less efficient in G2/M- than in G1-arrested cells. This may also explain the higher repair efficiency of the TS observed in G2/M-arrested cells, where the repair of the TS seems to take place at the expense of that of the NTS. Although the total amount of excised PDs from both TS and NTS appears similar in G1 and G2/M (∼60% in 3 h in wild-type cells), the repair strand bias is more pronounced in G2/M-arrested cells (Fig. 5). This could result from targeting most of the repair apparatus to the TS, increasing its repair rate. This phenomenon could be either target gene-dependent (GAL10) or cell cycle-dependent. However, since transcription of the GAL10 gene does not change throughout the cell cycle (24), it seems more likely that the difference in repair strand bias could be cell cycle- rather than gene-dependent. This dependency could be either direct or indirect through the action of the synchronization agent used (α-factor versus MBC). When an asynchronous cell population was UV-treated, an intermediate level of repair strand bias was observed (Fig. 5). The values are similar to those previously obtained in exponentially growing cells, using an indirect end-labeling technique (25). Livingstone-Zatchej et al. (25) have indeed shown that within 2 h of repair ∼80 and 20% of PDs were removed from the GAL10 TS and NTS, respectively. This shows that the treatment of cells with α-factor or MBC has no or only little effect on the excision of PDs.
Taken together, these results suggest that the difference in the level of repair strand bias shown here could be cell cycle-dependent. These data constitute the first observation of a relationship between the cell cycle and the transcription-coupled repair, and thus provide impetus for further studies to elucidate the precise reason for this discrepancy in strand-specific repair between G1- and G2/M-arrested cells.
In conclusion, our results show that RAD9-compromised cells are deficient in repair of photodimers, independently of their defect in cell cycle checkpoint. This defect is cell cycle independent since it was observed in both G1 and G2/M phases, although the effect is more pronounced in G2/M-arrested cells. This shows that, like in the Escherichia coli SOS response, the cell cycle checkpoint and DNA repair activation are coregulated. Future studies will define the precise role of the RAD9 cell cycle checkpoint gene in NER.
Acknowledgments
ACKNOWLEDGEMENTS
We would like to thank Dr M.C.Paterson and Dr M.A.Hannan for helpful discussions and comments on the manuscript. This work was supported by the King Faisal Specialist Hospital and Research Center, under the RAC proposal no. 990025.
References
- 1.Lindahl T. (1993) Instability and decay of the primary structure of DNA. Nature, 362, 709–715. [DOI] [PubMed] [Google Scholar]
- 2.Wood R.D. (1996) DNA repair in eukaryotes. Annu. Rev. Biochem., 65, 135–167. [DOI] [PubMed] [Google Scholar]
- 3.Lindahl T. and Wood,R.D. (1999) Quality control by DNA repair. Science, 286, 1897–1905. [DOI] [PubMed] [Google Scholar]
- 4.Friedberg E.C., Walker,G.C. and Siede,W. (1995) DNA Repair and Mutagenesis. American Society for Microbiology, Washington, DC.
- 5.Thoma F. (1999) Light and dark in chromatin repair: repair of UV-induced DNA lesions by photolyase and nucleotide excision repair. EMBO J., 18, 6585–6598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Citterio E., Vermeulen,W. and Hoeijmakers,J.H.J. (2000) Transcriptional healing. Cell, 101, 447–450. [DOI] [PubMed] [Google Scholar]
- 7.Friedberg E.C. (1996) Relationships between DNA repair and transcription. Annu. Rev. Biochem., 65, 15–42. [DOI] [PubMed] [Google Scholar]
- 8.Lowndes N.F. and Murguia,J.R. (2000) Sensing and responding to DNA damage. Curr. Opin. Genet. Dev., 10, 17–25. [DOI] [PubMed] [Google Scholar]
- 9.Weinert T. (1998) DNA damage and checkpoint pathways: molecular anatomy and interactions with repair. Cell, 94, 555–558. [DOI] [PubMed] [Google Scholar]
- 10.Weinert T.A. and Hartwell,L.H. (1988) The RAD9 gene controls the cell cycle response to DNA damage in Saccharomyces cerevisiae. Science, 241, 317–322. [DOI] [PubMed] [Google Scholar]
- 11.Weinert T.A. and Hartwell,L.H. (1990) Characterization of RAD9 of Saccharomyces cerevisiae and evidence that its function acts posttranslationally in cell cycle arrest after DNA damage. Mol. Cell. Biol., 10, 6554–6564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schiestl R.H., Reynolds,P., Prakash,S. and Prakash,L. (1989) Cloning and sequence analysis of the Saccharomyces cerevisiae RAD9 gene and further evidence that its product is required for cell cycle arrest induced by DNA damage. Mol. Cell. Biol., 9, 1882–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Siede W., Friedberg,A.S., Dianova,I. and Friedberg,E.C. (1994) Characterization of G1 checkpoint control in the yeast Saccharomyces cerevisiae following exposure to DNA-damaging agents. Genetics, 138, 271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Siede W., Friedberg,A.S. and Friedberg,E.C. (1993) RAD9-dependent G1 arrest defines a second checkpoint for damaged DNA in the cell cycle of Saccharomyces cerevisiae. Proc. Natl Acad. Sci. USA, 90, 7985–7989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vialard J.E., Gilbert,C.S., Green,C.M. and Lowndes,N.F. (1998) The budding yeast Rad9 checkpoint protein is subjected to Mec1/Tel1-dependent hyperphosphorylation and interacts with Rad53 after DNA damage. EMBO J., 17, 5679–5688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Emili A. (1998) MEC1-dependent phosphorylation of Rad9p in response to DNA damage. Mol. Cell ., 2, 183–189. [DOI] [PubMed] [Google Scholar]
- 17.Aboussekhra A., Vialard,J.E., Morrison,D.E., de la Torre-Ruiz,M.A., Cernakova,L., Fabre,F. and Lowndes,N.F. (1996) A novel role for the budding yeast RAD9 checkpoint gene in DNA damage-dependent transcription. EMBO J., 15, 3912–3922. [PMC free article] [PubMed] [Google Scholar]
- 18.de la Torre-Ruiz M.A., Green,C.M. and Lowndes,N.F. (1998) RAD9 and RAD24 define two additive, interacting branches of the DNA damage checkpoint pathway in budding yeast normally required for Rad53 modification and activation. EMBO J., 17, 2687–2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sherman F., Fink,G.R. and Hicks,J.B. (1986) Methods in Yeast Genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 20.Aboussekhra A. and Thoma,F. (1998) Nucleotide excision repair and photolyase preferentially repair the nontranscribed strand of RNA polymerase III-transcribed genes in Saccharomyces cerevisiae. Genes Dev., 12, 411–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wellinger R.E. and Thoma,F. (1997) Nucleosome structure and positioning modulate nucleotide excision repair in the non-transcribed strand of an active gene. EMBO J., 16, 5046–5056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tijsterman M., de Pril,R., Tasseron-de Jong,J.G. and Brouwer,J. (1999) RNA polymerase II transcription suppresses nucleosomal modulation of UV-induced (6-4) photoproduct and cyclobutane pyrimidine dimer repair in yeast. Mol. Cell. Biol., 19, 934–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Waters R., Zhang,R. and Jones,N.J. (1993) Inducible removal of UV-induced pyrimidine dimers from transcriptionally active and inactive genes of Saccharomyces cerevisiae. Mol. Gen. Genet., 239, 28–32. [DOI] [PubMed] [Google Scholar]
- 24.Spellman P.T., Sherlock,G., Zhang,M.Q., Iyer,V.R., Anders,K., Eisen,M.B., Brown,P.O., Botstein,D. and Futcher,B. (1998) Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridization. Mol. Biol. Cell, 9, 3273–3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Livingstone-Zatchej M., Meier,A., Suter,B. and Thoma,F. (1997) RNA polymerase II transcription inhibits DNA repair by photolyase in the transcribed strand of active yeast genes. Nucleic Acids Res., 25, 3795–3800. [DOI] [PMC free article] [PubMed] [Google Scholar]