

Abstract
We present a novel approach for characterizing surfaces utilizing super-resolution fluorescence microscopy with subdiffraction limit spatial resolution. Thermoplastic surfaces were activated by UV/O3 or O2 plasma treatment under various conditions to generate pendant surface-confined carboxylic acids (–COOH). These surface functional groups were then labeled with a photoswitchable dye and interrogated using single-molecule, localization-based, super-resolution fluorescence microscopy to elucidate the surface heterogeneity of these functional groups across the activated surface. Data indicated nonuniform distributions of these functional groups for both COC and PMMA thermoplastics with the degree of heterogeneity being dose dependent. In addition, COC demonstrated relative higher surface density of functional groups compared to PMMA for both UV/O3 and O2 plasma treatment. The spatial distribution of –COOH groups secured from super-resolution imaging were used to simulate nonuniform patterns of electroosmotic flow in thermoplastic nanochannels. Simulations were compared to single-particle tracking of fluorescent nanoparticles within thermoplastic nanoslits to demonstrate the effects of surface functional group heterogeneity on the electrokinetic transport process.
Graphical Abstract
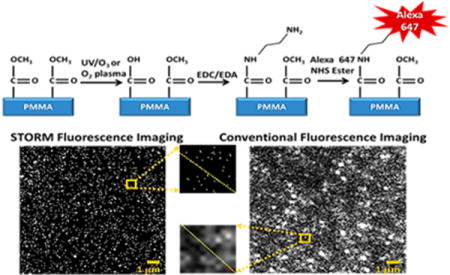
Thermoplastics are an attractive material for both microfluidics and nanofluidics.1–4 The interest in thermoplastics stems from the fact that both microscale and nanoscale structures can be produced with techniques that are conducive to high scale production of devices with high fidelity and high process yield rates.5–7 Indeed, thermoplastic-based microfluidic and nanofluidic devices have been used for a variety of applications, such as microarrays,8,9 solid-phase enzymatic reactors,10–12 solid-phase extractors for nucleic acids and proteins,13 affinity selection of biological cells,14–16 chromatography,17 and microchip electrophoresis.18,19
While the benefits of microchip electrophoresis have been well documented,20–22 new efforts have been devoted to reducing electrophoresis columns to nanometer sizes, because unique phenomena specific to nanoconfined domains can be exploited; thus, separations not possible in the microscale can be achieved using nanometer columns.23–28 For example, as the channel dimensions approach the thickness of the electrical double layer (EDL), EDL overlap results in nonplug, parabolic flow profiles. Furthermore, increased surface area-to-volume ratios result in surface interactions,29–35 giving rise to transverse electromigration (TEM).35 Because of EDL overlap, parabolic flow profiles can be generated, and with the action of TEM, separations can be undertaken that are not possible in microscale electrophoresis.29,31,32,36–38
Conventionally, nanoelectrophoresis is carried out using glass-based devices, which have a well characterized surface chemistry that is highly ordered and homogeneous. However, fabrication of such devices is costly and time-consuming, in many cases requiring deep UV lithography or ion beam milling of each device.39–41 In contrast, multiple thermoplastic nanocolumns can be replicated from a single master by nanoimprint lithography (NIL), reducing cost and simplifying fabrication.
In general, thermoplastics can be modified to increase their surface charge density and wettability, enabling the generation of electroosmotic flow (EOF).42 Thermoplastic devices can be activated by exposure to UV/O3 or O2 plasma,43 which forms oxygen-containing functionalities, including surface carboxylic acid groups (–COOH) as well as other oxygen-containing groups that are typically less abundant than the –COOH groups. Characterization of these activated surfaces has been demonstrated.14,44 But, previous measurements produce ensembled results averaged over relatively large areas (1 mm2)14 and, as such, did not address the spatial distribution of functional groups generated following activation of the polymer.14,44
The heterogeneity of surface functional groups following activation can have a profound impact on the electrophoretic performance of a nanoscale device. For example, heterogeneous distributions of surface charges can generate nonuniform zeta-potentials, which are known to cause recirculation in the EOF that can degrade separation efficiency.45,46 This issue is exacerbated when the column cross section is significantly reduced, such as is the case for nanoscale electrophoresis due to increased interaction with the heterogeneous surface.29,33,34
Assuming the Debye length is significantly smaller than the width of a fluidic channel and the surface charge density is uniform along both the axial and radial dimensions, the fluid velocity within a channel (υ) can be described by the Helmholtz-Smoluchowski equation:
| (1) |
where ε is the permittivity of the buffer, E is the applied electric field, is the average zeta potential, and η is the fluid viscosity. This equation describes the classic “plug-like” flow characteristic of microscale electrophoresis.45,47–50 However, eq 1 is not applicable for the majority of nanoscale electrophoretic separations because the Debye length approaches channel dimensions. Moreover, nonuniform distributions of surface charges modulate the zeta potential so that it can no longer be averaged.
To date, theoretical and experimental investigations into the effects of nonuniform surface charge have been explored only in a few cases.12,45,47,50,51 For example, Anderson and Idol studied EOF through pores with nonuniformly charged walls and found the mean fluid velocity to be accurately described by the Helmholtz equation when using the average zeta potential; however, recirculation of fluid was observed in regions where ζ deviated from the average.47
Ajdari explored alternating regions of positive and negative surface charges and showed the presence of steady convective recirculation in the EOF.50 Surface defects have also been shown to introduce nonuniform ζ with similar hydrodynamic contributions noted.52 Several other investigations45,48,51 relied on a step scheme in which ζ of one region was set to zero, while the downstream region had a higher ζ, thus generating recirculation at the junction, but plug-like EOF downstream.45,50
All of the aforementioned studies assumed that the surface charge density conforms to an equilibrium Boltzmann distribution, which assumes thermodynamic equilibrium where the ionic distributions are uniform along the channel wall and unaffected by fluid flow.51,53 However, this assumption is inaccurate in the case of nonuniform surface charge distribution. In these cases, it is necessary to adopt the Nernst-Plank model to account for the heterogeneity of surface charge. Fu et al. observed deviations from the typically used Boltzmann distribution; they also explored the effect of EDL overlap with nonuniform ζ. They showed that the velocity profiles in the regions of high and low ζ is parabolic due to EDL overlap.51
Investigations into the nonuniform spatial distribution of functional groups on surfaces with nm resolution has not been explored to our knowledge. The process of activating thermoplastics with UV/O3 or O2 plasma may introduce nonuniform surface charges. Hence, a thorough analysis of surface heterogeneity caused by activating amorphous thermoplastic surfaces is critical to understand the utility of thermoplastics as substrate materials for nanoscale electrophoresis and other applications. Many variables can affect the heterogeneity of surface confined –COOH groups, in particular, competitive chemical reactions giving rise to different products, differences in the tacticity of the polymer, and nanoscale roughness, which also has the potential to locally distort ζ.
To assess –COOH surface spatial distributions, it was necessary to precisely locate functional groups with nm resolution. Thus, we applied super-resolution microscopy with the –COOH groups labeled with a fluorescent reporter. Based on theoretical predictions, the –COOH surface density may be as high as 0.83 nmol/cm2 (five carboxyl groups/nm2) for a hexagonally ordered surface.54 Although polymers are not crystalline in nature, we can still expect the density of fluorescent reporters to be far below the resolution limit of conventional fluorescence due to diffraction.55 Super-resolution fluorescence microscopy allows for subdiffraction imaging using different approaches including Stochastic Optical Reconstruction Microscopy (STORM). Super-resolution microscopes have classically been used for imaging biological samples with a few demonstrations in synthetic samples,56 nanoparticles,57 or nanochannel solution ion distributions.58 We will show in this work the use of STORM microscopy for the analysis of thermoplastic surfaces activated by UV/O3 or O2 plasma and the subsequent coupling of a fluorophore to the pendant –COOH functional groups to examine the heterogeneity in the spatial distribution of these functional groups.
MATERIALS AND METHODS
Reagents and Materials
Poly(methyl methacrylate), PMMA, was purchased from Goodfellow (Berwyn, PA). Cyclic olefin copolymer (COC 8007 and 6017) were purchased from TOPAS Advanced Polymers (Florence, KY). Si ⟨100⟩ wafers were acquired from University Wafers (Boston, MA). Ethylenediamine (EDA), sodium dodecyl sulfate (SDS), dextrose, glucose oxidase (Aspergillus niger), Dulbecco’s phosphate buffered saline (1× DPBS, pH 7.4), catalase (bovine liver), monoethanolamine (MEA, pH 8.0–8.5), sodium bicarbonate buffer (pH 7.5), tripropylene glycol diacrylate (TPGA), trimethylolpropane triacrylate (TMPA), Irgacure 651 (photoinitiator), and potassium chloride (KCl) were purchased from Sigma-Aldrich (St. Louis, MO) and used as received. Alexa Fluor 647 NHS ester and 20 nm Nile red (535/575) carboxylate modified polystyrene spheres were secured from Life Technologies (Grand Island, NY). Antiadhesion (tridecafluoro-1,1,2,2-tetrahydrooctyl) tricholorosilane (T-Silane) was purchased from Gelest, Inc. (Morrisville, PA). Tris buffer (pH 8.0) was obtained from Fisher Scientific (Houston, TX). All dilutions were performed using 18 MΩ/cm Milli-Q water (EMD Millipore, Billerica, MA), with all measurements carried out at 25 °C.
Surface Activation and Modification
The protocol used for activating and modifying surfaces has previously been reported and validated.42 In brief, PMMA and COC (1 cm2) were sonicated for 30 min in 0.1% SDS and then exposed to either UV/O3 (31.1 mW/cm2, λ = 254 nm) or O2 plasma (50 W, 10 sccm) for various times to generate surface carboxyl groups with different densities. Covalent attachment of Alexa Fluor 647 NHS Ester to the surface confined –COOH groups was achieved by first generating an amine-terminated surface by incubation of the activated plastic in a solution containing 100 mg EDC and an excess of EDA (134 μL) in 2 mL of 0.1 M MES buffer (pH 4.8) for 20 min.42 The plastic was then removed from this solution, washed thoroughly with DI water, spotted (2 μL) with Alexa Fluor 647 NHS in 0.1 M sodium bicarbonate (pH 7.5), and incubated in the dark for 1 h. The samples were then thoroughly washed with DI water.
STORM Imaging
All imaging was performed on activated COC or PMMA thin films (100 μm) with the surface confined –COOH groups labeled with the fluorescent reporter. For STORM, the underlying mechanism for photoswitching between “on” and “off” states required the addition of a thiol containing molecule. For this reason, the buffer used for imaging was composed of 80% (v/v) 1× DPBS (pH 7.4), 10% (v/v) 1 M MEA, 10% (v/v) 50% glucose, and 1% (v/v) glucose oxidase/catalase oxygen scavenger. Imaging was performed using a Zeiss Axio Observer Z-1 inverted microscope with a 100× oil immersion objective (NA 1.46), an iXON EMCCD camera, and 405, 488, 561, and 640 nm wavelength lasers. For the purposes of these experiments, only the 405 and 650 nm laser were used. The laser power was precisely selected to balance photoswitching kinetics and photobleaching. To obtain the best resolution, one must optimize the number of photoswitching cycles as well as the time spent in the “on” or “off” state. In order to ensure that two dye molecules were not emitting at the same time within a diffraction limited area, the “off” time should be much greater than the “on” time. This was considered in our laser power selection.59,60 The image pixel size was 10 nm with a camera pixel size of 16 μm. Exposure times were 30 ms to ensure that single fluorescent events were captured with high signal-to-noise.
Each sample was imaged for 5 min (>10000 frames). Within this collection time, fluorophores cycle through several “on” and “off” states. Image frames capturing the blinking of surface confined fluorophores were then analyzed using Carl Zeiss Zen image processing software adopting the classical super-resolution image processing protocol. In brief, the Zen software performed a peak finding function in all frames based on minimum and maximum photon count thresholds to ensure that both noise and aggregated nonblinking molecules were excluded. Next, peak fitting algorithms were applied to determine the width and centroid position of each peak. Once all localizations were determined, drift correction and peak grouping functions were applied. One “on” event for a fluorophore can last for several frames with the collection time employed herein.
During the acquisition time, the molecule may appear to drift due to thermal expansion and mechanical vibration. The algorithm used for drift correction breaks the rendered STORM image into smaller, lower quality STORM images. Then, cross-correlation of subsequent images allowed a measurement of sample drift. This method is well documented and widely used.61,62 Furthermore, because a molecule may appear in several frames, several parameters were used to group these localizations to one molecule. These include maximum “on” time, defined as the maximum number of frames that peaks are allowed to be detected in order to be considered one molecule (5 frames), maximum off time, defined as the maximum number of frames that a peak can be missing and still considered one molecule (10 frames) as well as capture radius, defined as the pixels within which peaks of consecutive frames must lie in order to be considered as belonging to the same molecule (2 pixels).
Lastly, for accurate positioning, if the point spread function (PSF) of two different localizations overlapped and appeared as one distorted spot, these events were discarded. Further parameters such as photon count, point spread function diameter, and localization precision were used to render STORM images, as is classically done in super-resolution post processing. Conventional fluorescence images were collected using the same microscope without reconstruction postprocessing.
Control experiments were performed to determine nonspecific dye adsorption. Activated surfaces were incubated with just Alexa Fluor 647 NHS without the amine linker. Control experiments showed <1% nonspecific adsorption based on the total number of localizations.
COMSOL Simulations
Fluorophore centroids that were determined from the super-resolution images were mapped as single point charges on otherwise noncharged surfaces of a 50 nm × 50 nm × 1 μm (W × H × L) model nanochannel to approximate the effect of heterogeneous surface charges on the EOF. Geometries were constructed via a MATLAB code and imported into COMSOL Multiphysics (v 5.0) was used to solve for EOF profiles with a two ion buffer system (see Table S1 for modeling parameters). Notably, oxygen-containing functional groups other than –COOH groups, which were likely generated during activation, were not included in the model due to their unknown densities and positions. However, because these groups would be expected to be uncharged for the pH values used herein, they would not be expected to contribute to the EOF.
Nanoslit Fabrication
The fabrication of nanoslits in thermoplastics is detailed elsewhere.6,7 Briefly, a silicon master was fabricated by initially patterning two access microfluidic channels (55 μm wide, 12 μm deep, 1.5 cm long) into a Si ⟨100⟩ wafer using standard photolithography followed by anisotropic etching with 45% KOH. Next, 3 μm × 150 nm nanofluidic slits were patterned by FIB milling of the Si wafer using a Helios NanoLab 600 Dual Beam instrument (FEI, Hillsboro, OR).
An antiadhesion coating of T-Silane was applied to the Si master from the gas phase in a desiccator under vacuum for 2 h to facilitate demolding. A resin stamp was then made using UV-NIL from a UV-curable polymeric blend containing 68 wt % TPGA as the base, 28 wt % TMPA as the cross-linking agent, and 4 wt % Irgacure 651 as the photoinitiator that was coated onto a rigid COC back plate. The stamp was cured by exposure to 365 nm light (10 J/m2) through the COC back plate for 7 min in a CL-100 Ultraviolet Cross-linker (UVP, LLC, Upland, CA). The UV-cured resin was gently demolded from the Si master.
The stamp was imprinted into a 1.5 mm thick piece of COC by NIL with access holes used as reservoirs. NIL was performed at a pressure of 2888 kN/m2 for 120 s with the top and bottom plates maintained at a temperature of 95 °C using a Hex03 thermal imprinter (JenOptik AG, Jena, Germany). Pressure was applied after 30 s and was maintained during imprinting until the system was cooled to 45 °C. A 100 μm thick COC sheet of the same type as the substrate was used as the cover plate. Both the COC substrate and cover plate were preactivated with oxygen plasma. Thermal assembly was performed immediately at 70 °C for 900 s using 977 kN/m2 pressure.
Nanoelectrophoresis of Fluorescent Nanoparticles
All fluorescence imaging for the nanoelectrophoresis were performed using an Axiovert 35 inverted microscope (Carl Zeiss AG, Oberkochen, Germany) equipped with a 100× oil immersion objective (NA 1.3). For imaging, the optical system in Figure S1 was used. A Gaussian laser beam (Nd:VYAG; λex = 532 nm; P = 0.01–5 W; 2.2 mm beam diameter) was expanded with a 10× Keplerian beam expander to completely backfill the objective and the wings were knocked out with an iris to ensure uniform laser intensity in the field-of-view. The beam was focused using a lens into the back of a microscope objective to allow irradiation of the entire field of view and passed through a 532 nm laser line filter and dichroic filter. The emission signal was collected by the objective, passed through a dichroic filter, was spectrally selected using long-pass and bandpass filters, and was imaged onto an iXon3 897 EMCCD camera (Andor Technology Ltd., Belfast, United Kingdom) controlled by Metamorph software. Trackmate software in Fiji was used for data analysis.
All electrophoresis experiments were conducted in 1× PBS (pH 7.4). A 1.0 fM concentration of polystyrene microspheres was used to increase the likelihood of single particle occupancy within the nanoslit. Polymer nanoslits were primed with buffer and allowed to equilibrate prior to the electrophoresis. The fluorescent nanospheres were loaded into an injection reservoir and various field strengths were applied (100–300 V/cm) to electrokinetically drive the spheres into the nanoslits. Translocation events were captured and processed using the optical system described above.
RESULTS AND DISCUSSION
STORM versus Conventional Fluorescence
Typically, STORM is used to interrogate biological features with subdiffraction limit resolution.55 STORM accomplishes subdiffraction imaging by sequential activation and time-resolved localization of photoswitchable fluorophores. In STORM imaging, fluorophores were heavily biased toward the “off” state by the inclusion of a thiol (mercaptoethylamine) that disrupts the molecule’s conjugation, but when the fluorophore was exposed to UV light, the thiol was dissociated, conjugation was regenerated, and the molecule can reach the “on” state to generate fluorescence. By suppressing the probability of fluorescence, the centroids of individual fluorescent events can be precisely located with ~20 nm resolution, and the entire surface can be reconstructed by imaging for several minutes and merging the events that are observed throughout the tens of thousands of frames.
For our experiments, we sought to use STORM to probe thermoplastic surfaces that were exposed to UV/O3 irradiation or O2 plasma, which can generate surface-confined –COOH groups9 that are useful for generating EOF as well as immobilizing moieties such as small molecules42 and biomacromolecules.14 To visualize the surface confined –COOH groups by STORM, we conjugated these groups to a short diamine cross-linker and subsequently labeled the free primary amine with an NHS-ester fluorescent dye, allowing us to monitor the spatial distributions of the dye-labeled –COOH groups. We then captured >10000 frames as these surface confined fluorophores transitioned between “on” and “off” states. There frames were then imported into commercial image processing software and precise parameters were used to localize each fluorophore and render highly resolved STORM images.
Figure 1 compares STORM (a) and conventional fluorescence microscopy (c) imaging of 26 μm2 areas of UV/O3 treated COC. Specifically, we are imaging dye-labeled, surface-confined –COOH groups generated through UV/O3 activation. Also shown are line plots of a 1 μm2 subsection of these images. The conventional fluorescence image had several overlapping spots of various intensities, indicating that densely packed fluorophores could not be resolved (Figure 1d). The benefits of STORM imaging were immediately apparent with the centroid of each fluorophore resolved, even at the higher dose levels where the functional group spacing is low. For example, in the 1 μm2 subset images of Figure 1a the centroids of 41 different fluorescent events were localized by STORM, while an unknown number of fluorophores contributed to the conventional fluorescence image.
Figure 1.
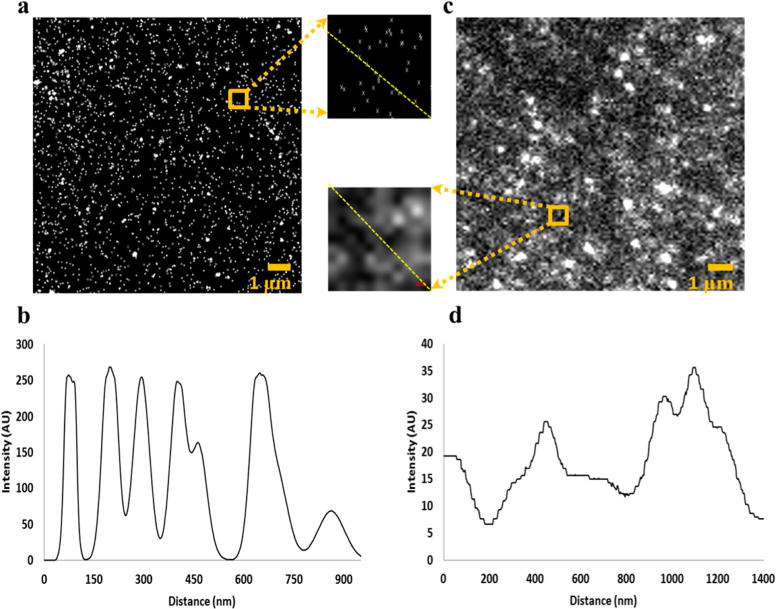
(a) STORM image of COC exposed to 1 min UV/O3 radiation (31.1 mW/cm2, λ = 254 nm) with 1 μm2 inset, and (b) line plot from top left to bottom right of (a) inset. (c) Conventional microscopy image of COC exposed to the same UV/O3 dosage also with accompanying 1 μm2 inset, and (d) line plot from dashed line in (c) inset.
However, not all 41 events in Figure 1a necessarily originated from different dye molecules. Each fluorescent molecule can cycle through on/off states multiple times throughout the 5 min imaging time; the dye’s position can shift slightly between cycles. It is unlikely that movement of the dye molecule relative to the surface would be significant because the diamine linker was only ~1 nm in length, thereby constraining the molecule’s movement. Rather, shifts in the fluorophore’s image can be attributed to drift of the microscope due to thermal expansion and mechanical vibrations. Furthermore, random errors of the fitting procedure, localization accuracy, and systematic errors of the microscope can also contribute to the inaccurate localization of molecules.
To determine the average number of blinking events for a given fluorophore, we examined individual localizations of the STORM images by hierarchal clustering analysis.63 By fitting the number of clustered blinking events to an exponential distribution (Figure S2), we determined a Poisson average of 7 blinks per fluorophore. These results were reproducible across various activated polymers (data not shown). Second, the spatial distribution of the clustered blinking events was fit with a Gaussian function (Figure S3); each molecule’s distribution of localization was characterized by a full width at half-maximum of ~40 nm. Two events can be resolved if they are separated by greater than the fwhm. We observed a localization accuracy (i.e., σ of the Gaussian function) of 17 nm. The localization accuracy was determined after all STORM postprocessing procedures, including drift correction; thus, this value represents the localization accuracy and drift efficiency. Since the experimentally measured localization accuracy is in agreement with the theoretical precision (σ = 20 nm, calculated from the photon count of 800–1000), the efficiency of drift correction was quite high and had little contribution to the overall error in localizations. Nevertheless, STORM enabled the localization of dye-labeled –COOH groups separated by >40 nm, which was not possible using conventional fluorescence microscopy.
Comparing Relative –COOH Density Using STORM
STORM imaging was used to investigate the relative changes in the density of dye-labeled –COOH groups on COC and PMMA surfaces that received various doses of UV/O3 irradiation or O2 plasma exposure. Representative images, where the centroids of blinking events were plotted, are shown in Figure 2a–e for COC and Figure 2f–j for PMMA exposed to UV/O3 irradiation for different times. While the absolute density of fluorophores could not be assessed due to multiple blinking events per fluorophore, relative changes in the surface density of dye-labeled –COOH groups could be deduced because the experimental conditions (buffer composition, light intensity, etc.) were the same throughout all STORM imaging. Thus, the dependence of –COOH surface density due to polymer, activation method and exposure time could be discerned, as shown in Figure 2k,l.
Figure 2.
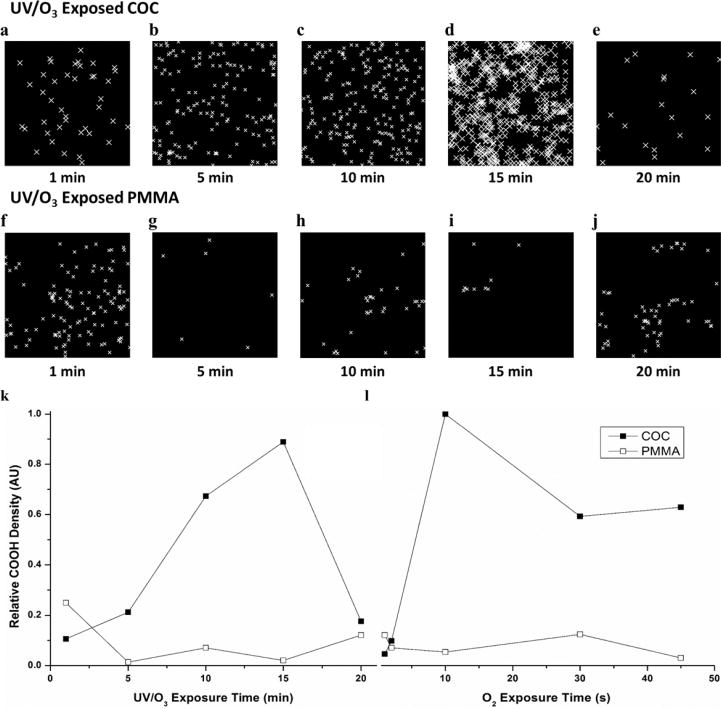
Representative STORM images of 1 μm2 (a–e) COC and (f–j) PMMA exposed to 1, 5, 10, 15, and 20 min UV/O3 radiation, respectively. Relative –COOH density vs exposure time for (k) UV/O3 and (l) O2 plasma-modified COC (closed squares) and PMMA (open circles). Lines are for visual purposes only. UV/O3 and O2 plasma exposure conditions were kept constant (see text for details). All total localizations were normalized to the greatest localization density, COC exposed to 10 s of O2 plasma.
There was an increase in –COOH relative density on COC surfaces as UV/O3 exposure increased up to 15 min. After 20 min exposure, there was a dramatic decrease in the relative –COOH surface density (Figure 2k, solid square). A similar trend was observed for COC exposed to O2 plasma with an increase observed up to 10 s exposure times and a drop in the relative density after 30 s exposures (Figure 2l, solid square). The apparent decrease in –COOH density for long exposure times for both UV/O3 and O2 plasma activation is most likely due to radical reactions occurring after –COOH generation, which can degrade the –COOH group and lead to effects such as –CO2 release, scissioning of the polymer chain, and etching of the surface.44,64 Chemical groups other than –COOH generated by these activation processes are not probed by our fluorescent reporter, which was specific for –COOH groups; however, the authors note that previous research has shown that –COOH functional groups are the dominate species following UV/O3 or plasma activation protocols performed on PMMA and COC thermoplastics.14
The density of –COOH functionalities on UV/O3 activated PMMA surfaces (Figure 2k, open square) was greatest for 1 min exposure with a decrease for longer exposure times until ~20 min, when it was observed that the –COOH relative density slightly increased. A similar trend was seen for O2 plasma exposed PMMA (Figure 2l, open square); the greatest –COOH relative densities were observed at 1 s exposure, and the –COOH relative surface density decreased from 2 to 10 s exposure, returned to a relatively high level after 30 s, and then decreased again after 45 s exposure. Two factors contributed to these results. First, –COOH generation is known to be more efficient on COC surfaces compared to PMMA, although –COOH formation dominates for both thermoplastics.14 Scissioning of PMMA polymer chains throughout UV/O3 exposure has been suggested to compete with –COOH formation and lead to extensive fragmentation and etching of the polymer, more so than for COC polymers.14 This provides an explanation for the relatively lower surface densities overall. Second, the PMMA contained impact modifiers, cross-linked poly(butyl acrylate),14 which may have a poorer propensity to undergo photooxidation to generate pendant –COOH groups. These impact modifiers can be observed by O2 plasma exposure in Figure S4, which showed surface roughness features on PMMA due to the differences in plasma etch rates between PMMA and poly(butyl acrylate).
Overall, two observations can be made from this data: (i) The maximum –COOH relative densities on PMMA substrates were achieved at the shortest activation times, 1 min UV/O3 and 1 s O2 plasma treatment. In the case of COC, longer exposure times were required to maximize the relative surface density of the –COOH groups. (ii) The greatest relative density of –COOH groups generated on PMMA by UV/O3 and O2 plasma were 25 and 10%, respectively, the densities observed on similarly treated COC substrates.
With our current localization accuracy of 17 nm and considering the maximum hexagonal packing density of fluorophores, the highest density that could be theoretically localized would be 4 × 1011 molecules/cm2, which is insufficient for a crystalline structure (~1 × 1014 molecules/cm2). However, the maximum number of localization density we encountered was ~5 × 1010 molecules/cm2. Thus, we have not reached the upper spatial resolution detection limit of our current STORM conditions.
Determination of Functional Group Heterogeneity Using STORM
The heterogeneous spatial distribution of –COOH groups can have a significant impact in applications such as nanoscale electrophoresis by creating recirculation in the flow field and thus, lowering the efficiency in the separation. The subdiffraction limit resolution afforded by STORM enabled us to observe these spatial distributions with accuracy not possible by traditional fluorescence microscopy. To determine the heterogeneity in the spatial distribution of –COOH groups on the polymer surface with nm resolution, the Euclidian distance between each localization determined through STORM was calculated; however, the Euclidean nearest-neighbor distance was confounded by blinking of the fluorophores in time. A given fluorophore will be detected multiple times, shifting slightly in position due to instrument drift and generating nearest-neighbor distances that do not sometimes reflect the true spatial distribution of dye-labeled –COOH groups. Thus, for each fluorescent event we excluded nearest-neighbors within the localization accuracy (17 nm) and assembled the distributions of nearest-neighbor distances for both thermoplastics under all activation conditions (Figure 3).
Figure 3.
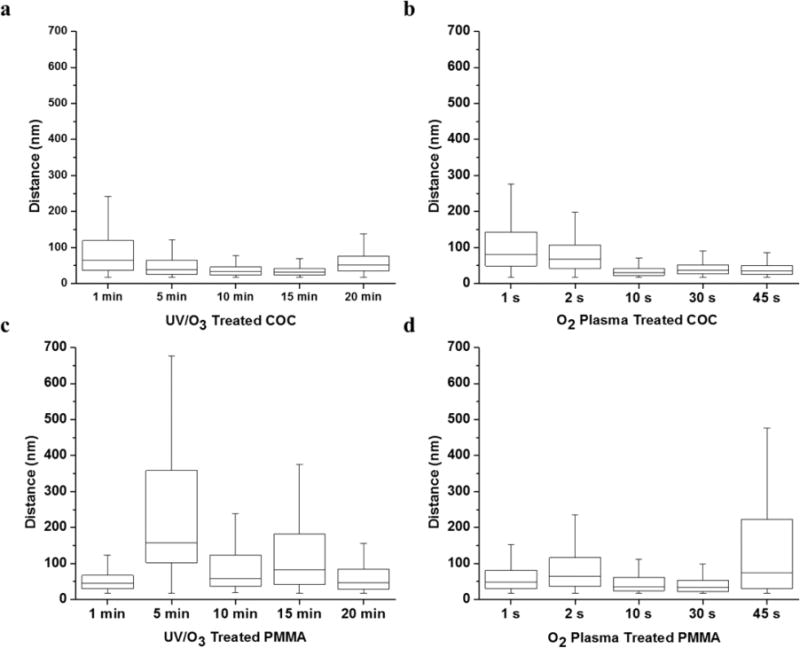
Distance between fluorophores in 1 μm2 subset images (see Figure 2) for (a) UV/O3 modified COC; (b) O2 plasma-modified COC; (c) UV/O3 modified PMMA; and (d) O2 plasma-modified PMMA. Error bars represent range; upper and lower edges of the boxes indicate the third and first quartiles, respectively, and the midline shows the median.
In general, irrespective of the polymer or activation source, increasing –COOH surface relative densities correlated, not surprisingly, with a decreased distance between functional groups. For example, at low doses of UV/O3 activation, the average nearest-neighbor distance between –COOH groups on COC was large with a median distance of 64 nm after 1 min exposure that decreased to 31 nm after 15 min exposure. The reduced spacing was mirrored by the increase in –COOH relative densities (Figure 2k), and a similar trend was observed for COC surfaces throughout the different O2 plasma exposure times. Not only did the median functional group distances decrease, but the variance in the distributions reduced as well. This was evident as the first and third quartiles approached the median spacing after 15 min UV/O3 exposure as well as a 4-fold decrease in the first and third quartile range from 1 min exposure to 15 min exposure.
Activated PMMA surfaces showed a broader spatial distribution of dye-labeled –COOH groups, although at short exposure times (1 min UV/O3 treatment and 1 s O2 plasma activation), the distributions were much narrower than the highly heterogeneous surfaces observed at longer activation conditions. For a PMMA surface activated by 1 min UV/O3 and 30 s O2 plasma exposures, the lowest median nearest-neighbor distances were 45 and 34 nm, respectively, and the distributions between the first and third quartiles ranged from 30 to 67 nm and 24–64 nm, respectively.
We note that in this analysis, we only approximated spatial distributions using the nearest-neighbor distance between dye-labeled –COOH groups that was greater than the 17 nm localization accuracy. At high –COOH densities, there appeared to be “patches” of polymer that were unmodified and these effects are not described by the nearest-neighbor analysis. This is especially apparent for COC, that was activated by 15 min UV/O3 irradiation (Figure 2d), where the unmodified patches were 100 × 100 nm in size. For PMMA, this may be attributed to the impact modifiers contained within the films (Figure S4). However, COC substrates are not known to contain such additives and we do not have sufficient data to source the origin of these unmodified patches.
COMSOL Simulations of EOF Flow with Nonuniform Surface Charge
In glass-based micro- and nanofluidic devices, the homogeneous surface distribution of silanol groups are known to generate uniform and stable EOF profiles.65 It is also known that the relatively low density of –COOH groups on activated thermoplastic surfaces reduces the EOF magnitude compared to glass.42 Here, we investigated if the spatial heterogeneity of the charged functional groups on the thermoplastic distort the uniformity of the EOF flow profile.
To this end, we first conducted three-dimensional computational fluid dynamics simulations of the EOF. The locations of –COOH groups on COC surfaces activated by 5 min of UV/O3 exposure, which were obtained via STORM imaging, were mapped directly onto the surfaces of a 50 × 50 × 500 nm (w × h × l) nanochannel. Each –COOH group was assigned as a point and given the charge of a single electron, and the EOF profiles generated by these point charges were simulated using a two ion buffer system. Polar functional groups, such as alcohols and other carbonyl-containing moieties, are also known to be generated by either UV/O3 or O2 plasma exposure on many thermoplastics; however, to a lesser extent than –COOH.14 We note that while these noncarboxylate functional groups could be generated using UV/O3 or O2 plasma activation, such as alcohols, aldehydes, and ketones, these groups would be uncharged at the pH values used in these experiments. Thus, their contribution to the EOF would not be present and would be determined only by the surface –COOH groups that we are monitoring. However, the reactions generating these noncharged functional groups can contribute to the observed –COOH surface heterogeneity.
As a reference, a nanochannel was modeled using a uniform charge density to mimic a silanol-based fused silica nanochannel (Figure 4a). One can observe a drop in the applied potential along the nanochannel’s length as well as the charged wall’s strong negative potential extending into the ionic solution. The resultant EOF profile approached a maximum longitudinal velocity of 40 μm/s in the channel’s center, but as the 6.9 nm Debye length was close to the nanochannel’s physical dimensions, the no-slip condition generated a parabolic flow profile (Figure 4b, left panel). Indeed, we are operating in a region with a κa29 value of 3.7, confirming some EDL overlap. Despite EOF nonuniformity along the axial dimension producing Taylor dispersion, the EOF is highly uniform along the channel’s length (Figure 4b, right panel, and Figure 4c). This is beneficial for electrophoretic separations as the resulting velocity streamlines are highly reproducible (Figure 4d) and uniform (i.e., no recirculation).
Figure 4.
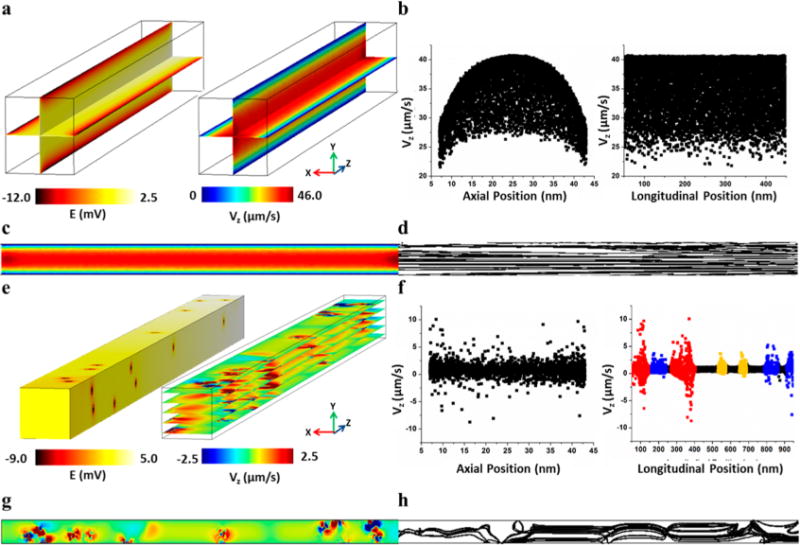
(a) COMSOL simulation showing the electric potential (left) and velocity magnitude (right) for a channel with uniform surface charge; (b) Velocity vs axial (right) and longitudinal (left) position to show the EOF flow profile for a channel with uniform surface charge; (c) One slice of the velocity magnitude of a uniform channel; (d) Streamline of the same velocity slice depicted in (c); (e) COMSOL simulation showing the electric potential (left) and velocity magnitude (right) where single point charges are mapped onto the nanochannel surfaces using the –COOH locations (centroids) obtained by STORM analysis of a COC surface exposed to 5 min UV/O3 activation. (f) Velocity vs axial (right) and longitudinal (left) position to show the EOF flow profile for the channel with nonuniform surface charge. The colors in the velocity vs Z position graph (right) represent an area in the channel with >5 (red), 3–4 (blue), and 1–2 (yellow) –COOH group(s) within 20 nm of each other; (g) One slice of the velocity profile to show fluid flow recirculation; (h) Streamline of the same velocity slice depicted in (e) to emphasize the fluid recirculation at areas with –COOH.
Unlike the fused silica nanochannel simulation, the UV/O3 activated thermoplastic nanochannel generated an irregular EOF profile. The positions of the negative point charges mapped from the STORM image can be seen in the electric potential plot in Figure 4e. Near these point charges, the longitudinal velocity profile is highly distorted, being positive within a Debye length of the point charge but also negative elsewhere, which is a strong indication of flow recirculation (Figure 4e,g). Because the –COOH distribution were heterogeneous along the channel’s longitudinal axis, there was a distorted velocity pattern along the channel’s longitudinal dimension (Figure 4f, left panel). The EOF’s longitudinal velocity could be correlated to the position of the deprotonated –COOH groups along the channel’s length (Figure 4f, right panel). Near each point charge, there are both positive and negative velocities, which scale in magnitude with the number of –COOH groups in proximity. The highest local velocities were 3, 7, and 10 μm/s in the regions with 1–2 (yellow), 3–4 (blue), and >5 (red) surface charges in close proximity. These phenomena were evident in the velocity streamline trace (Figure 4h), which indicated the trajectories’ of noncharged particles in the absence of diffusion. Also, in regions with no –COOH groups, there was a small (2 μm/s) forward velocity due to bulk flow of the buffer.
In summary, the fused silica-based simulation with uniform surface charge generated a nondistorted EOF profile throughout the device, although the flow was parabolic due to EDL overlap. Such parabolic flow can induce Taylor dispersion into an electrophoretic separation, but can offer unique electro-phoretic separations not possible without this EDL overlap, such as the free solution separation of double-stranded DNAs.66 The COC nanochannel, which had nonuniform surface charges that were mapped directly from the STORM images indicated a distorted EOF. However, even regions with relatively high localized EOF would not likely provide a noticeable effect on zonal dispersion resulting from recirculation because the particle’s electrophoretic mobility would likely dominate due to the small EOF observed due to the sparsely charged nature of the polymer channel walls.
This provides a unique opportunity for thermoplastic devices because of the ability to selectively and locally activate portions of a nanochannel by exposing to either O2 plasma or UV/O3 through a mask without creating global EOF. Also, by selecting the appropriate activating dose, the surface charge density can be controlled, which allows for control of the magnitude of the EOF without requiring surface passivation.
Nanoscale Electrophoresis
To empirically observe the potential effects of EOF distortion arising from a nonuniform distribution of –COOH groups on the nanocolumns’ wall, we performed single particle, nanoscale electrophoresis and simultaneously tracked the motion of a single fluorescent polystyrene nanoparticle (20 nm in diameter). A separate fluorescence microscope than the one used for STORM was operated in an epilumination format with a wide field-of-view and uniform laser excitation intensity profile across this field-of-view (see Figure S1), which enabled the precise tracking of individual nanoparticles through a nanoslit that was 3 μm wide and 150 nm deep fabricated in COC.7 The COC nanoslits were activated by 30 s of O2 plasma exposure then assembled with a thin COC cover plate thermally fusion bonded to the substrate. A nanoslit was chosen in place of a nanochannel here because nanoparticle movements within a 50 nm square nanochannel would occur below the diffraction limit and would require a rapid imaging super-resolution system for tracking the transport process. Similar phenomena, namely, fluid recirculation, were expected in the nanoslit and also, the nanoslit’s width (3 μm) enabled the detection of axial perturbations in nanoparticle motion that could be followed above the diffraction limit using a high framerate epillumination microscope (see Figure S1).
Figure 5a shows the distribution of travel times of the fluorescent nanoparticles in the COC nanoslit at two different electric field strengths. The migration of the negatively charged polystyrene nanoparticles was from cathode to anode (i.e., the nanoparticles’ electrophoretic mobility was greater than the EOF of the COC). This was anticipated based on the low EOF observed in thermoplastic devices42 as well as results from the fluid dynamics simulations (see Figure 4) that showed a small EOF due to the low surface charge density produced at the O2 plasma dosing conditions employed here (see Figure 2l).
Figure 5.
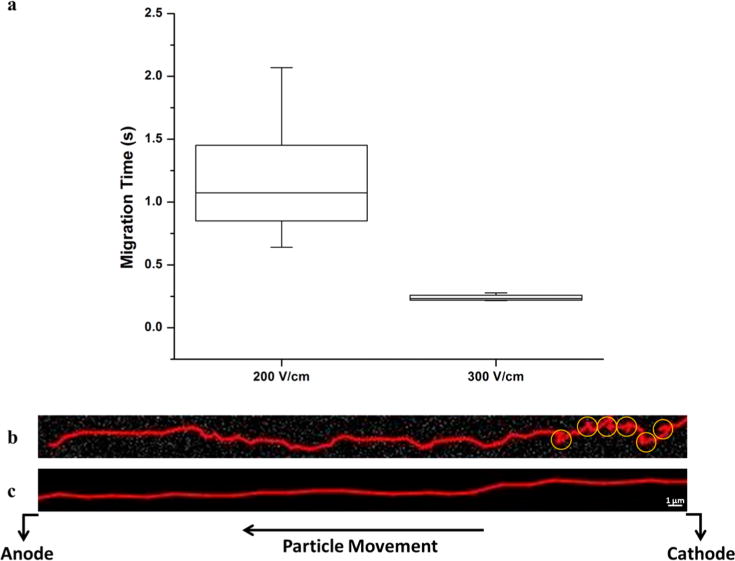
(a) Box plot comparing the minimum, first quartile, median, third quartile, and maximum migration time (s) for polystyrene beads at 200 and 300 V/cm migrating throughout the entire length (100 μm) of a COC nanoslit. (b) Trace of a single PS bead translocating a 3 μm × 150 nm × 100 μm (w × d × l) channel under a field strength of 200 V/cm. Yellow circles indicate regions of possible recirculation. (c) Trace of a single PS bead translocating a 3 μm × 150 nm × 100 μm (w × d × l) channel under a field strength of 300 V/cm. The depth of focus of our 100× objective was large enough to ensure that each PS bead remained in focus since our channel depth was 150 nm.
At 200 V/cm, the average migration time of the particles through the field-of-view (100 μm) was 1.2 ± 0.5 s, while the average migration time at 300 V/cm was 0.20 ± 0.02 s. Thus, a 50% increase in field strength generated a 600% decrease in the electrophoretic migration time. Further, the relative standard deviation of migration times was reduced from 40% at 200 V/cm to 8% at 300 V/cm, a 5-fold decrease (Figure 5a). We do note that some variance in migration times could be due to the differences in the sizes of the particles; however, this contribution to the overall standard deviation shown in the distribution of migration times for both applied fields would be the same. The relatively high variance noted at the lower electric field strength is most likely due to stick/slip motion. Sticking motions are typically seen at low electric fields, while slip dominates the motion at high electric fields.42 Stick/slip motion can be due to either solute/wall interactions or dielectrophoretic trapping induced by inhomogeneous electric fields caused by surface roughness. Thus, the zonal variance at 300 V/cm would be less due not only to reductions in the variance arising from longitudinal diffusion, but also reductions in stick/slip motion at the higher electric field.
We did observe transverse motion of the nanoparticles at 200 V/cm and some evidence of recirculation, while the application of 300 V/cm removed these artifacts (Figure 5b,c). While both the electrophoretic mobility and EOF should be directly proportional to the electric field strength, we suspect that stick/slip motion provided a nonlinear field dependence on the particles mobility, which we noted for the mobility of double-stranded DNA molecules migrating through polymer nano-channels.42
As these tracking dynamics are currently restricted to nanoslits due to diffraction limits, nanochannel columns (e.g., a 50 × 50 nm nanochannel) may show more particle-wall interactions and increased stick/slip motion. However, even higher electric fields could be used to mitigate stick/slip motion for nanocolumns while avoiding significant Joule heating and zonal dispersion that would classically be observed in microscale capillary electrophoresis columns.
CONCLUSIONS
Nanoelectrophoresis provides unique phenomena, such as EDL overlap and increased surface effects that can be taken advantage of to generate separations that are difficult to realize using microscale columns. However, factors such as surface charge density and heterogeneity must be understood to predict how nanoscale-induced properties can affect the performance of the analytical process imposed on the nanochannel, especially when using amorphous materials such as thermoplastics. Previous attempts to characterize thermoplastic surfaces were limited by averaging effects leading to an incomplete view of surface functional group heterogeneity following activation.
In this study, we demonstrated a novel approach for characterizing the surface charge density and heterogeneity using super-resolution fluorescence microscopy (STORM). STORM allowed for the precise localization of fluorophores (17 nm) conjugated to –COOH charged surface functional groups allowing for an assessment of charge heterogeneity. Our data indicated that control of the dose used for activation can lead to optimal functional group surface density with higher surface density correlated to lower surface charge heterogeneity.
Furthermore, we utilized COMSOL to reconstruct fluid flow profiles in nanochannels with a heterogeneous distribution of point charges mapped from the STORM images to understand the effects of surface charge heterogeneity on the EOF. The simulations indicated that there were regions of fluid recirculation at point charges leading to both positive and negative velocities; however, the overall EOF magnitude was significantly less than a uniform channel, such as glass, due to the decreased overall surface charge. Our electrophoretic results indicated, however, that when the electric field strength is high (≥300 V/cm), stick/slip motion can be significantly reduced meaning that any EOF-induced recirculation can be a minimal contribution to the overall zonal variance.
With the results presented herein, it should be feasible to consider the electrophoretic separation of single molecules in nanometer columns of short length (<100 μm) to produce high resolution separations of molecules in sub-10 ms time scales. This can provide the unique opportunity to employ nanoscale electrophoresis to affect the identification of single molecules through mobility matching. This strategy could potentially be envisioned for single-molecule DNA sequencing.67,68 The generation of single mononucleotides from intact DNAs could be produced from immobilized exonucleases that cleave intact double-stranded DNA molecules.11
Supplementary Material
Acknowledgments
The authors would like to thank the National Institutes of Health (R21HG006278; P41EB020594), the National Science Foundation (1507577) Research Fund (1.130090.01) of Ulsan National Institute of Science and Technology, and Roche for financial support of this work. C.E.O. thanks the National Science Foundation for support through a summer fellowship program (EAPSI), and J.M.J. thanks the ACS Division of Analytical Chemistry and the Society for Analytical Chemists of Pittsburgh for funding. Furthermore, the authors thank Dr. Mateusz Hupert for his SEM studies of PMMA as well as Dr. Franklin Uba for his assistance in the construction of the single molecule electrophoresis optical system and thoughtful discussions.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.5b04472.
Figures S1–S4 and Table S1 (PDF).
Notes
The authors declare no competing financial interest.
References
- 1.Nunes P, Ohlsson P, Ordeig O, Kutter J. Microfluid Nanofluid. 2010;9:145–161. [Google Scholar]
- 2.de Mello A. Lab Chip. 2002;2:31N–36N. doi: 10.1039/b203828p. [DOI] [PubMed] [Google Scholar]
- 3.Soper SA, Ford SM, Qi S, McCarley RL, Kelly K, Murphy MC. Anal Chem. 2000;72:642A–651 A. doi: 10.1021/ac0029511. [DOI] [PubMed] [Google Scholar]
- 4.Tsao C-W, DeVoe D. Microfluid Nanofluid. 2009;6:1–16. [Google Scholar]
- 5.Uba FI, Hu B, Weerakoon-Ratnayake K, Oliver-Calixte N, Soper SA. Lab Chip. 2015;15:1038–1049. doi: 10.1039/c4lc01254b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chantiwas R, Hupert ML, Pullagurla SR, Balamurugan S, Tamarit-Lopez J, Park S, Datta P, Goettert J, Cho Y-K, Soper SA. Lab Chip. 2010;10:3255–3264. doi: 10.1039/c0lc00096e. [DOI] [PubMed] [Google Scholar]
- 7.Chantiwas R, Park S, Soper SA, Kim BC, Takayama S, Sunkara V, Hwang H, Cho Y-K. Chem Soc Rev. 2011;40:3677–3702. doi: 10.1039/c0cs00138d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roy E, Stewart G, Mounier M, Malic L, Peytavi R, Clime L, Madou M, Bossinot M, Bergeron MG, Veres T. Lab Chip. 2015;15:406–416. doi: 10.1039/c4lc00947a. [DOI] [PubMed] [Google Scholar]
- 9.Soper SA, Hashimoto M, Situma C, Murphy MC, McCarley RL, Cheng Y-W, Barany F. Methods. 2005;37:103–113. doi: 10.1016/j.ymeth.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 10.Lee J, Soper SA, Murray KK. Rapid Commun Mass Spectrom. 2011;25:693–699. doi: 10.1002/rcm.4921. [DOI] [PubMed] [Google Scholar]
- 11.Oliver-Calixte NJ, Uba FI, Battle KN, Weerakoon-Ratnayake KM, Soper SA. Anal Chem. 2014;86:4447–4454. doi: 10.1021/ac5002965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen G, McCarley RL, Soper SA, Situma C, Bolivar JG. Chem Mater. 2007;19:3855–3857. [Google Scholar]
- 13.Battle KN, Jackson JM, Witek MA, Hupert ML, Hunsucker SA, Armistead PM, Soper SA. Analyst. 2014;139:1355–1363. doi: 10.1039/c3an02400h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jackson JM, Witek MA, Hupert ML, Brady C, Pullagurla S, Kamande J, Aufforth RD, Tignanelli CJ, Torphy RJ, Yeh JJ, Soper SA. Lab Chip. 2014;14:106–117. doi: 10.1039/c3lc50618e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Torphy RJ, Kamande JW, Tignanelli CJ, Moffit RA, Soper SA, Yeh J. J Surg Res. 2013;179:340. [Google Scholar]
- 16.Dharmasiri U, Njoroge SK, Witek MA, Adebiyi MG, Kamande JW, Hupert ML, Barany F, Soper SA. Anal Chem. 2011;83:2301–2309. doi: 10.1021/ac103172y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xia H, Murray K, Soper S, Feng J. Biomed Microdevices. 2012;14:67–81. doi: 10.1007/s10544-011-9586-7. [DOI] [PubMed] [Google Scholar]
- 18.Shadpour H, Musyimi H, Chen J, Soper SA. Journal of Chromatography A. 2006;1111:238–251. doi: 10.1016/j.chroma.2005.08.083. [DOI] [PubMed] [Google Scholar]
- 19.Njoroge SK, Witek MA, Battle KN, Immethun VE, Hupert ML, Soper SA. Electrophoresis. 2011;32:3221–3232. doi: 10.1002/elps.201100274. [DOI] [PubMed] [Google Scholar]
- 20.Lacher NA, Garrison KE, Martin RS, Lunte SM. Electrophoresis. 2001;22:2526–2536. doi: 10.1002/1522-2683(200107)22:12<2526::AID-ELPS2526>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 21.Dolník V, Liu S, Jovanovich S. Electrophoresis. 2000;21:41–54. doi: 10.1002/(SICI)1522-2683(20000101)21:1<41::AID-ELPS41>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 22.Kitagawa F, Otsuka K. J Pharm Biomed Anal. 2011;55:668–678. doi: 10.1016/j.jpba.2010.11.013. [DOI] [PubMed] [Google Scholar]
- 23.Pumera M, Wang J, Grushka E, Polsky R. Anal Chem. 2001;73:5625–5628. doi: 10.1021/ac015589e. [DOI] [PubMed] [Google Scholar]
- 24.Kinsella A, Minteer S. In: Molecular Biomethods Handbook. Walker J, Rapley R, editors. Humana Press; 2008. pp. 851–859. [Google Scholar]
- 25.Nilsson C, Birnbaum S, Nilsson S. Journal of Chromatography A. 2007;1168:212–224. doi: 10.1016/j.chroma.2007.07.018. [DOI] [PubMed] [Google Scholar]
- 26.Osiri JK, Shadpour H, Park S, Snowden BC, Chen Z-Y, Soper SA. Electrophoresis. 2008;29:4984–4992. doi: 10.1002/elps.200800496. [DOI] [PubMed] [Google Scholar]
- 27.Osiri J, Shadpour H, Soper S. Anal Bioanal Chem. 2010;398:489–498. doi: 10.1007/s00216-010-3914-2. [DOI] [PubMed] [Google Scholar]
- 28.Lin C-H, Lee G-B, Chen S-H, Chang G-L. Sens Actuators, A. 2003;107:125–131. [Google Scholar]
- 29.Piruska A, Gong M, Sweedler JV, Bohn PW. Chem Soc Rev. 2010;39:1060–1072. doi: 10.1039/b900409m. [DOI] [PubMed] [Google Scholar]
- 30.Schoch RB, Han J, Renaud P. Rev Mod Phys. 2008;80:839–883. [Google Scholar]
- 31.Yuan Z, Garcia AL, Lopez GP, Petsev DN. Electrophoresis. 2007;28:595–610. doi: 10.1002/elps.200600612. [DOI] [PubMed] [Google Scholar]
- 32.Pennathur S, Santiago J. Anal Chem. 2005;77:6782–6789. doi: 10.1021/ac0508346. [DOI] [PubMed] [Google Scholar]
- 33.Baldessari F, Santiago J. J Nanobiotechnol. 2006;4:12. doi: 10.1186/1477-3155-4-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Movahed S, Li D. Electrophoresis. 2011;32:1259–1267. doi: 10.1002/elps.201000564. [DOI] [PubMed] [Google Scholar]
- 35.Xuan X, Li D. Electrophoresis. 2006;27:5020–5031. doi: 10.1002/elps.200600031. [DOI] [PubMed] [Google Scholar]
- 36.Garcia A, Ista L, Petsev D, O’Brien M, Bisong P, Mammoli A, Brueck S, Lopez G. Lab Chip. 2005;5:1271–1276. doi: 10.1039/b503914b. [DOI] [PubMed] [Google Scholar]
- 37.Schoch RB, Bertsch A, Renaud P. Nano Lett. 2006;6:543–547. doi: 10.1021/nl052372h. [DOI] [PubMed] [Google Scholar]
- 38.Schoch RB, Renaud P. Appl Phys Lett. 2005;86:25311–25313. [Google Scholar]
- 39.Liang X, Chou S. Y. Nano Lett. 2008;8:1472–1476. doi: 10.1021/nl080473k. [DOI] [PubMed] [Google Scholar]
- 40.Menard LD, Ramsey JM. Nano Lett. 2011;11:512–517. doi: 10.1021/nl103369g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Menard LD, Ramsey JM. Anal Chem. 2013;85:1146–1153. doi: 10.1021/ac303074f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Uba FI, Pullagurla SR, Sirasunthorn N, Wu J, Park S, Chantiwas R, Cho Y-K, Shin H, Soper SA. Analyst. 2015;140:113–126. doi: 10.1039/c4an01439a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chan CM, Ko TM, Hiraoka H. Surf Sci Rep. 1996;24:1–54. [Google Scholar]
- 44.Wei S, Vaidya B, Patel AB, Soper SA, McCarley RL. J Phys Chem B. 2005;109:16988–16996. doi: 10.1021/jp051550s. [DOI] [PubMed] [Google Scholar]
- 45.Herr AE, Molho JI, Santiago JG, Mungal MG, Kenny TW, Garguilo MG. Anal Chem. 2000;72:1053–1057. doi: 10.1021/ac990489i. [DOI] [PubMed] [Google Scholar]
- 46.Datta R, Kotamarthi V. AIChE J. 1990;36:916–926. [Google Scholar]
- 47.Anderson JL. J Colloid Interface Sci. 1985;105:45–54. [Google Scholar]
- 48.Brotherton CM, Davis RH. J Colloid Interface Sci. 2004;270:242–246. doi: 10.1016/j.jcis.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 49.Keely CA, van de Goor TAAM, McManigill D. Anal Chem. 1994;66:4236–4242. [Google Scholar]
- 50.Ajdari A. Phys Rev Lett. 1995;75:755–758. doi: 10.1103/PhysRevLett.75.755. [DOI] [PubMed] [Google Scholar]
- 51.Fu LM, Lin JY, Yang RJ. J Colloid Interface Sci. 2003;258:266–275. doi: 10.1016/s0021-9797(02)00078-4. [DOI] [PubMed] [Google Scholar]
- 52.Long D, Stone HA, Ajdari A. J Colloid Interface Sci. 1999;212:338–349. doi: 10.1006/jcis.1998.6015. [DOI] [PubMed] [Google Scholar]
- 53.Chen L, Conlisk AT. Biomed Microdevices. 2009;11:251–258. doi: 10.1007/s10544-008-9231-2. [DOI] [PubMed] [Google Scholar]
- 54.Rasmussen JR, Stedronsky ER, Whitesides GM. J Am Chem Soc. 1977;99:4736–4745. [Google Scholar]
- 55.Bates M, Jones AS, Zhuang X. Cold Spring Harbor Protocols. 2013;2013:498–520. doi: 10.1101/pdb.top075143. [DOI] [PubMed] [Google Scholar]
- 56.Gramlich MW, Bae J, Hayward RC, Ross JL. Opt Express. 2014;22:8438–8450. doi: 10.1364/OE.22.008438. [DOI] [PubMed] [Google Scholar]
- 57.Lin H, Centeno SP, Su L, Kenens B, Rocha S, Sliwa M, Hofkens J, Uji-i H. ChemPhysChem. 2012;13:973–981. doi: 10.1002/cphc.201100743. [DOI] [PubMed] [Google Scholar]
- 58.Kazoe Y, Mawatari K, Sugii Y, Kitamori T. Anal Chem. 2011;83:8152–8157. doi: 10.1021/ac201654r. [DOI] [PubMed] [Google Scholar]
- 59.Bates M, Blosser TR, Zhuang X. Phys Rev Lett. 2005;94:108101. doi: 10.1103/PhysRevLett.94.108101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jones SA, Shim SH, He J, Zhuang X. Nat Methods. 2011;8:499–505. doi: 10.1038/nmeth.1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Huang B, Wang W, Bates M, Zhuang X. Science (Washington, DC, U S) 2008;(319):810–813. doi: 10.1126/science.1153529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Geisler C, Hotz T, Schönle A, Hell SW, Munk A, Egner A. Opt Express. 2012;20:7274–7289. doi: 10.1364/OE.20.007274. [DOI] [PubMed] [Google Scholar]
- 63.Shim SH, Xia C, Zhong G, Babcock H, Vaughan J, Huang B, Wang X, Xu C, Bi GQ, Zhuang X. Proc Natl Acad Sci USA. 2012;109:13978–13983. doi: 10.1073/pnas.1201882109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ozgen O, Aksoy EA, Hasirci V, Hasirci N. Surf Interface Anal. 2013;45:844–853. [Google Scholar]
- 65.Baker D. Capillary Electrophoresis. John Wiley and Sons, Inc; New York, NY: 1995. [Google Scholar]
- 66.Pennathur S, Baldessari F, Santiago JG, Kattah MG, Steinman JB, Utz PJ. Anal Chem. 2007;79:8316–8322. doi: 10.1021/ac0710580. [DOI] [PubMed] [Google Scholar]
- 67.Novak BR, Moldovan D, Nikitopoulos DE, Soper SA. J Phys Chem B. 2013;117:3271–3279. doi: 10.1021/jp309486c. [DOI] [PubMed] [Google Scholar]
- 68.Xia K, Novak BR, Weerakoon-Ratnayake KM, Soper SA, Nikitopoulos DE, Moldovan D. J Phys Chem B. 2015;119:11443–11448. doi: 10.1021/acs.jpcb.5b02798. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.