

Abstract
A strategy for acquiring structural information from sparsely isotopically labeled large proteins is illustrated with an application to the E. coli heat-shock protein, HtpG (high temperature protein G), a 145 kDa dimer. It uses 13C-alanine methyl labeling in a perdeuterated background to take advantage of the sensitivity and resolution of Methyl-TROSY spectra, as well as the backbone-centered structural information from 1H-13C residual dipolar couplings (RDCs) of alanine methyl groups. In all, 40 of the 47 expected crosspeaks were resolved and 36 gave RDC data. Assignments of crosspeaks were partially achieved by transferring assignments from those made on individual domains using triple resonance methods. However, these were incomplete and in many cases the transfer was ambiguous. A genetic algorithm search for consistency between predictions based on domain structures and measurements for chemical shifts and RDCs allowed 60% of the 40 resolved crosspeaks to be assigned with confidence. Chemical shift changes of these crosspeaks on adding an ATP analog to the apo-protein are shown to be consistent with structural changes expected on comparing previous crystal structures for apo- and complex- structures. RDCs collected on the assigned alanine methyl peaks are used to generate a new solution model for the apo-protein structure.
Keywords: Resonance assignments, Assignment program, Sparse labeling, Perdeuteration, Heat-shock protein, Hsp90, HtpG, protein structure, 1H-13C Ala methyl RDCs, Methyl-TROSY
Introduction
Problems attacked by structural biology researchers increasingly involve larger molecules and more complex molecular assemblies (Hennig and Sattler 2014). This is a significant issue for nuclear magnetic resonance (NMR) where applications have largely been limited to systems less than 50 kDa in molecular weight (Burmann and Hiller 2015; Rosenzweig and Kay 2016). Here we illustrate an approach that uses isotopic labeling in a single amino acid type (sparse labeling) to simplify spectra,13C labeling in a methyl group to improve sensitivity and resolution, and acquisition of data types particularly suited to sparse labeling to achieve resonance assignments and draw structural conclusions. We apply the approach to the 145 kDa dimer of high temperature protein G (HtpG), the E. coli homolog of Hsp90 heat shock proteins found in eukaryotes (Stechmann and Cavalier-Smith 2004).
Hsp90 proteins function as ATP dependent protein folding chaperones, binding to partially-folded client proteins, often in complex with co-chaperones, and facilitating completion of folding or simply preserving proteins until folding can be completed (Jackson 2013; Krukenberg et al. 2011). They have attracted considerable interest from the structural biology community, because the clients they serve are often involved in cell proliferation and migration, making Hsp90s a potential target for development of cancer therapeutics (Day et al. 2011; Miyata et al. 2013). Central questions revolve around the coupling between chaperone dynamics driven by the ATP hydrolysis cycle and client protein binding and remodeling.
HtpG is composed of three distinct domains, a C-terminal domain (CTD) that is responsible for dimerization, a middle-domain (MD) involved in client binding, and an N-terminal domain (NTD) that houses an ATP binding site. There are several crystal structures of full length Hsp90 proteins in apo, ADP, and non-hydrolysable ATP analog (AMPPNP, for example) forms. There is also a recent high resolution cryoEM structure of the human Hsp90 bound to a client kinase and an adaptor co-chaperone (Verba et al. 2016). These show substantial preservation of individual domain structures and the C-terminal domain contacts that make dimers the dominant species in all forms. However, there are large variations in inter-domain geometry. The apo HtpG crystal structure shows a V-shaped conformation (Shiau et al. 2006b), while ATP-like states have significant domain rotations resulting in a twisted, closed state (Ali et al. 2006; Krukenberg et al. 2008). The mitochondrial Hsp90 TRAP1 closed state is distinctly asymmetric (Lavery et al. 2014), while the yeast closed state in a co-chaperone complex is symmetric (Ali et al. 2006). Small angle X-ray scattering (SAXS) data (Krukenberg et al. 2008) and electron microscopy data (Southworth and Agard 2008) on the apo form suggest an even more open structure. Figure 1 illustrates the range of these structures. A model based on the SAXS data, the crystal structure of the apo form, and a homology model of HtpG built using the crystal structure of TRAP1 as a template have been superimposed by matching the last half of the C-terminal domains.
Figure 1.
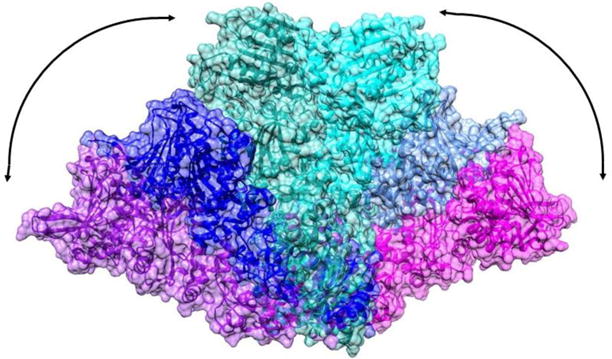
Motional range observed in Hsp90 dimers. The C-terminal domains have been matched for the SAXS model of apo-HtpG (purple-magenta), the apo-HtpG crystal structure (2IOQ, blue-light blue) and a homology model of AMPPNP-HtpG (green-cyan).
This level of variability most likely represents a subset of the conformational repertoire used to modulate client structure as well as interactions with other proteins. Recent cryo-EM structures that include co-chaperones directly highlight specific aspects of the Hsp90 conformational ensemble (Southworth and Agard 2011; Verba et al. 2016). While SAXS, cryo-EM, EPR and even fluorescent energy transfer studies (Schulze et al. 2016) provide valuable data on global structural forms, these are highly dynamic structures, and thus the landscape is likely further expanded through complex equilibria or multiple conformations in particular contexts. Moreover, many of the Hsp90 protein clients are themselves dynamic and/or partially folded abrogating study by crystallography and making even cryoEM challenging. It would clearly be desirable to have a broadly applicable monitor of inter-domain geometry and protein-protein interaction points that could be applied to samples in various states of client interaction, co-chaperone interaction, or ATP hydrolysis in solution.
NMR, even with just sparse labeled sites, can provide a powerful monitor of conformation change, contribute to descriptions of inter-domain geometry, and identify sites of protein and ligand interaction. Chemical shifts are easily measured and inherently dependent on local conformation in proteins and interactions involving surface contact between domains or other proteins. 15N-HSQC data collected on individual domains of an Hsp90 has, for example, been used to advantage in identifying client binding sites (Street et al. 2012) and co-chaperone interaction sites (Didenko et al. 2012; Retzlaff et al. 2010). Residual Dipolar Couplings (RDCs) are also easily measured from 15N-HSQC cross peaks. As they reflect average orientation of internuclear vectors, as well as the extent and anisotropy of motion, they can provide more specific information on inter-domain geometry (Lipsitz and Tjandra 2004; Prestegard et al. 2004).
There are some limitations to using these easily obtained data. First, there must be adequate sensitivity and resolution to detect a significant fraction of all the crosspeaks that could arise in HSQC spectra. Second, crosspeaks must be assigned to specific sites in the protein. The first condition can be met for many proteins by exploiting some unique properties of 1H-13C crosspeaks from methyl groups in HMQC spectra, a phenomenon known as Methyl-TROSY (Rosenzweig and Kay 2014; Tugarinov et al. 2003). The three identical methyl protons automatically provide additional sensitivity when spectra are detected through protons, and the cancellation of the dipolar interactions remaining in the 13C dimension of an HMQC spectrum leads to an unusually sharp and well resolved central peak of the HMQC crosspeak. This phenomenon is usually exploited by metabolic labeling of isoleucine (1), leucine and valine groups using precursors with 13C-labeled methyl groups (ILV-labeling) (Kay and Gardner 1997). A similar labeling strategy using just isoleucine has, in fact, been applied to Hsp90 systems, both at the isolated domain (Karagoz et al. 2011) and full length level (Karagoz et al. 2014). However, because of the number of degrees of freedom introduced by sidechain torsion angles, it is difficult to use RDC data coming from methyl groups of Ile, Leu or Val residues.
RDCs from 13C-labeled alanine methyls are, in principle, more easily interpreted because of their direct attachment to the polypeptide backbone of a protein and resulting lack of sidechain motional degrees of freedom (Godoy-Ruiz et al. 2010). Here we employ a method that exploits modulation of crosspeak intensity by spin-spin and dipolar coupling during the initial transfer of 1H magnetization in a Methyl-TROSY experiment (see description in the Methods section). Interpretation, however, still requires assignment to sites in structurally characterized domains, and assigning crosspeaks to specific sites for a protein as large as Hsp90 is a challenge. Most frequently, assignments are accomplished using backbone-directed experiments that exploit transfer of magnetization between directly bonded nuclear pairs in uniformly 13C and 15N labeled, perdeuterated, proteins using triple resonance experiments. This general strategy has been extended to samples that also include ILV-labeling (Tugarinov and Kay 2003), but increased line broadening and loss of sensitivity as molecular weight increases occurs. Some improvement is offered by the use of specifically designed isotopic precursors of isoleucine that facilitate correlation with assigned backbone resonances (Ayala et al. 2012), and there are approaches which facilitate transfer of assignments made on separately expressed protein domains by comparing data such as NOEs that can be collected on both single domains and full length proteins (Sprangers and Kay 2007). Assignments of isoleucine methyls in the Hsp90 example mentioned above were, in fact, made on isolated domains and then transferred to spectra of the full-length protein in cases where overlap allowed, but applications to very large proteins are still few in number. Here we seek an assignment strategy that can be directly applied to a large protein, like a full-length Hsp90, without the need to prepare samples having uniform isotopic labeling.
Recently we introduced a procedure for assigning 15N-HSQC crosspeaks without the aid of triple resonance experiments (Gao et al. 2017; Gao et al. 2016). This relies on a set of experiments easily acquired on sparsely labeled proteins using HSQC-based experiments (chemical shifts, 1H-15N RDCs, and 15N-HSQC-edited NOESY vectors) and prediction of these parameters using known domain structures. The data and predictions were combined using an assignment program, ASSIGN_SLP_1.1.2 (Gao et al. 2017), that uses a genetic algorithm available in MATLAB to search for the assignment giving the best match of prediction and experiment. The procedure was initially devised to allow assignment of glycosylated proteins, which are difficult to prepare in uniformly labeled forms for economic reasons, and it was tested on small and moderately sized non-perdeuterated proteins that were labeled with 15N in selected amino acid types. Here we extend ASSIGN_SLP_1.1.2 to determine assignments of 1H-13C Methyl-TROSY spectra, apply the procedure to a larger perdeuterated 13C-1H alanine methyl labeled protein, and use just the methyl chemical shifts from HMQC crosspeaks and 1H-13C RDCs to achieve assignment. Using simulated data, the procedure proves applicable to systems with as many as 50 unassigned peaks, provided a full set of accurate RDCs is available (we expect 47 peaks in 13C-alanine labeled HtpG). However, our real data on full-length HtpG is not sufficiently precise and there are a significant number of missing and overlapping crosspeaks. Therefore, we use a hybrid procedure in which individual domains of HtpG have been expressed in conventional double-labeled forms (uniform 13C and 15N labeling) to allow a subset of full-length crosspeaks to be associated with specific domains and provide triple resonance assignments for some methyl crosspeaks. The genetic algorithm procedure has been modified to take advantage of this additional data, and proceeding domain by domain it is able to extend alanine assignments well beyond those for peaks having unique overlaps between domains and full-length spectra.
With RDCs and domain assignments in hand it has been possible to assign, with a high level of confidence, 24 crosspeaks in the full length apo-protein and an additional 9 with a moderate level of confidence. Chemical shift perturbations of a subset of these crosspeaks on addition of AMPPNP to the apo-protein correlate well with regions close to the nucleotide binding site and regions of contact between domains seen in the ADP form of this protein (PDB ID 2IOP (Shiau et al. 2006b)). Using residue-specific RDCs we have also been able to generate a motionally-averaged structure for the apo form of HtpG that is distinct from that seen in the crystal structure. The procedures described offer future promise for application to Hsp90 systems and other complex systems of high molecular weight.
Experimental Methods
Materials
Standard laboratory chemicals were purchased from Sigma-Aldrich. Isotopically labeled alanine (3-13C, 2-2H alanine) was purchased from Cambridge Isotope Laboratories. Uniformly enriched glucose (U-13C, 2H) and 2H2O were purchased from Sigma Aldrich, as were 15N ammonium chloride and (U-13C) glucose for triple resonance experiments.
Protein preparation
Procedures for efficient labeling with 13C-labeled alanine have been previously described (Ayala et al. 2009; Godoy-Ruiz et al. 2010; Isaacson et al. 2007). In our case, the coding sequence, residues 1-624 (UniProtKB AC:P0A6Z3), was cloned into pET151DTopo with an N-terminal 6x-His tag. The protein was then expressed in E. coli BL21(DE3) as previously described (Tugarinov et al. 2006) with minor modifications. Briefly, an overnight culture in LB media grown at 37°C was used to inoculate a fresh starter culture in the morning (50 μL into 10 mL). Once the starter culture reached an OD600 of 0.6 (~3 hours), a 50 μL aliquot was used to inoculate 50 mL of M9/2H2O minimal media with (14NH4)2SO4 (1.25 g/L) as the sole nitrogen source. Once the OD600 reached 0.6 units, cells were pelleted at 4000 × g at room temperature and re-suspended in 1 L of M9/2H2O media and grown at 37°C to OD600 of 0.6 units (~11 hours). 100 mg of 13C-Ala (3-13C; 2-2H) was added to the culture 40 minutes prior to induction with 0.238 g of IPTG (1 mM). 100 mg of labeled alanine may not produce labeling at the highest levels, but proves adequate for the experiments conducted here. Cells were harvested after 4 hours of induction. The full-length protein was purified using Ni-NTA agarose, then cleaved with TEV protease to remove the His-tag prior to a final gel filtration step using a S200 16/60 (GE Healthcare) column. The yield was approximately 40 mg from 1 L culture.
The individual HtpG domains constituting the following sequence regions, residues 1 – 215, 230 – 495, and 511 – 624 for the N-terminal (NTD), middle (MD) and C-terminal (CTD) domains, respectively, were cloned, expressed, and purified following standard NESG protocols (Acton et al. 2011; Xiao et al. 2010). Briefly, the genes encoding these three HtpG domains from E. coli were amplified from genomic DNA and cloned into the pET15_NESG vector (Acton et al. 2011) in frame with an N-terminal affinity tag (MGHHHHHHHSHM), transformed into E. coli BL21(DE3) pMGK cells, and expressed overnight at 17°C in MJ9 minimal media (Jansson et al. 1996). Isotopically enriched samples were produced using either U-(15NH4)2SO4 and 5% U-13C-glucose/95% glucose or U-(15NH4)2SO4 and U-13C-glucose as the sole nitrogen and carbon sources. Proteins were purified using an ӒKTAxpress system (GE Healthcare) with a two-step protocol consisting of IMAC (HisTRAP HP) and gel filtration (HiLoad 26/60 Superdex 75) chromatography. Yields ranging 70–170 mg were obtained from 1 L of culture. The purities of these protein samples, estimated by SDS-PAGE analysis, were >95%. The molecular weights of the purified, isotope-enriched HtpG domains were verified by MALDI-TOF mass spectrometry.
Triple resonance data and assignments of HtpG domains
15N-HSQC, 13C-HSQC, HNCO, HNCACO, HNCA, HNCOCA, HNCACB, CBCACONH and HBHACONH data sets were collected on approximately 0.5 mM samples of single domain proteins prepared in 10 mM Tris buffer, pH 7.5, containing 100 mM NaCl. A 600 MHz Varian/Agilent NMR spectrometer operating at a sample temperature of 35 °C was used with standard Biopack sequences. To confirm backbone and alanine methyl assignments for the NTD, 15N-NOESY-HSQC and 13C-NOESY-HSQC data sets were collected using an 850 MHz Bruker Avance III NMR spectrometer with standard TopSpin 2.1 pulse sequences. For the CTD 15N-NOESY-HSQC data were used to confirm alanine assignments. Assignments were made with the aid of the PINE server (Bahrami et al. 2009) followed by manual validation using neighboring residue spectral characteristics. For the NTD backbone, all residues, including the 13 alanines, were assigned and validated. For the C-terminal domain resonances from 12 alanines were assigned and validated. However, because of the larger size and poor resolution for the MD, only one of the 22 alanine methyls was assigned. Had the middle domain been produced in a perdeuterated form, more assignments would likely be obtained. The NTD and CTD alanine assignments, along with raw NMR data on which they are based, have been deposited in the BMRB with accession numbers 27037 and 27038. These deposits will be updated as additional assignments are made.
NMR data on sparsely labeled HtpG
Methyl-TROSY data were collected on 100 μL, 216 μM samples of full length HtpG in Shigemi 5mm NMR tubes with and without the non-hydrolysable ATP analog, AMPPNP, using a 900 MHz Varian/Agilent NMR spectrometer at 37°C and a pulse sequence from Biopack. A higher pH buffer (pH 9.0, 25 mM Tris, 25 mM KCl, 5 mM MgCl2) was used in accordance with prior data on buffers compatible with stability of the AMPPNP bound form of the protein. Samples for RDC data were of similar concentration and buffer conditions, but also included 4.2% (v/v) C12E5 polyethylene glycol (PEG) bicelles or 12.5 mg/ml Pf1 bacteriophage from ASLA Biotech to achieve partial alignment. RDC data were collected using a 600 MHz Varian/Agilent spectrometer operating at 37 C and a pulse sequence (Figure 2) based on coupling constant modulation of methyl proton coherence during a constant time INEPT segment (points a-f) that replaces the initial 90X pulse in a previously published Methyl-TROSY experiment (Tugarinov et al. 2003).
Figure 2.
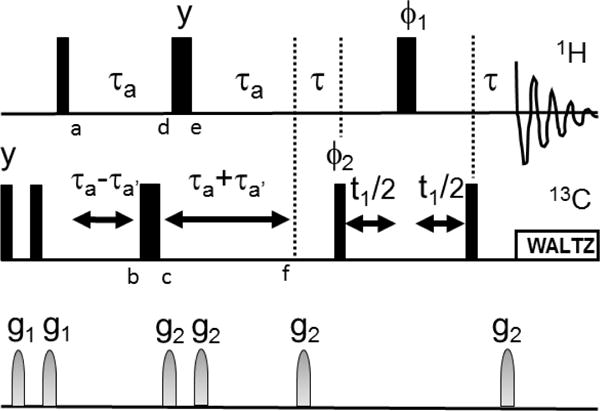
Pulse sequence for measurement of RDCs from Methyl-TROSY spectra. is 1/(2JCH) and a is selected to allow peak intensity to modulate through two zero crossings (~20 ms). a′ moves the position of the 13C 180 pulse to allow J modulation by 13C-1H couplings. Letters a-f inserted below pulses denote points in spin evolution discussed in the text. All rf pulses are in the x direction except as indicated. 1 and 2 are as in (Tugarinov et al. 2003). Gradients were of 500 ms duration and intensity settings 8000 and 4000 for g1 and g2 respectively, 100 increments were collected in the indirect dimension with a spectral width of 20 ppm.
Modulation of crosspeak intensity is a well-established technique for the measurement of RDCs for pairs of spin ½ nuclei (Tjandra and Bax 1997), and 4×4 product operators are commonly used to describe these techniques. However, for Nevertheless, the fact that methyl proton spin functions can be grouped into I = ½ and I = 3/2 sets (Ollerenshaw et al. 2003) suggests that application may offer insight into the behavior of two of the narrower methyl proton resonance components. Hence, we present a treatment for spin ½ coherence at the points labeled a-f in Figure 2 using 4×4 product operators. We will consider only the effects of scalar coupling; the 90-a-180-a segment effectively removes chemical shift evolution. The results are given by: −Iy, −Iy cos(J(a−a′)) + 2IxSz sin(J(a−a′)), −Iy cos(J(a−a′)) − 2IxSz sin(J(a−a′)), −Iy cos(J(a−2a′)) + 2IxSz sin (J(a−2a′)), −Iy cos(J(a−2a′)) + 2IxSz sin (J(a−2a′)) and −Iy cos(J(2a−2a′)) + 2IxSz sin(J(2a−2a′)) for the points a–f, respectively. A further evolution during results in −Iy cos(J(+2a−2a′)) + 2IxSz sin(J(+2a−2a′)). 2IxSz is converted to zero and two quantum coherence by the first 13C 90 pulse of the Methyl-TROSY sequence and the output of this sequence is modulated by sin(J(+2a−2a). From the above analysis it is clear that the first delay of the Methyl-TROSY could be deleted for a small improvement in sensitivity. However, data were collected with the sequence in Figure 2. The sequence can be more rigorously verified by use of the density propagators described by (Ollerenshaw et al. 2003) or by use of programs such as SPINACH (Hogben et al. 2011).
An example of the data collected is given in Figure 3. Nine (+2a−2a) delays were used beginning at 4.0 ms and ending at 17.3 ms. The data were collected with an average recycle time of 1.2 s and 32 transients at each delay. Data sets for both isotropic (open symbols) and aligned (closed symbols) are shown. The data were fit to the sin(J(+2a −2a) or sin((J+RDC)(+2a −2a) using a Perl script that calls the non-linear least squares routine in gnuplot. The RDCs were extracted by taking the difference of isotropic (J) and aligned couplings (J+RDC), −18 Hz in the example given.
Figure 3.
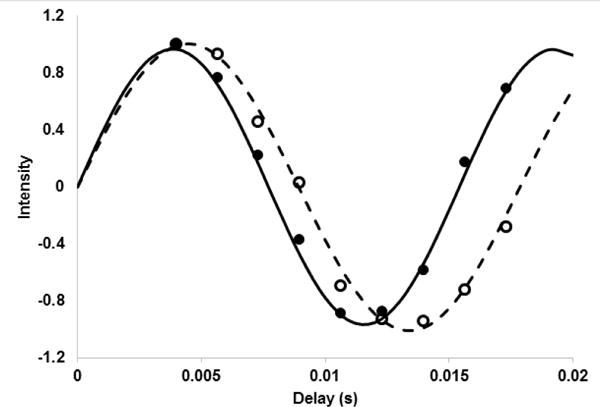
J-modulated intensities for crosspeak 7 (A543, 18.4 ppm, 1.39 ppm). Open symbols are for a sample aligned in phage and closed symbols are for an isotropic sample. The lines are reproduced from best fit parameters of 112.0 Hz and 129.7 Hz.
Assignments of sparsely-labeled HtpG
The program ASSIGN_SLP_1.1.2, which is based on a genetic algorithm search for the assignments best matching measured and predicted NMR data (chemical shifts, RDCs, NOEs), was modified to achieve assignments of full length HtpG. This modified version is available at the website: http://tesla.ccrc.uga.edu/software/. The numbers of observable NOEs are naturally reduced in a perdeuterated protein and few NOEs between the well-dispersed 13C-1H-labeled alanine methyls were observed. Hence, only RDCs and chemical shifts were used. The lack of NOEs, combined with the increased protein size, made it necessary to use data coming from partial assignments of crosspeaks from individual domains. Normally one would expect to transfer assignments based on an exact match of crosspeak positions in domain and full-length spectra, but because of changes in chemical shifts that arise from domain-domain interactions and pH differences, overlap of crosspeaks is not exact. Therefore, a list of possible matches was generated by considering all assigned domain crosspeaks within a generous chemical shift radius of a full length crosspeak (0.12 ppm 1H shift, 1.2 ppm 13C shift). These lists were transformed to a user input constraint matrix in which a zero indicated an acceptable assignment and a one indicated an unacceptable assignment. Penalties were assigned based on the occurrence of assignments carrying a one or zero (~10 for ones and 0 for zeros) and added to an overall assignment score. As in the original description of the program (Gao et al. 2017), score contributions for agreement of measured and predicted chemical shifts (calculated using the program PPM_ONE) (Li and Brüschweiler 2015) were represented as root-mean-square-deviations (RMSDs) normalized to 1 for deviations equal to estimated errors. Similarly, score contributions for RDCs came from RMSDs of measured versus predicted values, normalized and adjusted for information content. The predicted RDCs, however, must be recalculated for each assignment. Therefore, procedures paralleling those in the REDCAT program (Valafar and Prestegard 2004) are incorporated directly in ASSIGN_SLP_1.1.2. To facilitate use of the program, raw RDC values were corrected for methyl rotation and use of a C to C vector, as opposed to an actual C-H vector, in the back-calculation.
Because RDCs depend on domain orientations, and we are not confident that the relative orientations of domains in the apo crystal structure are preserved in solution, we approached assignments domain by domain. Because many of the crosspeaks seen in spectra of the single domain constructs cannot be unambiguously associated with just a single crosspeak from spectra of the full length protein, it was frequently the case that assignment tasks involved more crosspeaks than alanine sites. It is also possible to have more alanine sites than crosspeaks in certain assignment tasks, because crosspeaks can be unobservable due to motional broadening or overlap. Our implementation of the genetic algorithm requires an equal number of sites and crosspeaks. Therefore, data for extra sites or crosspeaks, as well as missing data, were designated with 999, and contributions to scores were omitted whenever a 999 occurred.
ASSIGN_SLP_1.1.2 outputs a list of possible assignments that includes those with a total score less than a user-entered cut-off. It is recommended that this be set to approximately 1.5 times the number of data types (in our case there are four data types, two chemical shifts (1H and 13C) and RDCs from two alignment media). Because the normalized scores would be one at the limit of estimated error for each data type, a score of 6 would correspond to solutions with all observables deviating from predictions by approximately 1.5 times standard error. The completely correct assignment having all crosspeaks paired with the correct site is nearly always in this output, but it is not necessarily the top score assignment. However, interest is really in which sites can be assigned to a particular crosspeak with high confidence, as opposed to identifying a completely correct set of assignments. Therefore, we have devised a criterion for selecting these high confidence sites (Gao et al. 2017). Based on test cases with known assignments, a site that is assigned to the same crosspeak more than 50% of the time in the list using the aforementioned cut-off, is an assignment made with high confidence (~95% confidence limit). Additional sites with a particular assignment simply being the most frequent (usually 2 times the next most frequent) are considered to be ones made with moderate confidence.
Homology modeling of HtpG-AMPPNP
A homology model for the HtpG dimer in the presence of AMPPNP was constructed using the UCSF Chimera program (Pettersen et al. 2004). The zebra fish mitochondrial Hsp90, TRAP1, (PDB code 4IPE) was selected as a template (Lavery et al. 2014). The 36% identical sequences were aligned using Clustal Omega (Sievers et al. 2011) using default parameters as provided in the Chimera interface. Modeling was done via the web version of MODELLER (Sali and Blundell 1993) again using default parameters as provided in the Chimera interface.
Modeling of the solution apo-HtpG structure
Using RDC data from each domain, the program REDCAT (Valafar and Prestegard 2004) was used to extract a set of principal order parameters and Euler angles that relate the principal alignment frame to the original coordinate frame. In each case, the highest resolution domain structure for the apo form was used (2IOR, 2GQ0 and 1SF8 for the NTD, MD and CTD, respectively). The Euler angles were used to rotate each domain into its principal alignment frame and the domains were assembled by translating the domains (and their three 180 rotational equivalents) to find the best option for covalent linkage.
Results and Discussion
Assignment of HtpG
Figure 4 shows a Methyl-TROSY spectrum of full length HtpG 13C-1H labeled in all alanine methyl groups. The sensitivity is high for a system of this size (~145 kDa as a dimer). The spectrum was acquired in approximately 1 hr on a 100 μL, 216 μM sample. There are approximately 40 resolvable crosspeaks in the region where alanine methyl crosspeaks fall. This is 85% of the possible 47 peaks expected based on the expression construct. While it is possible to have alanine methyls scramble to valine and leucine (Ayala et al. 2009), under the conditions of our expression protocol the level of scrambling appears to be small. The appearance of crosspeaks from individual domains assigned to alanine methyls in similar regions, and in similar numbers, supports this contention. RDCs were collected for each of the resolved peaks. There were a few cases where the fit to a single modulated and decaying exponential was unacceptable due to low signal to noise or possibly multiple modulations in the case of overlapping peaks. In all, 30 and 37 RDCs were found acceptable for the phage and peg alignments, respectively. These, along with errors estimated from fitting, are included in Supplemental Table 1. The chemical shifts of crosspeaks and the 13C-1H methyl RDCs were compared to predictions from PPM_ONE (Li and Brüschweiler 2015) and REDCAT (Valafar and Prestegard 2004), respectively, using the highest-resolution crystal structure available for each HtpG domain, i,e., 2IOR for NTD, 1SF8 for CTD and 2GQ0 for MD (Harris and Shiau 2004; Shiau et al. 2006b).
Figure 4.
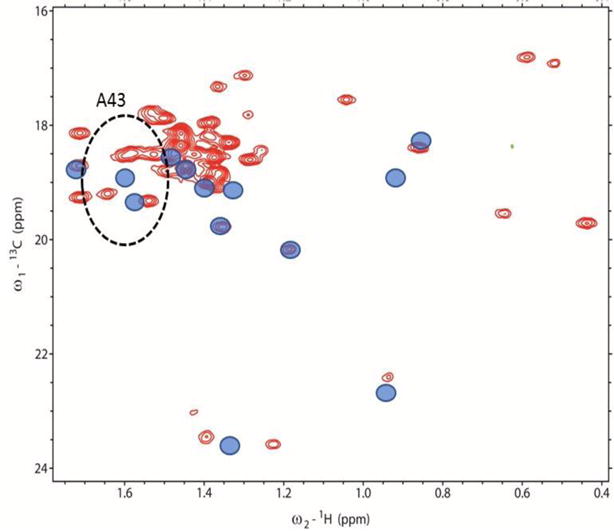
Methyl-TROSY spectrum of 13C-1H-alanine labeled and perdeuterated HtpG. Blue circles representing chemical shifts of crosspeaks of the separately expressed N-terminal domain are superimposed. The dotted ellipse centered at the single-domain shifts of A43 (radii 1.2 and 0.12 ppm) encloses 4 crosspeaks (red contours) judged to possibly belong to A43 in full length HtpG.
We started the search for assignments using the domain with the highest number of triple resonance assignments (NTD). Also shown in Figure 4 are symbols (blue circles) at the chemical shifts of alanine methyls taken from HNCACB and 13C-HSQC spectra of the isolated NTD. An ellipse is drawn around the point corresponding to A43 at a radii corresponding to estimated uncertainties in position due to separation of domains and differences in pH (0.12 ppm in the proton dimension and 1.2 ppm in the 13C dimension). This shows that four crosspeaks seen in the full-length protein (red contours) can potentially be assigned to the methyl resonance of residue A43. A similar analysis was done for each of the other Ala methyl resonances assigned in the NTD. In some cases the number of possible crosspeaks is reduced from the number found in the ellipse due to unique assignments of a crosspeak to other domains. Table 1 shows the final list of crosspeaks in the full-length protein that could possibly be assigned to each of the residues in the NTD. It also shows the isolated domain chemical shifts, final assignments and a confidence estimate. Initially, 5 residues were uniquely assigned to a crosspeak and the others had degeneracies ranging from 2 to 7. The program ASSIGN_SLP_1.1.2 was then used to search for the best assignment of the 13 sites in the N terminus to the 22 crosspeaks appearing in the various lists of degeneracies. Using the program twelve crosspeaks were assigned with high confidence and one with moderate confidence.
Table 1.
Crosspeak Assignments for the NTD of full-length apo-HtpG.
| Possible crosspeaks | Residue | 13C shift | 1H shift | Assignment | Confidence |
|---|---|---|---|---|---|
| 14,15,20,21,24,28,29 | 39 | 18.7 | 1.45 | 20 | moderate |
| 16 | 42 | 18.9 | 1.74 | 16 | high |
| 9,11,19,22 | 43 | 19.1 | 1.63 | 19 | high |
| 26,35 | 50 | 18.4 | 0.86 | 26 | high |
| 12,13,18 | 98 | 19.2 | 1.36 | 13 | high |
| 14,15,20,21,24,27,28 | 114 | 18.9 | 1.43 | 24 | high |
| 26,35 | 130 | 18.9 | 0.93 | 35 | high |
| 4 | 134 | 23.0 | 0.95 | 4 | high |
| 5 | 143 | 20.3 | 1.21 | 5 | high |
| 6 | 144 | 19.9 | 1.37 | 6 | high |
| 9,11 | 157 | 19.5 | 1.60 | 11 | high |
| 2 | 165 | 23.8 | 1.38 | 2 | high |
| 12,13,15,18,21 | 205 | 19.2 | 1.40 | 12 | high |
We then attempted assignments for the next most highly assigned domain, the CTD. Examining overlap of crosspeaks between the isolated domain and the full length protein in a manner similar to that described above, only 2 sites could be uniquely assigned. The other 10 sites had numbers of possible cross peaks ranging from 2 to 7. Three of the crosspeaks in the list of possible assignments were then eliminated based on their high confidence assignment to sites in the NT. Application of the program resulted in 6 additional high confidence assignments of crosspeaks to sites in the CTD and four moderate confidence assignments.
The middle domain had only one definitive assignment, and the remaining crosspeaks had lists of 1 to 7 crosspeaks associated with each of the remaining twelve sites. ASSIGN_SLP_1.1.2 yielded six high confidence assignments and four moderate confidence assignments. The complete list of 13C-1H-methyl assignments for single domain and full-length proteins is included in Supplemental Table 1. While the assignments are far from complete, they allow identification of some residues undergoing shifts on conversion from apo to AMPPNP forms of HtpG and a limited analysis of the changes in domain-domain orientations on this conversion.
Chemical shift perturbations on AMPPNP addition
Figure 5 shows a superposition of Methyl-TROSY spectra of apo and AMPPNP forms of HtpG. AMPPNP was added at 5 mM concentration and heated at 37 °C for 1 hour to assure complete conversion to the nucleotide bound form (Krukenberg et al. 2009). Assigned peaks that shift or disappear are labeled with residue numbers. It is useful to look at these residues in the context of where they lie in the respective structures. There is no structure of the AMPPNP form of HtpG. However, there is a crystal structure of the AMPPNP form of the mitochondrial Hsp90, TRAP1 (Lavery et al. 2014), and SAXS data suggest that the overall conformations of AMPPNP forms of TRAP1 and HtpG are similar. We have made a homology model of the AMPPNP form of HtpG using the TRAP1 structure as a template.
Figure 5.
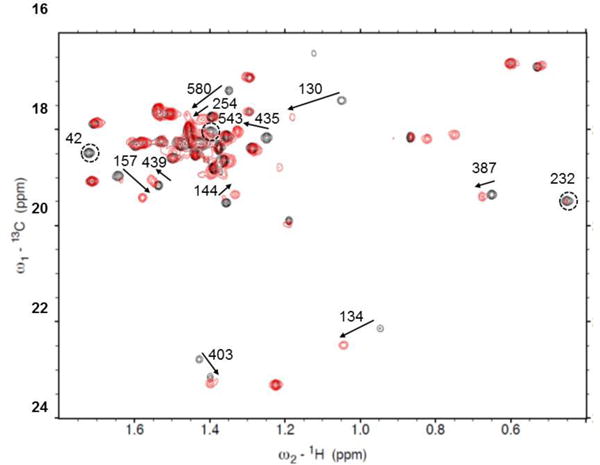
Crosspeak shifts on adding AMPPNP to apo-HtpG (black-apo, red-AMPPNP). Arrows show shifts, circles show peaks disappearing. Correlations are based on minimal differences in peak positions and in some cases may be ambiguous.
Figure 6A shows ribbon diagrams for superimposed NTDs of the crystal structure of the apo form (2IOQ) and the homology model of the AMPPNP form. The perturbed alanine residues are colored in red. There are significant structural differences throughout this domain, several perturbed residues are close to the nucleotide binding site (residues A42, A130, A134). A232 and A254 are in the linker between the NTD and the MD, where major differences in the modeled structure are seen.
Figure 6.
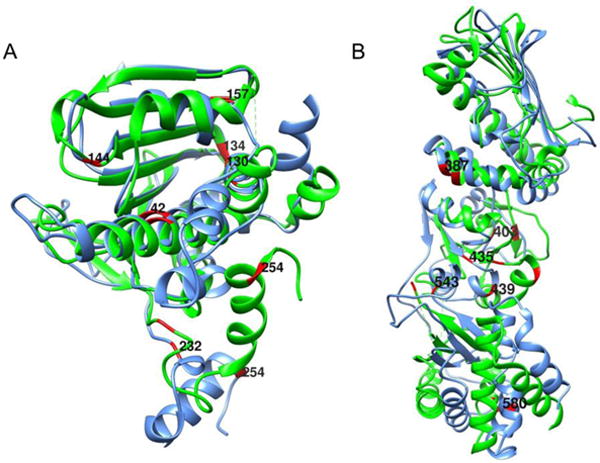
Superimposed ribbon structures of the N-terminal (A) and middle plus C-terminal (B) domains of apo (green) and AMPPNP (blue) forms of HtpG. 13C-1H-labeled alanines with resonances that differ in chemical shift are shown in red.
Figure 6B shows ribbon diagrams for a superposition of the MD and CTD for the two structures with perturbed alanines in red. A435, A439 and A543 are close to the junction between the MD and CTD where substantial structural differences are predicted. A387 is in a region of less structural difference, but this is near a predicted area of contact between monomers in the AMPPNP dimer structure. A580 in the CTD is near the dimerization interface of both structures and in a region that shows some structural variation, as predicted by our model. A403, in the MD, is isolated from inter-domain contacts, but still shows some predicted structural variation. Hence, most perturbations of chemical shifts can be rationalized based on a structural comparison of apo crystal structures and a homology model of the AMPPNP structure. This supports the validity of the assignments, and demonstrates the ability of chemical shift to report on regions of structural change in proteins.
Inter-domain structure of apo-HtpG
Hsp90 proteins clearly sample a range of conformations as evidenced by different crystal, SAXS and EM structures. Internal structures of individual domains might be expected to be better preserved, and there is even some data to suggest that certain inter-domain contacts may be preserved. In the pair of structures for apo and ADP forms of HtpG CTD and MD, orientations are nearly identical (Shiau et al. 2006a). Matching C carbons in the MD plus CTD (residues 233–624) the overall alignment is 2.2Å. They align even closer in the GRP94 apo and AMPPNP structures (Dollins et al. 2007). RDCs provide one means of assessing the conservation of domain-domain orientations. When sufficient data are available (>>5) for a rigid segment, a best set of order parameters can be determined and used to back-calculate RDCs from a trial structure and compare those to measured RDCs for all sites. A Q factor (Bax 2003) gives a measure of how well the crystal structure compares to that in solution. The amount of data we have for some of the domains is marginal, so we initially tried fitting a 2 domain segment. Using 18 alanine methyl RDCs from the phage alignment, spread over the MD and CTD of apo-HtpG, a Q factor of 0.81 was obtained. This is not very good agreement. Some assessment of the level of agreement can be obtained by comparing Q factors obtained for individual domains to that for combined domains, but this must be done using the same number of RDCs. Using 8 randomly picked alanine methyl RDCs, 4 from each domain, we found an average Q factor of 0.4. This can be compared to Q factors of the individual domains using similar numbers of RDCs; Q factors of the MD and CTD are 0.2 and 0.2. The larger Q for the combined domains clearly suggests that even the MD - CTD orientations of the apo form of HtpG seen in the crystal structure are not well maintained in solution.
In principle, the principal order parameters obtained for each domain, or their combination in a generalized degree of order (GDO) (Prestegard et al. 2000), can be used as a direct indicator of internal motion; if the structure was flexible, and ordering occurred principally through interactions with one domain, GDOs would be smaller for non-interacting domains. The GDOs determined for phage alignment are 0.0018, 0.0020 and 0.0023 for the NTD, MD, and CTD, respectively, showing no significant indication of preferential alignment and allowing no clear conclusion about internal domain motions. However, the GDOs for PEG alignment are 0.0039, 0.0036 and 0.0007 indicating possible preferential alignment by the NTD or MD in this medium, and the existence of some internal motion reducing average alignment of the CTD.
It is also possible to use RDC data on a domain by domain basis to determine an average structure of the apo form in solution. An order tensor determined for each domain can be converted to a set of principle order parameters and Euler angles that can be used to transform coordinates in the original frame to those in a principal alignment frame. For a rigid molecule all domains would share one alignment frame. Hence, once aligned, domains can be assembled by translation to optimize inter-domain connectivities. Because RDCs are insensitive to rotation about any of the principal frame axes by 180°, four possible orientations of each added domain need to be examined in this assembly process, but usually only one of these will allow reasonable linkages between domains. A similar procedure can be used when there is inter-domain motion, but domain positions must then be viewed as a representation of an average structure. A structure of the apo form assembled in this way from PEG alignment data is shown in Figure 7A. For the NTD to MD connection, only a 180 z rotation of the MD produces an acceptable linkage.
Figure 7.
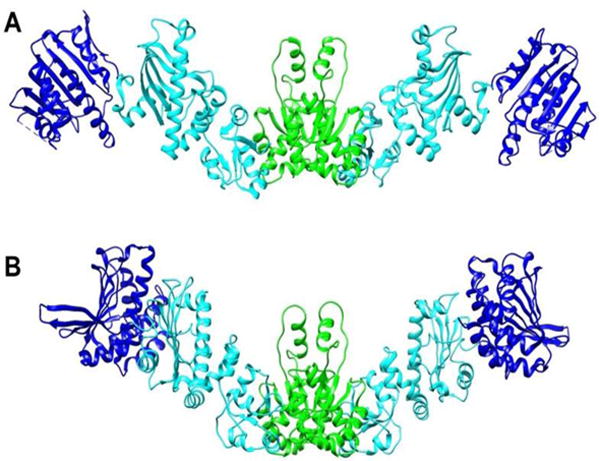
Models of the apo-HtpG dimer. A) Domain orientations were obtained by RDC analysis. B) A model based on SAXS data obtained in solution at high pH (Krukenberg et al. 2008). NTD, MD and CTD are colored blue, cyan and green respectively.
For the MD to CTD connections both zero and 180 x rotations produce acceptable structures. Because of the symmetry of the CTD dimer and its orientation in the principal alignment frame, these rotations actually produce the pair of MDs as seen in the dimer structure. The relative orientations for domains are well defined by RDCs. However, their translational positions depend entirely on acceptable covalent connections between domains. This is not a serious problem for the NTD to MD connection, because there are no missing residues between the structures used to model this pair of domains. For the MD to CTD more than 15 residues are missing and there is a significant lack of translational definition.
There is a SAXS model for apo-HtpG as it exists in solution at high pH (Krukenberg et al. 2008). It is depicted in Figure 7B. To aid comparison we have oriented CTDs similarly and translated the NTD-MD pair of the RDC model to match as best as possible the MD in the SAXS model. It is apparent that the MD domains are oriented similarly in the two models, while the orientation of the NTD differs resulting in a more extended structure. However, one must use caution in interpreting these models. We do see evidence of inter-domain motion, so these represent structures subject to averaging processes that are quite dependent on the source of data. Also, RDC data available in this case are quite minimal (8 or 9 RDCs per domain), and therefore, results are more prone to error than those of typical applications. However, the observation of an apo structure that is more extended in solution than those in crystal structures of apo and nucleotide-bound forms is certainly well-supported by these data.
Conclusion
We have clearly demonstrated the ability to achieve assignments for resonances for a large protein system isotopically labeled only in the methyl carbons of alanines. Use of a 13C methyl group presents advantages in terms of sensitivity and resolution because of Methyl-TROSY effects (Rosenzweig and Kay 2014). The labeling of just a single amino acid provides additional simplification of spectra by limiting the number of crosspeaks. While only partial assignments have been achieved with confidence using a combination of chemical shift and RDC data, the number of assignments proves adequate to identify many regions of structural change on adding an ATP analog to the apo form of HtpG. RDCs, although limited in number, have also allowed the construction of a new model for the solution form of the apo protein. It is likely that similar data collected in the presence of a client protein could provide insight into the mechanism of this important heat-shock protein.
It would clearly be advantageous to assign a more complete set of resonances, and possibly to do this without resorting to supplemental information from individually expressed domains. This should be possible with the addition of additional data types. 15N-filtered NOEs have proven very valuable in the assignment of HSQC crosspeaks in other applications (Gao et al. 2017; Gao et al. 2016). 13C-filtered NOEs for methyl groups would have similar value. These were not available here because of perdeuteration and the lack of sufficient methyl-methyl contacts for alanines in HtpG. However, partial deuteration has been shown to preserve resolution to some extent, while providing significant number of inter-residue NOEs (Kay and Gardner 1997; Nietlispach et al. 1996). Also, there is a variety of other information, including pseudo contact shifts (PCS) (Allegrozzi et al. 2000; Schmitz et al. 2012; Yagi et al. 2013) and paramagnetic relaxation enhancements (PREs) (Battiste and Wagner 2000; Clore 2011) that might be collected on appropriately tagged proteins. All of this bodes well for future applications of sparse labeling approaches and applications of alanine methyl RDCs to structural characterization of large and glycosylated proteins.
Supplementary Material
Acknowledgments
We gratefully acknowledge financial support from the National Institute of General Medical Sciences (NIGMS Protein Structure Initiative grant U54GM094597 (GTM), biotechnology resource grant P41GM103390 and grant R01GM033225 (JHP)) and from the HHMI (DAA). Molecular graphics and structural analyses were performed with the UCSF Chimera package. Chimera is developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIGMS P41-GM103311). Manuscript content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Supporting Information
Two tables, one showing residual dipolar coupling data along with full-length apo-HtpG alanine assignments and the other showing assignment of alanine resonances for the individual domains.
References
- Acton TB, et al. Preparation of protein samples for nmr structure, function, and small-molecule screening studies. In: Kuo LC, editor. Fragment-based drug design: Tools, practical approaches, and examples, vol 493. Methods in enzymology. 2011. pp. 21–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali MMU, et al. Crystal structure of an hsp90-nucleotide-p23/sba1 closed chaperone complex. Nature. 2006;440:1013–1017. doi: 10.1038/nature04716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allegrozzi M, Bertini I, Janik MBL, Lee YM, Lin GH, Luchinat C. Lanthanide induced pseudocontact shifts for solution structure refinements of macromolecules in shells up to 40 å from the metal ion. J Am Chem Soc. 2000;122:4154–4161. [Google Scholar]
- Ayala I, Hamelin O, Amero C, Pessey O, Plevin MJ, Gans P, Boisbouvier J. An optimized isotopic labelling strategy of isoleucine-gamma(2) methyl groups for solution nmr studies of high molecular weight proteins. Chemical Communications. 2012;48:1434–1436. doi: 10.1039/c1cc12932e. [DOI] [PubMed] [Google Scholar]
- Ayala I, Sounier R, Use N, Gans P, Boisbouvier J. An efficient protocol for the complete incorporation of methyl-protonated alanine in perdeuterated protein. J Biomol NMR. 2009;43:111–119. doi: 10.1007/s10858-008-9294-7. [DOI] [PubMed] [Google Scholar]
- Bahrami A, Assadi A, Markley JL, Eghbalnia H. Probablilistic interaction networ of evidence algorithm and its application to complete labeling of peak lsts from protein nmr spectrscopy. PLoS Computational Biology. 2009;5:e1000307. doi: 10.1371/journal.pcbi.1000307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battiste JL, Wagner G. Utilization of site-directed spin labeling and high resolution heteronuclear nuclear magnetic resonance for global fold determination of large proteins with limited nuclear overhauser effect data. Biochemistry. 2000;39:5355–5365. doi: 10.1021/bi000060h. [DOI] [PubMed] [Google Scholar]
- Bax A. Weak alignment offers new nmr opportunities to study protein structure and dynamics. Protein Sci. 2003;12:1–16. doi: 10.1110/ps.0233303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burmann BM, Hiller S. Chaperones and chaperone-substrate complexes: Dynamic playgrounds for nmr spectroscopists. Progress in Nuclear Magnetic Resonance Spectroscopy. 2015;86–87:41–64. doi: 10.1016/j.pnmrs.2015.02.004. [DOI] [PubMed] [Google Scholar]
- Clore GM. Exploring sparsely populated states of macromolecules by diamagnetic and paramagnetic nmr relaxation. Protein Sci. 2011;20:229–246. doi: 10.1002/pro.576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day JEH, et al. Targeting the hsp90 molecular chaperone with novel macrolactams. Synthesis, structural, binding, and cellular studies. Acs Chemical Biology. 2011;6:1339–1347. doi: 10.1021/cb200196e. [DOI] [PubMed] [Google Scholar]
- Didenko T, Duarte AMS, Karagoz GE, Rudiger SGD. Hsp90 structure and function studied by nmr spectroscopy. Biochimica Et Biophysica Acta-Molecular Cell Research. 2012;1823:636–647. doi: 10.1016/j.bbamcr.2011.11.009. [DOI] [PubMed] [Google Scholar]
- Dollins DE, Warren JJ, Immormino RM, Gewirth DT. Structures of grp94-nucleotide complexes reveal mechanistic differences between the hsp90 chaperones. Mol Cell. 2007;28:41–56. doi: 10.1016/j.molcel.2007.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Q, Chalmers GR, Moremen KM, Prestegard JH. Nmr assignments of sparsely labeled proteins using a genetic algorithm. J Biomol NMR. 2017;67:283–294. doi: 10.1007/s10858-017-0101-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Q, et al. Structural aspects of heparan sulfate binding to robo1-ig1-2. ACS Chemical Biology. 2016;11:3106–3113. doi: 10.1021/acschembio.6b00692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godoy-Ruiz R, Guo CY, Tugarinov V. Alanine methyl groups as nmr probes of molecular structure and dynamics in high-molecular-weight proteins. J Am Chem Soc. 2010;132:18340–18350. doi: 10.1021/ja1083656. [DOI] [PubMed] [Google Scholar]
- Harris SF, Shiau AK. The crystal structure of the carboxy-terminal dimerization domain of htpg, the escherichia coli hsp90, reveals a potential substrate binding site. Structure. 2004;12:1087–1097. doi: 10.1016/j.str.2004.03.020. [DOI] [PubMed] [Google Scholar]
- Hennig J, Sattler M. The dynamic duo: Combining nmr and small angle scattering in structural biology. Protein Sci. 2014;23:669–682. doi: 10.1002/pro.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogben HJ, Krzystyniak M, Charnock GTP, Hore PJ, Kuprov I. Spinach - a software library for simulation of spin dynamics in large spin systems. J Magn Reson. 2011;208:179–194. doi: 10.1016/j.jmr.2010.11.008. [DOI] [PubMed] [Google Scholar]
- Isaacson RL, Simpson PJ, Liu M, Cota E, Zhang X, Freemont P, Matthews S. A new labeling method for methyl transverse relaxation-optimized spectroscopy nmr spectra of alanine residues. J Am Chem Soc. 2007;129:15428–+. doi: 10.1021/ja0761784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson SE. Hsp90: Structure and function. In: Jackson S, editor. Molecular chaperones, vol 328. Topics in current chemistry. 2013. pp. 155–240. [DOI] [PubMed] [Google Scholar]
- Jansson M, Li YC, Jendeberg L, Anderson S, Montelione GT, Nilsson B. High-level production of uniformly n-15- and c-13-enriched fusion proteins in escherichia coli. J Biomol NMR. 1996;7:131–141. doi: 10.1007/BF00203823. [DOI] [PubMed] [Google Scholar]
- Karagoz GE, et al. Hsp90-tau complex reveals molecular basis for specificity in chaperone action. Cell. 2014;156:963–974. doi: 10.1016/j.cell.2014.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karagoz GE, et al. N-terminal domain of human hsp90 triggers binding to the cochaperone p23. Proc Natl Acad Sci USA. 2011;108:580–585. doi: 10.1073/pnas.1011867108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay LE, Gardner KH. Solution nmr spectroscopy beyond 25 kda. Curr Opin Struct Biol. 1997;7:722–731. doi: 10.1016/s0959-440x(97)80084-x. [DOI] [PubMed] [Google Scholar]
- Krukenberg KA, Bottcher UMK, Southworth DR, Agard DA. Grp94, the endoplasmic reticulum hsp90, has a similar solution conformation to cytosolic hsp90 in the absence of nucleotide. Protein Sci. 2009;18:1815–1827. doi: 10.1002/pro.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krukenberg KA, Forster F, Rice LM, Sali A, Agard DA. Multiple conformations of e-coli hsp90 in solution: Insights into the conformational dynamics of hsp90. Structure. 2008;16:755–765. doi: 10.1016/j.str.2008.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krukenberg KA, Street TO, Lavery LA, Agard DA. Conformational dynamics of the molecular chaperone hsp90. Q Rev Biophys. 2011;44:229–255. doi: 10.1017/s0033583510000314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavery LA, Partridge JR, Ramelot TA, Elnatan D, Kennedy MA, Agard DA. Structural asymmetry in the closed state of mitochondrial hsp90 (trap1) supports a two-step atp hydrolysis mechanism. Mol Cell. 2014;53:330–343. doi: 10.1016/j.molcel.2013.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Brüschweiler R. Ppm_one: A static protein structure based chemical shift predictor. J Biomol NMR. 2015;62:403–409. doi: 10.1007/s10858-015-9958-z. [DOI] [PubMed] [Google Scholar]
- Lipsitz RS, Tjandra N. Residual dipolar couplings in nmr structure analysis. Annu Rev Biophys Biomol Struct. 2004;33:387–413. doi: 10.1146/annurev.biophys.33.110502.140306. [DOI] [PubMed] [Google Scholar]
- Miyata Y, Nakamoto H, Neckers L. The therapeutic target hsp90 and cancer hallmarks. Curr Pharm Des. 2013;19:347–365. doi: 10.2174/138161213804143725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nietlispach D, et al. An approach to the structure determination of larger proteins using triple resonance nmr experiments in conjunction with random fractional deuteration. J Am Chem Soc. 1996;118:407–415. doi: 10.1021/ja952207b. [DOI] [Google Scholar]
- Ollerenshaw JE, Tugarinov V, Kay LE. Methyl trosy: Explanation and experimental verification. Magnetic Resonance in Chemistry. 2003;41:843–852. doi: 10.1002/mrc.1256. [DOI] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. Ucsf chimera - a visualization system for exploratory research and analysis. Journal of Computational Chemistry. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Prestegard JH, Al-Hashimi HM, Tolman JR. Nmr structures of biomolecules using field oriented media and residual dipolar couplings. Q Rev Biophys. 2000;33:371–424. doi: 10.1017/s0033583500003656. [DOI] [PubMed] [Google Scholar]
- Prestegard JH, Bougault CM, Kishore AI. Residual dipolar couplings in structure determination of biomolecules. Chemical Reviews. 2004;104:3519–3540. doi: 10.1021/cr030419i. [DOI] [PubMed] [Google Scholar]
- Retzlaff M, et al. Asymmetric activation of the hsp90 dimer by its cochaperone aha1. Mol Cell. 2010;37:344–354. doi: 10.1016/j.molcel.2010.01.006. [DOI] [PubMed] [Google Scholar]
- Rosenzweig R, Kay LE. Bringing dynamic molecular machines into focus by methyl-trosy nmr. In: Kornberg RD, editor. Annual review of biochemistry, vol 83. Annual review of biochemistry. 2014. pp. 291–315. [DOI] [PubMed] [Google Scholar]
- Rosenzweig R, Kay LE. Solution nmr spectroscopy provides an avenue for the study of functionally dynamic molecular machines: The example of protein disaggregation. J Am Chem Soc. 2016;138:1466–1477. doi: 10.1021/jacs.5b11346. [DOI] [PubMed] [Google Scholar]
- Sali A, Blundell TL. Comparative protein modeling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- Schmitz C, Vernon R, Otting G, Baker D, Huber T. Protein structure determination from pseudocontact shifts using rosetta. J Mol Biol. 2012;416:668–677. doi: 10.1016/j.jmb.2011.12.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze A, Beliu G, Helmerich DA, Schubert J, Pearl LH, Prodromou C, Neuweiler H. Cooperation of local motions in the hsp90 molecular chaperone atpase mechanism. Nat Chem Biol. 2016;12:628–+. doi: 10.1038/nchembio.2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiau AK, Harris SF, Southworth DR, Agard DA. Structural analysis of e-coli hsp90 reveals dramatic nucleotide-dependent conformational rearrangements. Cell. 2006a;127:329–340. doi: 10.1016/j.cell.2006.09.027. [DOI] [PubMed] [Google Scholar]
- Shiau AK, Harris SF, Southworth DR, Agard DA. Structural analysis of e. Coli hsp90 reveals dramatic nucleotide-dependent conformational rearrangements. Cell. 2006b;127:329–340. doi: 10.1016/j.cell.2006.09.027. [DOI] [PubMed] [Google Scholar]
- Sievers F, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using clustal omega. Mol Syst Biol. 2011;7 doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southworth DR, Agard DA. Species-dependent ensembles of conserved conformational states define the hsp90 chaperone atpase cycle. Mol Cell. 2008;32:631–640. doi: 10.1016/j.molcel.2008.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southworth DR, Agard DA. Client-loading conformation of the hsp90 molecular chaperone revealed in the cryo-em structure of the human hsp90:Hop complex. Mol Cell. 2011;42:771–781. doi: 10.1016/j.molcel.2011.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprangers R, Kay LE. Quantitative dynamics and binding studies of the 20s proteasome by nmr. Nature. 2007;445:618–622. doi: 10.1038/nature05512. [DOI] [PubMed] [Google Scholar]
- Stechmann A, Cavalier-Smith T. Evolutionary origins of hsp90 chaperones and a deep paralogy in their bacterial ancestors. J Eukaryot Microbiol. 2004;51:364–373. doi: 10.1111/j.1550-7408.2004.tb00580.x. [DOI] [PubMed] [Google Scholar]
- Street TO, Lavery LA, Verba KA, Lee CT, Mayer MP, Agard DA. Cross-monomer substrate contacts reposition the hsp90 n-terminal domain and prime the chaperone activity. J Mol Biol. 2012;415:3–15. doi: 10.1016/j.jmb.2011.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tjandra N, Bax A. Measurement of dipolar contributions to (1)j(ch) splittings from magnetic-field dependence of j modulation in two-dimensional nmr spectra. J Magn Reson. 1997;124:512–515. doi: 10.1006/jmre.1996.1088. [DOI] [PubMed] [Google Scholar]
- Tugarinov V, Hwang PM, Ollerenshaw JE, Kay LE. Cross-correlated relaxation enhanced h-1-c-13 nmr spectroscopy of methyl groups in very high molecular weight proteins and protein complexes. J Am Chem Soc. 2003;125:10420–10428. doi: 10.1021/ja030153x. [DOI] [PubMed] [Google Scholar]
- Tugarinov V, Kanelis V, Kay LE. Isotope labeling strategies for the study of high-molecular-weight proteins by solution nmr spectroscopy. Nature Protocols. 2006;1:749–754. doi: 10.1038/nprot.2006.101. [DOI] [PubMed] [Google Scholar]
- Tugarinov V, Kay LE. Ile, leu, and val methyl assignments of the 723-residue malate synthase g using a new labeling strategy and novel nmr methods. J Am Chem Soc. 2003;125:13868–13878. doi: 10.1021/ja030345s. [DOI] [PubMed] [Google Scholar]
- Valafar H, Prestegard JH. Redcat: A residual dipolar coupling analysis tool. J Magn Reson. 2004;167:228–241. doi: 10.1016/j.jmr.2003.12.012. [DOI] [PubMed] [Google Scholar]
- Verba KA, Wang RYR, Arakawa A, Liu YX, Shirouzu M, Yokoyama S, Agard DA. Structural biology atomic structure of hsp90-cdc37-cdk4 reveals that hsp90 traps and stabilizes an unfolded kinase. Science. 2016;352:1542–1547. doi: 10.1126/science.aaf5023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao R, et al. The high-throughput protein sample production platform of the northeast structural genomics consortium. Journal of Structural Biology. 2010;172:21–33. doi: 10.1016/j.jsb.2010.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi H, Pilla KB, Maleckis A, Graham B, Huber T, Otting G. Three dimensional protein fold determination from backbone amide pseudocontact shifts generated by lanthanide tags at multiple sites. Structure. 2013;21:883–890. doi: 10.1016/j.str.2013.04.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.