

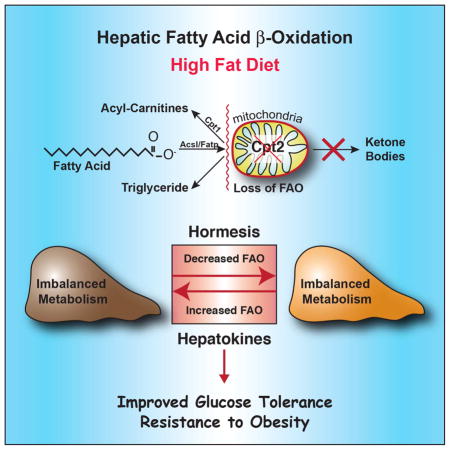
SUMMARY
The liver has a large capacity for mitochondrial fatty acid β-oxidation, which is critical for systemic metabolic adaptations such as gluconeogenesis and ketogenesis. To understand the role of hepatic fatty acid oxidation in response to a chronic high fat diet (HFD), we generated mice with a liver-specific deficiency of mitochondrial long chain fatty acid β-oxidation (Cpt2L−/− mice). Paradoxically, Cpt2L−/− mice were resistant to HFD-induced obesity and glucose intolerance with an absence of liver damage, although they exhibited serum dyslipidemia, hepatic oxidative stress and systemic carnitine deficiency. Feeding a HFD induced hepatokines in mice with a loss of hepatic fatty acid oxidation that enhanced systemic energy expenditure and suppressed adiposity. Additionally, the suppression in hepatic gluconeogenesis was sufficient to improve HFD-induced glucose intolerance. These data show that inhibiting hepatic fatty acid oxidation results in a systemic hormetic response that protects mice from HFD-induced obesity and glucose intolerance.
eTOC
Lee et al. show that contrary to expectations, the loss of hepatic fatty acid oxidation (FAO) confers resistance to weight gain and adiposity in response to a high fat diet. Additionally, they show that loss of heptic FAO, and consequently hepatic gluconeogenesis, protects mice from high fat diet-induced glucose intolerance.
INTRODUCTION
The metabolism, storage and flux of lipids in the liver plays a central role in diet-induced obesity, diabetes and nonalcoholic fatty liver disease (NAFLD). However, the detailed mechanisms governing the shift from normal physiology to pathophysiology are poorly understood, particularly with respect to the role of lipid metabolism. The liver represents a metabolic hub for the uptake and distribution of lipids via free fatty acids, lipoproteins and de novo lipogenesis. Consequently, an imbalance between hepatic lipid uptake/anabolism and export/catabolism in chronic states of over-nutrition has been implicated in the etiology of many obesity-related diseases. (Mashek, 2013; Samuel et al., 2004; Savage et al., 2007; Szendroedi et al., 2011). The liver has a large capacity for mitochondrial fatty acid β-oxidation and represents a major catabolic pathway in hepatocytes. Inborn errors in fatty acid oxidation often present with serious metabolic disease that center on hepatic function. Additionally, many chronic diseases exhibit disruptions in hepatic mitochondrial fatty acid oxidation (Samuel et al., 2004; Savage et al., 2007).
Long chain fatty acids are first activated in the cytosol to long chain acyl-CoAs via long chain acyl-CoA synthetases. In order to traverse the mitochondrial membrane and gain access to the mitochondrial matrix, long chain acyl-CoAs depend on carnitine and carnitine palmitoyltransferases. Activated long chain acyl-CoAs are converted to long chain acylcarnitines via carnitine palmitoyltransferase 1 (Cpt1) located on the mitochondrial outer membrane. Long chain acylcarnitines can then translocate into the mitochondrial matrix via the carnitine:acylcarnitine translocase, where they are finally converted back to long chain acyl-CoAs via carnitine palmitoyltransferase 2 (Cpt2) (Kerner and Hoppel, 2000; McGarry and Brown, 1997). Cpt1 is encoded by multiple genes that exhibit differential tissue specificity (Kerner and Hoppel, 2000). However, Cpt2 is encoded by a single gene. Long chain acyl-CoAs generated by Cpt2 can then be processed through β-oxidation to provide the carbon substrate for ketogenesis (acetyl-CoA) and mitochondrial bioenergetics (ATP, NADH, FADH2) to facilitate gluconeogenesis. (Chen et al., 1999; Kerner and Hoppel, 2000; McGarry and Brown, 1997; Soling et al., 1968). Previously, we generated a mouse model with a conditional loss-of-function of Cpt2 specifically in the liver (referred to herein as Cpt2L−/− mice) that are deficient in hepatic mitochondrial long chain fatty acid β-oxidation. Cpt2L−/− mice were relatively normal under carbohydrate-fed conditions, but fasting resulted in hepatic steatosis and serum dyslipidemia with an absence of circulating ketone bodies. Feeding a ketogenic diet to Cpt2L−/− mice resulted in severe hepatomegaly, liver damage and death with a complete absence of adipose triglyceride stores (Lee et al., 2016b). As expected, hepatic fatty acid oxidation was a critical adaptation to fasting.
To understand the role of hepatic fatty acid β-oxidation in response to a chronic high fat diet (HFD), we challenged Cpt2L−/− and control mice to a long-term high fat feeding. Unexpectedly, Cpt2L−/− mice were resistant to HFD-induced obesity and glucose intolerance with an absence of liver damage, although they exhibited serum dyslipidemia, hepatic oxidative stress and systemic carnitine deficiency. The suppression in adiposity and obesity was associated with a HFD-dependent increase in whole body energy expenditure that was normalized by lowering energy expenditure via thermoneutral housing. Additionally, the deficiency in fatty acid oxidation suppressed hepatic glucose production, resulting in greatly improved glucose tolerance. These data demonstrate the central role of hepatic fatty acid oxidation to systemic energy homeostasis and highlight a surprisingly beneficial role of inhibiting hepatic fatty acid oxidation to systemic metabolic dysfunction.
RESULTS
Mice with a loss of hepatic fatty acid oxidation are resistant to high fat diet-induced obesity
Previously, we showed that Cpt2L−/− mice were relatively normal under carbohydrate-fed conditions, but the exposure of the liver to a high burden of free fatty acids via prolonged fasting or a ketogenic diet elicited profound cellular and physiological adaptations (Lee et al., 2016b). Since hepatic fatty acid oxidation is central to systemic energy balance, we were interested in how feeding a chronic lipid-rich diet to Cpt2L−/− mice would affect systemic metabolic dysfunction. Therefore, we placed Cpt2L−/− and littermate control (Cpt2lox/lox) mice on a high fat (HFD) or low fat diet (LFD) for 16 weeks. Although Cpt2L−/− mice have a genetically imposed deficiency in energy expenditure, both male and female Cpt2L−/− mice were paradoxically resistant to HFD-induced body weight gain. (Figures 1A, 1A′). The loss of hepatic fatty acid oxidation did not affect body size as there was no genotypic change in femur length. The suppression in body weight in HFD-fed Cpt2L−/− mice was due largely to a reduction of fat mass (Figures 1B, 1B′). Cpt2L−/− mice fed a LFD for 16 weeks did not exhibit changes in body weight, although both male and female mice had a small suppression in fat mass (Figure S1). The suppression in fat mass in male mice was mainly due to a decrease in iWAT (Figure 1C). Histological examination of iWAT demonstrated smaller adipocytes in HFD-fed Cpt2L−/− mice compared to control mice (Figures 1D, 1E). These data show that a loss of hepatic fatty acid oxidation paradoxically results in resistance to HFD-induced obesity and adiposity.
Figure 1. Liver-specific deficiency in fatty acid oxidation is protective against HFD-induced body weight gain and adiposity.

(A) Body weight and femur lengths of male Cpt2lox/lox and Cpt2L−/− mice fed a HFD for 16wks (n=12).
(B) Body compositions of male Cpt2lox/lox and Cpt2L−/− mice fed a HFD for 16wks measured by EchoMRI (n=12).
(C) Wet weights of iWAT and gWAT unilateral depots, liver (left lobe) and kidney for male Cpt2lox/lox and Cpt2L−/− mice fed a HFD for 16wks (n=12).
(D) H&E stained sections of iWAT from male Cpt2lox/lox and Cpt2L−/− mice fed a HFD for 16wks. Scale bar, 100 μm.
(E) Adipocyte size distribution from H&E stained sections of iWAT from Cpt2lox/lox and Cpt2L−/− mice fed a HFD for 16wks.
(F) Serum metabolites in male Cpt2lox/lox and Cpt2L−/− mice fed a HFD for 16wks (n=6).
(G) Serum lucose and insulin in male Cpt2lox/lox and Cpt2L−/− mice fed a HFD for 16wks (n=6).
(H) Serum leptin and adiponectin in male Cpt2lox/lox and Cpt2L−/− mice fed a HFD for 16wks (n=6).
(I) Blood acylcarnitines of male Cpt2lox/lox and Cpt2L−/− mice following a 16-week chronic high fat feeding (n=5–8).
(J) Urine acylcarnitines of male Cpt2lox/lox and Cpt2L−/− mice following a 16-week chronic high fat feeding (n=6). Urine acylcarnitine values were normalized to creatinine values.
Data are expressed as mean ± SEM. *p<0.05; **p<0.01; ***p<0.001.
To further understand the systemic response to a loss of hepatic fatty acid oxidation under HFD-fed conditions, we examined circulating metabolites in Cpt2lox/lox and Cpt2L−/− mice. As expected, Cpt2L−/− mice exhibited suppressed β-hydroxybutyrate and increased NEFA (Figure 1F). Additionally, Cpt2L−/− mice had greatly improved glucose and insulin levels more indicative of LFD-fed mice (Figure 1G). Consistent with decreased adiposity and improved glucose homeostasis, leptin was lower and adiponectin was unchanged in Cpt2L−/− mice. (Figure 1H). Previously, we had shown significant alterations in serum acylcarnitines in Cpt2L−/− mice in response to a 24hr fast or feeding a ketogenic diet (Lee et al., 2016b). HFD-fed Cpt2L−/− mice exhibited a dramatic suppression of free carnitine and acetyl-carnitine in both blood (Figure 1I) and urine (Figure 1J). Other short chain fatty acids were also significantly lower in HFD-fed Cpt2L−/− mice. Interestingly, Cpt2L−/− mice exhibited more medium chain monounsaturated acylcarnitines in their urine, suggesting a cell nonautonomous effect on the kidney (Figure 1J). These data show that the loss of hepatic fatty acid oxidation alters systemic lipid metabolism and improves glucose homeostasis following chronic high fat feeding.
High fat diet elicits oxidative stress and a hormetic response in Cpt2L−/− mice
The loss of hepatic fatty acid oxidation would be predicted to potentiate HFD-induced hepatic steatosis. Surprisingly, 16wks of HFD feeding did not exacerbate hepatic steatosis or liver triglyceride (TAG) content in Cpt2L−/− livers (Figures 2A, 2B). Interestingly, although Cpt2L−/− livers did not exhibit potentiated steatosis, there was a loss of metabolic zonation evident in control livers (Figure 2A). A potential explanation for the resistance to weight gain in Cpt2L−/− mice could be liver failure. However, other than reduced adiposity, Cpt2L−/− mice were overtly indistinguishable from littermate controls. Additionally, there was no difference in serum ALT activity, a measure of liver damage (Figure 2C). These data show that the loss of hepatic fatty acid oxidation does not result in potentiated fatty liver or liver damage following chronic high fat feeding.
Figure 2. Loss of hepatic fatty acid oxidation induces oxidative stress.

(A) H&E stained sections of liver from male Cpt2lox/lox and Cpt2L−/− mice fed a HFD for 16wks.
(B) Liver TAG content of male Cpt2lox/lox and Cpt2L−/− mice fed a HFD for 16wks (n=6).
(C) Measure of liver damage via serum ALT activity in male Cpt2lox/lox and Cpt2L−/− mice fed a HFD for 16wks (n=6).
(D) TBARS assay measuring lipid peroxidation in liver of male Cpt2lox/lox and Cpt2L−/− mice fed a HFD for 16wks (n=6).
(E) Total glutathione in livers of Cpt2lox/lox and Cpt2L−/− mice fed a HFD for 16wks (n=7).
(F) Western blot of Hsp27 in livers of Cpt2lox/lox and Cpt2L−/− mice fed a HFD for 16wks.
(G) Gene expression of fatty acid synthesis genes in livers of Cpt2lox/lox and Cpt2L−/− mice fed a HFD for 16wks (n=6).
(H) Western blots of proteins in fatty acid synthesis in livers of Cpt2lox/lox and Cpt2L−/− mice fed a HFD for 16wks. All blots were normalized to Hsc70.
(I) Total saponified fatty acid concentration in Liver and iWAT of Cpt2lox/lox and Cpt2L−/− mice fed a HFD for 16wks (n=5–6). See also Table S2.
(J) Enrichment of fatty acids by chain length in Liver and iWAT of Cpt2lox/lox and Cpt2L−/− mice fed a HFD for 16wks (n=5–6). See also Table S2.
Defects in mitochondrial oxidative metabolism can increase oxidative stress. Therefore, we determined markers of oxidative stress in Cpt2L−/− and control HFD-fed mice. The livers of Cpt2L−/− mice exhibited increased lipid peroxidation and decreased glutathione content indicative of increased oxidative burden (Figures 2D, 2E). Serum lipid peroxidation was not affected (Figure S2A). Consistent with an increase in oxidative stress, the Hsp27 protein, which is induced upon oxidative stress, was increased in Cpt2L−/− livers (Figure 2F). Next, we profiled genes involved in oxidative stress via PCR array. Although there were few significant transcriptional changes in genes that are involved in the oxidative stress pathway, we observed a substantial reduction in Scd1 expression (Figure S2B). In a larger cohort, we examined the expression of other genes in fatty acid biosynthesis. Cpt2L−/− livers showed a significant suppression of fatty acid biosynthetic genes Acly, Acc, Fasn, Srebp1 and Scd1 (Figure 2G). These changes were confirmed at the protein level for Acly, Acc, Fasn, and Scd1 (Figure 2H). Additionally, we assayed the expression of peroxisomal and autophagy related genes (Lee et al., 2014; Zhang et al., 2007). We observed small transcriptional changes in Cyp4a1, Lc3b and Gabarap1 (Figure S2C). To further understand the impact of a deficiency of fatty acid oxidation on fatty acid metabolism, we analyzed the fatty acid composition of Cpt2L−/− liver and iWAT. Cpt2L−/− liver exhibited a small increase in total fatty acids (Figure 2I, Table S2). Interestingly, the liver was enriched in medium chain fatty acids and depleted in C20–C22 length fatty acids (Figure 2J). Although we have not observed changes in very long chain fatty acid oxidation (C24:0) in Cpt2L−/− livers (Lee et al., 2016b), these data suggest that in the absence of mitochondrial oxidation, long chain fatty acid chain shortening in the peroxisome can be accelerated.
Although Cpt2L−/− livers exhibited increased oxidative stress and suppressed biosynthetic genes following chronic high fat feeding, the expression of mitochondrial proteins was not changed between Cpt2L−/− and Cpt2lox/lox mice (Figure 3A). On the other hand, genes involved in fatty acid oxidation, such as Acot1, Acot2, Acox1, Cpt1a, Acadm, Acadl, and Hadha, as well as canonical Pparα target genes Pdk4 and Ehhadh and stress-induced genes such as Atf3 were increased in Cpt2L−/− livers (Figure 3B) albeit to a lesser degree than fasting or a ketogenic diet (Lee et al., 2016b). In addition to the fatty acid oxidative genes being upregulated, there was an increase in mRNA expression of the hepatokines Igfbp1, Gdf15 and Fgf21 (Figure 3C). This was reflected in an increase in serum levels of Igfbp1, Gdf15 and Fgf21, suggesting that these hepatokines are contributing to physiological adaptations in response to a loss of mitochondrial fatty acid oxidation (Figure 3D). Due to the reported role of Igfbp1 and Fgf21 in bone we measured aspects of bone quality (Wang et al., 2015). The increase in these hepatokines did not affect long bone growth (Figure 1A, 1B) or bone structure (Figure S3). These data suggest that in response to the high lipid burden from a HFD, the liver is not only attempting to compensate for the loss of fatty acid oxidation by up-regulating oxidative programming, but also seeking to increase catabolism in peripheral tissues by way of secreted hepatokines. These data are consistent with a beneficial hormetic response.
Figure 3. Loss of hepatic fatty acid oxidation induces hepatokines.

(A) Western blot for mitochondrial complexes and Hsc70 in livers of Cpt2lox/lox and Cpt2L−/− mice fed a HFD for 16wks.
(B) Gene expression of fatty acid metabolism and Pparα target genes in livers of Cpt2lox/lox and Cpt2L−/− mice fed a HFD for 16wks (n=6).
(C) Liver mRNA of igfbp1, gdf15, and fgf21 in Cpt2lox/lox and Cpt2L−/− mice fed a HFD for 16wks (n=6).
(D) Serum concentrations of Igfbp1, Gdf15 and Fgf21 in Cpt2lox/lox and Cpt2L−/− mice fed a HFD for 16wks (n=6).
Data are expressed as mean ± SEM. *p<0.05; **p<0.01; ***p<0.001.
Loss of fatty acid oxidation suppresses hepatic gluconeogenesis
To probe further into the role of hepatic fatty acid oxidation in the metabolic alterations elicited by chronic high fat feeding, we performed 1H-NMR based steady-state metabolomics analysis on serum, liver and kidney of Cpt2L−/− and Cpt2lox/lox mice fed a HFD for 16wks. Consistent with the putative requirement of fatty acid oxidation on gluconeogenesis, the liver exhibited a >6-fold suppression in glucose as well as a suppression in many gluconeogenic substrates such as lactate, alanine and glutamine (Figure 4A). Consistent with an improvement in blood glucose, Cpt2L−/− serum showed a decrease in glucose and suppressed β-hydroxybutyrate (Figure 4B). These improvements were mirrored in the kidney metabolome (Figure 4C). These data suggest that the reduction in serum and hepatic glucose levels in Cpt2L−/− mice resulted from reduced hepatic gluconeogenesis. To investigate directly whether the loss of Cpt2 inhibited hepatic gluconeogenesis as expected by the steady-state data as well as its putative role, we traced the incorporation of 1-13C-lactate into glucose in 24hr fasted Cpt2L−/− and Cpt2lox/lox mice by a combination of NMR and mass spectrometry. 1-13C-Lactate carbon enrichment was observed in the third (C3) and fourth (C4) position of glucose as previously described (Wolfe, 1992). Comparison of 13C-Glucose chemical shifts with BMRB 13C glucose reference showed expected C enrichments in C3 and C4 of glucose and incorporation at C3 and C4 were lower in Cpt2L−/− mice compared to Cpt2lox/lox mice (Figure 4D). To further validate the NMR results, we profiled 1-13C-lactate-derived isotopologues of glucose (from m+0 to m+2) using HPLC/QTOF (Figure 4E). MS analysis of stable isotope distribution supported the NMR results; total glucose and the percentages of 1-13C-lactate derived carbons in glucose isotopologues were significantly lower in Cpt2L−/− mice. These data confirm the putative requirement for fatty acid oxidation on the de novo production of glucose in the liver.
Figure 4. Metabolic alterations upon the loss of hepatic fatty acid oxidation.

(E) 1H-NMR in liver of Cpt2lox/lox and Cpt2L−/− mice fed a high fat diet for 16 weeks (n=6).
(F) 1H-NMR in serum of Cpt2lox/lox and Cpt2L−/− mice fed a high fat diet for 16 weeks (n=6).
(G) 1H-NMR in kidney of Cpt2lox/lox and Cpt2L−/− mice fed a high fat diet for 16 weeks (n=6).
(H) NMR analysis of 1-13C-lactate incorporation into glucose in 24hr fasted mice (n=3).
(I) Isotopomer analysis of 1-13C-lactate incorporation into glucose in 24hr fasted mice (n=3).
(J) Total bile acids in serum of Cpt2lox/lox and Cpt2L−/− mice fed a HFD for 9wks (n=4–7).
(K) Bile acid metabolic genes in ileum of Cpt2lox/lox and Cpt2L−/− mice fed a HFD for 9wks (n=4).
Data are expressed as mean ± SEM. *p<0.05; **p<0.01; ***p<0.001.
Cpt2L−/− mice exhibit altered bile acid metabolism
One of the most surprising results from the metabolomics analysis was that glycine was significantly reduced in the livers of Cpt2L−/− mice measured via 1H-NMR (Figure 4A); however, serum and kidney glycine were significantly elevated (Figure 4B, 4C). Glycine is often referred to as a conditionally essential amino acid. That is, de novo production cannot keep up with demand because it is at the crossroads of many pathways. One potential sink for hepatic glycine is glutathione, which is indeed suppressed in HFD-fed Cpt2L−/− livers (Figure 2E). Another potential sink is in the production of bile acids and consistent with this notion, there was an increase in serum bile acids in HFD-fed Cpt2L−/− mice compared to Cpt2lox/lox mice (Figure 4F). Because of the increase in total bile acids, we examined bile acid metabolic genes in the liver and ileum to determine if the loss of fatty acid oxidation altered the transcriptional programming of bile acid metabolism. There were no significant changes in genes involved in bile acid metabolism in the livers of Cpt2L−/− and Cpt2lox/lox mice (Figure S4). Fgf15, a bile acid responsive gene derived from the ileum, has been shown to positively affect whole body energy expenditure (Potthoff et al., 2012). However, Fgf15 was suppressed in the ileum of Cpt2L−/− mice as well as other bile acid responsive genes such as ibabp (Figure 4G). These data suggest that while bile acid metabolism is altered in Cpt2L−/− mice, Fgf15 is likely not responsible for the resistance to HFD-induced obesity in this model.
Cpt2L−/− mice exhibit improved glucose tolerance and insulin sensitivity
Previously we have shown that Cpt2L−/− mice are able to maintain euglycemia during a 24hr fast (Lee et al., 2016b). Here we have shown that fatty acid oxidation is required for hepatic gluconeogenesis, and mice fed a HFD for 16wks exhibit marked improvements in glucose and insulin. To understand the role of hepatic fatty acid oxidation on systemic glucose metabolism, we first performed ipGTT on chow-fed 9wk old mice. Cpt2L−/− mice cleared blood glucose modestly more efficiently than Cpt2lox/lox mice during the ipGTT, suggesting that the mice were more insulin sensitive (Figure 5A). To further determine the role of hepatic fatty acid oxidation on gluconeogenesis and insulin sensitivity, we performed hyperinsulinemic-euglycemic clamp studies on Cpt2L−/− and control mice. As expected, these studies showed impaired endogenous glucose production during the hyperinsulinemic-euglycemic clamp (Figure 5B). These data demonstrate an increased ability of insulin to suppress endogenous glucose production in these mice (Figure 5B). However, insulin-stimulated glucose uptake in skeletal muscle and adipose were not affected (Figure S5). These data are consistent with the idea that inhibiting hepatic fatty acid oxidation and therefore hepatic gluconeogenesis may be sufficient to improve systemic glucose handling.
Figure 5. Loss of hepatic fatty acid oxidation improves glucose tolerance and insulin sensitivity.

(A) Intraperitoneal glucose tolerance test (ipGTT) including area under the curve in male Cpt2lox/lox and Cpt2L−/− mice fed a chow diet (n=8–11).
(B) Hyperinsulinemic-euglycemic clamp showing glucose infusion rate (GIR), endogenous glucose production (EGP) and insulin suppression of EGP (n=7–9).
(C) Body weight of male Cpt2lox/lox and Cpt2L−/− mice fed a high fat diet for 4 wks (n=11–12).
(D) ipGTT including area under the curve in male Cpt2lox/lox and Cpt2L−/− mice fed a high fat diet for 4 wks (n=11–12).
(E) Serum insulin levels measured at baseline and glucose stimulated (15min) conditions in Cpt2lox/lox and Cpt2L−/− mice fed a high fat diet for 4 wks (n=6).
(F) Intraperitoneal insulin tolerance test (ipITT) including area above curve in male Cpt2lox/lox and Cpt2L−/− mice fed a high fat diet for 5 wks (n=11–12).
Data are expressed as mean ± SEM. *p<0.05; **p<0.01; ***p<0.001.
Feeding a HFD to Cpt2L−/− mice for 16wks resulted in substantial improvements in glucose and insulin. However, the suppression in adiposity in Cpt2L−/− mice represents a significant confounder in those experiments. Therefore, we fed a new cohort of mice a HFD for 4wks, which is sufficient to generate glucose intolerance but before apparent changes in body weight or composition (Figures 1A, 1B, 4C). Cpt2L−/− mice exhibited an improvement in ipGTT compared to control mice fed a HFD for 4wks (Figure 5D). Also, basal and glucose-induced serum insulin was markedly improved in Cpt2L−/− mice (Figure 5E). Finally, Cpt2L−/− mice exhibited improved ipITT following the short-term HFD challenge (Figure 5F). Together these data support the idea that suppressed hepatic glucose production is sufficient to improve systemic glucose dysfunction.
The loss of hepatic fatty acid oxidation enhances whole body energy expenditure in response to high fat feeding
Decreased body weight, adiposity and increased systemic catabolism upon a HFD in Cpt2L−/− mice led us to determine whole body bioenergetics in Cpt2lox/lox and Cpt2L−/− mice. In our previous studies, we found no differences in metabolic parameters between 9wk-old Cpt2lox/lox and Cpt2L−/− mice fed a chow diet, even following a 24hr fast (Lee et al., 2016b). Again, because altered body weight and adiposity are major confounders to energy expenditure, we fed Cpt2lox/lox and Cpt2L−/− mice a HFD for 4wks before significant changes in body weight were observed. Cpt2L−/− mice exhibited an increase in energy expenditure in both the light and dark phases (Figure 6A, 6B). To determine if a loss of hepatic fatty acid oxidation resulted in increased brown or beige fat, we measured thermogenic genes in interscapular brown adipose tissue (iBAT) and inguinal white adipose tissue (iWAT). Cpt2L−/− iBAT showed no difference in the expression of these genes, and Cpt2L−/− iWAT exhibited a significant suppression of Ucp1, Dio2 and Fgf21 (Figure S6). These data suggest a brown/beige adipocyte-independent increase in energy expenditure. There was no change in body weight or lean mass, but Cpt2L−/− mice exhibited a small decrease in fat mass even at this time point (Figure 6B). Interestingly Cpt2L−/− mice also displayed a small increase in food intake; however, there was no change in the respiratory exchange ratio (Figure 6B), suggesting that between Cpt2lox/lox and Cpt2L−/− mice, there was no preference in whole body fatty acid or glucose utilization in a genotype-specific manner. These data suggest the increased energy expenditure could be driving the resistance to HFD-induced obesity seen in Cpt2L−/− mice.
Figure 6. Loss of hepatic fatty acid oxidation increases energy expenditure in response to high fat feeding.

(A) Energy expenditure under dark and light cycles of female Cpt2lox/lox and Cpt2L−/− mice after 4 wks on high fat diet (n=10–13).
(B) Metabolic parameters of female Cpt2lox/lox and Cpt2L−/− mice fed a HFD for 4wks (n=10–13).
Data are expressed as mean ± SEM. *p<0.05; **p<0.01; ***p<0.001.
Thermoneutral housing normalizes body weight gain and adiposity in Cpt2L−/− mice
We next sought to determine the contribution of the HFD-induced increase in energy expenditure in Cpt2L−/− mice to body weight gain and adiposity. Standard housing conditions (21°C) impose a mild cold stress to mice, thereby increasing their energy expenditure as well as food intake (Lee et al., 2016a). In order to suppress energy expenditure, we housed Cpt2lox/lox and Cpt2L−/− female mice in a thermoneutral environment (30°C) and fed a HFD for 16wks. Under standard housing conditions, Cpt2L−/− mice exhibited resistance to HFD-induced obesity (Figure 1A, 1A′); however, housing mice at a thermoneutral environment eliminated this difference (Figure 7A). Under standard housing conditions, female Cpt2L−/− mice exhibited a suppression in both iWAT and gWAT. The size of both of these depots were normalized at thermoneutrality while the increase in liver weight of Cpt2L−/− mice was not affected by ambient temperature (Figure 7B). Hepatic triglycerides and insulin were not changed between groups (Figure S7). Finally, we examined the effect of thermoneutrality on the hepatic genes that we have shown to be affected by a loss of Cpt2. There was an amelioration of hepatic Igfbp1 expression by thermoneutral housing, but Atf3, Gdf15 and Fgf21 continued to be upregulated in Cpt2L−/− livers regardless of ambient temperature. Pparα target genes in the liver, such as Pdk4, Acot1, Acot2 and Ehhadh, were suppressed by thermoneutral housing but were still increased in Cpt2L−/− livers compared to control (Figure 7C). These data suggest that the resistance to HFD-induced obesity in mice with a loss of hepatic fatty acid oxidation is mediated by an increase in systemic energy expenditure.
Figure 7. Loss of hepatic fatty acid oxidation at thermoneutrality normalizes body weight during high fat feeding.

(A) Body weight of female Cpt2lox/lox and Cpt2L−/− mice fed a high fat diet at thermoneutrality (30°C) for 16 weeks (n=11–12).
(B) Wet weights of iWAT and gWAT unilateral depots, liver (left lobe), kidney and heart of female Cpt2lox/lox and Cpt2L−/− mice fed a HFD for 16wks at 21°C (n=12–14) or 30°C (n=11–12).
(C) Gene expression in livers of female Cpt2lox/lox and Cpt2L−/− mice fed a HFD for 16wks at 21°C or 30°C (n=6).
Data are expressed as mean ± SEM. *p<0.05; **p<0.01; ***p<0.001.
DISCUSSION
The oxidation of long chain fatty acids in hepatocytes represents a major energetic pathway not only for the liver but also for the animal as a whole. Hepatic fatty acid oxidation is at the crossroads of important biological adaptations, particularly during times of increased lipid burden. Therefore, even small changes in hepatic fatty acid oxidation are often invoked as a mechanism to explain the phenotypes of models of obesity, diabetes and NAFLD. The appeal of this idea is that if a defect in hepatic fatty acid oxidation can exacerbate the metabolic syndrome then enhancing hepatic fatty acid oxidation could represent an effective therapy. In contrast to this rational extrapolation, mice with a hepatic-specific deficiency in mitochondrial fatty acid oxidation (Cpt2L−/− mice) are resistant to—instead of prone to—the major pathophysiologic features elicited by a HFD, including obesity and glucose intolerance, even though the mice have a genetically-imposed deficiency in hepatic energy expenditure.
The response of Cpt2L−/− mice to a HFD is similar to models of increased hepatic oxidative metabolism in general or fatty acid oxidation in particular (An et al., 2004; Orellana-Gavalda et al., 2011; Perry et al., 2013; Stefanovic-Racic et al., 2008). Although at face value these results are seemingly at odds, this biphasic response is most aptly defined as hormesis (Yun and Finkel, 2014). We have previously shown that Cpt2L−/− mice fasted for 24hr or fed a ketogenic diet enlist a host of adaptations in an attempt to maintain systemic energy homeostasis during these short-term challenges (Lee et al., 2016b). Here, we observe a similar phenomenon whereby the liver induces hepatokines in response to the chronic feeding of a lipid-rich diet. Whole body KO mice of Acadvl and a skeletal muscle-specific KO of Cpt1b are similarly resistant to HFD-induced sequelae (Vandanmagsar et al., 2016; Wicks et al., 2015; Zhang et al., 2010). Other models of impaired mitochondrial oxidative metabolism are also resistant to HFD-induced obesity and glucose intolerance, presumably via a hormetic response as well (Chung et al., 2017; Pospisilik et al., 2007). Similarly, part of the mechanism of action of the anti-diabetic drug metformin may be a mild inhibition of mitochondrial electron transport chain (Owen et al., 2000). The benefits of altered substrate utilization may be the generation of a stress response via an imbalance between substrate flux and cellular demand rather than an elimination of excess nutrients per se. Defining the similarities between a gain and loss of hepatic substrate oxidation may identify the most effective biological interventions to treat the metabolic syndrome.
The failure to suppress hepatic glucose production is a hallmark of insulin resistance and type II diabetes. Although the transcriptional and hormonal control of gluconeogenesis in hepatocytes are important regulators of hepatic glucose metabolism, the systemic regulation and delivery of substrates for hepatic gluconeogenesis represents another important mode of regulation. That is, the availability of substrates to generate intramitochondrial acetyl-CoA and NADH is sufficient to drive inappropriate glucose production (Perry et al., 2015; Titov et al., 2016). Defects in hepatic lipid metabolism and oxidative stress have been implicated in the etiology of insulin resistance. Our data suggest that the inability of Cpt2L−/− mice to support hepatic gluconeogenesis is sufficient to protect mice from glucose intolerance following a HFD by lowering fasting blood glucose even though the rate of glucose disposal is similar. Consistent with this idea are mice with a hepatic-specific deletion in glucose-6-phosphatase, which are resistant to HFD-induced obesity and glucose intolerance (Abdul-Wahed et al., 2014; Mutel et al., 2011). Additionally, the improvement in glucose handling in Cpt2L−/− mice that exhibit high hepatic oxidative stress suggest that hepatic oxidative stress is not sufficient to drive glucose intolerance and supports the idea that inhibiting gluconeogenesis is an effective strategy to improve glucose levels in diabetes. Finally, Cpt2L−/− mice are extremely carnitine-deficient, similar to human inborn errors of fatty acid oxidation. The liver is one of the major organs responsible for synthesizing carnitine, and previous studies have shown that carnitine supplementation is beneficial for glucose tolerance (Noland et al., 2009; Power et al., 2007; Ringseis et al., 2012). It is not clear if carnitine administration to people with diabetes will ultimately prove successful; however, our results show that carnitine deficiency does not contribute to insulin resistance in our model.
One of the most surprising results from the current study is that Cpt2L−/− mice do not exhibit potentiated hepatic steatosis following chronic high-fat feeding. Fasting or a ketogenic diet greatly increases fatty liver in Cpt2L−/− mice (Lee et al., 2016b). However, Cpt2L−/− mice fed a chronic HFD did not exhibit potentiated liver TAG. Fasting imparts an immediate delivery of free fatty acids from adipose tissue to the liver. Adipose lipolysis generates fatty acids in excess of need, and surplus fatty acids are repackaged and exported in lipoproteins through the circulation. The difference between fasting and dietary lipids may simply be the kinetics as the slower yet continuous delivery of dietary lipids may be exported efficiently to non-hepatic tissues. Humans with fatty liver disease do not exhibit suppressed fatty acid oxidation but instead have enhanced hepatic fatty acid oxidation, although it is not clear if the rate is still insufficient for supply (Koliaki et al., 2015).
In conclusion, we have generated mice with a hepatocyte-specific deficiency of mitochondrial long chain fatty acid β-oxidation. These mice are paradoxically resistant to HFD-induced obesity and glucose intolerance in the absence of exacerbated liver disease. Although Cpt2L−/− mice have several hallmarks of diabetic liver, including increased hepatic oxidative stress and systemic carnitine deficiency, they are protected from glucose intolerance. The notion of increasing substrate oxidation in brown adipose tissue, skeletal muscle or liver as a means of losing body weight may simply prove unrealistic in the face of complex systemic physiological adaptations (Lee et al., 2016a; Lee et al., 2015; Rodriguez et al., 2014; Vandanmagsar et al., 2016; Wicks et al., 2015). However, harnessing the systemic components responsible for hormesis may represent a path towards rational therapies for obese and diabetic patients.
EXPERIMENTAL PROCEDURES
Animals
Cpt2 lox/lox and Cpt2L−/− mice were previously described (Lee et al., 2016b). Mice were housed in ventilated racks with a 14hr-light/10hr-dark cycle and fed a standard chow diet (2018SX Teklad global). For diet studies, Cpt2 lox/lox and Cpt2L−/− mice were either fed a 60% high fat diet (D12492, Research Diets) or a low fat diet (D12450J, Research diets) starting at 6wks of age and continuing for 16wks. At the end of the diet study, fat and lean mass were measured via magnetic resonance imaging analysis (QNMR EchoMRI100; Echo Medical Systems). Analysis of metabolites were done for free glycerol and TAG (Sigma), β-hydroxybutyrate (StanBio), total cholesterol, NEFA (Wako), Bile acids (BioVision), TBARS and Glutathione (Cayman) and ALT activity (Sigma). Serum insulin (Crystal Chem), Fgf21, Gdf15, Igfbp1 and Adiponectin (R&D Systems) were measured by ELISA. Total saponified fatty acids were quantified by GCMS as we have previously described (Bowman et al., 2017; Reamy and Wolfgang, 2011). Acylcarnitines were quantified as previously described (Lee et al., 2016b). Urine acylcarnitine levels were normalized to creatinine levels in urine (Cayman). Glucose and insulin tolerance tests were performed as described (Lee et al., 2015). To determine whole body bioenergetics, mice were fed a 60% HFD at 9 weeks of age and continued for 4 weeks prior to the experiment. At 13 weeks of age, body fat and lean mass of Cpt2 lox/lox and Cpt2L−/− mice were determined via magnetic resonance imaging. Then these mice were then individually housed in Oxymax indirect calorimetry cages (Columbus Instruments). Indirect calorimetry and metabolic cage studies were normalized to lean mass as described (Ellis et al., 2013). Hyperinsulinemic-Euglycemic studies were performed as described (Kim, 2009). All procedures were performed in accordance with the NIH’s Guide for the Car and Use of Laboratory Animals and under the approval of the Johns Hopkins Medical School Animal Care and Use Committee.
NMR Spectroscopy
Steady-state 1H NMR Spectroscopy was done as previously described(Gonzalez-Hurtado et al., 2017; Selen et al., 2015). 13C NMR spectra were collected using a zgpg30 pulse program on a 500MHz Bruker Avance III (Bruker, Germany). Each sample was tuned, matched and shimmed using TopShim before the acquisition of the spectrum. 65536 data points were collected with spectra width of 29762 Hz. 16000 scans were collected for each 13C spectrum, with acquisition time of 1.1sec and a relaxation delay of 1.5 sec. Proton decoupling was performed using a standard WALTZ16 pulse sequence. Mestrenova was used to analyze 13C and 2D HSQC spectra. 1D 13C spectra were zero filled to 128K data points. 0.1mM dioxane was used as internal reference and the chemical shift was calibrated by referencing dioxane signal to 67.4 ppm. 1D 13C spectra were manually phase corrected and spline function was applied for the baseline correction (Mestrenova, Spain). The peak height of dioxane was used to normalize the peak height of the carbon signals of glucose. BMRB 13C glucose reference spectrum (500MHz, Bruker Avance) was used to profile the glucose peaks in the spectra. 2D 1H, 13C- heteronuclear single quantum coherence spectroscopy (HSQC) NMR spectra were acquired using 500MHz Bruker Avance. 2D data collected with 16 scans using Bruker pulse program ‘hsqcetgp’ 2D 1H, 13C-HSQC spectra were used for further verification of the peaks of interest in 13C spectra.
Steady-State Metabolite Analysis with HPLC-QTOF
Frozen 50–100mg Cpt2 lox/lox and Cpt2L−/− liver tissue were homogenized in 600ul ice-cold methanol and centrifuged at 12,000rpm for 15 minutes. Supernatant was transferred to a new tube and the pellet was extracted with methanol-water (80%–20%) followed by a second centrifuge. Supernatants were combined and left in a speed-vac for overnight drying. Pellet was used for protein quantification. Dried materials were reconstituted in 200ul acetonitrile-water (80%–20%). Chromatographic separation (HPLC) was performed with Supelco Discovery HS F5-3 column (15cm*2.1mm, 3um). The HPLC system (Agilent 1200 series) was operated at the flow rate 0.5 mL/min with solvent A (water with 0.1% formic acid), and solvent B (acetonitrile). The LC parameters were set as follows; auto sampler temperature 4C and injection volume 4ul. Gradient conditions were; 0–15min; 100% A, 15–18 min; 70% A, 19–26 min; 50% A, 27–35min; 0% A, 35.10–50 min; 100% A. Metabolomics experiment was performed on an Agilent QTOF LC/MS system (Agilent Tech 6520). Data were collected in negative mode. Data analysis performed using Agilent Profinder Software.
Real time PCR
RNA was isolated from all tissues using the RNeasy Mini Kit (QIAGEN). Using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems), we reverse transcribed 1–2μg of total RNA. The cDNA was diluted to 2 ng/μl and amplified by primers in a 20-μl reaction using SsoAdvanced SYBR Green Supermix (BioRad). We calculated mRNA using a 2^deltaCT relative to the average of the housekeeping genes cyclophilinA and rpl22 expression. All primers and gene information were previously reported (Lee et al., 2015) or provided in Table S1.
Western blots
Liver homogenates from 16-week HFD or acute insulin injected Cpt2 lox/lox and Cpt2L−/− mice were prepared using 1x RIPA buffer with protease and phosphatase inhibitors. The protein concentration was measured using the Pierce BCA Protein Assay kit (Thermo Scientific). We used 30μg of protein on an SDS-PAGE and then transferred it to a nitrocellulose (Protran BA 83, Whatman) membrane. We then blocked with 3% BSA-TBST (Tris-buffered saline with Tween20). The membranes were probed with antibodies Pcx (Cell Signaling), Acly (Cell Signaling), Total Acc (Cell Signaling), Fasn (BD Biosciences), Scd1 (Cell Signaling), Hsp27 (Cell Signaling), Mitoprofile (Abcam) and Hsc70 (Santa Cruz Biotechnology) diluted 1:1000 with 3% BSA in TBST. Hsc70 used the appropriate Cy3 fluorescent secondary antibody, and the other primary antibodies used corresponding secondary antibodies conjugated to horseradish peroxidase. Images were collected using an Alpha Innotech FluorChemQ and presented with minimal image processing.
Statistical analysis
Data were analyzed with the assistance of Prism. Significance was determined using an unpaired two-tailed t test for single variable experiments and two-way ANOVA with Bonferroni post hoc correction for multiple variable experiments.
Supplementary Material
Highlights.
Loss of hepatic fatty acid oxidation (FAO) confers resistance to obesity.
Loss of hepatic FAO results in beneficial hormesis.
The suppression of FAO is sufficient to improve diet-induced glucose intolerance.
Resistance to obesity results from increased energy expenditure.
Acknowledgments
We would like to thank Natasha Zachara for essential reagents, Susan Aja for metabolic cage studies and Caitlyn Bowman for helpful discussion. We would like to thank Ann Moser at the Kennedy Krieger Institute for assistance in GC/MS analysis of fatty acids. This work was supported in part by a National Institutes of Health grant R01NS072241 and American Diabetes Association grant #1-16-IBS-313 to M.J.W. The bone phenotyping was performed at the Baltimore Diabetes Research Center and funded by NIH grant DK079637. Part of this study was performed at the National Mouse Metabolic Phenotyping Center at UMass Medical School and funded by NIH grant U2C-DK093000.
Footnotes
Conflict of interest
The authors declare that they have no conflicts of interest with the contents of this article.
Author contributions
J.L., J.C, E.S.A. and R.C.R. conducted the experiments and analyzed the results. E.S.A. conducted the NMR experiments and analyzed the results. E.S.A, L.Z. and T.H. conducted the isotopologue labeling experiment. M.J.W. conceived the idea for the project. J.L. J.C. and M.J.W. wrote the paper with input and approval of all authors.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abdul-Wahed A, Gautier-Stein A, Casteras S, Soty M, Roussel D, Romestaing C, Guillou H, Tourette JA, Pleche N, Zitoun C, et al. A link between hepatic glucose production and peripheral energy metabolism via hepatokines. Mol Metab. 2014;3:531–543. doi: 10.1016/j.molmet.2014.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An J, Muoio DM, Shiota M, Fujimoto Y, Cline GW, Shulman GI, Koves TR, Stevens R, Millington D, Newgard CB. Hepatic expression of malonyl-CoA decarboxylase reverses muscle, liver and whole-animal insulin resistance. Nature medicine. 2004;10:268–274. doi: 10.1038/nm995. [DOI] [PubMed] [Google Scholar]
- Bowman CE, Rodriguez S, Selen Alpergin ES, Acoba MG, Zhao L, Hartung T, Claypool SM, Watkins PA, Wolfgang MJ. The Mammalian Malonyl-CoA Synthetase ACSF3 Is Required for Mitochondrial Protein Malonylation and Metabolic Efficiency. Cell Chem Biol. 2017 doi: 10.1016/j.chembiol.2017.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Iqbal N, Boden G. The effects of free fatty acids on gluconeogenesis and glycogenolysis in normal subjects. J Clin Invest. 1999;103:365–372. doi: 10.1172/JCI5479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung HK, Ryu D, Kim KS, Chang JY, Kim YK, Yi HS, Kang SG, Choi MJ, Lee SE, Jung SB, et al. Growth differentiation factor 15 is a myomitokine governing systemic energy homeostasis. The Journal of cell biology. 2017;216:149–165. doi: 10.1083/jcb.201607110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis JM, Wong GW, Wolfgang MJ. Acyl coenzyme A thioesterase 7 regulates neuronal fatty acid metabolism to prevent neurotoxicity. Mol Cell Biol. 2013;33:1869–1882. doi: 10.1128/MCB.01548-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Hurtado E, Lee J, Choi J, Selen Alpergin ES, Collins SL, Horton MR, Wolfgang MJ. The Loss Of Macrophage Fatty Acid Oxidation Does Not Potentiate Systemic Metabolic Dysfunction. Am J Physiol Endocrinol Metab. 2017 doi: 10.1152/ajpendo.00408.2016. ajpendo 00408 02016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerner J, Hoppel C. Fatty acid import into mitochondria. Biochim Biophys Acta. 2000;1486:1–17. doi: 10.1016/s1388-1981(00)00044-5. [DOI] [PubMed] [Google Scholar]
- Kim JK. Hyperinsulinemic-euglycemic clamp to assess insulin sensitivity in vivo. Methods in molecular biology (Clifton, NJ. 2009;560:221–238. doi: 10.1007/978-1-59745-448-3_15. [DOI] [PubMed] [Google Scholar]
- Koliaki C, Szendroedi J, Kaul K, Jelenik T, Nowotny P, Jankowiak F, Herder C, Carstensen M, Krausch M, Knoefel WT, et al. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab. 2015;21:739–746. doi: 10.1016/j.cmet.2015.04.004. [DOI] [PubMed] [Google Scholar]
- Lee J, Choi J, Aja S, Scafidi S, Wolfgang MJ. Loss of Adipose Fatty Acid Oxidation Does Not Potentiate Obesity at Thermoneutrality. Cell Rep. 2016a;14:1308–1316. doi: 10.1016/j.celrep.2016.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Choi J, Scafidi S, Wolfgang MJ. Hepatic Fatty Acid Oxidation Restrains Systemic Catabolism during Starvation. Cell Rep. 2016b;16:201–212. doi: 10.1016/j.celrep.2016.05.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Ellis JM, Wolfgang MJ. Adipose fatty acid oxidation is required for thermogenesis and potentiates oxidative stress-induced inflammation. Cell Rep. 2015;10:266–279. doi: 10.1016/j.celrep.2014.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JM, Wagner M, Xiao R, Kim KH, Feng D, Lazar MA, Moore DD. Nutrient-sensing nuclear receptors coordinate autophagy. Nature. 2014;516:112–115. doi: 10.1038/nature13961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashek DG. Hepatic fatty acid trafficking: multiple forks in the road. Adv Nutr. 2013;4:697–710. doi: 10.3945/an.113.004648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGarry JD, Brown NF. The mitochondrial carnitine palmitoyltransferase system. From concept to molecular analysis. Eur J Biochem. 1997;244:1–14. doi: 10.1111/j.1432-1033.1997.00001.x. [DOI] [PubMed] [Google Scholar]
- Mutel E, Gautier-Stein A, Abdul-Wahed A, Amigo-Correig M, Zitoun C, Stefanutti A, Houberdon I, Tourette JA, Mithieux G, Rajas F. Control of blood glucose in the absence of hepatic glucose production during prolonged fasting in mice: induction of renal and intestinal gluconeogenesis by glucagon. Diabetes. 2011;60:3121–3131. doi: 10.2337/db11-0571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noland RC, Koves TR, Seiler SE, Lum H, Lust RM, Ilkayeva O, Stevens RD, Hegardt FG, Muoio DM. Carnitine insufficiency caused by aging and overnutrition compromises mitochondrial performance and metabolic control. The Journal of biological chemistry. 2009;284:22840–22852. doi: 10.1074/jbc.M109.032888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orellana-Gavalda JM, Herrero L, Malandrino MI, Paneda A, Sol Rodriguez-Pena M, Petry H, Asins G, Van Deventer S, Hegardt FG, Serra D. Molecular therapy for obesity and diabetes based on a long-term increase in hepatic fatty-acid oxidation. Hepatology. 2011;53:821–832. doi: 10.1002/hep.24140. [DOI] [PubMed] [Google Scholar]
- Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J. 2000;348(Pt 3):607–614. [PMC free article] [PubMed] [Google Scholar]
- Perry RJ, Camporez JP, Kursawe R, Titchenell PM, Zhang D, Perry CJ, Jurczak MJ, Abudukadier A, Han MS, Zhang XM, et al. Hepatic acetyl CoA links adipose tissue inflammation to hepatic insulin resistance and type 2 diabetes. Cell. 2015;160:745–758. doi: 10.1016/j.cell.2015.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry RJ, Kim T, Zhang XM, Lee HY, Pesta D, Popov VB, Zhang D, Rahimi Y, Jurczak MJ, Cline GW, et al. Reversal of hypertriglyceridemia, fatty liver disease, and insulin resistance by a liver-targeted mitochondrial uncoupler. Cell metabolism. 2013;18:740–748. doi: 10.1016/j.cmet.2013.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pospisilik JA, Knauf C, Joza N, Benit P, Orthofer M, Cani PD, Ebersberger I, Nakashima T, Sarao R, Neely G, et al. Targeted deletion of AIF decreases mitochondrial oxidative phosphorylation and protects from obesity and diabetes. Cell. 2007;131:476–491. doi: 10.1016/j.cell.2007.08.047. [DOI] [PubMed] [Google Scholar]
- Potthoff MJ, Kliewer SA, Mangelsdorf DJ. Endocrine fibroblast growth factors 15/19 and 21: from feast to famine. Genes Dev. 2012;26:312–324. doi: 10.1101/gad.184788.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Power RA, Hulver MW, Zhang JY, Dubois J, Marchand RM, Ilkayeva O, Muoio DM, Mynatt RL. Carnitine revisited: potential use as adjunctive treatment in diabetes. Diabetologia. 2007;50:824–832. doi: 10.1007/s00125-007-0605-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reamy AA, Wolfgang MJ. Carnitine palmitoyltransferase-1c gain-of-function in the brain results in postnatal microencephaly. Journal of neurochemistry. 2011;118:388–398. doi: 10.1111/j.1471-4159.2011.07312.x. [DOI] [PubMed] [Google Scholar]
- Ringseis R, Keller J, Eder K. Role of carnitine in the regulation of glucose homeostasis and insulin sensitivity: evidence from in vivo and in vitro studies with carnitine supplementation and carnitine deficiency. Eur J Nutr. 2012;51:1–18. doi: 10.1007/s00394-011-0284-2. [DOI] [PubMed] [Google Scholar]
- Rodriguez S, Ellis JM, Wolfgang MJ. Chemical-genetic induction of Malonyl-CoA decarboxylase in skeletal muscle. BMC Biochem. 2014;15:20. doi: 10.1186/1471-2091-15-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel VT, Liu ZX, Qu X, Elder BD, Bilz S, Befroy D, Romanelli AJ, Shulman GI. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J Biol Chem. 2004;279:32345–32353. doi: 10.1074/jbc.M313478200. [DOI] [PubMed] [Google Scholar]
- Savage DB, Petersen KF, Shulman GI. Disordered lipid metabolism and the pathogenesis of insulin resistance. Physiol Rev. 2007;87:507–520. doi: 10.1152/physrev.00024.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selen ES, Bolandnazar Z, Tonelli M, Butz DE, Haviland JA, Porter WP, Assadi-Porter FM. NMR Metabolomics Show Evidence for Mitochondrial Oxidative Stress in a Mouse Model of Polycystic Ovary Syndrome. J Proteome Res. 2015;14:3284–3291. doi: 10.1021/acs.jproteome.5b00307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soling HD, Willms B, Friedrichs D, Kleineke J. Regulation of gluconeogenesis by fatty acid oxidation in isolated perfused livers of non-starved rats. Eur J Biochem. 1968;4:364–372. doi: 10.1111/j.1432-1033.1968.tb00220.x. [DOI] [PubMed] [Google Scholar]
- Stefanovic-Racic M, Perdomo G, Mantell BS, Sipula IJ, Brown NF, O’Doherty RM. A moderate increase in carnitine palmitoyltransferase 1a activity is sufficient to substantially reduce hepatic triglyceride levels. Am J Physiol Endocrinol Metab. 2008;294:E969–977. doi: 10.1152/ajpendo.00497.2007. [DOI] [PubMed] [Google Scholar]
- Szendroedi J, Phielix E, Roden M. The role of mitochondria in insulin resistance and type 2 diabetes mellitus. Nat Rev Endocrinol. 2011;8:92–103. doi: 10.1038/nrendo.2011.138. [DOI] [PubMed] [Google Scholar]
- Titov DV, Cracan V, Goodman RP, Peng J, Grabarek Z, Mootha VK. Complementation of mitochondrial electron transport chain by manipulation of the NAD+/NADH ratio. Science. 2016;352:231–235. doi: 10.1126/science.aad4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandanmagsar B, Warfel JD, Wicks SE, Ghosh S, Salbaum JM, Burk D, Dubuisson OS, Mendoza TM, Zhang J, Noland RC, et al. Impaired Mitochondrial Fat Oxidation Induces FGF21 in Muscle. Cell Rep. 2016;15:1686–1699. doi: 10.1016/j.celrep.2016.04.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Wei W, Krzeszinski JY, Wang Y, Wan Y. A Liver-Bone Endocrine Relay by IGFBP1 Promotes Osteoclastogenesis and Mediates FGF21-Induced Bone Resorption. Cell Metab. 2015;22:811–824. doi: 10.1016/j.cmet.2015.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicks SE, Vandanmagsar B, Haynie KR, Fuller SE, Warfel JD, Stephens JM, Wang M, Han X, Zhang J, Noland RC, et al. Impaired mitochondrial fat oxidation induces adaptive remodeling of muscle metabolism. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:E3300–3309. doi: 10.1073/pnas.1418560112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe RR. Radioactive and Stable Isotope Tracers in Biomedicine: Principles and Practice of Kinetic Analysis. Wiley; 1992. [Google Scholar]
- Yun J, Finkel T. Mitohormesis. Cell Metab. 2014;19:757–766. doi: 10.1016/j.cmet.2014.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Christianson J, Liu ZX, Tian L, Choi CS, Neschen S, Dong J, Wood PA, Shulman GI. Resistance to high-fat diet-induced obesity and insulin resistance in mice with very long-chain acyl-CoA dehydrogenase deficiency. Cell Metab. 2010;11:402–411. doi: 10.1016/j.cmet.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Liu ZX, Choi CS, Tian L, Kibbey R, Dong J, Cline GW, Wood PA, Shulman GI. Mitochondrial dysfunction due to long-chain Acyl-CoA dehydrogenase deficiency causes hepatic steatosis and hepatic insulin resistance. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:17075–17080. doi: 10.1073/pnas.0707060104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.