

Summary
Human breast cancers that exhibit high proportions of immune cells and elevated levels of proinflammatory cytokines predict poor prognosis. Here, we demonstrate that treatment of human MCF-7 breast cancer cells with pro-inflammatory cytokines results in ERα-dependent activation of gene expression and proliferation, in the absence of ligand or presence of 4OH-tamoxifen (TOT). Cytokine activation of ERα and endocrine resistance is dependent on phosphorylation of ERα at S305 in the hinge domain. Phosphorylation of S305 by IKKβ establishes an ERα cistrome that substantially overlaps with the estradiol (E2)-dependent ERα cistrome. Structural analyses suggest that S305-P forms a charge-linked bridge with the C-terminal F domain of ERα that enables inter-domain communication and constitutive activity from the N-terminal coactivator-binding site, revealing the structural basis of endocrine resistance. ERα therefore functions as a transcriptional effector of cytokine-induced IKKβ signaling, suggesting a mechanism through which the tumor microenvironment controls tumor progression and endocrine resistance.
eTOC Blurb
Stender et al. show that inflammatory cytokines activate unliganded ERα though kinase-dependent phosphorylation. This phosphorylation causes structural changes on ERα that lead to transcriptional activation, endocrine resistance, and enhanced invasiveness of breast cancer cells.
Introduction
Approximately 75% of breast tumors express estrogen receptor alpha (ERα), and breast cancer patients with ERα+ tumors generally receive endocrine therapy targeting either estrogen (E2) production with aromatase inhibitors or ER activity with Selective ERα Modulators (SERMs) such as TOT. Unfortunately up to 50% of breast cancer patients will fail endocrine treatments, resulting in a recurrent, endocrine-resistant tumor (Clarke et al., 2015). Several mechanisms have been proposed to contribute to an endocrine-resistant state, including activation of growth factor and kinase pathways (e.g., Her2/neu, MAPK), amplification of transcriptional co-activator proteins (e.g., SRC3), mutations in the ligand-binding domain of ERα, mutations in enzymes that convert TOT to its active metabolite, and constitutive activation of other transcription factors such as NF-κB (Musgrove and Sutherland, 2009).
ERα is a member of the nuclear receptor superfamily, which has the prototypical domain structure, lettered A–F domains (Figure S1A), of a centrally located DNA binding domain, a carboxy-terminal ligand binding domain (LBD) containing a protein interaction site called activation function-2 (AF2), and an amino-terminal transcriptional activation function domain (AF1)(Carson-Jurica et al., 1990). ERα can be activated through ligand binding to the LBD or through kinase-dependent phosphorylation in multiple domains (Bruce et al., 2014). Upon activation, ERα recruits transcriptional co-activators and corepressors, components of the basal transcriptional machinery, and the RNA polymerase II complex to regulatory regions of target genes (Métivier et al., 2003). These regulatory sites typically contain either a full or half estrogen response element (ERE) at distant enhancers, or occur through protein-protein interactions with other transcription factors including FOXA1 (Hurtado et al., 2011), AP1 (Kushner et al., 2000), Sp1 (Porter et al., 1997), RUNX1 (Stender et al., 2010) and p65/RELA (Pradhan et al., 2012).
The presence of tumor-associated macrophages (TAMs) in breast tumors is positively correlated with poor prognosis and low survival rates (Leek et al., 1996). Current concepts posit that during tumor initiation, activated macrophages create an inflammatory environment that is mutagenic and promotes tumor growth. As tumors progress to malignancy, macrophages exhibiting features of so-called alternative activation stimulate angiogenesis, enhance tumor cell migration and invasion, and suppress anti-tumor immunity (Chanmee et al., 2014). Proinflammatory cytokines, including interleukin 1 beta (IL1β) and tumor necrosis factor alpha (TNFα) are released from innate immune cells such as macrophages, and increase the invasiveness and metastasis of ER+ breast cancer cells, while their levels correlate with increasing disease severity (Baumgarten and Frasor, 2012). Further, these cytokines activate NF-κB (Oeckinghaus et al., 2011), which is associated with failure of both endocrine and chemotherapies (Sas et al., 2012). Repressing kinases upstream of NF-κB activity can restore sensitivity to ERα antagonists in cell-based models of resistance (deGraffenried et al., 2004; Zhu et al., 2006). Endocrine resistance from inflammatory signaling could thus occur through kinasemediated phosphorylation events that directly control ERα activity, and/ or through genomic cross-talk from ERα interaction with RelA/p65 NF-κB, which can occur at both ERE enhancers and inflammatory promoters, and depending on the context lead to either repression or activation (Franco et al., 2015).
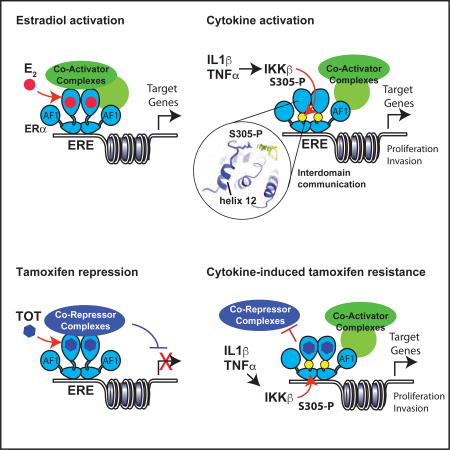
Defining the structural mechanisms for endocrine resistance has been hampered by a lack of understanding of how the ligand is “read” by amino acids in the LBD pocket and how this chemical information is transduced into biological signals involving multiple domains of ERα. With E2 bound ERα, the steroid receptor coactivators, SRC1/SRC2/SRC3 can bind independently to AF1 and AF2 (Webb et al., 1998), bridging an inter-domain interaction that enhances activity (Figure 1A) (Metivier et al., 2001). Proximally to the ligand, the last helix in the LBD, helix 12, can adopt different ligand-induced conformations to control protein recruitment to the AF2 surface, including coactivators and corepressors (Figure 1A–B) (Shang and Brown, 2002; Shiau et al., 1998). However, with TOT, agonist activity is through recruitment of coactivators to Nterminal AF1 (Figure 1C) (McInerney and Katzenellenbogen, 1996; Nwachukwu et al., 2016a) and it is also controlled by the C-terminal F domain (Montano et al., 1996), a flexible ~50 amino extension past helix 12. While the competition between coactivators and corepressors has been well studied with respect to TOT activity (Fig 1B), the structural basis for these interdomain signaling circuits is unknown.
Figure 1. Inflammatory cytokines regulate E2-dependent target genes through ERα.

(A) Diagram of the hormone activation conformer of ERα where both AF1 and AF2 contribute to gene activation.
(B) Diagram of TOT-bound ERα actively repressing transcription via recruitment of corepressors to multiple domains of ERα.
(C) Diagram of TOT- bound ERα activating gene expression through coactivator recruitment to AF1.
(D) Heat map for mRNA-Seq expression of the 801 transcripts regulated in MCF7 cells treated with E2, IL1β, or TNFα. The mRNA expression is shown for MCF7 cells treated with Veh, E2, E2 + ICI, IL1β, IL1β + ICI, TNFα, and TNFα + ICI for 3 hours.
(E) Gene ontology analysis for genes that are regulated by E2, IL1β, and TNFα.
(F) Cancer cell extravasation assay using the CAM assay. MCF7 WT and MCF7 ERα null cells were treated with Veh, IL1β or TNFα. Values are expressed as mean ± SEM. *p<0.05, (Student’s t-test) compared to MCF7 WT ERα cells.
(G) QPCR analysis for MYC mRNA in MCF7 cells treated with siRNA for control (CTL) or ERα (ESR1) and stimulation with vehicle (Veh), E2, IL1β or TNFα. Values are expressed as mean ± SEM.*p<0.05 compared to siCtl, E2. **p<0.05 compared to siCtl, IL1β. ***p<0.05 compared to siCtl, TNFα.
(H) MCF-7 cells were transferred to steroid free media and treated with a dose curve of TOT and assayed for proliferation after 5 days. Mean +/−SD (N=4).
(I) MCF-7 cells were treated with a dose curve of IL1β + TOT and assayed for proliferation after 5 days. Mean +/− SD (N=4).
Given the essential nature of the ERα and NF-κB signaling pathways in the development and progression of breast cancer and the presence of IL1β and TNFα in breast tumors, we sought to identify the global impact of IL1β and TNFα on ERα-dependent transcription and the ERα and NF-κB p65 cistromes in breast cancer cells. We discovered that pro-inflammatory cytokine treatments activate the unliganded and TOT-bound ERα through an IKKβ-dependent phosphorylation of ERα at S305, and this is sufficient to drive ERα-dependent proliferation. Structural analyses demonstrate that phosphorylation of S305 widens the AF2 surface to permit coactivator binding, explaining how phosphorylation activates the apo-ERα in the absence of an agonist through both AF1 and AF2. However, TOT binding induces a helix-12 conformation that occludes the AF2 surface and in doing so brings the F domain near the hinge domain. Our structural model suggests S305-P forms hydrogen bonds with the F domain that stabilize helix-12 in this conformation, block AF2 interaction with both coactivators and corepressors, and enable constitutive activity through AF1 and endocrine resistance. Importantly, cytokine treatment remodels ERα cistromes largely independently of NF-κB, revealing that in this context ERα drives an integral ― and independent — transcriptional response pathway to cytokine-induced IKKβ signaling to control breast cancer proliferation.
Results
Inflammatory cytokines regulate E2-dependent target genes through ERα
To define the transcriptional outcomes of activating hormone-dependent and pro-inflammatory signaling pathways in human breast cancer cells, we performed deep sequencing (mRNA-Seq) of polyadenylated mRNA isolated from MCF-7 cells treated with 10 nM estradiol (E2), 10 ng/ml TNFα, or 10 ng/ml IL1β for 3 hours. From two replicate samples, we identified ~800 mRNAs that are significantly stimulated (2 fold, p<0.05) by either E2, IL1β, or TNFα treatments (Figure 1D). Unsupervised k-means clustering of the expression data partitions the genes into two main clusters: (1) genes activated by E2 treatments, and (2) genes activated by the cytokine treatments (Figure 1D). The E2-regulated cluster can be further broken into a cluster of genes selectively regulated by E2 treatment, and genes regulated by E2 and one or both of the proinflammatory cytokines. Of the E2 regulated genes, approximately 22% were induced > 2-fold by one or both cytokines (Figure S1B). The proportions of E2-induced genes induced by cytokines were similar using cutoffs of 1.5-fold and 4-fold induction (not shown), indicating that this relationship is not dependent on specific thresholds of gene activation. To define mRNAs in which inflammatory signaling activates gene expression through ERα-dependent mechanisms, we incubated MCF-7 cells with the ERα full antagonist and ERα degrader, Fulvestrant (ICI 182,780, ICI), for 24 hours prior to E2 or cytokine treatments (Wijayaratne and McDonnell, 2001). ICI treatment blocked induction of nearly all (269/285) of the E2-regulated genes, while also inhibiting 10% (41/401) of all IL1β targets and 8.5% (44/512) of the TNFα targets (Figure 1D and S1B–C). Nearly all of the cytokine-induced genes that were sensitive to ICI treatment were also induced by E2 in an ICI-sensitive manner (Figure 1D). Gene ontology analysis for the E2-regulated genes demonstrates an enrichment for genes associated with early E2 response organ morphogenesis, ion transport, epithelial cell proliferation, while the inflammatory cytokine gene signature was enriched for inflammatory response, epithelial mesenchymal transition, cell migration and invasion, and the early E2 response (Figure 1E).
To determine if IL1β and TNFα treatments confer invasive qualities onto wild-type MCF-7 cells, cancer cell extravasation rates, a key step of the metastatic cascade, were measured in vivo using the chorioallantoic membrane of chick embryos (Kim et al., 2016). Consistent with the gene ontology analysis, IL1β and TNFα treatments increased the extravasation of wild-type MCF-7 cells, while knockout of the ERα using CRISPR-Cas9 technology significantly decreased the cytokine-dependent invasion phenotype in MCF-7 cells in the chick chorioallantoic membrane assay (Figure 1F). To further confirm the requirement of ERα we targeted it with two specific siRNA oligos, which compromised the ability of E2, IL1β, or TNFα to stimulate Myelocytomatosis Viral Oncogene Homolog (MYC) gene expression in MCF-7 cells (Figure 1G) or mRNAs for Peptidoglycan Recognition Protein 2 (PGLYRP2), and Transforming Growth Factor α (TGFα)(Figure S1D). With TOT treatment, 10 nM was sufficient to inhibit growth of MCF-7 cells (Figure 1H). Remarkably, less than 1 ng/ml of I L1 β is sufficient to drive proliferation of MCF-7 cells in the presence of TOT, while higher doses of IL1β stimulated robust growth (Figure 1I). Collectively these results demonstrate that stimulation of at least a subset of the common mRNA transcripts by inflammatory cytokines occurs through hormone-independent activation of the ERα.
Inflammatory cytokines activate the ERα cistrome
To understand how cytokines regulate ERα interaction with DNA and whether it requires crosstalk with NF-κB binding, we performed chromatin immunoprecipitation (ChIP) assays coupled with next-generation sequencing (ChIP-Seq) for ERα and p65 in MCF-7 cells. We observed ~15,000 ERα peaks from two experiments in MCF-7 cells treated with E2, IL1β, or TNFα(Figure 2A – B). ERα was recruited to ~90% of these genomic locations in the presence of E2 but nearly 40% of these sites in the presence of cytokines, indicating considerable overlap between E2-driven and cytokine-driven ERα cistromes (Figure 2A–B and S2A–B). We performed Global Run-on sequencing (GRO-Seq) and identified significant enhancer activity at the strongest 10% of ERα binding sites (200/2,180) in MCF-7 cells treated with E2 (p<2.8E-44), IL1β (p<2.9E-13), and TNFα (p<1.5E-10) compared to vehicle treatment (Figure 2C), indicating cytokines not only influence ER recruitment, but also enhance transcriptional activity at specific enhancer regions. In order to understand epigenetic features at E2 preferential and common ERα enhancers, we analyzed available datasets for chromatin accessibility and active chromatin features (He et al., 2012; Li et al., 2013). We found that compared to E2-preferential ERα binding sites, the common enhancers have significantly higher basal enhancer accessibility, as indicated by DNase I hypersensitivity (p<1.5E-119), H3K4me2 (p<2.0E-92), and H3K27ac (p<9.8E-172) (Figure 2D–E).
Figure 2. Inflammatory cytokines activate the ERα cistrome.

(A) Heat map for 15,213 ERα ChIP-Seq peaks in MCF-7 cells treated with vehicle, E2, IL1β, TNFα.
(B) Venn diagram of ERα cistrome in MCF-7 cells treated with E2, IL1β, and TNFα.
(C) Boxplot of the GRO-Seq signal at distal top 10% of E2-ERα binding sites in MCF-7 cells treated with Veh, E2, TNFα, and IL1β.
(D) Boxplot for DNase hypersensitivity (GSE33216) at E2 preferential ERα peaks and E2+Cytokine ERα peaks.
(E) Boxplot for H3K4me2 (top, GSE33216) or H3K27ac (bottom, GSE45822) at E2 preferential ERα peaks and E2+Cytokine ERα peaks.
(F) Venn diagram for the ERα and p65 cistromes in MCF-7 cells treated with E2 or IL1β.
(G) Top, De novo motif analysis for the E2 preferential cistome (n=8,296) identified in figure 2A, or bottom, for ERα binding sites identified in the presence of E2 and either TNFα, IL1β, or both TNFα and IL1β treatments (n=5,442).
Cytokine-induced ERα cistrome is largely NF-κB independent
To determine whether this cytokine-driven ERαa cistrome was through p65-mediated tethering to NF-κB response elements (κBRE), we performed ChIP-seq for p65 in MCF-7 cells. Minimal overlap was observed with E2 treatment as ERα and p65 shared only 294 peaks, which accounts for only 2% of the total ERα binding in response to E2. (Figure 2F). However, the IL1β or TNFα treatments resulted in ER/p65 overlap at 1,436 (23% of the IL1β-dependent ER peaks) and 687 (23.5% of TNFα-dependent ER peaks) genomic locations, respectively (Figure 2F and S2C).
Motif analyses identified an enrichment of NF-κB response elements (κBRE) with cytokine, but not E2-induced cistromes (Figure 2G). Of the sites co-bound by ERα and p65, 20% contain both and ERE and κBRE motifs, 9% contain a κBRE only, further supporting that the cytokine-driven ERα cistrome is largely NF-κB independent. To further investigate whether p65 recruitment is necessary for recruitment of ERα, we compared the IL1(β-dependent ERα ChlP-Seq profiles at genomic locations lacking nearby p65 and at genomic locations exhibiting co-recruitment of p65 within 200 bp. This analysis indicates that ERoc is recruited more robustly to places of co-occupancy with p65, but also demonstrates that co-occupancy with p65 is not required for ERα binding (Figure S2D). Furthermore, IL1(β-induced binding of ERα at genomic locations lacking nearby p65 binding is associated with a similar increase in eRNA, as is observed at ERα binding sites with nearby p65 (Figure S2E). Therefore, while NF-κB likely facilitates ERα binding at a subset of its cistrome, IL1(β and TNFα treatments induce ERα binding and transcriptional activity at thousands of locations in the genome without a requirement for co-recruitment of the NF-κB member, p65.
Phosphorylation of ERα at S305 is required for cytokine-dependent ERα activation
To identify phosphorylation events that might activate ERα in the absence of hormone, we compared ERα-S118, an ERK target (Gori et al., 2011), and ERα-S305 phosphorylation, known to induce TOT resistance by PKA (Michalides et al., 2004). We observed robust phosphorylation of ERoc at S305 upon IL1(β and TNFα treatments, but not E2 treatment (Figure 3A). To define a role for S305 in the MCF7 cellular background context, we utilized CRISPR-Cas9 technology to knockout the endogenous ERα protein (Figure 3B) by targeting the ESR1 genomic locus with three sgRNAs, and then subsequently expressed either the WT or the S305A mutant ERα at near endogenous levels (Figure 3B). Treatment of the WT and S305A expressing MCF-7 cells with E2 induced the mRNA of three target genes identified from our RNA-seq data, PGLYRP2, MYC, and TGFA (Figure 3C). In contrast, IL1β and TNFα treatments induced the mRNA expression of these target genes in WT expressing cells, but not in S305A expressing cells. Therefore, S305 is required for inflammatory cytokines to activate endogenous gene expression. To further support a role for S305 in cytokine-mediated ERα activation, IL1β, but not E2, treatment of MCF-7 cells results in recruitment of phosphorylated ERα-S305 to the PGLYRP2, MYC and TGFA enhancers (Figure 3D). Together, these data indicate that cytokinedependent ERα transcriptional activation and genomic recruitment requires phosphorylation of ERα at S305.
Figure 3. Inflammatory cytokine treatments increase S305 phosphorylation on ERα.

(A) Western blot analysis for ERα-S118-P, ERα-S305P, and ERα in MCF-7 cells treated with Veh, E2, IL1β or TNFα.
(B) Western blot analysis for ERα and Histone H3 in MCF-7 cells (ERα+), CRISPR-cas9 knock out ERα MCF7 cells (ER-), and CRISPR-cas9 ESR1 MCF7 cells transfected with WT or S305A.
(C) QPCR analysis for the indicated mRNAs in MCF7 cells expressing WT or S305A estrogen receptors treated with Veh, E2, IL1β or TNFα. Values are expressed as mean ± SEM.*p<0.05, (Student’s t-test) compared to MCF7 WT ERα cells.
(D) ChIP-PCR of ERα-S305p recruitment in MCF-7 cells to the indicated genomic locus in the presence of Veh, E2 or IL1β. Values are expressed as mean ± SEM.*p<0.05, (Student’s t-test) compared to Veh sample.
IKKβ phosphorylates ERα at S305 in response to cytokine treatments
To define potential kinases responsible for the cytokine-dependent phosphorylation of S305, we treated MCF-7 cells with IL1β in the presence of inhibitors for Protein Kinase A (PKA) and p21-activated Kinase 1 (PAK1), both kinases known to phosphorylate ERα at S305 (Michalides et al., 2004; Rayala et al., 2006) and Inhibitor of Nuclear Factor κB kinase (IKK), a kinase robustly activated by TNFα and IL1β treatments. IL1β-dependent S305 phosphorylation was not affected by PAK1 inhibition, but was partially reduced with the PKA inhibitor, and significantly reduced with IKK inhibition (Figure 4A – B). As the IKK inhibitor targets both IKKα and IKKβ, we next treated MCF-7 cells with siRNA oligonucleotides specific for either IKKα or IKKβ, and assessed whether IL1β-dependent phosphorylation of S305 was affected. Knockdown of IKKβ, but not IKKα, eliminated the IL1β-dependent phosphorylation of ERα at S305 (Figure 4C). Since IL1β treatments specifically recruited ERα phosphorylated at S305 to genomic loci (Figure 3D), we tested whether inhibition of IKK signaling would impair the IL1β dependent ERα cistrome. Indeed, treatment of MCF-7 cells with the IKK inhibitor reduces the overall IL1β-dependent ERα cistrome by approximately 80% (Figure 4D). The reduction of IL1β-dependent ERα recruitment in the presence of IKK inhibition was confirmed in independent chromatin immunoprecipitation experiments at the PGLYRP2, MYC, and TGFA genomic loci (Figure 4E and Figure S3A). Furthermore, treatment of MCF-7 cells with the IKK inhibitor reduced IL1β-dependent activation of PGLYRP2, MYC and TGFA mRNA (Figure 4F and S3B). The IKK inhibitor also abolished the ability of TNFα or IL1β to prevent TOT-dependent repression of both MYC and PGLYRP2 mRNA expression (Figure 4G and S3C) in MCF7 cells expressing WT ERα. Therefore, inhibiting the activity of IKK, and subsequently blocking the cytokine-dependent phosphorylation of ERα-S305, restores TOT sensitivity even in the presence of inflammatory cytokines.
Figure 4. IKKβ phosphorylation of S305 ERα is required for cytokine-dependent ERα activation.

(A) Western blot analysis for ERα-S305-P and ERα in MCF-7 cells treated with Veh, or IL1β in the absence or presence of inhibitors for PKA, PAK1and IKKα/β.
(B) Quantification of two independent western blot analysis as performed in figure 4A. Values are expressed as mean ± SEM.*p<0.05, (Student’s t-test) compared to IL1β sample.
(C) Western blot analysis for ERα-S305-P and total ERα in MCF-7 cells treated with siRNA for Ctl, IKKα, or IKKβ and treated with Veh or IL1β.
(D) Histogram of ERα ChIP-Seq signal in the presence of Veh, IL1β, IKK7, or a combination ofIL1β andIKK7.
(E) ChIP-PCR of ERα recruitment to the MYC genomic locus in the presence of Veh, IL1β, or a combination of IL1β and IKK7. Values are expressed as mean ± SEM. *p<0.05, (Student’s t-test) IL1β + IKK7 compared to IL1β sample.
(F) Quantitative real-time PCR data for MYC mRNA in MCF-7 cells treated with Veh, IL1β, or a combination of IL1β and IKK7. Values are expressed as mean ± SEM.*p<0.05, (Student’s t-test) IL1β +IKK7 compared to IL1β sample.
(G) Same as F, but cells were also treated with TOT or TNFα, as indicated.
Inflammatory cytokines induce TOT resistance through S305 phosphorylation
To define the mechanism through which the NF-κB signaling pathway induces endocrine resistance in ER+ breast cancer we examined the roles of ERα and S305. IL1β treatment prevented the effects of TOT in MCF7 WT ERα cells, while not affecting the anti-proliferative effects of TOT in MCF7 cells expressing the S305A mutation (Figure 5A). To assess the global role of pro-inflammatory cytokines on TOT-dependent repression of ERα activity, we performed RNA-Seq on mRNA isolated from MCF7 cells treated with vehicle, E2, E2+TOT and E2+TOT with the addition of either IL1β or TNFα for 3 hours. Of the 285 E2-regulated transcripts identified in Figure 1C, we discovered that 71% (202/285) were inhibited more than 50% upon addition of TOT (Figure 5B – D and S4A). Remarkably, addition of either IL1β or TNFα prevented TOT repression on 85% (171/285) of the TOT-sensitive genes (Figure 5B–C). In MCF7 cells expressing WT ERα, the target genes PGLYRP2 and MYC were activated by E2 and repressed upon addition of TOT (Figure 5D–E and S4B), while IL1β and TNFα overrode TOT suppression of these genes. As with proliferation, the pro-inflammatory cytokines did not relieve TOT-dependent repression of ERα target genes in MCF7 cells engineered to only express the ERα-S305A mutation (Figure 5E, S4B). IL1β was also sufficient to drive ERα to the MYC gene in the presence of TOT and stimulate recruitment of SRC3 and Pol II (Figure 5F), effects also found at the PGLYRP2 and TFGA genes (Figure S4C). This suggests a mechanism by which cytokine control of ERα genomic location, coactivator recruitment, and induction of proliferative genes such as MYC drives TOT resistance.
Figure 5. Phosphorylation of ERα at S305 is required for cytokine-dependent ERα activation.

(A) Cell proliferation assay for MCF7 cells expressing either WT or S305A estrogen receptors. Cells were treated with Veh, E2, E2 + TOT, E2 + TOT + IL1β for 6 days. Values are expressed as mean ± SEM. *p<0.05, (Student’s t-test) compared to MCF7 WT ERα cells.
(B) Heat map for mRNA-Seq expression of the 202 transcripts regulated in MCF7 cells treated with E2 that are sensitive to TOT. The mRNA expression is shown for MCF7 cells treated with Veh, E2, E2 + TOT, E2 + TOT + IL1β, and E2 + TOT + TNFα.
(C) Pie graphs showing the percentage of E2 regulated genes sensitive to TOT treatment. The genes sensitive to TOT treatments are further stratified based on whether addition of IL1β or TNFα, restores expression mRNA levels to near E2 induced levels.
(D) QPCR data for MYC mRNA in MCF-7 cells expressing either ERα or ERα-S305A treated with Veh, E2, E2 + TOT, and E2+ TOT+ IL1β. Values are expressed as mean ± SEM. *p<0.05, (Student’s t-test) ERα compared to ERα S305 sample.
(E) UCSC genome browser image for mRNA-Seq expression at the MYC genomic locus.
(F) ChIP-PCR of ERα, Pol II, or SRC3 recruitment to the MYC genomic locus in the presence of Veh, E2, I L1 β, and TOT, or E2 + TOT + IL1β. Values are expressed as mean ± SEM. *p<0.05, (Student’s t-test) compared to Veh treatment. #p<0.05, (Student’s t-test) compared to E2 treatment.
Structural basis for S305-mediated control of ERα activity
S305 lies in the hinge domain PTM cassette, 299KRSKKNS305 that is subject to acetylation, sumoylation, ubuiquitination at the lysine residues (Cui et al., 2004; Sentis et al., 2005). Phosphorylation at S305 blocks acetylation of K303, while K302 and K303 control receptor degradation (Berry et al., 2008). This region of the hinge domain, just before helix 1 in the LBD, has been disordered in crystal structures to date. However, helix 1 is located adjacent to the AF2 surface, which is important for both agonist and SERM activity. Thus S305 could modulate AF2 structure to control co-regulator binding. After solving more than 150 crystal structures of the ERα-LBD bound to different ligands (Nwachukwu et al., 2016b), we obtained only one that showed clear electron density for the PTM cassette 299KRSKKNS305 main chain, and the side chains starting at Ser301. The ERα-LBD was crystallized with the LxxLL coactivator peptide and the ligand, (8R,9S,13S,14S,17S)-13-methyl-17-(phenylamino)-7,8,9,11,12,13,14,15,16,17-decahydro-6H–cyclopenta[a]phenanthren-3-ol, a phenylamine-derivative of estradiol that we call PA-E2. The structure was solved by molecular replacement with data extending to 2.2 Å (PDB:5U2B, Table 1). However, this was also first time we have observed that the LxxLL coactivator peptide was absent despite being in the crystallization buffer, raising the question of whether the PTM cassette conformation was responsible for rejection of the coactivator peptide.
Table 1.
Crystallographic Data Collection and Refinement Statistics
| PA-E2 | OBHS | |
|---|---|---|
| Data Collection | ||
| Wavelength (Å) | 1.0331 | 1.0750 |
| Space group | P 1 | P 1 21 1 |
| Cell dimensions | ||
| a, b, c (Å) | 60.58, 64.44, 135.41 | 54.70, 81.36, 58.36 |
| α, β, γ (°) | 83.11, 75.20, 61.99 | 90.00, 111.07, 90.00 |
| Resolution range (Å) | 2.22 (2.30 – 2.22)* | 1.86 (1.87–1.86) |
| Rmerge | 0.028 (0.383) | 0.050 (0.739) |
| I/σ(I) | 18.40 (2.31) | 18.0 (2.2) |
| Completeness (%) | 97.75 (91) | 96.9 (96.4) |
| Multiplicity | 2.0 (2.0) | 3.6 (3.6) |
| Refinement | ||
| No. of reflections | 84,038 (8,089) | 38,270 (1,946) |
| Rwork / Rfree | 0.2192 / 0.2447 | 0.169 / 0.215 |
| No. of atoms | 11,791 | 4,435 |
| Protein | 10,800 | 3,984 |
| Ligands | 133 | 104 |
| Water | 858 | 347 |
| B-factors | ||
| Protein | 52.72 | 45 |
| Ligands | 40.7 | 26.3 |
| Water | 49.17 | 44.5 |
| R.m.s. deviations | ||
| Bond lengths (Å) | 0.002 | 0.01 |
| Bond angles (°) | 0.58 | 0.98 |
The hinge domain PTM cassette was ordered across the top of the LBD on the opposite side from the ligand, binding across h10 and the h3-h5 3-turn loop, which forms part of the AF2 surface (Figure 6A–B). The PTM cassette docking site on the ERα LBD corresponds to an allosteric control site in the androgen receptor, called binding function-3 (BF-3), where small molecule ligands inhibit activity by altering the shape of the AF2 surface (Figure 6C) (Estébanez-Perpiñá et al., 2007). ERα-S305 was buried adjacent to the AF2 surface, forming a tight 2.4 Å hydrogen bond with D369 in the h3-h4 loop. (Figure 6B). K302 lay across the surface between h4 and h10, and made a 3.3 Å salt bridge with E471, whose side chain is typically disordered, and with the backbone carbonyl of D369 on the h3-h4 loop (Figure 6B). E471 is further stabilized by a hydrogen bond with T371 on helix 5 located on the opposite side from the AF2 surface (Figure 6B). K303 extended away from the surface, forming a weak salt bridge with E470 (Figure 6B). Importantly, phosphorylation of S305 would require the PTM cassette to rotate into solution, also breaking the electrostatic interactions with K302 and K303.
Figure 6. S305 ERα modulates the AF2 surface.

(A) Crystal structure of the PA-E2-bound ERα LBD showing helix-1 (h1) and the PTM cassette of the hinge domain (residues 299-310) in coral, the PTM cassette-interacting region of h10 in yellow, and the coactivator-binding site, AF2 in cyan.
(B) A closer view of panel A showing the network of hydrogen bonds (green dashed lines) that reinforces LBD interaction with the PTM cassette of the hinge domain.
(C) Structure of the testosterone-bound AR LBD in complex with a BF3-binding compound (PDB 2YLP, PMID: 22047606). The hinge domain-interacting region of the ERα (panel B) corresponds to the BF3-binding site of AR.
(D) Hinge domain interaction with the LBD alters the AF2 surface. Structures of the PA-E2-bound ERα LBD with (cyan) or without (gray) an SRC2 peptide (green) were superposed.
(E) U2OS cells were transfected with either WT-ERα or ΔAB-ERα, lacking AF-1, and a 3xERE-luc reporter. The next day cells were treated with dose curves of TOT or ICI. Data is mean ± SEM, n=3
(F) Transient transfection in MCF7 ER- cells with WT or ΔAF1 estrogen receptors and 3ERE-Luiciferase, and treated with Veh, E2, IL1β, E2 + TOT, E2 + TOT + IL1β. Values are expressed as mean ± SEM. *p<0.05, (Student’s t-test) compared to MCF7 WT ERα cells.
S305 displays bi-directional signaling with AF2
To understand the impact of the PTM cassette binding to BF-3, we soaked SRC2/ERα-LBD crystals with PA-E2 and obtained a 2.4 Å structure (PDB: 5TLD) as part of a meta-analysis of 142 structures (Nwachukwu et al., 2016b). It is notable that in the presence of the peptide, the PTM cassette was disordered, as was the side-chain of E471. Superpositioning of the structures revealed that E471 in structure 1, which again was stabilized by K302 (Figure 6B), induced a slight shift in T371 and more dramatic rotation of h5 (Figure 6B). In structure 2, V376 in helix 5 forms close Van der Waals interactions with L690 and L694 in the SRC2 peptide. However, the rotation of h5 in structure 1 repositioned V376 to clash with SRC2 (Figure 6D). Thus interaction of the PTM cassette with BF-3 inhibits coactivator binding through allosterically narrowing the AF2 surface, suggesting that phosphorylation of S305 activates apo-ERα by widening the AF2 surface. Further, this communication is bi-directional, as this model suggests coactivator binding drives the PTM cassette off of BF-3 and into solution. Importantly, we previously demonstrated that phosphorylation of S305 by PKA enhances E2-mediated interaction of ERα with the fragment of SRC3 that binds AF2 (Likhite et al., 2006). However, in the in vitro binding assay there was no interaction of the SRC3 fragment with apo ERα, suggesting that AF1 may also play a role. Further, TOT agonist activity is mediated by solely through AF1 in various cell types and in vivo (Figure 6E and S5A) (McInerney and Katzenellenbogen, 1996; Nwachukwu et al., 2016a). To test the domain requirements for cytokine-mediated activation of ERα we transfected MCF-7 ERα(-) cells with WT or domain mutant ERα, ΔAF1 (Figure 6F). IL1β-dependent TOT resistance required an intact AF-1, shown by the reduced activation with the ΔAB-ER. With apo-ERα there was less difference between WT and ΔAB-ER, suggesting that both AF1 and AF2 contribute to IL1β-induced activity. These results demonstrate that endocrine resistance derives from inter-domain communication (Figure 6F) that is controlled by S305-ERα phosphorylation (Figure 5).
Structural basis of S305-mediated control of tamoxifen agonist activity
How AF1 is regulated by TOT in the LBD and S305 in the hinge domain is not known. The LBD contains a molecular switch that controls recruitment of coactivators and corepressors to the LBD (Figure 7A – B). When bound to agonists, helix-12 is stabilized to form one side of AF2 allowing coactivator recruitment (Figure 7A)(Shiau et al., 1998). A repressive conformer was visualized by deleting helix 12, revealing a longer groove that accommodates the three helical turn CoRnR motif found in SMRT and NCoR corepressors (Figure 7B)(Heldring et al., 2007), which we call the CoRnR groove. This binding interaction is conserved, as seen in the PPARα/SRMT structure, where h12 docks loosely to the side of the CoRnR groove to allow corepressor binding (Xu et al., 2002), demonstrating that helix-12 must be displaced from the CoRnR groove for active repression.
Figure 7. S305 ERα controls tamoxifen resistance through inter-domain communication.

(A) Surface of the ERα LBD bound to an agonist, with h12 colored blue, and an LxxLL peptide from SRC2 colored yellow. 3UUD.PDB
(B) The ERα LBD Δh12 shown as surface bound to a phage display derived corepressor peptide, colored red.
(C) Model of S305-P making a salt bridge to R548 was generated from the TOT structure, 3ERT.pdb using VMD, as described in the methods.
(D) The phosphate was removed from S305 in the model generated in Figure 7C, and both models were used for molecular dynamics simulations.
(E) The S305 and S305-P models were subject to steered molecular dynamics simulations, where an increasing force was applied to h12 to pull it off of the CoRnR groove.
(F) Transient transfection in MCF7 ER- cells with WT or F domain mutant estrogen receptors and 3ERE-Luiciferase, and treated with Veh, E2, IL1β, E2+TOT, E2+TOT+IL1β. Values are expressed as mean ± SEM. *p<0.05, (Student’s t-test) compared to Veh treatment
(G) Structure of ERα with h12 trapped in the conformer seen with TOT. The CoRnR groove is shown as gray ribbons and h12 is shown as cyan α-carbon trace. Two point mutants are shown. From the PDB_REDO (Joosten et al., 2014) version of 3os8.pdb.
(H) HepG2 cells transfected with a 3xERE-driven luciferase reporter and ERα WT or Leu372Ser/Leu536Ser expression plasmids, were stimulated with TOT or ICI.
(I) Model of full-length ERα showing how p-305 orients the F domain towards the hinge, DNA – binding domain (DBD), and AF1.
(J) Structure of the ERα-LBD showing how the agonist conformer orients the beginning of the F-domain (PDB: 4ZNY).
Importantly, TOT, and all other SERM/ERα structures with intact helix-12 show that the ligand side chain flips helix-12 onto the CoRnR groove (Shiau et al., 1998), blocking both corepressors and coactivators to the LBD (Fig. 7C), leading to the question of how TOT can regulate AF1 activity. AF1 was discovered because it is constitutively active when fused to Gal4 DNA binding domain (Tora et al., 1989), demonstrating that the LBD is required for active repression of AF1. This led us to the hypothesis that what we have been calling the antagonist conformation with helix-12 in docked in the CoRnR groove (Brzozowski et al., 1997) is in fact the SERM resistance conformer, or SERM agonist conformer. In this conformer, S305-P is adjacent to the LBD/F domain juncture, a site that includes three basic residues in amino acids 547–550 just after helix-12, suggesting that S305-P forms a salt bridge with these amino acids in the F-domain, stabilizing helix-12 to block both coactivator and corepressor binding to AF-2, and enabling constitutive AF-1 activity. Thus we are hypothesizing that the structural models shown in Figure 7A – C drive the molecular circuits depicted in Figure 1A–C, respectively.
Using the TOT structure as a starting model, we performed molecular dynamics (MD) simulations with ERα amino acids 298–554 and phosphorylated S305, and identified a potential electrostatic interaction with R548 (Figure 7C). We then deleted the phosphate and compared the two models in MD simulation, but this did not show differences in fluctuation of helix-12 (Figure 7D). However, we observed significant differences in steered molecular dynamics, which measured the work required to pull h12 off of the surface, demonstrating that S305-P does indeed stabilize h12 docking into the CoRnR groove to block binding of corepressors and coactivators (Figure 7E). To validate this model further, we mutated H547, R548, and H550 to alanine, the amino acids in the F domain in proximity to the hinge (Figure 7C), which we hypothesize to form stabilizing salt bridges with S305. While the WT ERα showed TOT agonist activity in presence of IL1β, the triple mutant was only activated by E2, and TOT was now repressive in the presence of IL1β (Fig. 7F), demonstrating that the salt bridge between the hinge and F domains was required for TOT resistance.
To test more generally whether the conformer seen in the published SERM crystal structures and Figure 7C represents the SERM agonist conformer, we introduced two point mutants that stabilize helix-12 docked into the CoRnR groove. These mutations are used for improving crystallization by limiting conformational heterogeneity (Bruning et al., 2010; de Savi et al., 2015). Mutation of L372 to Arg or Ser was rationally designed to add a hydrogen bond between helix-12 and helix-5. Though the mutant L372S side chain was disordered in our published structures, we did visualize S372R forming a salt bridge to D545 in helix-12 (Figure 7G). L536S shows the serine h-bonding with the amide backbone of h12, a helix-capping interaction that facilitates h12 stability (Figure 7G). We previously demonstrated that in cells these conformational trapping mutants block NCoR corepressor binding to ERα (Bruning et al., 2010), as predicted by our model. To test whether this mutant also induced agonist activity, we tested it in MCF-7 ERKO cells (Figure 7H) and HepG2 cells (Figure S5B), which display TOT agonist activity. By transfecting a ranging quantity of plasmid amounts, we show that the conformational trapping mutant ERα L372S/L536S stimulated activity with vehicle and TOT, but responded normally to E2 (Figure 7H). Importantly, the increased activity was not evident with the ΔAB-ERα (Figure S5C), demonstrating that the conformer seen in SERM crystal structures and Figure 7C represents the endocrine resistance conformation. This supports the model that the molecular circuit shown in Figure 1B is determined by helix-12 docking in the SERM agonist conformer to block corepressors and allow AF1 activity.
These data suggest structural models for how the ligands control AF1 and the F domain, and how S305 phosphorylation impacts this process. H12 is in a dynamic equilibrium between different conformers, and when in the SERM agonist conformer, the F domain is extended towards the hinge, DNA binding domain and AF1, adjacent to the PTM cassette in the hinge, including S305 (Figure 7I). We also obtained structures of the LBD where we also visualized the beginning of the F domain in the agonist conformation, where it docked against helix-11 in the other subunit of the dimer (Figure 7J and S5D) (PDB: 4ZN9 and 5U2D). P552 is located just after h12, and docked against helix-11 N523 and H524 in the dimer partner (Figure S5E). The proline induced a sharp turn so that the following amino acids are directed near each other, in a parallel orientation facing directly away from the hinge/DBD/AF1 domains. These results suggest that the distinct helix-12 conformers mediate ligand- or phosphorylation-driven transitions in inter-domain communication. The full agonist helix-12 conformer, as part of the AF2 surface, promotes F domain–LBD interactions across ERα dimer partners. In contrast, the SERM agonist helix-12 conformer, docked in the CoRNR groove, promotes F domain–hinge domain interactions within ERα subunits, blocks coregulator recruitment to the LBD, and enables AF1 activity.
Discussion
Here, we evaluated the broad impact of IL1β and TNFα on global transcriptional responses of human MCF-7 cells and the binding and activity of ERα. We provide compelling evidence that ERα acts as an independent effector of the cytokine-mediated transcriptional response, accounting for ~10% of the overall transcriptional programs induced by IL1β and TNFα. We further demonstrate that cytokine-dependent activation of ERα is driven largely by ERα-S305 phosphorylation by IKKβ, an upstream activator of NF-κB transcription. Importantly, these effects were sufficient to induce growth of breast cancer cells in the absence of E2, conditions mimicking those of aromatase-inhibitor treatment, endocrine therapy that blocks production of E2, and antagonized the anti-proliferative effects of TOT.
Nuclear receptors (NRs) function as allosteric scaffold proteins, where the different domains comprise a molecular circuit board (Good et al., 2011) such that PTMs or binding events with ligands, DNA, and other proteins alter binding activity at distal sites in the receptors. This circuitry enables the defining signaling features of NRs, including tissue-, pathway-, and gene-selective outcomes, and a full range of graded transcriptional responses by different ligands. While AF-1 and AF-2 can synergize by interacting independently with the same coregulator complexes (Kraus et al., 1995; Metivier et al., 2001; Webb et al., 1998) (Figure 1A), here we define how SERMS such as TOT are “read” by ERα as an agonist (Figure 1C and Figure 7C). While the antagonist conformation was originally defined as helix-12 docking into the CoRnR groove to block coactivator binding (Brzozowski et al., 1997; Shiau et al., 1998), we show that it represents the SERM agonist or SERM resistant conformer by virtue of blocking all protein interactions, including corepressors, and enabling constitutive AF1 activity (Figure 7B–C). There is a dynamic equilibrium between the states 1) AF2-off/AF1-on versus 2) AF2-actively repressive/AF1-off, which drive selectivity as tissue-specific coactivator and corepressor complexes compete for binding (Figure 1B – C), and suggesting one mechanism for how amplification and overexpression of coactivators is oncogenic in breast cancer by driving the AF1-on state (Good et al., 2011). In addition to modulation by coregulator levels, we now show that inflammatory cytokines alter this equilibrium through ERα-S305 phosphorylation to form a salt bridge with basic residues at the LBD/ F domain interface, associated with recruitment of SRC3 to proliferative genes such as MYC. This suggests that the F domain may play a role in inter-domain communication, but also acts to keep helix-12 docked in the SERM agonist conformer in the context of IKKβ activity. Thus the transcriptional response integrates multi-domain signals from PTM status, ligand-specific conformers, and the ensemble of coregulators to drive cytokine driven-endocrine resistance.
Importantly, S305 phosphorylation also drives TOT resistance in response to other kinase and growth factor signaling pathways, suggesting a common structural mechanism for treatment resistance (Bentin Toaldo et al., 2015; Bostner et al., 2010). Our work suggests a two-step model for developing inhibitors for treatment resistant disease, where efficacy requires that the SERM side chain 1) destabilize helix 12 from the CoRnR grove, and 2) interact with and promote corepressor binding. This can be visualized in structures, where raloxifene directly contacts and slightly shifts helix-12 compared to TOT-ERα (Brzozowski et al., 1997; Shiau et al., 1998), explaining its lower uterotrophic activity. However, the raloxifene side chain also directly contacts the CoRnR box motif in that structure (Heldring et al., 2007). This competition between helix-12 and corepressor explains why such small changes to the side chain produce widely different agonist/antagonist profiles during medicinal chemistry campaigns (Grese et al., 1997) as the ligand—and S305 phosphorylation—modulate the ratio of helix-12/corepressor binding to the CoRnR groove to determine the agonist/antagonist profile of the ligand.
IL1β treatment, but not E2 treatment, specifically recruits the ERα-S305-P species to ERα enhancer regions (Figure 3D), demonstrating that it can also regulate sequence specific DNA binding. Previous studies have established that phosphorylation of ERα-S305 is sufficient for ERα to interact with the coactivator, SRC1, which contributes to a TOT-resistant state in breast cancer cells (Michalides et al., 2004; Zwart et al., 2007). Further, cytokine-dependent ERα recruitment appears to take advantage of open chromatin, which may be driven by differences in conformational states of ERα as well as the associated coregulators. Importantly, phosphorylation of S305 would be expected to have some overlapping functions driven by IKKB or PKA, but occur in very different signaling and transcriptional contexts, such as activation of NF-κB versus CREB, which is downstream of β-adrenergic/PKA signaling. The increased invasiveness of cytokine-treated cancer cells is for example a defining phenotype of NF-κB signaling, and may require both ERα- and NF-κB-mediated transcription.
While >75% of cytokine-induced ERα-mediated DNA binding events appear to be NF-κB independent, several studies have demonstrated that molecular crosstalk between the E2 and NF-κB signaling pathways are important for breast cancer. Classically, the convergence of these two pathways promotes protein-protein interactions between ERα and NF-κB family members, resulting in mutual trans-repression of target gene expression (Bodine et al., 1999; Feldman et al., 2007; Nwachukwu et al., 2014), which has been linked to gender-based repression of liver cancer, where ERα can repress the tumorigenic effects of IL6 (Naugler et al., 2007) For several genes, we have also shown that ERα and the NF-κB factor, p65, work cooperatively to up-regulate genes associated with tumor aggressiveness (Franco et al., 2015; Pradhan et al., 2012). Thus the important roles of inflammation in regulating reproductive physiology occur at multiple levels, with IKKβ-mediated phosphorylation of ERα defining an important new component that is coopted in cancer to drive endocrine resistance.
STAR Methods
Cell culture and transient transfections
MCF-7, U2OS, and HEPG2 cells were cultured as previously reported (Stender et al., 2007). Cells were cultured in minimal essential media (MEM) containing 5% calf serum with antibiotics. Prior to treatments, cells were cultured in phenol-red free media containing 5% charcoal-dextran stripped calf serum for at least 3 days. The ERα S305A and F-Domain mutants were generated using standard site-directed mutagenesis protocols. For transfections, cells were plated in 48-well plates and transfected when approximately 80% confluent. Transfections were performed with 0.2 µg of the 3ERE-TK-luciferase, and 0.2 µg ERα expression vectors using X-tremeGene reagent (Roche). After incubation for 8 h at 37° C in a 5% CO2 incubator, the cells were washed one time with medium containing 5% charcoal dextran-treated calf serum and then replaced with 1 ml medium plus serum. Cells were treated with the indicated ligand or 0.1% ethanol control for 24 h at 37° C and cell lysates were then harvested using reporter lysis buffer (Promega) and analyzed using the Luciferase Assay system (Promega) on a MLX Microtiter Plate Luminometer (Dynex Technologies).
Quantitation of cancer cell extravasation
Cells were plated to reach 70% confluency and then were pre-treated with IL1β (1 µM final), TNFα (0.1 nM final), or vehicle (DMSO) overnight. After pre-treatment, cells were washed with PBS, trypsinized, and diluted in PBS to reach a concentration of 1E6 cells/mL. Cells were injected into the chorioallantoic membrane of embryonic day 13 (D13) chick embryos and cancer cell extravasation rates were quantified as described previously (Kim et al., 2016). For each cell line and respective treatment (N>8/cell line + treatment), >200 cells were enumerated at T=0 hrs post injection within a user-defined field of view (FOV) which was placed on the CAM of each embryo. At T=24 hrs post-injection, the number of cancer cells that had extravasated into the stromal space of the CAM within each FOV was enumerated. Extravasation rates (%) were calculated by: extravasated cells within the FOV at T=24 hrs/all cells present in the FOV at T=0 hrs.
Proliferation
Cell proliferation assays were performed using Cell Titer Non-Radioactive Cell Proliferation Assay (Promega) as before (Stender et al., 2007). Cells were incubated in phenol-red media containing 5% charcoal-dextran stripped calf serum for at least 3 days prior to treatments and MTT incorporation was assessed after 6 days. In cases of co-stimulation of cells with IL1β and TOT, a 30 minute incubation with IL1β was necessary for the observed effects.
Western blot analysis
Cell protein lysates were separated in SDS polyacrylamide gels and transferred to nitrocellulose membranes. Blots were incubated in blocking buffer (5% milk in Tris buffered saline with 0.5% Tween) and then with specific antibodies for ERα (sc-543, Santa Cruz Biotechnology), ERα-S118-P (2511, Cell Signaling), ERα-S305-P (05–922R, Millipore), Histone H3 (Active Motif, 39763) followed by detection using horseradish peroxidase-conjugated secondary antibodies with Supersignal West Femto Detection Kit (Pierce), as described by the manufacturer.
RNA purification and quantitative real-time PCR
RNA was purified and quantified as previously described (Stender et al., 2007). RNA was either reverse transcribed into cDNA for quantitative real-time PCR using gene-specific primers or used for next-generation library preparation. The primer sequences used in this study are supplied as a supplemental table.
siRNA transfections
MCF-7 cells were plated at a density of 150,000 cells/well in 6 well plates in phenol-red free media. Two days later cells were transfected with 50 nM siRNA for control, ERα, IKKα, or IKKβ using 4uL Dharmafect (Dharmacon) per well. Transfections were incubated for 48 hours before being treated with indicated treatments. Oligonucleotides were ordered from Dharmacon: IKKα (MU-003473-02-0002), IKKβ (MU-003503-0-0002) and ERα: Oligo1 (UCAUCGCAUUCCUUGCAAA), Oligo2 (UUUGUUACUCAUGUGCCUGAT).
Generation of engineered MCF7 cells using CRISPR-CAS9
Three CRISPR-Cas9 guides targeting the open reading frame of the human ESR1 genomic locus were cloned into the pLentiCRISPRv2 (Addgene 52961) after BSMBI digestion. The three guide sequences are as follows: Guide1: GTAGACCTGCGCGTTGGCGG, Guide 2: GTCGCCTTTCCTGCAGCCCCA, Guide 3: GCACCATTGATAAAAACAGG. Lentivirus pools for all three guides were generated using 3rd generation lentiviral systems and MCF7 cells were transduced for 48 hours prior to puromycin selection. After two weeks of puromycin selection, the knockdown efficiency was assessed by Western blot analysis using an ERα specific antibody. These cells were then transduced with lentivirus preps for LVX-hESR1-P2A–mRuby made with either WT or S305A estrogen receptors and mRuby positive cells were sorted using conventional cell sorting techniques to generate cell populations expressing only the WT or S305A estrogen receptors.
Chromatin immunoprecipitation assays
Chromatin immunoprecipitation assays were performed as described before with some revisions (Stender et al., 2010). MCF-7 cells were crosslinked with 2 mM disuccinimidyl glutarate for 30 minutes prior to 10 minutes of 1% formaldehyde. The antibodies used in these studies were: ERα (sc-543, Santa Cruz Biotechnologies); p65 (sc-372, Santa Cruz Biotechnology); RNA Pol II (sc-900, Santa Cruz Biotechnology); SRC3 (sc-9119, Santa Cruz Biotechnology); and ERα-S305-P (05–922R, Millipore). For the precipitations protein A Dynabeads (10003D, Invitrogen) were coated with antibody prior to pulldown and excess antibody was washed away. Pulldowns occurred while rotating for 16 hours at 4C. Beads were then washed with with TSE I (20 mM Tris/HCl pH 7.4@20°C, 150 mM NaCl, 0.1% SDS, 1% Triton X-100, 2 mM EDTA), twice with TSE III (10 mM Tris/HCl pH 7.4@20°C, 250 mM LiCl, 1% IGEPAL CA-630, 0.7% Deoxycholate, 1 mM EDTA), and twice with TE followed by elution from the beads using elution buffer (0.1 M NaHCO3, 1% SDS). Elutions were subsequently de-crosslinked overnight at 65° C and DNA was purified using ChIP DNA Clean and Concentrator (Zymo Research) and DNA was either used for qPCR or prepared into libraries for high-throughput sequencing. The primer sequences used in this study are supplied as a supplemental table.
Preparation of next-generation sequencing libraries
Libraries were prepared from 2–3 biological replicates per condition. Global run-on, RNA-Seq, and ChIP-Seq were prepared as previously described (Kaikkonen et al., 2013). Sequencing libraries were prepared using magnetic beads similar to described previously using barcoded adapters (NextFlex, Bioo Scientific)(Garber et al., 2012).
High throughput sequencing and analysis
ChIP fragments were sequenced for 36 or 50 cycles on an Illumina Genome Analyzer II or HiSeq 2000, respectively, according to the manufacturer’s instructions. RNA-Seq and GRO-Seq results were trimmed to remove A-stretches originating from the library preparation. Each sequence tag returned by the Illumina Pipeline was aligned to the hg19 assembly using ELAND allowing up to 2 mismatches. Only tags that mapped uniquely to the genome were considered for further analysis. Peak finding and downstream analysis was performed using HOMER, a software suite created for analysis of highthroughput sequencing data (Heinz et al., 2010). Detailed instructions for analysis can be found at http://biowhat.ucsd.edu/homer/. Data visualization was performed using Microsoft Excel, JavaTreeGraph and software packages available in R. Gene ontology analysis was performed using Metascape.org (Tripathi et al., 2015).
Protein expression
Human ERα-Y537S LBD amino acids 298–554 in the MCSG7 vector was expressed in E. coli as a fusion with 8xHis and TEV cleavage site. A glycerol stock was used to streak LB agar plates, and the next morning, the colonies were washed and collected into a starter culture or dispensed directly into 2 liter soda bottles containing 1 liter of LB/ampicillin and shaken at 37°C. When the optical density of the culture reached ~0.6, the temperature was lowered to 26°C and 1 mM IPTG was added. After 6 hours the cultures were collected on ice. Note that protein induction at lower temperature overnight did not produce protein that yielded crystals, which we attribute to a small amount of truncated protein. The bacterial pellet was lysed by sonication. The protein was induced in BL21 (DE3) cells, and purified with immobilized nickel affinity chromatography. The eluted protein was mixed with a 1:30 ratio (by weight) of his-tagged TEV protease and dialyzed overnight in 20 mM Tris pH 8, 50 mM NaCl, 50 mM β-mercaptoethanol, and 10% glycerol. The next day, the solution was passed through nickel-NTA beads (Qiagen) to remove uncut ERα, the cut tags, and the TEV protease. The flow-through was diluted 2× in H2O and subjected to ion exchange chromatography with a Q-FF column (GEHealth). The protein was eluted in 175–185 mM NaCl, 20 mM Tris pH 8.0, 50 mM β-mercaptoethanol, and 10% glycerol, and then concentrated to 0.3 mM (10 mg/ml). The concentrated protein was aliquoted and mixed with 1 mM ligands and 1–2 mM GRIP peptide, and incubated overnight. The next day, the protein-ligand slurries were centrifuged at maximum speed for 15 minutes in a 4 °C microcentrifuge, and the supernatant used to set up crystal trials.
Crystallization
The LBD solution (1 mM) was mixed with 1.5 mM receptor-interacting peptide of steroid receptor coactivator 2 (SRC2) and either PA-E2, OBHS, or the soaking compound 3-methyl-6-phenyl-3H–imidazo[4,5-b]pyridin-2-amine (PhIP, 3–5 mM), and incubated overnight at 4°C. The mixture was centrifuges at 15,000g and the supernatant subject to vapor diffusion in hanging drop format at room temperature. For soaked structures of PA-E2 and OBHS, 5 mM ligand was added to crystals and data collected 3–4 weeks later. The cocrystalized structure with the soaked structures with OBHS (PDB: 4ZNY) and PA-E2 (PDB:5TLD) have been previously published (Nwachukwu et al., 2016b; Smith et al., 2011).
Structure determination
X-ray diffraction data was collected at the Stanford Synchrotron Radiation Lightsource, beam-line 11-1. Diffraction data was reduced using HKL-2000 software (Minor et al., 2006). The data was then analyzed to solve the structure of LBD complex by molecular replacement and automated rebuilding, using the PHENIX software suite (Adams et al., 2011; Bruning et al., 2010), and the 1.8Å structure of the ERα-Y537S LBD (PBD: 2B1V) (Hsieh et al., 2006) as a starting model. Data that was categorized at strongly anisotropic (http://services.mbi.ucla.edu/anisoscale/) (Strong et al., 2006) was truncated before refinement. Atomic coordinates and restraints for ligands were generated using ChemBioDraw® software (PerkinElmer Inc. Waltham, MA) and the PHENIX module, eLBOW (Moriarty et al., 2009). Ligands were docked into the LBDs using Coot (Emsley et al., 2010). The structures were then subject to 256 distinct refinement strategies using extensive combinatorial refinement (ExCoR) (Nwachukwu et al., 2013). Visualization of an ensemble of the three structures with the lowest Rfree was used to guide rebuilding. The final round of refinement was performed with a single refinement strategy for each structure using default settings in phenix.refine, including XYZ, B-factor, and occupancy refinement, TLS with groupings chosen by phenix.find_tls_groups, optimize X-ray/Stereochemistry and Xray/ADP weights, and refinement of waters. Images were generated with CCP4mg (McNicholas et al., 2011).
Structure modeling and molecular dynamics simulations
The missing N-terminal loop and C-terminal F domain were modeled for both phosphorylated and unphosphorylated states with MODELLER v9.12 (Eswar et al., 2007). The monomer antagonist-bound crystallographic structure (PDB Code: 3ERT) was employed as the template. Only the structure of residues 298–307 and 546–554 was modeled while all other parts were kept the same as the template. One initial model per state was generated, which was then subjected to 100 runs of loop refinement. Homodimer structures were then constructed via symmetry operation on monomer models using the Bio3D package(Grant et al., 2006; Skjærven et al., 2014). Molecular dynamics (MD) simulations were performed with AMBER12 (Case et al., 2012) and corresponding force field ff99SB (Rezácová et al., 2008). Additional parameters for phosphorylated serine (p-S305) were taken from Homeyer et al. (Homeyer et al., 2006). The GAFF force field(Wang et al., 2004) and the AM1-BCC partial charge model (Jakalian et al., 2002) were employed for the antagonist 4-hydroxytamoxifen (OHT). The structure models were employed as the starting conformation for proteins. Coordinates for ligand were directly extracted from the antagonist-bound structure (PDB 3ERT). In all systems, Arg and Lys were protonated while Asp and Glu were deprotonated. The protonation states for His residues were determined based on an inspection of the residues local environment and their pKa values as calculated by PROPKA3 (Olsson et al., 2011). The phosphate group of p-S305 is un-protonated. Simulation structures were solvated in a truncated cubic box of pre-equilibrated TIP3P water molecules, which extended 12 Å in each dimension from the surface of the solute. Sodium (Na+) or chloride (Cl−) counter-ions were added to neutralize the systems. Energy minimization was performed in four stages, with each stage employing 500 steps of steepest decent followed by 1500 steps of conjugate gradient. First, minimization for solvent only was performed with fixed positions of protein and ligand atoms. Second, side-chain and ligand were relaxed with backbone still fixed. Third, all protein and ligand atoms were relaxed with fixed solvent. Fourth, all atoms were free to move without any restraint. Following minimization, 10ps of MD simulation was performed to heat the system from 0K to 300K under constant-volume periodic boundary conditions. A further 1ns of equilibration simulation was performed at constant temperature (T=300K) and constant pressure (P=1bar). Subsequent 5×40-ns production phase MD were then performed under the same conditions as equilibration, where 5× means five independently performed simulation replicates with the same initial conformation but distinct initial atomic velocities. For both energy minimization and MD simulations, the particle-mesh Ewald summation method was adopted to treat long-range electrostatic interactions. In addition, an 8Å cutoff was used to truncate the shortrange nonbonded Van de Waals’ interactions. Additional operational parameters for MD included a 2-fs time step, removal of the center-of-mass motion every 1000 steps and update of the nonbonded neighbor list every 25 steps. All hydrogen atoms were constrained using the SHAKE algorithm.
In addition to conventional MD simulations, 5×5-ns steered MD (SMD) simulations were performed to measure the energy needed to pull H12 away from its initial position under distinct phosphorylation states. For each replicate, a randomly generated conformational snapshot from equilibration process was adopted as the initial structure of SMD. Distance between atoms I358:Cα (H3) and L544:Cα (H12) was restrained (with a force constant 5,000 kcal·mol−1·Å−2) to a reference value, which was then uniformly changed from the initial distance value (7–8Å) to 20Å. The work performed on the system was then calculated by integrating external forces over this distance during the simulation.
Data and Software Accessibility
The high-throughput data used for these studies has been deposited in the Gene Expression Omnbius (GEO) repository GSE67295. Raw data for Western blots can be found at https://data.mendeley.com/datasets/9xfzwrttjv/1
Supplementary Material
Highlights.
ERα is a signaling effector of pro-inflammatory cytokines in breast cancer cells
IL1β and TNFα increase breast cancer invasiveness dependent on ERα
Cytokines activate unliganded ERα via IKKβ phosphorylation of S305
Phospho-S305 results in structural changes of ERα and tamoxifen resistance
Acknowledgments
We thank Gregory Fonseca for critical comments and Leslie Van Ael for assistance with preparation of the manuscript. These studies were supported by NIH grants R01 DK091183, R01 DK015556 (to JAK), R01 CA17390 and P30 DK063491. S.S. is supported by the Frenchman’s Creek Women for Cancer Research. The BallenIsles Men’s Golf Association supports J.C.N.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions:
Conceptualization, J.D.S, J.F., J.C.N., C.L., K.W.N., and C.K.G.; Methodology, J.D.S., H.S.L., J.C.N., X.Y., B.J.G., K.W.N., and C.K.G; Formal Analysis, J.D.S., J.C.N., X.Y, B.J.G., T.I., K.W.N., and C.K.G.; Investigation, J.D.S, Y.K., I.K., T.S., M.Y., J.C.N., S.S., J.N., T.I., R.E., X.Y., B.J.G., X.Y., and R.E.; Data Curation, J.D.S., and J.C.N.; Writing - Original Draft, J.D.S., C.K.G., J.F., and C.K.G.; Writing - Review & Editing, J.D.S, J.F., J.C.N., J.A.K., K.W.N., and C.K.G.; Visualization, J.D.S., K.W.N., J.C.N., S.S., X.Y., B.J.N., and C.K.G.; Supervision, C.K.G., K.W.N., J.F., J.A.K., T.I., and B.J.G.; Funding Acquisition, C.K.G., and K.W.N.
References
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Echols N, Headd JJ, Hung LW, Jain S, Kapral GJ, Grosse Kunstleve RW, et al. The Phenix software for automated determination of macromolecular structures. Methods. 2011;55:94–106. doi: 10.1016/j.ymeth.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgarten SC, Frasor J. Minireview: Inflammation: an instigator of more aggressive estrogen receptor (ER) positive breast cancers. Mol. Endocrinol. 2012;26:360–371. doi: 10.1210/me.2011-1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentin Toaldo C, Alexi X, Beelen K, Kok M, Hauptmann M, Jansen M, Berns E, Neefjes J, Linn S, Michalides R, et al. Protein Kinase A-induced tamoxifen resistance is mediated by anchoring protein AKAP13. BMC Cancer. 2015;15:588. doi: 10.1186/s12885-015-1591-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry NB, Fan M, Nephew KP. Estrogen receptor-alpha hinge-region lysines 302 and 303 regulate receptor degradation by the proteasome. Mol. Endocrinol. 2008;22:1535–1551. doi: 10.1210/me.2007-0449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodine PV, Harris HA, Komm BS. Suppression of ligand-dependent estrogen receptor activity by bone- resorbing cytokines in human osteoblasts. Endocrinology. 1999;140:2439–51. doi: 10.1210/endo.140.6.6612. [DOI] [PubMed] [Google Scholar]
- Bostner J, Skoog L, Fornander T, Nordenskjöld B, Stål O. Linköping University Post Print Estrogen Receptor-alpha Phosphorylation at Serine 305 , Nuclear p21-Activated Kinase 1 Expression, and Response to Tamoxifen in Postmenopausal Breast Cancer. Clin. Cancer Res. 2010;5:1624–1633. doi: 10.1158/1078-0432.CCR-09-1733. [DOI] [PubMed] [Google Scholar]
- Bruce MC, McAllister D, Murphy LC. The kinome associated with estrogen receptor-positive status in human breast cancer. Endocr. Relat Cancer. 2014;21:R357–R370. doi: 10.1530/ERC-14-0232. [DOI] [PubMed] [Google Scholar]
- Bruning JBJ, Parent AA, Gil G, Zhao M, Nowak J, Pace MC, Smith CL, Afonine PV, Adams PD, Katzenellenbogen Ja, et al. Coupling of receptor conformation and ligand orientation determine graded activity. Nat. Chem Biol. 2010;6:837–843. doi: 10.1038/nchembio.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brzozowski AM, Pike AC, Dauter Z, Hubbard RE, Bonn T, Engström O, Ohman L, Greene GL, Gustafsson JA, Carlquist M. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature. 1997;389:753–758. doi: 10.1038/39645. [DOI] [PubMed] [Google Scholar]
- Carson-Jurica MA, Schrader WT, O’Malley BW. Steroid receptor family: structure and functions. Endocr. Rev. 1990;11:201–220. doi: 10.1210/edrv-11-2-201. [DOI] [PubMed] [Google Scholar]
- Chanmee T, Ontong P, Konno K, Itano N. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers (Basel) 2014;6:1670–1690. doi: 10.3390/cancers6031670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke R, Tyson JJ, Dixon JM. Endocrine resistance in breast cancer -An overview and update. Mol. Cell. Endocrinol. 2015;418:220–234. doi: 10.1016/j.mce.2015.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Y, Zhang M, Pestell R, Curran EM, Welshons WV, Fuqua SAW. Phosphorylation of estrogen receptor a blocks its acetylation and regulates estrogen sensitivity. Cancer Res. 2004;64:9199–9208. doi: 10.1158/0008-5472.CAN-04-2126. [DOI] [PubMed] [Google Scholar]
- deGraffenried LA, Chandrasekar B, Friedrichs WE, Donzis E, Silva J, Hidalgo M, Freeman JW, Weiss GR. NF-κB inhibition markedly enhances sensitivity of resistant breast cancer tumor cells to tamoxifen. Ann. Oncol. 2004;15:885–890. doi: 10.1093/annonc/mdh232. [DOI] [PubMed] [Google Scholar]
- De Savi C, Bradbury RH, Rabow AA, Norman RA, de Almeida C, Andrews DM, Ballard P, Buttar D, Callis RJ, Currie GS, et al. Optimization of a Novel Binding Motif to ( E )-3-(3,5-Difluoro-4-((1 R ,3 R )-2-(2-fluoro-2-methylpropyl)-3-methyl-2,3,4,9-tetrahydro-1 H -pyrido[3,4- b ]indol-1-yl)phenyl)acrylic Acid (AZD9496), a Potent and Orally Bioavailable Selective Estrogen Recepto. J. Med Chem. 2015;58:8128–8140. doi: 10.1021/acs.jmedchem.5b00984. [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr. Sect. D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estébanez-Perpiñá E, Arnold LA, Nguyen P, Rodrigues ED, Mar E, Bateman R, Pallai P, Shokat KM, Baxter JD, Guy RK, et al. A surface on the androgen receptor that allosterically regulates coactivator binding. Proc. Natl. Acad. Sci. U. S A. 2007;104:16074–16079. doi: 10.1073/pnas.0708036104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswar N, Webb B, Marti-Renom MA, Madhusudhan MS, Eramian D, Shen M, Pieper U, Sali A. Comparative protein structure modeling using Modeller. 2007 doi: 10.1002/0471140864.ps0209s50. [DOI] [PubMed] [Google Scholar]
- Feldman I, Feldman GM, Mobarak C, Dunkelberg JC, Leslie KK. Identification of proteins within the nuclear factor-κ B transcriptional complex including estrogen receptor-α. Am. J. Obstet Gynecol. 2007;196:394.e1–11. doi: 10.1016/j.ajog.2006.12.033. discussion 394.e11-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco HL, Nagari A, Kraus WL. TNFα signaling exposes latent estrogen receptor binding sites to alter the breast cancer cell transcriptome. Mol. Cell. 2015;58:21–34. doi: 10.1016/j.molcel.2015.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garber M, Yosef N, Goren A, Raychowdhury R, Thielke A, Guttman M, Robinson J, Minie B, Chevrier N, Itzhaki Z, et al. A High-Throughput Chromatin Immunoprecipitation Approach Reveals Principles of Dynamic Gene Regulation in Mammals. Mol. Cell. 2012;47:810–822. doi: 10.1016/j.molcel.2012.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good MC, Zalatan JG, Lim WA. Scaffold Proteins: Hubs for Controlling the Flow of Cellular Information. Science (80-) 2011;332:680–686. doi: 10.1126/science.1198701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gori I, Pellegrini C, Staedler D, Russell R, Jan C, Canny GO. Tumor necrosis factor α activates estrogen signaling pathways in endometrial epithelial cells via estrogen receptor α. Mol. Cell. Endocrinol. 2011;345:27–37. doi: 10.1016/j.mce.2011.06.043. [DOI] [PubMed] [Google Scholar]
- Grant BJ, Rodrigues APC, ElSawy KM, McCammon JA, Caves LSD. Bio3d: An R package for the comparative analysis of protein structures. Bioinformatics. 2006;22:2695–2696. doi: 10.1093/bioinformatics/btl461. [DOI] [PubMed] [Google Scholar]
- Grese TA, Sluka JP, Bryant HU, Cullinan GJ, Glasebrook AL, Jones CD, Matsumoto K, Palkowitz AD, Sato M, Termine JD, et al. Molecular determinants of tissue selectivity in estrogen receptor modulators. Pharmacology. 1997;94:14105–14110. doi: 10.1073/pnas.94.25.14105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He HH, Meyer CA, Chen MW, Jordan VC, Brown M, Liu XS. Differential DNase I hypersensitivity reveals factor-dependent chromatin dynamics. Genome Res. 2012;22:1015–1025. doi: 10.1101/gr.133280.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple Combinations of Lineage-Determining Transcription Factors Prime cis-Regulatory Elements Required for Macrophage and B Cell Identities. Mol. Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heldring N, Pawson T, McDonnell D, Treuter E, Gustafsson JÅ, Pike ACW. Structural insights into corepressor recognition by antagonist-bound estrogen receptors. J. Biol. Chem. 2007;282:10449–10455. doi: 10.1074/jbc.M611424200. [DOI] [PubMed] [Google Scholar]
- Homeyer N, Horn AHC, Lanig H, Sticht H. AMBER force-field parameters for phosphorylated amino acids in different protonation states: Phosphoserine, phosphothreonine, phosphotyrosine, and phosphohistidine. J. Mol. Model. 2006;12:281–289. doi: 10.1007/s00894-005-0028-4. [DOI] [PubMed] [Google Scholar]
- Hsieh RW, Rajan SS, Sharma SK, Guo Y, DeSombre ER, Mrksich M, Greene GL. Identification of ligands with bicyclic scaffolds provides insights into mechanisms of estrogen receptor subtype selectivity. J. Biol. Chem. 2006;281:17909–17919. doi: 10.1074/jbc.M513684200. [DOI] [PubMed] [Google Scholar]
- Hurtado A, Holmes KA, Ross-Innes CS, Schmidt D, Carroll JS. FOXA1 is a key determinant of estrogen receptor function and endocrine response. Nat Genet. 2011;43:27–33. doi: 10.1038/ng.730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakalian A, Jack DB, Bayly CI. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002;23:1623–1641. doi: 10.1002/jcc.10128. [DOI] [PubMed] [Google Scholar]
- Kaikkonen MU, Spann NJ, Heinz S, Romanoski CE, Allison KA, Stender JD, Chun HB, Tough DF, Prinjha RK, Benner C, et al. Remodeling of the enhancer landscape during macrophage activation is coupled to enhancer transcription. Mol Cell. 2013;51:310–325. doi: 10.1016/j.molcel.2013.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Williams KC, Gavin CT, Jardine E, Chambers AF, Leong HS. Quantification of cancer cell extravasation in vivo. Nat. Protoc. 2016;11:937–948. doi: 10.1038/nprot.2016.050. [DOI] [PubMed] [Google Scholar]
- Kraus WL, McInerney EM, Katzenellenbogen BS. Ligand-dependent, transcriptionally productive association of the amino- and carboxyl-terminal regions of a steroid hormone nuclear receptor. Proc. Natl. Acad. Sci. U. S. A. 1995;92:12314–12318. doi: 10.1073/pnas.92.26.12314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushner PJ, Agard DA, Greene GL, Scanlan TS, Shiau AK, Uht RM, Webb P. Estrogen receptor pathways to AP-1. J. Steroid Biochem. Mol Biol. 2000;74:311–317. doi: 10.1016/s0960-0760(00)00108-4. [DOI] [PubMed] [Google Scholar]
- Leek RD, Lewis CE, Whitehouse R, Greenall M, Clarke J, Harris AL. Association of macrophage infiltration with angiogenesis and prognosis in invasive breast carcinoma. Cancer Res. 1996;56:4625–4629. [PubMed] [Google Scholar]
- Li W, Notani D, Ma Q, Tanasa B, Nunez E, Chen AY, Merkurjev D, Zhang J, Ohgi K, Song X, et al. Functional roles of enhancer RNAs for oestrogen-dependent transcriptional activation. Nature. 2013;498:516–520. doi: 10.1038/nature12210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Likhite VS, Stossi F, Kim K, Katzenellenbogen BS, Katzenellenbogen Ja. Kinase-Specific Phosphorylation of the Estrogen Receptor Changes Receptor Interactions with Ligand, Deoxyribonucleic Acid, and Coregulators Associated with Alterations in Estrogen and Tamoxifen Activity. Mol. Endocrinol. 2006;20:3120–3132. doi: 10.1210/me.2006-0068. [DOI] [PubMed] [Google Scholar]
- McInerney EM, Katzenellenbogen BS. Different regions in activation function-1 of the human estrogen receptor required for antiestrogen- and estradiol-dependent transcription activation. J. Biol. Chem. 1996;271:24172–24178. doi: 10.1074/jbc.271.39.24172. [DOI] [PubMed] [Google Scholar]
- McNicholas S, Potterton E, Wilson KS, Noble MEM. Presenting your structures: The CCP4mg molecular-graphics software. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011;67:386–394. doi: 10.1107/S0907444911007281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metivier R, Penot G, Flouriot G, Pakdel F. Synergism Between ER Transactivation Function 1 (AF-1) and AF-2 Mediated by Steroid Receptor Coactivator Protein-1: Requirement for the AF-1 -Helical Core and for a Direct Interaction Between the N- and C-Terminal Domains. Mol. Endocrinol. 2001;15:1953–1970. doi: 10.1210/mend.15.11.0727. [DOI] [PubMed] [Google Scholar]
- Métivier R, Penot G, Hübner MR, Reid G, Brand H, Koš M, Gannon F. Estrogen recepαtor- directs ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoter. Cell. 2003;115:751–763. doi: 10.1016/s0092-8674(03)00934-6. [DOI] [PubMed] [Google Scholar]
- Michalides R, Griekspoor A, Balkenende A, Verwoerd D, Janssen L, Jalink K, Floore A, Velds A, Van ‘t, Veer L, Neefjes J. Tamoxifen resistance by a conformational arrest of the estrogen receptor α after PKA activation in breast cancer. Cancer Cell. 2004;5:597–605. doi: 10.1016/j.ccr.2004.05.016. [DOI] [PubMed] [Google Scholar]
- Minor W, Cymborowski M, Otwinowski Z, Chruszcz M. HKL-3000: The integration of data reduction and structure solution - From diffraction images to an initial model in minutes. Acta Crystallogr. Sect. D Biol. Crystallogr. 2006;62:859–866. doi: 10.1107/S0907444906019949. [DOI] [PubMed] [Google Scholar]
- Montano MM, Ekena K, Krueger KD, Keller AL, Katzenellenbogen BS. Human estrogen receptor ligand activity inversion mutants: Receptors that interpret antiestrogens as estrogens and estrogens as antiestrogens and discriminate among different antiestrogens. Mol. Endocrinol. 1996;10:230–242. doi: 10.1210/mend.10.3.8833652. [DOI] [PubMed] [Google Scholar]
- Moriarty NW, Grosse-Kunstleve RW, Adams PD. Electronic ligand builder and optimization workbench (eLBOW): A tool for ligand coordinate and restraint generation. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009;65:1074–1080. doi: 10.1107/S0907444909029436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musgrove EA, Sutherland RL. Biological determinants of endocrine resistance in breast cancer. Nat. Rev. Cancer. 2009;9:631–643. doi: 10.1038/nrc2713. [DOI] [PubMed] [Google Scholar]
- Naugler WE, Sakurai T, Kim S, Maeda S, Kim K, Elsharkawy AM, Karin M. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science. 2007;317:121–124. doi: 10.1126/science.1140485. [DOI] [PubMed] [Google Scholar]
- Nwachukwu JC, Southern MR, Kiefer JR, Afonine PV, Adams PD, Terwilliger TC, Nettles KW. Improved crystallographic structures using extensive combinatorial refinement. Structure. 2013;21:1923–1930. doi: 10.1016/j.str.2013.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nwachukwu JC, Srinivasan S, Bruno NE, Parent AA, Hughes TS, Pollock JA, Gjyshi O, Cavett V, Nowak J, Garcia-Ordonez RD, et al. Resveratrol modulates the inflammatory response via an estrogen receptor-signal integration network. Elife. 2014;3:e02057. doi: 10.7554/eLife.02057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nwachukwu JC, Srinivasan S, Zheng Y, Wang S, Min J, Dong C, Liao Z, Nowak J, Wright NJ, Houtman R, et al. Predictive features of ligand-specific signaling through the estrogen receptor. Mol. Syst. Biol. 2016a;12:864. doi: 10.15252/msb.20156701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nwachukwu JC, Srinivasan S, Bruno NE, Nowak J, Wright NJ, Minutolo F, Rangarajan ES, Izard T, Yao X-Q, Grant BJ, et al. Systems Structural Biology Analysis of Ligand Effects on ERα Predicts Cellular Response to Environmental Estrogens and Anti-hormone Therapies. Cell Chem. Biol. 2016b;24:35–45. doi: 10.1016/j.chembiol.2016.11.014. [DOI] [PubMed] [Google Scholar]
- Oeckinghaus A, Hayden MS, Ghosh S. Crosstalk in NF-κB signaling pathways. Nat Immunol. 2011;12:695–708. doi: 10.1038/ni.2065. [DOI] [PubMed] [Google Scholar]
- Olsson MHM, SØndergaard CR, Rostkowski M, Jensen JH. PROPKA3: Consistent treatment of internal and surface residues in empirical p K a predictions. J. Chem. Theory Comput. 2011;7:525–537. doi: 10.1021/ct100578z. [DOI] [PubMed] [Google Scholar]
- Porter W, Saville B, Hoivik D, Safe S. Functional synergy between the transcription factor Sp1 and the estrogen receptor. Mol. Endocrinol. 1997;11:1569–1580. doi: 10.1210/mend.11.11.9916. [DOI] [PubMed] [Google Scholar]
- Pradhan M, Baumgarten SC, Bembinster LA, Frasor J. CBP mediates NF-κB-dependent histone acetylation and estrogen receptor recruitment to an estrogen response element in the BIRC3 promoter. Mol. Cell Biol. 2012;32:569–575. doi: 10.1128/MCB.05869-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayala SK, Talukder AH, Balasenthil S, Tharakan R, Barnes CJ, Wang RA, Aldaz M, Khan S, Kumar R. P21-activated kinase 1 regulation of estrogen receptor-α activation involves serine 305 activation linked with serine 118 phosphorylation. Cancer Res. 2006;66:1694–1701. doi: 10.1158/0008-5472.CAN-05-2922. [DOI] [PubMed] [Google Scholar]
- Rezácová P, Borek D, Moy SF, Joachimiak A, Otwinowski Z. Crystal structure and putative function of small Toprim domain-containing protein from Bacillus stearothermophilus. Proteins. 2008;70:311–319. doi: 10.1002/prot.21511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sas L, Lardon F, Vermeulen PB, Hauspy J, Van Dam P, Pauwels P, Dirix LY, Van Laere SJ. The interaction between ER and NFκB in resistance to endocrine therapy. Breast Cancer Res. 2012;14:212. doi: 10.1186/bcr3196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sentis S, Le Romancer M, Bianchin C, Rostan M-C, Corbo L. Sumoylation of the estrogen receptor alpha hinge region regulates its transcriptional activity. Mol. Endocrinol. 2005;19:2671–2684. doi: 10.1210/me.2005-0042. [DOI] [PubMed] [Google Scholar]
- Shang Y, Brown M. Molecular determinants for the tissue specificity of SERMs. Science. 2002;295:2465–2468. doi: 10.1126/science.1068537. [DOI] [PubMed] [Google Scholar]
- Shiau AK, Barstad D, Loria PM, Cheng L, Kushner PJ, Agard DA, Greene GL. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell. 1998;95:927–937. doi: 10.1016/s0092-8674(00)81717-1. [DOI] [PubMed] [Google Scholar]
- Skjærven L, Yao X-Q, Scarabelli G, Grant BJ. Integrating protein structural dynamics and evolutionary analysis with Bio3D. BMC Bioinformatics. 2014;15:399. doi: 10.1186/s12859-014-0399-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J, Morgan JR, Zottoli SJ, Smith PJ, Buxbaum JD, Bloom OE. Regeneration in the era of functional genomics and gene network analysis. Biol. Bull. 2011;221:18–34. doi: 10.1086/BBLv221n1p18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stender JD, Frasor J, Komm B, Chang KCN, Kraus WL, Katzenellenbogen BS. Estrogen-regulated gene networks in human breast cancer cells: involvement of E2F1 in the regulation of cell proliferation. Mol. Endocrinol. 2007;21:2112–2123. doi: 10.1210/me.2006-0474. [DOI] [PubMed] [Google Scholar]
- Stender JD, Kim K, Charn TH, Komm B, Chang KCN, Kraus WL, Benner C, Glass CK, Katzenellenbogen BS. Genome-wide analysis of estrogen receptor alpha DNA binding and tethering mechanisms identifies Runx1 as a novel tethering factor in receptor-mediated transcriptional activation. Mol. Cell. Biol. 2010;30:3943–3955. doi: 10.1128/MCB.00118-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strong M, Sawaya MR, Wang S, Phillips M, Cascio D, Eisenberg D. Toward the structural genomics of complexes: crystal structure of a PE/PPE protein complex from Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 2006;103:8060–8065. doi: 10.1073/pnas.0602606103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tora L, White J, Brou C, Tasset D, Webster N, Scheer E, Chambon P. The human estrogen receptor has two independent nonacidic transcriptional activation functions. Cell. 1989;59:477–487. doi: 10.1016/0092-8674(89)90031-7. [DOI] [PubMed] [Google Scholar]
- Tripathi S, Pohl MO, Zhou Y, Rodriguez-Frandsen A, Wang G, Stein DA, Moulton HM, Dejesus P, Che J, Mulder LCF, et al. Meta- and Orthogonal Integration of Influenza “oMICs” Data Defines a Role for UBR4 in Virus Budding. Cell Host Microbe. 2015;18:723–735. doi: 10.1016/j.chom.2015.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Wolf RM, Caldewell JW, Kollman PA, Case DA. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004;56531 doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- Webb P, Nguyen P, Shinsako J, Anderson C, Feng W, Nguyen MP, Chen D, Huang S-M, Subramanian S, McKinerney E, et al. Estrogen Receptor Activation Function 1 Works by Binding p160 Coactivator Proteins. Mol Endocrinol. 1998;12:1605–1618. doi: 10.1210/mend.12.10.0185. [DOI] [PubMed] [Google Scholar]
- Wijayaratne AL, McDonnell DP. The Human Estrogen Receptor-α is a Ubiquitinated Protein Whose Stability is Affected Differentially by Agonists, Antagonists, and Selective Estrogen Receptor Modulators. J. Biol. Chem. 2001;276:35684–35692. doi: 10.1074/jbc.M101097200. [DOI] [PubMed] [Google Scholar]
- Xu HE, Stanley TB, Montana VG, Lambert MH, Shearer BG, Cobb JE, McKee DD, Galardi CM, Plunket KD, Nolte RT, et al. Structural basis for antagonist-mediated recruitment of nuclear co-repressors by PPARalpha. Nature. 2002;415:813–817. doi: 10.1038/415813a. [DOI] [PubMed] [Google Scholar]
- Zhu P, Baek SH, Bourk EM, Ohgi KA, Garcia-Bassets I, Sanjo H, Akira S, Kotol PF, Glass CK, Rosenfeld MG, et al. Macrophage/cancer cell interactions mediate hormone resistance by a nuclear receptor derepression pathway. Cell. 2006;124:615–629. doi: 10.1016/j.cell.2005.12.032. [DOI] [PubMed] [Google Scholar]
- Zwart W, Griekspoor A, Berno V, Lakeman K, Jalink K, Mancini M, Neefjes J, Michalides R. PKA-induced resistance to tamoxifen is associated with an altered orientation of ERalpha towards co-activator SRC-1. EMBO J. 2007;26:3534–3544. doi: 10.1038/sj.emboj.7601791. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.